

Abstract
Acute kidney injury (AKI) is a growing problem with untoward economic and medical consequences. Anticancer drug toxicity remains an important and increasing cause of AKI. Importantly, drug-induced AKI affects all nephron segments—vasculature, glomerulus, tubules, and interstitium. Recent studies have increased insight into the subcellular mechanisms of drug-induced AKI that include direct cellular toxicity and immune-mediated effects. Identification of patients with high-risk cancer before drug exposure may allow prevention or at least a reduction in the development and severity of nephrotoxicity. Recognition of drug-induced AKI and rapid discontinuation (or dose reduction) of the offending agents, when appropriate, are critical to maximizing kidney function recovery. Preventive measures require understanding patient and drug-related risk factors coupled with correcting risk factors, assessing baseline kidney function before initiation of therapy, adjusting the drug dosage and avoiding use of nephrotoxic drug combinations.
Keywords: acute interstitial nephritis, acute kidney injury, acute tubular necrosis, drug nephrotoxicity, glomerulopathy, onco-nephrology, thrombotic microangiopathy
Acute kidney injury (AKI) is a growing problem with untoward economic and medical consequences.1 Furthermore, AKI is associated with progressive chronic kidney disease (CKD), leading to high mortality rates.2 Importantly, drug-induced AKI (DI-AKI) accounts for 19% to 26% of cases with AKI among hospitalized patients.3 Over the last decade, drug development has produced numerous life-saving medications for patients with cancer, which are unfortunately complicated by nephrotoxicity. We will review the underlying mechanisms and clinical syndromes of AKI induced by underrecognized and emerging anticancer therapies that involve various nephron segments.
Phenotype of Drug-Induced Acute Kidney Injury
Based on the International Serious Adverse Event Consortium report, drug-induced kidney disease can be classified by the 4 following clinical phenotypes: AKI, glomerular disease, nephrolithiasis/crystalluria, and tubular dysfunction.3 DI-AKI is then further characterized as acute tubular necrosis, acute interstitial nephritis, and osmotic nephrosis. Primary and secondary criteria need to be met to qualify as AKI (as well as the other 3 phenotypes). To provide insight into the mechanism, adverse drug reactions are classified into type A and B reactions. Type A reactions are dose-dependent toxicities that are alleviated by drug withdrawal or dose reduction, whereas type B reactions are idiosyncratic and require drug withdrawal for resolution.3 Acute, subacute, and chronic timing of drug exposure are also considered for nephrotoxic drugs. Notably, drugs that cause glomerular and tubular injury, as well as crystalluria/nephrolithiasis may be associated with AKI, which ultimately takes precedence in phenotyping drug-induced kidney injury. As such, we will classify drug-induced AKI into the following categories: (i) acute vascular disease, (ii) acute glomerular disease, (iii) acute tubular injury/necrosis, and (iv) acute interstitial nephritis. Table 1 contains numerous drug examples of these 4 categories of kidney injury.
Table 1.
Categories of drug-induced acute kidney injury
| Category of kidney injury | Clinical-pathologic diagnosis | Mechanisms of kidney injury | Drug examples |
|---|---|---|---|
| Acute vascular disease | TMA | Direct endothelial injury | Gemcitabine, mitomycin C, calcineurin inhibitors, others |
| VSMC dysfunction (VEGF deficiency) | Anti-angiogenesis drugs | ||
| Antibody mediated | IFN, thienopyridines, quinine | ||
| Vasculitis | Immune-mediated injury | Biologic agents, cocaine adulterated with levamisole, PTU, carbimazole, methimazole, hydralazine, minocycline, allopurinol, febuxostat, penicillamine, sulfasalazine, sofosbuvir in a kidney transplant recipient | |
| Acute glomerular disease | |||
| Epithelial cells (podocytes) | MCD | Direct cellular injury | Pamidronate, IFN, TKIs, lithium, NSAIDs |
| FSGS (including collapsing form) | TKIs, sirolimus, pamidronate, anabolic steroids, lithium, IFN | ||
| Endothelial cells | TMA | (See above) | (See above) |
| ANCA-associated vasculitis | Necrotizing crescentic GN | Immune-mediated injury | (See above) |
| Endocapillar lesions | Class III/IV/V LN | ||
| High risk | TNF-α inhibitors, procainamide, hydralazine | ||
| Moderate risk | Quinidine | ||
| Low risk | TNF-α inhibitors, carbamazepine, PTU, methyldopa, captopril, acebutolol, chlorpromazine, isoniazid, minocycline | ||
| Subepithelial space | Membranous nephropathy | Subepithelial IC deposits | Gold therapy, penicillamine and bucillamine, captopril, NSAIDs |
| Acute tubular disease | Acute tubular necrosis | Direct tubular cell injury | Antineoplastic, antimicrobial, antiviral, antifungal drugs, NSAIDs, calcineurin inhibitors |
| Tubular epithelial vacuolization | Antineoplastic agents, Deferasirox, IVIg | ||
| Intratubular obstruction with or without inflammation | Methotrexate, orlistat, antibiotics, anticoagulants | ||
| Acute interstitial disease | Acute interstitial nephritis | Type 4 hypersensitivity | Antimicrobial agents, PPIs, H2 antagonists, NSAIDs, others |
| Antibody mediated | Immune checkpoint inhibitors |
ANCA, anti-neutrophilic cytoplasmic antibody; FSGS, focal segmental glomerulosclerosis; GN, glomerulonephritis; IFN, interferon; LN, lupus nephritis; MCD, minimal change disease; NSAIDs, nonsteroidal anti-inflammatory drugs; PPIs, proton pump inhibitors; PTU, propylthiouracil; SLE, systemic lupus erythematosus; TKIs, tyrosine kinase inhibitors; TMA, thrombotic microangiopathy; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor; VSMC, vascular smooth muscle cell.
