

Abstract
The LysR member of bacterial transactivators, OxyR, governs transcription of genes involved in the response to H2O2 and organic (alkyl) hydroperoxides (AHP) in the Gram-negative pathogen, Pseudomonas aeruginosa. We have previously shown that organisms lacking OxyR are rapidly killed by <2 or 500 mM H2O2 in planktonic and biofilm bacteria, respectively. In this study, we first employed a bioinformatic approach to elucidate the potential regulatory breadth of OxyR by scanning the entire P. aeruginosa PAO1 genome for canonical OxyR promoter recognition sequences (ATAG-N7-CTAT-N7-ATAG-N7-CTAT). Of >100 potential OxyR-controlled genes, 40 were strategically selected that were not predicted to be involved in the direct response to oxidative stress (e.g., catalase, peroxidase, etc.) and screened such genes by RT-PCR analysis for potentially positive or negative control by OxyR. Differences were found in 7 of 40 genes when comparing an oxyR mutant vs. PAO1 expression that was confirmed by ß-galactosidase reporter assays. Among these, phnW, encoding 2-aminoethylphosphonate:pyruvate aminotransferase, exhibited reduced expression in the oxyR mutant compared to wild-type bacteria. Electrophoretic mobility shift assays indicated binding of OxyR to the phnW promoter and DNase I footprinting analysis also revealed the sequences to which OxyR bound. Interestingly, a phnW mutant was more susceptible to t-butyl-hydroperoxide (t-BOOH) treatment than wild-type bacteria. Although we were unable to define the direct mechanism underlying this phenomenon, we believe that this may be due to a reduced efficiency for this strain to degrade t-BOOH relative to wild-type organisms because of modulation of AHP gene transcription in the phnW mutant.
Introduction
Pseudomonas aeruginosa (PA) is an important human pathogen that is currently ranked fifth by the World Health Organization in overall infections [1]. Burn and cancer chemotherapy patients, chronic alcoholics, the immunocompromised (e.g., HIV infection) and patients suffering from cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) lung disease are particular prone to highly problematic or fatal infections by this organism [2,3].
The response to such infections, especially in the airway, is mediated by macrophages and subsequently neutrophils, both of which are capable of triggering an oxygen-dependent antimicrobial respiratory burst. This burst involves the production of reactive oxygen intermediates (ROI) including superoxide (O2-), hydrogen peroxide (H2O2), the hydroxyl radical (HO.), peroxy acids and organic peroxides (e.g., lipid). The organism must also face production of reactive nitrogen intermediates (RNI), the most reactive being peroxynitrite (ONOO-), a product of nitric oxide (NO, generated by host NO synthases) and O2-. The defensive response to H2O2 and organic peroxides is governed by the global transactivator, OxyR [4]. PA OxyR triggers transcriptional activation of classical antioxidant genes encoding catalases A and B (KatA [5], KatB [6]), periplasmic alkyl hydroperoxide reductases B (AhpB) [6] and cytoplasmic AhpCF (6) but not organic hydroperoxide reductase, Ohr [7]. Ohr is specifically inducible by organic hydroperoxide but is not part of the OxyR regulon [7].
In this study, we first used a bioinformatics approach to identify genes controlled by OxyR based upon a canonical OxyR promoter recognition sequence ATAG-N7-CTAT-N7-ATAG-N7-CTAT. Of over 100 genes identified as being putatively under OxyR control, our goal was to identify those that would not be predicted to be involved in the classical oxidative stress response to identify the physiological breadth of the OxyR regulon. Here, we identified the phnW gene, encoding 2-aminoethylphosphonate:pyruvate aminotransferase, whose promoter region matched 12 of 16 bp of the aforementioned OxyR promoter recognition sequence. Surprisingly, PhnW was found to play a role in resistance to t-butyl hydroperoxide (t-BOOH), but not H2O2 nor cumene hydroperoxide (CHP). However, PhnW did play an indirect role in t-BOOH degradation in vivo, yet the purified protein did not appear to possess direct degradation or modification properties when exposed to t-BOOH in vitro.
Materials and methods
Bacterial strains, plasmids and planktonic growth conditions
The bacteria used in this study (listed in Table 1) were grown in Luria-Bertani broth (L-broth) supplemented with appropriate antibiotic(s) when required for selective pressure. Aerobic cultures were grown at 37°C with shaking at 200 rpm at a 1/10 volume to total Erlenmeyer flask ratio. Media were solidified with 1.5% Bacto-agar. Frozen bacterial stocks were stored at -80°C in a 1:1 mixture of 30% glycerol and stationary-phase bacterial suspension.