Risk Factors for Nephrotoxicity
DI-AKI most often occurs in at-risk hosts. There are innate patient risk factors that enhance drug nephrotoxicity. Whenever possible, these nephrotoxic risk factors should be corrected before prescribing a potentially nephrotoxic drug. The presence of nonmodifiable risk factors should prompt a risk-benefit analysis. Renal handling of medications and the innate toxicity of the drug further increase risk for the development of DI-AKI.4, 5, 6, 7
Innate Patient Risk Factors
A number of underlying patient characteristics predispose to drug-induced nephrotoxicity. Among them are age and sex. Older age and female sex, common characteristics in patients with cancer, are associated with reduced muscle mass (lower serum creatinine with higher estimated glomerular filtration rate [GFR]) and reduced total body water (increased serum drug concentration), which promote nephrotoxicity via excess drug dosing higher, potentially toxic serum drug concentrations. Hypoalbuminemia also increases potential nephrotoxicity by increasing the unbound fraction of drug in the serum, exposing cells to higher concentrations. Additional variables that predispose to DI-AKI include both multiple comorbidities (cancer, diabetes mellitus, hypertension, etc.) and polypharmacy, which is a particular problem in patients with cancer and the elderly.4, 5, 6, 7
The prevalence of kidney disease is increasing in patients with cancer. Importantly, underlying acute and CKD further increase risk for drug-related nephrotoxicity. Yet there are numerous other risk factors to consider. Both true (vomiting, diarrhea, diuretics) and effective (congestive heart failure, cirrhosis, nephrosis) intravascular volume depletion increase nephrotoxic drug risk by virtue of a lower GFR (prerenal azotemia) and increased drug exposure to tubular cells (slow drug and/or metabolite transport through the tubules). Hypoalbuminemia associated with nephrotic syndrome or hepatic failure also increases risk by increasing free drug concentrations. Acute hepatic failure and advanced cirrhosis are particularly high-risk factors for DI-AKI due to the severe prerenal physiology associated with advanced liver dysfunction, the reduced muscle mass masking underlying AKI and/or CKD, hypoalbuminemia, and hyperbilirubinemia (which may cause bile cast nephropathy). All of these factors make the patient with severe liver failure highly susceptible to DI-AKI.4, 5, 6, 7
Finally, a number of other comorbidities increase the risk for DI-AKI. Multiple myeloma, lymphoma and leukemias, renal cell cancer (and other malignancies), diabetes mellitus, sepsis, and various acid-base disturbances are often associated with enhanced drug nephrotoxicity. Patients with diabetes often have unrecognized kidney disease, which is a major risk factor for DI-AKI. Sepsis by virtue of its associated systemic and renal hemodynamic alterations as well as the synergistic effect between endotoxin and toxic drugs increases the risk for DI-AKI. Certain acid-base disturbances (acid or alkaline urine) favor intrarenal crystal formation with some drugs, while patients with underlying multiple myeloma, which manifest various forms of kidney disease (cast nephropathy, AL amyloidosis, light chain deposition disease, uric acid nephropathy, etc.) in nearly 50% of patients, are at an increased risk of DI-AKI.4, 5, 6, 7
Drug-Related Risk Factors
Numerous medications are inherently nephrotoxic.4, 5, 6, 7 The route of drug administration (arterial vs. venous vs. oral) affects nephrotoxicity based on the achieved maximum peak drug concentration. The dose and duration of potentially nephrotoxic drugs also lends the risk for kidney injury. Higher doses and prolonged durations of treatment with drugs commonly employed in patients with cancer, the aminoglycosides and amphotericin, have been shown to increase nephrotoxicity. Specific drug combinations may result in synergistic nephrotoxicity, with examples such as cephalosporins plus aminoglycosides, vancomycin plus aminoglycosides, or cephalosporins plus acyclovir, all widely used in patients with cancer receiving chemotherapy.
The nephrotoxic potential of drugs also relates to the pharmacological compound itself, which is often heightened in the kidney microenvironment. For example, renal tubular epithelial cells in the collecting duct and loop of Henle are at a greater risk for drug injury due to their high metabolic activity and, as a result, the relatively hypoxic microenvironment where they reside.4 Another example is a drug with molecular characteristics that favor crystal precipitation within tubular lumens in the setting of sluggish urine flow rates and low urinary pH.4 Crystalline nephropathy from methotrexate and its metabolites is such a drug.
In addition, the kidney is also a metabolic organ. It oxidizes drugs via cytochrome p450 and other enzyme systems located within the renal parenchyma into various metabolites. A number of these metabolites may damage the kidney via various mechanisms including oxidative stress and the formation of injurious reactive oxygen species.4
The pathway of drug excretion by the kidneys is also a risk factor for nephrotoxicity. Approximately 32% of the top 200 prescribed drugs in the United States in 2010 are eliminated by the kidneys with more than 25% of the absorbed dose excreted unchanged in urine.8, 9 Renal excretion of many anticancer drugs (cisplatin, ifosfamide, methotrexate, etc.) requires transport via peritubular capillaries to allow access to the basolateral surface of renal tubular epithelial cells.8, 9 Once at the basolateral membrane, these drugs are transported into cells by organic anion and organic cation transporters, respectively, and subsequently excreted into tubular lumens via multiple efflux transporters (multidrug resistance proteins, multidrug and toxin extrusion protein, peptide transporter, and P-glycoprotein) on the apical membrane (Figure 1). Any dysfunction or inhibition of renal efflux drug transporters may result in abnormal drug accumulation in renal tubular cells, leading to drug-induced nephrotoxicity. Genetic polymorphisms that affect the function of these transporters may explain differences in patient susceptibility to drug nephrotoxicity. In contrast, other drugs such as the aminoglycosides enter the renal tubular epithelial cell via the megalin-cubilin transport pathway. Cationic aminoglycosides, via attraction to the anionic apical membrane charge of proximal tubular cells (due to anionic phospholipids) and volume depletion, enhance aminoglycoside uptake and increase nephrotoxic risk. Pinocytosis of drugs such as sucrose-containing i.v. Ig and hydroxyethyl starch at proximal tubular cell apical membrane surfaces is associated with accumulation of these nonmetabolizable substances and tubular cell swelling and AKI.4
Figure 1.

Proximal tubular cell with various basolateral transporters and apical efflux transporters. MATE, multidrug and toxic compound extrusion; MRP, multidrug resistant protein transporter; OAT, organic anion transporter; OCT, organic cation transporter; P-gp, p-glycoprotein; PEPT, peptide transporter; SGLT2, sodium glucose cotransporter-2.
Mechanisms at Specific Nephron Segments
Acute Vascular Disease
Thrombotic microangiopathy (TMA) represents the most severe form of vascular endothelial cell injury that occurs systemically and within the renal parenchyma.10 This form of vascular injury is commonly associated with severe AKI. In general, injury can be conceptualized into 2 types of vascular toxicity: type I, which is associated with irreversible injury, and type II, which is more often a reversible form of dysfunction.11
Clinically, like other causes of TMA, drug-induced TMA is characterized by microangiopathic hemolytic anemia, thrombocytopenia with variable degrees of end organ injury, including AKI (Table 1). However, renal-limited TMA may also occur with certain drugs. Pathologically, endothelial swelling and necrosis, vascular and glomerular thrombosis, mesangiolysis, glomerular basement membrane duplication with cellular interposition, mucoid intimal edema, and fibrin deposition are observed. When evaluating a patient with clinical evidence for TMA, a drug-induced etiology should always be considered. The most common culprits are the anticancer agents including the anti-angiogenesis drugs and conventional chemotherapeutic agents such as gemcitabine and mitomycin C.12, 13, 14, 15 Other drugs associated with TMA include interferon, calcineurin inhibitors, and antiplatelet agents such as thienopyridines, which are widely used in hypercoagulable patients with cancer.12, 13, 14, 15 Drugs may promote this form of vascular endothelial injury through a diverse number of mechanisms. Direct endothelial damage by agents such as mitomycin and gemcitabine, and induction of autoantibodies to ADAMTS-13 and antiplatelet antibodies, as noted with ticlopidine, are possible mechanisms of TMA.12, 13, 16 In addition, when ADAMTS-13 deficiency or complement factor H mutations are present, drugs may be a second hit that triggers TMA.14 A recently observed mechanism causing TMA is disturbed angiogenesis resulting from inhibition of the vascular endothelial growth factor pathway by various drugs.11
It is worth noting that the vascular toxicity profile of the new anti-angiogenic agents differs from what is seen with conventional chemotherapy-induced vascular toxicity. A type I profile of vascular injury promotes sustained vascular toxicity with endothelial injury and apoptosis occurs with chemotherapeutic agents such as mitomycin C, gemcitabine, cisplatin, bleomycin, and vincristine.11, 17 In contrast, type II represents a transient vascular toxicity with reversible endothelial and vascular smooth muscle cell dysfunction. This form of TMA is observed with bevacizumab and other anti-angiogenic drugs.17
Acute Glomerular Disease
AKI from drug-induced glomerular disease can be broadly classified into 2 specific forms: direct cellular injury and immune-mediated injury (Table 1). Drug-related direct glomerular cell injury involving the endothelial and visceral epithelial (podocyte) cells is an important cause of AKI. TMA, minimal change disease (MCD), and focal segmental glomerulosclerosis are some of the lesions noted.11, 18 A number of medications are associated with immune-mediated glomerular injury, 2 of which include lupus-like kidney lesions and anti-neutrophilic cytoplasmic antibody–related pauci-immune vasculitis.