Table 1. Bacterial strains and plasmids used in this study.
| Strains and plasmids | Genotype or description | Ref or source |
|---|---|---|
| E. coli DH5-αMC | F-ⱷ80dlacZΔM15 endA1 recA1 hsdR17(rK- mK-) supE44 thi-1 gyrA96 Δ(lacZYA-argF)U169 | Invitrogen |
| oxyR | P. aeruginosa oxyR mutant with removal of antibiotic resistance cassette | This study |
| oxyR/poxyR | P. aeruginosa oxyR mutant containing pUCP20-oxyR | This study |
| ohr ahpC | P. aeruginosa ohr and ahpC double mutant | This study |
| pQF50 | Broad-host-range lacZ transcriptional fusion, CbR | [8] |
| pEX100T | Vector to construct mutants in P. aeruginosa, broad- host-range, CbR | [9] |
| miniCTX | Site-specific integration-proficient plasmid for P. aeruginosa, TcR | [9] |
| pUCP20 | Pseudomonas shuttle vector, CbR | [10] |
| PA phnW::Gm | P. aeruginosa phnW mutant containing Gm cassette | This study |
| pEX100T phnW::Gm | pEX100T with Gm integrated in phnW | This study |
| pQF50 phnW-lacZ | pQF50 with upstream sequence of phnW containing OxyR putative binding domain fusion with lacZ | This study |
| pUCP20 phnW | pUCP20 containing complete sequence of phnW gene | This study |
| PA phnW::Gm/pphnW | P. aeruginosa phnW mutant containing pUCP20 phnW | This study |
| oxyR phnW::Gm | P. aeruginosa oxyR and phnW double mutant | This study |
| oxyR phnW::Gm/pphnW | P. aeruginosa oxyR and phnW double mutant containing pUCP20 phnW | This study |
| ohr ahpC/pphnW | P. aeruginosa ohr and ahpC double mutant containing pUCP20 phnW | This study |
| SM10 | thi-1 thr leu tonA lacY supE recA::RP4-2-Tc::Mu (Kmr) | This study |
Manipulation of recombinant DNA and genetic techniques
Transcription of the phnW gene was studied by using pQF50-based promoter fusion assays [8]. A phnW::Gm mutant was constructed using the pEX100T allelic exchange system and 6% sucrose counter-selection [11]. Complementation of this mutant was performed using pUCP19, an Escherichia-Pseudomonas shuttle vector [9]. All techniques were performed based upon standard cloning protocols [9,11,12]. All PCR products were amplified using pfu DNA polymerase (BRL) using an MJ research thermal cycler and the nucleotide sequence confirmed using the Cincinnati Children’s Hospital Medical Center DNA sequencing and Genotyping Facility.
Analysis of potential OxyR-regulated genes in PA PAO1
Potential OxyR-binding sites identified in the PA PAO1 genome were based on similarity to the well characterized Escherichia coli OxyR-regulated promoter sequences (ATAG-N7-CTAT-N7-ATAG-N7-CTAT) [6]. The DNA motif search tool in the Pseudomonas Genome Database was used to scan the PAO1 genome [12]. Candidate sites were further screened based on the type of genomic region that the site was found in intergenic versus putative open reading frames and whether proteins encoded by the downstream gene were predicted to be secreted past or embedded within the cytoplasmic membrane [12]. As predicted, representatives of the OxyR-regulated promoters under oxidizing conditions (e.g., H2O2, CHP, t-BOOH exposure) were the well-established PA OxyR-controlled genes katB, ahpB and ahpCF genes, respectively [6,13] (Table 2).
Table 2. The bioinformatic results of OxyR-dependent genes candidate and some well-known OxyR-dependent genes.