Direct Glomerular Cell Injury
Drug-induced vascular endothelial cell injury often extends into the glomerular capillaries, resulting in glomerular TMA. In fact, as described above in the pathology of TMA, glomerular endothelial cell necrosis, glomerular thrombosis and fibrin deposition, mesangiolysis, and glomerular basement membrane duplication are noted. Drug-induced TMA is often associated with severe AKI and variable systemic findings.
Podocyte injury can occur with a number of drugs including interferon, pamidronate, nonsteroidal anti-inflammatory drugs, and sirolimus. AKI, nephrotic syndrome, and histologic lesions targeting the podocyte (MCD or focal segmental glomerulosclerosis) complicate interferon therapy.18 Similarly, high-dose pamidronate damages podocytes and causes both MCD and focal segmental glomerulosclerosis with nephrotic proteinuria and acute and chronic kidney injury.19 CKD and dialysis requiring end-stage renal disease not uncommonly complicate pamidronate therapy despite drug discontinuation and steroid administration. Nonsteroidal anti-inflammatory drugs, which are commonly employed to treat pain in patients with cancer, are associated with numerous clinical renal syndromes. With nonsteroidal anti-inflammatory drug therapy, the podocytopathy MCD is the most commonly observed glomerular lesion, whereas membranous glomerulopathy may also develop.20 Sirolimus therapy may be complicated by proteinuria, occasionally nephrotic range, with a de novo focal segmental glomerulosclerosis lesion.21
Immune-Mediated Glomerular Injury
Drug-induced autoimmunity, which develops in <1% of drug-exposed patients, may cause renal lesions that mimic systemic lupus erythematosus or vasculitis. Moreover, kidney involvement only occurs in approximately 5% of such patients. In general, drug withdrawal is associated symptom resolution. However, immunosuppressive therapy may be required to suppress the inflammatory process when severe kidney injury develops.22, 23
Drug-induced autoimmunity is classified as type B or idiosyncratic reaction, which is unrelated to the drug mechanism of action. Rather, activation of the adaptive immune system by the drug or metabolites may underlie the immune injury. This autoimmune process was initially described following reports of vasculitis and a lupus-like syndrome with exposure to hydralazine, procainamide, and sulfadiazine. Subsequently, tumor necrosis factor-α inhibitors were also linked to autoantibody formation and immune-mediated glomerular disease.24 Autoantibodies are produced in a majority of patients developing immune-mediated glomerular disease from hydralazine and procainamide, which is more commonly observed in slow acetylators suggesting a relationship to higher drug exposure. With the anti–tumor necrosis factor-α agents, switch from T helper cell 1 to T helper cell 2 cytokine production leads to autoantibody production.25
Drug-induced immune-mediated glomerulopathies consisting of anti-neutrophilic cytoplasmic antibody–related pauci-immune vasculitis (Figure 2) and lupus-like renal lesions are examined as a background before evaluating biologic agents.22 A large retrospective analysis of drug-associated vasculitis in 250 patients with myeloperoxidase-positive anti-neutrophilic cytoplasmic antibody–associated vasculitis–examined common medication exposure.26 Thirty patients with the highest anti-myeloperoxidase antibody titers were examined for exposure to 11 candidate medications. Of note, 18 of the 30 patients (60%) were exposed to hydralazine (n = 10), propylthiouracil (n = 3), penicillamine (n = 2), allopurinol (n = 2), or sulfasalazine (n = 1). Kidney involvement was noted in 9 of 10 patients with hydralazine, with 5 exhibiting pauci-immune necrotizing glomerulonephritis. Kidney involvement was noted in 4 of 8 patients exposed to propylthiouracil, penicillamine, allopurinol, or sulfasalazine. Thus, drug-associated vasculitis should be entertained with a history of drug exposure, high-titer anti-myeloperoxidase antibodies, and the presence of other autoantibodies for perinuclear anti-neutrophilic cytoplasmic antibody (elastase or lactoferrin).24
Figure 2.
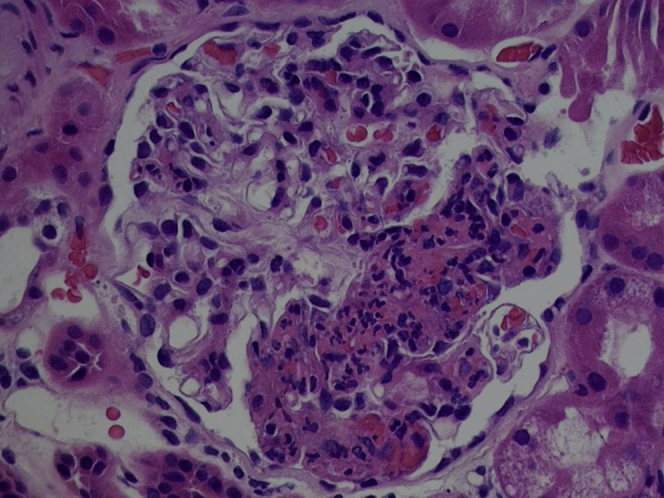
Histology demonstrating focal segmental necrotizing glomerulonephritis.