| Gene Name | Gene Product | Matching bases | Motif Location | Distance to gene | Motif Sequence—separated into 4-mers | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATAG | Spacer | CTAT | Spacer | ATAG | Spacer | CTAT | |||||
| PA3236 | probable glycine betaine-binding protein precursor (BetX) | 13 | 3624730–3624766 | 187 | ATCG | GCCTGTC | CAAT | CGAGTGC | ATAG | AGCGCTT | CGAT |
| PA0848 | probable alkyl hydroperoxide reductase (AhpB) | 13 | 927016–927052 | 95 | ATAG | GCTGACT | CTAT | CGTGCGA | ATTG | AAATTCG | ACAT |
| PA0846 | probable sulfate uptake protein | 13 | 924922–927147 | 2094 | ATGT | CGAATTT | CAAT | TCGCACG | ATAG | AGTCAGC | CTAT |
| PA1310 | 2-aminoethylphosphonate:pyruvate aminotransferase (PhnW) | 12 | 1420198–1420162 | 128 | ATCG | GCCTGGC | TTAT | GGCTGGC | ATCG | GAACAAA | CAAT |
| PA0139 | alkyl hydroperoxide reductase subunit C (AhpC) | 12 | 158120–158084 | 101 | ATAG | ATTTAGG | TAAT | CAGTGAA | ATGG | TCTAAAT | CAAT |
| PA2957 | probable transcriptional regulator | 10 | 3317470–3317506 | 32 | ATCG | AAGCCGC | GTAT | TATGCCT | ATTC | AGCACGA | AAAT |
| PA1097 | transcriptional regulator (FleQ) | 10 | 1187514–1187478 | 73 | ATAA | GCAGCCA | GCAT | TTGGCCA | CTAG | TTAAGTC | AAAT |
| PA1898 | quorum-sensing control repressor (QscR) | 10 | 2068974–2068938 | 516 | ATCG | GCACGGA | CAAT | GAAATGC | CTGG | TCGAATT | AAAT |
| PA1423 | probable chemotaxis transducer (BdlA) | 10 | 1549778–1549814 | 191 | ATAT | TTCCGAC | GAAT | GGCGGTA | ATTT | GTTTCAC | CCAT |
| PA5095 | probable permease of ABC transporter | 10 | 5738331–5738295 | 974 | ATCT | GGAGCGT | CGAA | CCATGCG | ATAC | AAGAAGT | CGAT |
| PA4613 | Catalase (KatB) | 9 | 5171801–5171837 | 75 | ATTG | AAAAACC | TAAT | CGCGCCG | GTGA | GGAATAT | CAAT |
| PA2999 | Na+-translocating NADH:ubiquinone oxidoreductase subunit Nrq1 (NqrA) | 9 | 3357556–3357592 | 57 | ATCA | TCGCGCG | CAAC | GTACTGA | ATTG | GCACGAT | TGAT |
| PA1003 | Transcriptional regulator (MvfR) | 9 | 1087437–1087473 | 342 | ATTC | GACGAAA | AGAA | AATCCGG | ATAT | TTACCGG | TTAT |
| PA0192 | probable TonB-dependent receptor | 9 | 218860–218824 | 312 | ATGC | CTTTGTC | CAAT | ATTTCCA | ATGC | CGGTAAA | GCAT |
| PA1648 | probable oxidoreductase | 9 | 1795990–1796026 | 103 | ATGA | TTCGCCG | CCAT | CAGTCTT | ATCA | GCGGGCT | GGAT |
| PA4403 | secretion protein (SecA) | 9 | 4937680–4937644 | 1028 | ATGG | TGCCGGC | TCAG | AAAAATG | ATGT | GCATGAC | TTAT |
| PA3242 | probable lauroyl acyltransferase | 9 | 3630801–3630765 | 160 | ATGA | GCCAAGC | CGAC | CTCCTCG | ATCA | AGACCCC | GTAT |
| PA0594 | peptidyl-prolyl cis-trans isomerase (SurA) | 9 | 654104–654068 | 296 | ATAT | GGCAACG | AAAA | CGACATC | ATCA | AGCAGCA | CGAT |
Semiquantitative expression of OxyR-dependent genes by RT-PCR
Total RNA from exponential phase PA and oxyR mutant bacteria was purified using a RiboPure-Bacteria kit as specified by the manufacturer (Ambion). After treatment with DNase I, 1 μg of total RNA was converted to cDNA by using random primers and reverse transcriptase (Promega). Then, 1 μl of cDNA from either PA PAO1 or oxyR mutant bacteria was used as a template to amplify the genes of interest that were not of the classic antioxidant variety (e.g., catalase/peroxidase) as well as omlA, the latter of which is a constitutively expressed gene and is used frequently as a reliable, internal control [14] using Taq DNA polymerase. After designated cycles of the RT-PCR were completed, amplified products was analyzed by electrophoresis on 1.2% agarose gels after sampling every 5 cycles between 15–45 cycles at 55°C. Band intensities were determined by using AlphaEase FC StandAlone software (Alpha Innotech). Then, the optimum PCR cycles were used to amplify a 253-bp phnW fragment using both PA PAO1 and oxyR mutant bacteria, respectively.
ß-galactosidase assays
All bacteria containing transcriptional phnW-lacZ fusions were grown in L-broth to exponential phase and exposed to 250 μM of t-BOOH for 30 min. Cell-free extracts from selected bacterial strains harboring the phnW-lacZ fusions were then used to assay ß-galactosidase activity [15]. All assays were performed at least in triplicate, and the values recorded as the mean +/- standard error.
Electrophoretic mobility shift assay (EMSA)
A 199-bp DNA fragment upstream of the phnW gene containing the putative OxyR binding domain was amplified using Pfu DNA polymerase. The accuracy of this DNA fragment was confirmed by DNA sequencing analysis. The EMSA was performed following the DIG gel shift protocol, 2nd generation (Roche). Briefly, the DNA fragment was 3’-end-labeled with terminal transferase and DIG-11-ddUTP. Then, 0.8 ng of DNA labeled probe was added to the binding reaction with increasing amounts of OxyR protein using bovine serum albumin as a competitor to demonstrate OxyR specific binding. The reaction mixture was then separated by electrophoresis on 6% non-denaturing polyacrylamide gels. The protein-DNA probe complex was transferred to positively charged nylon membranes by electroblotting followed by signal detection using chemiluminescence.
DNase I footprinting analysis
A radioactively labeled DNA fragment harboring putative OxyR recognition sequences was prepared by PCR. The reaction was initiated using a PA1310FW primer (5´-CAGGCGCTCGGCGGCGCG-3´) and a PA1310RV primer (5´-CTTCGCGGTCCGGGCGGT-3´) generated a 199-bp DNA fragment to identify the OxyR binding motif upstream of the phnW gene. Labeled probe (50 pmol) was added to binding buffer [20 mM Tris-HCl, pH 7.0, 50 mM KCl, 1 mM EDTA, 5% glycerol, 50 μg ml-1 BSA, 5 μg ml-1 calf thymus DNA, 0.5 μg ml-1 poly-(dI-dC)]. After the addition of purified OxyR (500 and 1000 nM), the reaction was incubated at 25°C for 15 min. Subsequently, 25 μ l of 0.2 mM Mg2+-0.1 mM Ca2+-0.5 U of DNase I was added to the binding reaction, and incubated for an additional 1 min. Finally, 200 μl of stop solution (20 mM EDTA, pH 8.0, 1.0% SDS and 0.2 M NaCl) was added. The mixture was then extracted with phenol-chloroform and the DNA precipitated with 95% ethanol at -20°C. The pellets were resuspended in sequencing buffer and loaded onto a 5% denaturing polyacrylamide sequencing gel. After transfer of the sequencing gel to PVDF membranes, the OxyR-specific footprint was assessed by autoradiography.
Filter paper disk assay for t-BOOH sensitivity
An exponential phase bacterial suspension was added to 5 ml of fresh L-broth containing 0.8% low-melting-point agarose (SeaPlaque) to a final O.D.600 nm of ~0.01. The suspensions were then distributed evenly on the LB agar surface. After the agarose solidified, filter paper disks (7 mm) impregnated with 10 μl of either 0.3 M or 0.5 M t-BOOH were placed on the top-agar surface and the plates were incubated at 37°C for 24 hr. The zones of killing were measured and the results are presented as the average from at least 3 independent experiments +/- standard error (SE).
t-BOOH degradation assay
The ability of PA and selected isogenic mutants to degrade t-BOOH was investigated as previously described [16]. Briefly, overnight cultures of all PA bacteria (wild type, mutant or complemented strains) were inoculated into L-broth to a final O.D.600 of 0.1 and allowed to grow at 37°C with shaking for ~3 hr, at which point various cells were diluted into fresh L-broth at an O.D.600 of 0.4. Then, 200 μM of t-BOOH was added to each culture. The remaining amount of t-BOOH was determined at 0, 6, 9 and 12 min, respectively. At each time point, 1 ml of untreated and treated cells were collected by centrifugation to clarify the bacteria. An aliquot (100 μl) was added to 400 μl of 25 mM sulfuric acid followed by 500 μl of freshly prepared reaction buffer (200 μM ferrous ammonium sulfate, 200 μM xylenol orange, and 25 mM sulfuric acid). The reaction was incubated for 10 min at room temperature and the optical density of the solution measured at 540 nm. The concentration of remaining t-BOOH in the culture was calculated using a standard curve that was repeated for each set of experiments. To investigate the potential ability of purified PhnW to degrade or modify t-BOOH, PhnW (see below for purification details) was added to a solution containing 200 μM of t-BOOH and 100 μM of the reductant, dithiothreitol (DTT). Finally, the reaction mixture was incubated at 37°C and investigated for the degradation of t-BOOH as mentioned above at 15, 30 and 60 min, respectively.