Biologic strategies that target key cytokines in specific inflammatory diseases include a number of drugs. Inhibitors of IL (interleukin)-1 (anakinra), IL-6 (tocilizumab), tumor necrosis factor-α (adalimumab, infliximab), IL-12/IL-23 (ustekinumab), and IL-17 (secukinumab) are some examples. Biologic drug use has been linked with the paradoxical development of systemic and organ-specific autoimmune processes including adverse kidney disorders. Although rare, these adverse autoimmune effects may lead to kidney failure and death. A systematic review described 26 cases of adverse kidney effects of 707 patients treated with these drugs.27 Various pathological findings were observed including MCD, mesangial glomerulonephritis with or without immune-complex renal vasculitis, membranoproliferative glomerulonephritis, necrotizing (or not) crescentic glomerulonephritis, and class III/IV lupus nephritis. On the basis of clinical manifestations and renal histology, patients were classified into the following: (i) glomerulonephritis-associated with systemic vasculitis (41.3%), (ii) drug-induced lupus-like glomerulonephritis (13.9%), and (iii) isolated autoimmune renal disorders (44.8%). The biologic drug most frequently associated with autoimmune kidney disease was etanercept (15 cases, 51.7%), followed by adalimumab (9 cases, 31.0%) and infliximab (3 cases, 10.3%). Other drugs associated with drug-induced lupus included tocilizumab and abatacept (1 patient each; 3.4% each). End-stage renal disease was reported in 3 patients with glomerulonephritis-associated with systemic vasculitis and 1 patient with isolated autoimmune renal disorders, and 1 death was reported in glomerulonephritis-associated with systemic vasculitis. Worse prognosis was associated with glomerulonephritis-associated with systemic vasculitis and lack of biologic withdrawal.28 When these adverse kidney effects occur, the biologic drug must be discontinued and patients should be treated according to clinical manifestations and kidney biopsy findings.27
Acute Tubular Injury/Necrosis
Acute tubular injury (Figure 3) is histologically characterized by necrotic cell death and inflammation. Several different pathways of regulated necrosis are involved in this process including necroptosis, mitochondrial permeability transition-induced regulated necrosis, parthanatos, and ferroptosis.28 The mechanisms of drug-induced tubular renal disease include many of these but need further evaluation to more definitively identify which processes apply to the drug associated with acute tubular injury (Table 1).
Figure 3.
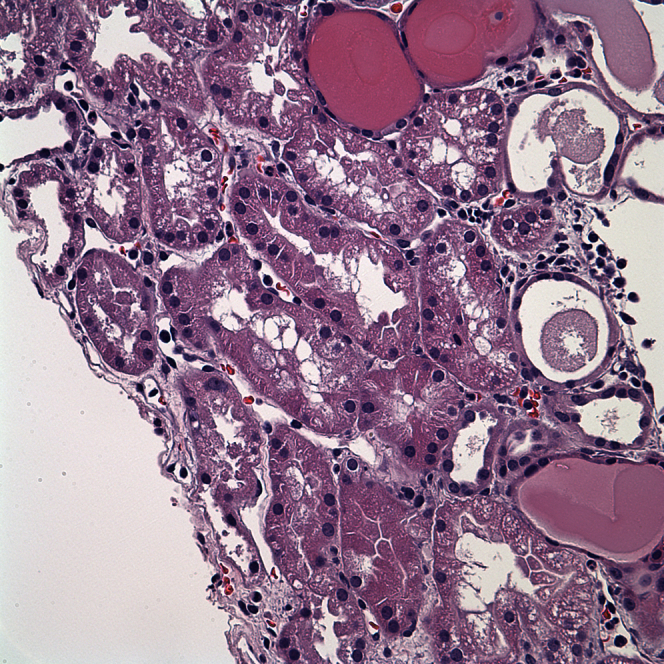
Histology demonstrating acute tubular injury. Note the tubular vacuoles, apical blebbing, tubular cell dropout, and proteinaceous casts.
Acute renal tubular cell toxicity is, at least in part, dose dependent and the cause of acute and chronic kidney injury that develops after exposure to classic conventional chemotherapeutic drugs and new anticancer drugs. The nephrotoxicity of some of the newer therapeutic agents will be our focus.
Crizotinib
Anaplastic lymphoma kinase 1 is a member of the insulin receptor tyrosine kinase family, which the small molecule inhibitor crizotinib therapeutically targets. Crizotinib received accelerated FDA approval for treatment of lung cancers harboring the activating echinoderm microtubule-associated protein-like 4–anaplastic lymphoma kinase gene fusion, which is present in approximately 2% to 7% of non–small cell lung cancers. An eGFR decrease of nearly 25% was observed after exposure to crizotinib.29 Most trial patients recovered kidney function on drug discontinuation. Unfortunately, kidney biopsies were not performed in this trial and the potential histopathology was not determined.30 Biopsy-proven acute tubular injury associated with crizotinib treatment has been reported in only a few cases.30, 31 However, the true relationship between crizotinib and AKI remains unclear as this drug is associated with a form of pseudo-AKI, which appears to be due to drug-induced reduced tubular creatinine secretion. In addition to AKI, crizotinib has also been associated with an increased risk of development and progression of kidney cysts. Edema formation likely related to mesenchymal epithelial transition growth factor pathway inhibition and electrolyte abnormalities are rare drug complications.32
Vemurafenib and Dabrafenib
BRAFV600E is one of the most common oncogenic mutations in human cancers. This mutation induces constitutive activation of the mitogen-activated protein kinase signaling pathway, which increases cancer cell proliferation, differentiation, and survival. Malignant melanoma frequently has a B-RAF V600 mutation that can be effectively targeted by the selective B-RAF inhibitors vemurafenib and dabrafenib.33 Although effective anticancer agents, these drugs may also produce nephrotoxicity. A GFR decline at 1 and 3 months of therapy occurred in 15 of 16 patients, which was complicated by persistent kidney injury after 8 months of follow-up. Based on clinical data, acute interstitial nephritis (AIN) was the presumed cause of AKI in 4 patients treated with vemurafenib.34 Three of 4 patients recovered kidney function after drug discontinuation, although no biopsy data were available.
Vemurafenib was associated with AKI (primarily Kidney Disease Improving Global Outcomes [KDIGO] stage 1 within 3 months of drug exposure) in 60% of 74 patients treated with this drug. Kidney biopsy revealed tubulointerstitial injury in 2 patients. Kidney function recovered within 3 months of drug discontinuation.34 One case series included a patient (of 8) who had a kidney biopsy demonstrating ATN as a potential mechanism of renal injury.35 Although the mechanism of kidney injury is unknown, these drugs may interfere with the downstream mitogen-activated protein kinase pathway, increasing susceptibility to ischemic tubular injury. A review of Food and Drug Administration Adverse Event Reporting System data for both agents revealed a substantial number of nephrotoxic effects. A total of 132 cases of AKI in patients receiving vemurafenib therapy and 13 cases in patients receiving dabrafenib therapy were reported.36 Metabolic disturbances from these drugs have also been described in Food and Drug Administration Adverse Event Reporting System.37 Kidney biopsies are required to elucidate the mechanism behind the nephrotoxicity of the BRAF inhibitors.
mTOR Inhibitors
The “mammalian target of rapamycin” (mTOR) is a serine/threonine protein kinase at the nexus of various intracellular signaling pathways. This kinase forms part of 2 different protein complexes (mTORC1-rapamycin sensitive, mTORC2-rapamycin insensitive). mTOR inhibitor family members such as everolimus and temsirolimus are prescribed at higher doses to treat renal cell carcinoma, refractory hormone receptor-positive breast cancer, other solid tumors, and even lymphomas. A potential complication is AKI, which developed in 15% of patients with renal cell carcinoma after everolimus treatment.37 A series of 4 patients with biopsy-documented ATN attributed to the mTOR inhibitors everolimus, temsirolimus, or combined TORC1/TORC2 inhibitor were described; 3 recovered kidney function after drug withdrawal.38
Pemetrexed
Pemetrexed disodium is a folate analog metabolic inhibitor that is FDA approved for the treatment of advanced or metastatic nonsquamous non–small cell lung cancer and malignant pleural mesothelioma.39 Reversible AKI after drug discontinuation has been reported in pemetrexed clinical trials; however, irreversible AKI and interstitial fibrosis associated have also been reported despite both drug discontinuation and treatment with oral prednisone and regular vitamin supplementation.31 One of 11 biopsied patients developed end-stage renal disease, whereas the remainder had stable CKD, some with persistent tubular defects. Renal histopathology available in the pemetrexed-associated renal insufficiency case reports includes ATN31 with mild-to-moderate interstitial fibrosis and, in a minority of cases, mild interstitial inflammation. In a single-center study evaluating for drug-related interstitial nephritis, pemetrexed was implicated in 5 of 56 cases.40
Crystalline Nephropathy
Crystalline nephropathy is a kidney disease that is characterized by the deposition of insoluble crystals within the tubules.41, 42 A number of drugs are associated with this disorder including methotrexate, oral sodium phosphate purgatives, ethylene glycol, ascorbic acid, orlistat, acyclovir, sulfadiazine, amoxicillin, and the fluoroquinolone antibiotics (Table 2).41, 42 Diagnosis is often made by visualizing characteristic drug crystals in the urine sediment (especially crystalline casts) or intratubular crystals (with surrounding cellular reaction) on kidney biopsy specimens. Prevention of and treatment for drug-induced crystalline nephropathy are general supportive measures including volume expansion with isotonic solutions (with urinary alkalinization for certain drugs) and discontinuation of the causative drug. The prognosis of renal function recovery is generally favorable, but late recognition of this complication, prolonged drug exposure, and the inability to correct the coexisting volume depletion may lead to irreversible kidney damage with interstitial fibrosis and tubular atrophy.41, 42
Table 2.
Drug-induced crystalline nephropathy
| Type of crystal | |
|---|---|
| Acyclovir | Birefringent needle-shaped |
| Amoxicillin | Birefringent needle-shaped |
| Indinavir | Plate-like, fan-shaped, sun burst |
| Atazanavir | Needle-like crystals |
| Ciprofloxacin | Needles, sheaves stars, birefringent |
| Methotrexate | Crystalline, Birefringent compact or needle-shaped golden, arranged in annular structures. Positive on methenamine silver and negative Von Kossa and alizarin red stains |
| Orlistat | Calcium oxalate (poorly birefringent, eight-faced bipyramid, “mail envelope”) |
| Sodium phosphate | Calcium phosphate (amorphous, granular, white) |
| Sulfadiazine | Shocks of wheat or shell-shaped rosettes |
| Triamterene | Birefringent colored spheres |
The pathogenesis of kidney injury occurring in crystalline nephropathies was initially thought to be due primarily to tubular obstruction after intratubular crystal precipitation. However, severe and extensive tubular obstruction is required to cause AKI, and experimental evidence casts doubt on this as the sole mechanism.43, 44 In fact, crystal-induced inflammasome-mediated inflammation offers a new explanation for kidney injury from intratubular crystal deposition.45 Several crystalline substances, including monosodium urate and calcium oxalate, are well-characterized activators of the NLRP3 (NOD-like receptor family, pyrin containing domain-3) inflammasome.45, 46 Furthermore, inflammasome underlies the development of AKI and CKD.45, 46, 47 Further studies examining the role of NLRP3 inflammasome in the pathogenesis of drug-induced crystal nephropathy are required.
Acute Interstitial Nephritis
In developed countries, medications are the most common cause of AIN, representing more than 70% of all cases. Although certain agents are more common, virtually all classes of drugs have been implicated in causation of AIN.48, 49, 50 The classical presentation, primarily associated with antibiotics and manifested as hypersensitivity reactions (skin rash, eosinophilia, and fever), has been largely replaced by oligosymptomatic presentations such as an unexplained rise in serum creatinine concentration. Serum and urine studies as well as renal imaging are also unreliable. As a result, kidney biopsy is frequently required to make a definitive diagnosis of AIN. This disorder is characterized histologically as an inflammatory cell infiltrate (focal or diffuse) associated with tubulitis and tubular injury. Glomeruli and vessels are typically spared. Treatment includes identifying and discontinuing the culprit drug and potentially a course of corticosteroids. Early identification and treatment is important to enhance renal recovery and avoid CKD. When identified late, drug-induced AIN can progress to a chronic interstitial nephritis triggered by fibroblast activation and manifested as interstitial fibrosis and tubular atrophy.
Several mechanisms are postulated to cause drug-induced AIN. Included are type 4 hypersensitivity reaction with T cells playing a dominant role and a rare antibody-mediated reaction where drugs induce antibodies to the tubular basement membrane or drug-related immune complexes deposit within the renal interstitium.51, 52, 53
Immune Checkpoint Inhibitors
Targeted therapies have changed the landscape of cancer patient therapy in the field of oncology. The immune checkpoint inhibitors (CPIs) are an addition therapy in the oncologist’s arsenal to combat cancer. They are considered among the most innovative and promising agents in the treatment of cancer. In contrast to first-generation immunotherapies, namely, cytokines-based agents, CPIs have fewer adverse effects.54
Cytotoxic T-lymphocyte antigen 4 and programmed cell death-1 are 2 essential components of immunotherapy. Ipilimumab and tremelimumab (anti-cytotoxic T-lymphocyte antigen 4 blocking antibodies), and pembrolizumab and nivolumab (antibodies targeting programmed cell death-1 receptors) are FDA approved for several malignancies. These effective anticancer agents are unfortunately complicated by immunological end-organ injury involving the lungs and gastrointestinal tract, various endocrine organs, and the kidneys. Acute interstitial nephritis is the major kidney lesion induced by these drugs and is due to severe inflammatory cell infiltrates with or without granuloma that occur within the renal parenchyma. The mechanism of injury is presumably the result of boosted cell-mediated immunity.55
Thirteen patients developed AKI in a median time 3 months (21–245 days) after exposure to the immune CPIs.56 Renal features included pyuria in 8 patients, as well as low-grade proteinuria (median 0.48 g/g; 0.12–0.98 g/g) and median peak serum creatinine concentration of 4.5 mg/dl (3.6–7.3 mg/dl). AKI events (4.9%) were more commonly observed in patients on combined drug therapy as compared with those on monotherapy (ipilimumab 2.0%, nivolumab 1.9%, and pembrolizumab 1.4%). AIN was observed in 12 patients (3 with granuloma formation), whereas 1 patient had TMA. Among the 12 patients with AIN, glucocorticoid treatment in 10 patients resulted in a complete (n = 2) or partial (n = 7) kidney function recovery. Despite glucocorticoids, 4 patients required hemodialysis; however, only 2 required chronic dialysis. No kidney function improvement was seen in the remaining 2 patients with AIN who did not receive glucocorticoid treatment. Six cases of biopsy-proven AIN (Figure 4) after therapy with nivolumab and pembrolizumab for lung cancer were described.57 AKI developed at anywhere from 3 to 18 months after CPI exposure. AIN may have developed from loss of tolerance to other drugs that were previously well tolerated before CPI administration. There were no autoimmune examination findings or consistent diagnostic laboratory tests noted. Kidney function improved back to or near baseline after CPI and potential cooffending drug discontinuation, as well as steroid initiation in 5 of 6 patients. Although no patient required hemodialysis, 1 patient did develop recurrent AKI after CPI rechallenge.