Purification of PhnW
The phnW coding region was amplified from PA genomic DNA using Pfu DNA polymerase with primer phnW F-pET BamHI (5-ACTGGATCCATGAGCACTGCCGAACGCGCACCCAT-3´) and primer phnW R-pET NotI(5´-TATGCGGCCGCGATCTCGAGGACTTCCAGTTCGCGCAGC-3´). The expected PCR product which is ~1.1 kb was digested with BamHI and NotI and ligated within compatible sites of the expression vector, pET23a, creating pET-phnW. E. coli BL21(DE3)/pLysE containing pET-phnW was grown in L-broth supplemented with 100 μg/ml ampicillin at 37°C with shaking until the O.D.600 of the cells reached 0.6–0.8. Then, 1 mM IPTG was added and incubated for 3 hr to induce the expression of PhnW. The cell pellet was kept at -20°C until use. To initiate purification of this protein, the cell pellets were resuspended in sonication buffer (50 mM NaH2PO4, 20 mM Tris-HCl pH 8.0 and 100 mM NaCl and 20 mM imidazole). After sonication with a Sonic Dismembrator Model 500 for 15 min on ice, the supernatant was separated from cell debris by centrifugation at 13,000 x g for 30 min at 4°C. The supernatant was loaded on a Ni-NTA agarose column pre-equilibrated with sonication buffer. Non-specific binding proteins were removed with 10 column volumes (CVs) of washing buffer (sonication buffer with increasing imidazole concentration to 50 mM). PhnW was eluted from the Ni-NTA agarose column using 5 CVs of elution buffer (sonication buffer with increasing the imidazole concentrations to 100 mM). The fractions containing the PhnW protein were pooled and dialyzed against 50 mM Tris, pH 8.0, 5% glycerol, 0.05 M NaCl and 0.1 mM EDTA.
Use of KEGG (Kyoto Encyclodedia of Genes and Genomes) pathway database for identification of the potential role of PhnW in cellular reducing power
The KEGG Pathway Database (http://www.genome.jp/kegg/pathway.html) was used to identify where PhnW lies in fundamental PA metabolism. First, after clicking on the “Organism” bar, we typed in “Pseudomonas” and scrolled down to PA strain PAO1 (codename “pae”). We then scrolled down to section 1.6 (Metabolism of other amino acids) and selected subsection 00440 for “phosphonate and phosphinate metabolism.” The phnW gene is coined PA1310 in the PA PAO1 genome (www.pseudomonas.com) and we subsequently entered phnW in the open box and hit return. An elegant pathway map is given with the PhnW enzymatic reaction presented in red font with the enzyme commission number of 2.6.1.37 in S2 Fig.
Results
Analysis of putative PA genes under OxyR control using a bioinformatics analysis
The entire PA PAO1 genome was first screened for OxyR-binding domains using the OxyR DNA recognition sequence ATAG-N7-CTAT-N7-ATAG-N7-CTAT [6]. The results are listed in Table 2. The bases listed are those that are conserved in the OxyR promoter sequences for the experimentally confirmed katB, ahpB and ahpCF that we published 16 years ago [6] and more recently using ChIP-chip analyses which also includes the bdlA (biofilm dispersion locus, [17]) and PA3236 genes [18]. We were particularly interested in the promoter regions of potentially OxyR-regulated genes that were not predicted to be involved in the classical response to oxidative stress (such as genes encoding catalases A [5] and B [6], alkyl hydroperoxide reductases (Ahps, [6]), organic hydroperoxide reductase (Ohr), overlapping regulon genes (Ssp), and membrane proteins involved is the Ssp response. The optimal score for the OxyR box analysis was 13/16, for the upstream regions of the betX and ahpB genes, the latter of which is periplasmic, known to be controlled by OxyR, and involved in protection against both H2O2 and alkyl hydroperoxides [6]. The promoters of two other known OxyR-controlled genes included the katB-ankB operon and ahpCF that received scores of 9/16 and 12/16, respectively. In contrast to a recent study by Wei et. al. [19] who identified genes under OxyR control when organisms were exposed to H2O2, we found that only 8/40 of the genes matched from bioinformatic studies in the aforementioned study. These genes were betX, ahpB, ahpC, bdlA, katB, nadA, mscL and mvfR, respectively.
Semi–quantitative expression of genes culled from bioinfomatic predictions by reverse transcriptase polymerase chain reaction (RT-PCR)
Our first experiment to corroborate our bioinformatic predictions that OxyR either positively or negatively controls transcription of specific genes was by performing RT-PCR from exponential phase bacteria. Genes that matched 9–13 of 16 nucleotides of the OxyR binding motif used in our bioinformatic search that did not encode predicted hypothetical proteins (totaling 40 genes), were amplified and compared between an oxyR mutant and wild-type bacteria. Our results revealed that 3/40 genes could not be amplified (even with 1 μg of RNA). Duplicate results of the RT-PCR analysis revealed differences in gene expression of 15 genes compared between wild-type bacteria and the oxyR mutant. These included mvfR, qscR, PA2957, PA0846, betX, PA0192, PA1097, PA1423, phnW, PA1648, PA3242, PA4403, PA2999, PA0594 and PA5095 (Table 2). Among this group, the RT-PCR results of the phnW gene, encoding 2-aminoethylphosphonate:pyruvate aminotransferase, revealed lower gene expression levels in the oxyR mutant when compared to wild-type bacteria under non-inducing conditions (Fig 1), which was also confirmed transcriptionally by ß-galactosidase activity assays (Fig 2). The upstream sequence that is considered a putative OxyR binding domain of phnW matched 12 of 16 bases. Two sets of control genes were used; the omlA gene which is a constitutively expressed internal control [14], and a positive control gene, ahpC, which is a known member of the PA OxyR regulon [6].
Fig 1. Semi-quantitative expression of PA phnW.
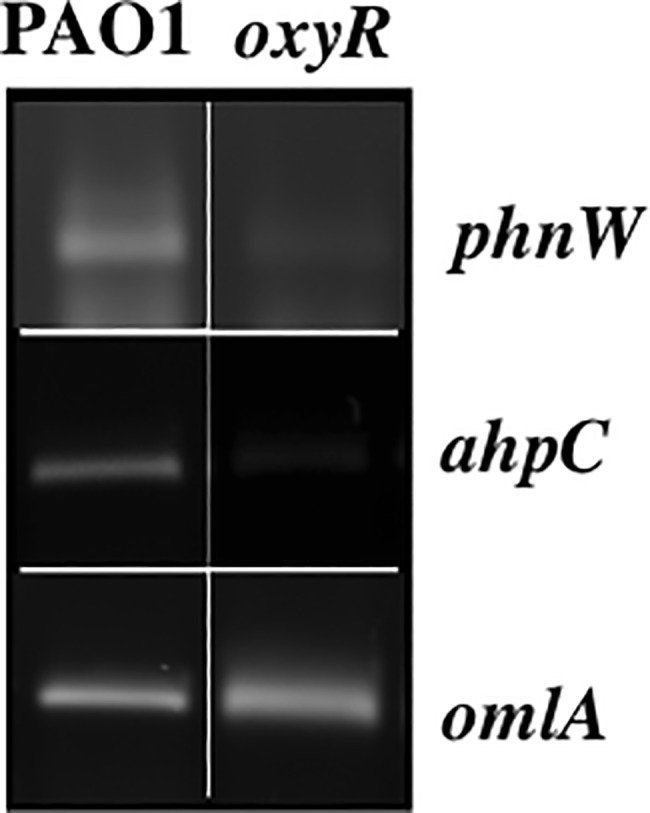
Total RNA was isolated from exponential phase PA PAO1 or its isogenic oxyR mutant. Then, 1 μl of cDNA was used to amplify the phnW promoter region with specific primers. The omlA gene was used as an internal constitutive control [14] and ahpC was used as a positive gene under OxyR control [6].
Fig 2. Determination of phnW expression levels in wild-type and mutant strains when exposed to t-BOOH.

All bacteria which contained a phnW-lacZ transcriptional fusion plasmid (pQF-phnW) were grown to exponential phase and exposed to 250 μM t-BOOH. ß-galactosidase activity assays were reported as the mean +/- standard error compared to untreated bacteria. The assays were performed using three independent experiments. The white bars represent untreated bacteria while the gray bars represent t-BOOH treated organisms, respectively.
Confirmation of OxyR regulation of phnW by transcriptional ß-galactosidase reporter fusion analyses
From our previous experiments using RT-PCR, only 4/15 genes (phnW, bdlA, bexT and qscR) that showed lower expression in the oxyR mutant were further evaluated by transcriptional ß-galactosidase reporter assays. Herein, we focused only on the phnW gene. The upstream region of the phnW gene which contains the OxyR binding domain was cloned into pQF50, a transcriptional fusion vector [8], and then transformed into the following strains: PAO1, oxyR, oxyR-attB oxyR, oxyR-attB oxyR C199S, as well as ahpC, ohr or ahpC ohr double mutant bacteria. As a reminder, AhpCF and Ohr in PA are two of the major proteins involved in the cytoplasmic detoxification of organic hydroperoxides [6,7,13]. The results of the ß-galactosidase activity assays confirmed our RT-PCR results that phnW gene expression is lower in the oxyR mutant than in wild-type bacteria (Figs 1 and 2). Our results also showed complementation of the phnW mutant in both cell backgrounds harboring either OxyR or OxyR C199S (constitutively OxyR reduced form), the latter of which is unable to sense oxidative stress [5]. Expression of phnW was also ~30% lower in the ahpC mutant while it was 80% lower in the oxyR mutant when compared to wild-type levels. Still, phnW expression could be induced to approximately parental levels by exposure to 250 μM of t-BOOH, but not in ohr or ahpC ohr mutant bacteria (Fig 2). However, exposure of ahpC ohr mutant bacteria to 250 μM t-BOOH caused a slight reduction (less than 10%) in phnW expression (Fig 2).