Figure 4.
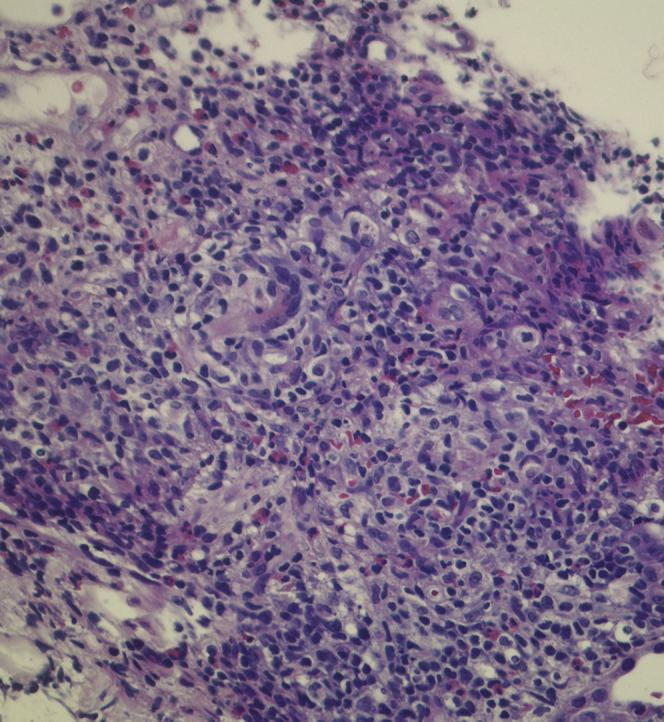
Histology demonstrating acute interstitial nephritis. Note the interstitial inflammatory infiltrate composed of lymphocytes, plasma cells, and eosinophils.
Lenalidomide
Lenalidomide is a derivative of thalidomide that is successfully employed to treat patients with multiple myeloma or myelodysplastic syndrome. Lenalidomide has been rarely associated with kidney injury, with 2 case reports noting AIN on kidney biopsy.58 One of these patients developed eosinophilia and systemic allergic symptoms.59, 60 It is currently unclear whether this adverse renal effect is significant and a true drug complication.
Future Directions for Predicting Drug-Induced AKI
Cell- and Biomarkers-Based Assays
In current times, routine biomarkers such as serum creatinine and blood urea nitrogen are employed to assess kidney function and detect kidney injury. However, these biomarkers increase late in the course of injury and only after substantial kidney injury occurs, when the damage may be irreversible. They also may increase when there is no structural kidney injury (functional decline in GFR). Over more than a decade, aggressive research has identified several early and prognostic biomarkers of AKI. However, sensitive and specific biomarkers are also needed to detect drug nephrotoxicity at earlier stages. Initial work is underway. In addition, biomarkers that identify when the medication is associated with a functional decline in GFR (not true injury as seen with an ACE inhibitor) or “pseudo-injury” from a decrease in tubular creatinine secretion via drug competition for the organic cation transporter system (as seen with trimethoprim) will provide more insight.
Significant progress has been achieved in identifying protein biomarkers of true kidney damage (β-2-microglobulin, clusterin, neutrophil gelatinase-associated lipocalin, IL-18, kidney injury molecule-1, albumin, urinary total protein, and trefoil factor 3) (Table 3).61 Work is underway to see if these biomarkers prove useful in clinical drug trials, and can be translated to the clinic for standard patient care.
Table 3.
Cell-based and biomarker-based assays for predicting nephrotoxicity
| Region specificity | Biomarkers |
Cells used for in vitro high-throughput toxicity prediction | |
|---|---|---|---|
| Traditional | Novel | ||
| Glomerulus | SCr, BUN, Pro/Alb/MiAlb | CysCa, A1M, B2Ma | PCL (podocyte) |
| Proximal tubule | LMWP | KIM-1a, CysCa, A1M, B2Ma, clusterina, TFF3a, NGAL, NAG, αGST, IL-6,8,18, M-CSF, TNF-α, osteopontin | Proximal tubule epithelia: LLC-RK1, LLC-PK1, NRK-52E, OK, HK-2, hRPTEC, hRPTEC/TERT1 |
| Distal tubule | Clusterina, TFF 3a, NGAL, GST-μ/π, H-FABP, osteopontin | MDCK | |
| Collecting duct | αGST | ||
| Renal papilla | KIM-1, clusterin | ||
A1M, α1-microglobulin; AP, alkaline phosphatase; B2M, β2-microglobulin; BUN, blood urea nitrogen; CysC, cystatin C; GST, glutathione S-transferase; H-FABP, heart-type fatty acid-binding protein; IL, interleukin; KIM-1, kidney injury molecular-1; LMWP, low molecular weight proteinuria; M-CSF, macrophage colony-stimulating factor; NAG, N-acetyl-β-d-glucosaminidase; NGAL, neutrophil gelatinase-associated lipocalin; Pro/Alb/MiAlb, proteinuria/albuminuria/microalbuminuria; SCr, serum creatinine; TFF3, trefoil factor 3; TNF-α, tumor necrosis factor-α.
Urinary kidney safety biomarkers endorsed by the European Medicines Agency and the US FDA.
MicroRNAs and Kidney-on-a-Chip Technology
Improved model systems to predict DI-AKI are crucially needed in drug development. Two future approaches toward DI-AKI management are based on microRNAs62 and kidney-on-a-chip technology.63 Hopefully, these newer technologies can improve our understanding and management of drug-related kidney injury.
Conclusions
Drug nephrotoxicity remains an important problem for patients with cancer and clinicians caring for this group. As new anticancer medications are released into clinical practice, recognition of their adverse consequences, in particular DI-AKI, is clearly required. It is therefore incumbent on the onco-nephrology community to remain aware of and vigilant for the potential nephrotoxic effects of the ever growing list of drugs employed. Clinicians should be cognizant that DI-AKI affects all nephron segments and there are various subcellular mechanisms that underlie nephrotoxicity. Identification of high-risk individuals before drug exposure is required to prevent or reduce the development and severity of nephrotoxicity. Future studies should evaluate whether the use of early markers of tubular damage can further elucidate the mechanisms of drug nephrotoxicity. This would allow the design of more-rational prevention and treatment strategies to reduce the incidence and severity of DI-AKI.
Disclosure
All the authors declared no competing interests.