Electrophoretic mobility shift assay (EMSA) of the phnW promoter region
Next, we assessed the binding of purified OxyR to a 199-bp DNA fragment upstream of the phnW gene that contained the putative OxyR binding domain by the EMSA. First, OxyR binding to this fragment was abolished by an unlabeled phnW competing DNA fragment (see CP in Fig 3). To investigate the specific binding of OxyR on the putative OxyR-binding site upstream of phnW, the unrelated protein (BSA) or unrelated DNA (pUCP20) was added to the reaction. Our results showed no change in the mobility shift between the phnW fragment and OxyR protein (see UP and UD in Fig 3, respectively). These data revealed the specific binding of OxyR on the putative OxyR binding sequence upstream of the phnW gene.
Fig 3. Electrophoretic mobility shift assay (EMSA) to indicate OxyR binding to the promoter region upstream of phnW.

Purified OxyR was added to 0.8 ng of DIG-nonradioactive labeled 199-bp DNA fragment of phnW containing the putative phnW OxyR binding domain in the binding buffer and separated on a polyacrylamide gel as described in the materials methods section. The binding reaction consisted of a labeled phnW fragment and various quantities of OxyR protein. UP is the addition of 2 μg of unrelated protein (BSA) to the binding reaction; F is free probe; the addition of increasing concentrations of OxyR (100, 250, 500 nM) to labeled phnW probe are listed; CP is the addition of 125-fold excess of unlabeled phnW DNA to the binding reaction; UD, the addition of 125-fold excess of unrelated DNA (pUCP20 plasmid) to the binding reaction. The positions of free and bound phnW probe are shown on the left (see arrows).
DNase I footprinting analysis of the phnW promoter
We next determined the precise location of the OxyR binding domain within the phnW promoter that was based upon our bioinformatic search using a DNase I footprinting analysis. Various concentrations of purified OxyR protein were used. The footprint pattern showed that OxyR at 500 nM binds to these upstream sequences (Fig 4A). This putative OxyR binding domain is shown in bold, italic, and lower case nucleotide sequences in Fig 4B. We also performed a computer-based promoter prediction derived from 500-bp upstream of the phnW gene using the BDGP: Neural Network Promoter Prediction program (prokaryotic mode, reverse strand “no” command) and discovered a weak promoter score of 0.49; a perfect promoter score for this particular program is 1.0 (http://www.fruitfly.org/seq_tools/promoter.html). Such a low score indicated that the phnW gene is not a prioritized gene from the overall regulatory scheme in PA.
Fig 4. Localization of the OxyR binding domain of the phnW promoter by DNase I footprinting analysis.

(A). The 199-bp upstream sequence of phnW containing a putative OxyR domain was used to treat with DNase I in the presence of OxyR at 0, 500 and 1000 nM. The digested DNA was analyzed on a 5% denaturing polyacrylamide gel, followed by autoradiography. The sequence to which OxyR binds on this fragment was identified. (B). The upstream sequences of phnW that are underlined are the primers used in this study. The bold letters indicate the OxyR binding domain within the phnW upstream sequence while the lower case bold letters indicate the putative OxyR binding site based upon a match to the consensus ATAG-N7-CTAT-N7-ATAG-N7-CTAT sequence used to search for OxyR-dependent gene candidates. The underlined and bold ATG indicates the translational initiation codon of the phnW gene.
Sensitivity of a PA phnW mutant to t-BOOH and partial rescue of t-BOOH resistance in ohr ahpC mutant bacteria
Sensitivity to t-BOOH of the isogenic phnW mutant relative to a number of mutants, complemented and wild-type strains was next examined. Our preliminary results indicated that the phnW mutant is only susceptibility to t-BOOH, and not to CHP and H2O2, respectively. Our results showed that the phnW mutant (lane 2 vs. lane 1 in Fig 5A and 5B) was more susceptible to t-BOOH compared to wild-type and complemented bacteria (lane 3 vs. lane 1 in Fig 5A and 5B) in the exponential but not the stationary growth phase (lane 1 vs. lane 3 in Fig 5C and 5D). In contrast, using stationary phase organisms, the oxyR phnW mutant was slightly more sensitive to t-BOOH than the oxyR mutant (lane 4 vs. lane 5 in Fig 5C and 5D), while provision of the phnW gene to the oxyR mutant provided some measure of protection against t-BOOH (lane 6 vs. lane 4 in Fig 5C and 5D). Moreover, provision of the phnW gene in trans could also protect a strain lacking AhpC and Ohr proteins (ohr ahpC mutant), that have previously been shown to be highly susceptible to organic hydroperoxides including t-BOOH [7]. Collectively, our results show that PhnW helps to partially protect the ohr ahpC double mutant from t-BOOH killing (lane 2 vs. lane 1 in Fig 5E and 5F).