References
- 1.Koyner J.L., Cerdá J., Goldstein S.L. Acute Kidney Injury Advisory Group of the American Society of Nephrology. The daily burden of acute kidney injury: a survey of U.S. nephrologists on World Kidney Day. Am J Kidney Dis. 2014;64:394–401. doi: 10.1053/j.ajkd.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 2.Chawla L.S., Eggers P.W., Star R.A., Kimmel P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371:58–66. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehta R.L., Awdishu L., Davenport A. Phenotype standardization for drug-induced kidney disease. Kidney Int. 2015;88:226–234. doi: 10.1038/ki.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perazella M.A. Renal vulnerability to drug toxicity. Clin J Am Soc Nephrol. 2009;4:1275–1283. doi: 10.2215/CJN.02050309. [DOI] [PubMed] [Google Scholar]
- 5.Kane-Gill S.L., Goldstein S.L. Drug-induced acute kidney injury: a focus on risk assessment for prevention. Crit Care Clin. 2015;31:675–684. doi: 10.1016/j.ccc.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Schetz M., Dasta J., Goldstein S., Golper T. Drug-induced acute kidney injury. Curr Opin Crit Care. 2005;11:555–565. doi: 10.1097/01.ccx.0000184300.68383.95. [DOI] [PubMed] [Google Scholar]
- 7.Pazhayattil G.S., Shirali A.C. Drug-induced impairment of renal function. Int J Nephrol Renovasc Dis. 2014;7:457–468. doi: 10.2147/IJNRD.S39747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrissey K.M., Stocker S.L., Wittwer M.B. Renal transporters in drug development. Annu Rev Pharmacol Toxicol. 2013;53:503–529. doi: 10.1146/annurev-pharmtox-011112-140317. [DOI] [PubMed] [Google Scholar]
- 9.International Transporter Consortium. Giacomini K.M., Huang S.M., Tweedie D.J. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.George J.N., Nester C.M. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371:1847–1848. doi: 10.1056/NEJMc1410951. [DOI] [PubMed] [Google Scholar]
- 11.Izzedine H., Perazella M.A. Thrombotic microangiopathy, cancer, and cancer drugs. Am J Kidney Dis. 2015;66:857–868. doi: 10.1053/j.ajkd.2015.02.340. [DOI] [PubMed] [Google Scholar]
- 12.Glezerman I., Kris M.G., Miller V. Gemcitabine nephrotoxicity and hemolytic uremic syndrome: report of 29 cases from a single institution. Clin Nephrol. 2009;71:130–139. doi: 10.5414/cnp71130. [DOI] [PubMed] [Google Scholar]
- 13.Hunt D., Kavanagh D., Drummond I. Thrombotic microangiopathy associated with interferon beta. N Engl J Med. 2014;370:1270–1271. doi: 10.1056/NEJMc1316118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orvain C., Augusto J.F., Besson V. Thrombotic microangiopathy due to acquired ADAMTS13 deficiency in a patient receiving interferon-beta treatment for multiple sclerosis. Int Urol Nephrol. 2014;46:239–242. doi: 10.1007/s11255-013-0401-7. [DOI] [PubMed] [Google Scholar]
- 15.Izzedine H., Massard C., Spano J.P. VEGF signalling inhibition-induced proteinuria: mechanisms, significance and management. Eur J Cancer. 2010;46:439–448. doi: 10.1016/j.ejca.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Zakarija A., Bennett C. Drug-induced thrombotic microangiopathy. Semin Thromb Hemost. 2005;31:681–690. doi: 10.1055/s-2005-925474. [DOI] [PubMed] [Google Scholar]
- 17.Herrmann J. Tyrosine kinase inhibitors and vascular toxicity: impetus for a classification system? Curr Oncol Rep. 2016;18:33. doi: 10.1007/s11912-016-0514-0. [DOI] [PubMed] [Google Scholar]
- 18.Markowitz G.S., Nasr S.H., Stokes M.B., D'Agati V.D. Treatment with IFN-{alpha}, -{beta}, or -{gamma} is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010;5:607–615. doi: 10.2215/CJN.07311009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perazella M.A., Markowitz G.S. Bisphosphonate nephrotoxicity. Kidney Int. 2008;74:1385–1393. doi: 10.1038/ki.2008.356. [DOI] [PubMed] [Google Scholar]
- 20.Pirani C.L., Valeri A., D'Agati V., Appel G.B. Renal toxicity of nonsteroidal anti-inflammatory drugs. Contrib Nephrol. 1987;55:159–175. doi: 10.1159/000413416. [DOI] [PubMed] [Google Scholar]
- 21.Letavernier E., Bruneval P., Mandet C. High sirolimus levels may induce focal segmental glomerulosclerosis de novo. Clin J Am Soc Nephrol. 2007;2:326–333. doi: 10.2215/CJN.03751106. [DOI] [PubMed] [Google Scholar]
- 22.Radhakrishnan J., Perazella M.A. Drug-induced glomerular disease: attention required! Clin J Am Soc Nephrol. 2015;10:1287–1290. doi: 10.2215/CJN.01010115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao X., Chang C. Diagnosis and classification of drug-induced autoimmunity (DIA) J Autoimmun. 2014;48–49:66–72. doi: 10.1016/j.jaut.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Hogan J.J., Markowitz G.S., Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300–1310. doi: 10.2215/CJN.01910215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin R.L. Drug-induced lupus. Expert Opin Drug Saf. 2015;14:361–378. doi: 10.1517/14740338.2015.995089. [DOI] [PubMed] [Google Scholar]
- 26.Choi H.K., Slot M.C., Pan G. Evaluation of antineutrophil cytoplasmic antibody seroconversion induced by minocycline, sulfasalazine, or penicillamine. Arthritis Rheum. 2000;43:2488–2492. doi: 10.1002/1529-0131(200011)43:11<2488::AID-ANR16>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 27.Piga M., Chessa E., Ibba V. Biologics-induced autoimmune renal disorders in chronic inflammatory rheumatic diseases: systematic literature review and analysis of a monocentric cohort. Autoimmun Rev. 2014;13:873–879. doi: 10.1016/j.autrev.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Linkermann A. Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. 2016;89:46–57. doi: 10.1016/j.kint.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 29.Brosnan E.M., Weickhardt A.J., Lu X. Drug-induced reduction in estimated glomerular filtration rate in patients with ALK-positive non-small cell lung cancer treated with the ALK inhibitor crizotinib. Cancer. 2014;120:664–674. doi: 10.1002/cncr.28478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gastaud L., Ambrosetti D., Otto J. Acute kidney injury following crizotinib administration for non-small-cell lung carcinoma. Lung Cancer. 2013;82:362–364. doi: 10.1016/j.lungcan.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Perazella M.A., Izzedine H. New drug toxicities in the onco-nephrology world. Kidney Int. 2015;87:909–917. doi: 10.1038/ki.2015.30. [DOI] [PubMed] [Google Scholar]
- 32.Izzedine H., El-Fekih R.K., Perazella M.A. The renal effects of ALK inhibitors. Invest New Drugs. 2016;34:643–649. doi: 10.1007/s10637-016-0379-y. [DOI] [PubMed] [Google Scholar]
- 33.Rizzo D., Ruggiero A., Amato M. BRAF and MEK inhibitors in pediatric glioma: new therapeutic strategies, new toxicities. Expert Opin Drug Metab Toxicol. 2016;12:1397–1405. doi: 10.1080/17425255.2016.1214710. [DOI] [PubMed] [Google Scholar]
- 34.Teuma C., Perier-Muzet M., Pelletier S. New insights into renal toxicity of the B-RAF inhibitor, vemurafenib, in patients with metastatic melanoma. Cancer Chemother Pharmacol. 2016;78:419–426. doi: 10.1007/s00280-016-3086-7. [DOI] [PubMed] [Google Scholar]
- 35.Launay-Vacher V., Zimner-Rapuch S., Poulalhon N. Acute renal failure associated with the new BRAF inhibitor vemurafenib: a case series of 8 patients. Cancer. 2014;120:2158–2163. doi: 10.1002/cncr.28709. [DOI] [PubMed] [Google Scholar]
- 36.Jhaveri K.D., Sakhiya V., Fishbane S. Nephrotoxicity of the BRAF inhibitors vemurafenib and dabrafenib. JAMA Oncol. 2015;1:1133–1134. doi: 10.1001/jamaoncol.2015.1713. [DOI] [PubMed] [Google Scholar]
- 37.Ha S.H., Park J.H., Jang H.R. Increased risk of everolimus-associated acute kidney injury in cancer patients with impaired kidney function. BMC Cancer. 2014;14:906. doi: 10.1186/1471-2407-14-906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Izzedine H., Escudier B., Rouvier P. Acute tubular necrosis associated with mTOR inhibitor therapy: a real entity biopsy-proven. Ann Oncol. 2013;24:2421–2425. doi: 10.1093/annonc/mdt233. [DOI] [PubMed] [Google Scholar]
- 39.Hughes A., Calvert P., Azzabi A. Phase I clinical and pharmacokinetic study of pemetrexed and carboplatin in patients with malignant pleural mesothelioma. J Clin Oncol. 2002;20:3533–3544. doi: 10.1200/JCO.2002.10.073. [DOI] [PubMed] [Google Scholar]
- 40.Airy M., Raghavan R., Truong L.D., Eknoyan G. Tubulointerstitial nephritis and cancer chemotherapy: update on a neglected clinical entity. Nephrol Dial Transplant. 2013;28:2502–2509. doi: 10.1093/ndt/gft241. [DOI] [PubMed] [Google Scholar]
- 41.Herlitz L.C., D'Agati V.D., Markowitz G.S. Crystalline nephropathies. Arch Pathol Lab Med. 2012;136:713–720. doi: 10.5858/arpa.2011-0565-RA. [DOI] [PubMed] [Google Scholar]
- 42.Yarlagadda S.G., Perazella M.A. Drug-induced crystal nephropathy: an update. Expert Opin Drug Saf. 2008;7:147–158. doi: 10.1517/14740338.7.2.147. [DOI] [PubMed] [Google Scholar]
- 43.Mulay S.R., Evan A., Anders H.J. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol Dial Transplant. 2014;29:507–514. doi: 10.1093/ndt/gft248. [DOI] [PubMed] [Google Scholar]
- 44.Isaka Y., Takabatake Y., Takahashi A. Hyperuricemia-induced inflammasome and kidney diseases. Nephrol Dial Transplant. 2016;31:890–896. doi: 10.1093/ndt/gfv024. [DOI] [PubMed] [Google Scholar]
- 45.Knauf F., Asplin J.R., Granja I. NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int. 2013;84:895–901. doi: 10.1038/ki.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hutton H.L., Ooi J.D., Holdsworth S.R., Kitching A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 2016;21:736–744. doi: 10.1111/nep.12785. [DOI] [PubMed] [Google Scholar]
- 47.Anders H.J., Muruve D.A. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22:1007–1018. doi: 10.1681/ASN.2010080798. [DOI] [PubMed] [Google Scholar]
- 48.Michel D.M., Kelly C.J. Acute interstitial nephritis. J Am Soc Nephrol. 1998;9:506–515. doi: 10.1681/ASN.V93506. [DOI] [PubMed] [Google Scholar]
- 49.Schwarz A., Krause P.H., Kunzendorf U. The outcome of acute interstitial nephritis: risk factors for the transition from acute to chronic interstitial nephritis. Clin Nephrol. 2000;54:179–190. [PubMed] [Google Scholar]
- 50.Clarkson M.R., Giblin L., O'Connell F.P. Acute interstitial nephritis: clinical features and response to corticosteroid therapy. Nephrol Dial Transplant. 2004;19:2778–2783. doi: 10.1093/ndt/gfh485. [DOI] [PubMed] [Google Scholar]
- 51.Paueksakon P., Fogo A.B. Drug-induced nephropathies. Histopathology. 2017;70:94–108. doi: 10.1111/his.13064. [DOI] [PubMed] [Google Scholar]
- 52.Davidman M., Olson P., Kohen J. Iatrogenic renal disease. Arch Intern Med. 1991;151:1809–1812. [PubMed] [Google Scholar]
- 53.Border W.A., Lehman D.H., Egan J.D. Antitubular basement-membrane antibodies in methicillin-associated interstitial nephritis. N Engl J Med. 1974;291:381–384. doi: 10.1056/NEJM197408222910803. [DOI] [PubMed] [Google Scholar]
- 54.Garrett M.D., Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308–316. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 55.Izzedine H, Mateus C, Boutros C, et al. Renal effects of immune checkpoint inhibitors [e-pub ahead of print]. Nephrol Dial Transplant. http://dx.doi.org/10.1093/ndt/gfw382. Accessed January 19, 2017. [DOI] [PubMed]
- 56.Cortazar F.B., Marrone K.A., Troxell M.L. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016;90:638–647. doi: 10.1016/j.kint.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shirali A.C., Perazella M.A., Gettinger S. Association of acute interstitial nephritis with programmed cell death 1 inhibitor therapy in lung cancer patients. Am J Kidney Dis. 2016;68:287–291. doi: 10.1053/j.ajkd.2016.02.057. [DOI] [PubMed] [Google Scholar]
- 58.González Rodríguez A.P. Management of the adverse effects of lenalidomide in multiple myeloma. Adv Ther. 2011;28(suppl 1):1–10. doi: 10.1007/s12325-010-0104-8. [DOI] [PubMed] [Google Scholar]
- 59.Lipson E.J., Huff C.A., Holanda D.G. Lenalidomide-induced acute interstitial nephritis. Oncologist. 2010;15:961–964. doi: 10.1634/theoncologist.2010-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shaaban H., Layne T., Guron G. A case of DRESS (drug reaction with eosinophilia and systemic symptoms) with acute interstitial nephritis secondary to lenalidomide. J Oncol Pharm Pract. 2014;20:302–304. doi: 10.1177/1078155213502569. [DOI] [PubMed] [Google Scholar]
- 61.US FDA. Review of Qualification Data for Biomarkers of Nephrotoxicity Submitted by the Predictive Safety Testing Consortium. www.fda.gov. EMEA/FDA guideline on detection of drug-induced nephrotoxicity (2009). http://www.ema.europa.eu. Accessed January 19, 2017.
- 62.Pavkovic M., Riefke B., Ellinger-Ziegelbauer H. Urinary microRNA profiling for identification of biomarkers after cisplatin-induced kidney injury. Toxicology. 2014;324:147–157. doi: 10.1016/j.tox.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 63.Wilmer M.J., Ng C.P., Lanz H.L. Kidney-on-a-chip technology for drug-induced nephrotoxicity screening. Trends Biotechnol. 2016;34:156–170. doi: 10.1016/j.tibtech.2015.11.001. [DOI] [PubMed] [Google Scholar]