Fig 5. Sensitivity of bacteria to t-BOOH.

Bacteria from the aerobic exponential phase (A,B). PAO1, phnW::Gm and phnW::Gm/pphnW) or (E,F). ohr ahpC and ohr ahpC/pphnW) or stationary phase (C,D). PAO1, oxyR/poxyR, phnW::Gm, oxyR phnW::Gm, oxyR and oxyR/pphnW) were used to determine sensitivity to t-BOOH at 0.3 M (exponential) or 0.5 M (stationary), respectively. The experiments were independent and repeated at least three times. The values shown are means +/- standard error.
PhnW influences the cellular degradation rate of t-BOOH
To determine whether PhnW influences the degradation rate of t-BOOH, a xylenol orange–iron reaction assay that allows for precise quantification of organic hydroperoxides was employed [20]. The rate of t-BOOH (200 μM) degradation by exponential phase, aerobic bacteria was measured in selected bacteria and the results were expressed as the percent of t-BOOH remaining in the media after 12 min incubation (Fig 6). Wild-type bacteria (white bar) rapidly degraded t-BOOH, as did an oxyR mutant containing pUCP-oxyR (white bar with dots) or a phnW mutant containing pUCP-phnW (dark gray bar). As expected, the oxyR mutant revealed the slowest t-BOOH degradation rate (light gray bar) compared with all test strains. At t-BOOH exposure times from 0 to 9 min, the phnW mutant showed no difference in the rate of t-BOOH degradation compared to that of oxyR/poxyR, phnW::Gm/pphnW and wild-type bacteria, but clear differences were evident in t-BOOH degradation at about 12 min after exposure (Fig 6). The phnW mutant degraded only 55% of the t-BOOH compared to the 80% degradation observed in wild-type bacteria at the end of assay (12 min). The ability of purified PhnW to degrade t-BOOH also investigated. However, Purified PhnW did not show a direct ability to degrade t-BOOH in vitro.
Fig 6. Time course of t-BOOH degradation by various PA strains.

The rate of PA t-BOOH degradation, an organic hydroperoxide, was investigated using xylenol orange–iron reaction system as described in the materials and methods section. The exponential phase of PAO1 (white bar), oxyR (light gray bar), oxyR/poxyR (dotted bar), phnW::Gm/pphnW (black bar) and phnW::Gm (dark gray bar) cells were used in this study in the present of 200 μM of t-BOOH and measured the rate of degradation of t-BOOH in each cell at difference time points. The percentage of t-BOOH remaining was reported after 12 min of incubation. The experiments were independently repeated at least three times and typical results are shown.
Discussion
The major response of PA and other Gram-negative bacteria including E. coli, S. typhimurium and PA to H2O2 and alkyl hydroperoxides is governed by the major LysR family member and trans-activator, OxyR [6,20,21,22]. The typical oxidative activation mechanism of OxyR requires formation of an inter-molecular disulfide bridge between amino acid residues 199 and 208 mediated by H2O2 or organic hydroperoxides [5]. Still, another new mechanism of OxyR-mediated gene activation anaerobic S-nitrosylation in E. coli, events that are distinct from OxyR-mediated gene activation under oxidative stess conditions [23]. However, the main dominant paradigm for bacterial responses to the aforementioned oxidants involves traditional antioxidant enzymatic machinery that includes but are not limited to catalases, peroxidases and alkyl hydroperoxide reductases. Other forms of OxyR-mediated gene regulation include the activation of regulatory RNAs including OxyS [24]. We have studied the role of PA OxyR and various antioxidants under its control for more than 17 years [5,6,17,22]. However, in this work, we wished to identify PA OxyR-dependent genes that would not be predicted to play a classical role in the oxidative stress response. First, we used the DNA motif ATAG-N7-CTAT-N7-ATAG-N7-CTAT that is the E. coli OxyR consensus binding sequence to search the PA genome database for genes that are possibly regulated by PA OxyR. When compared to a previous study by Wei et al., [18], who identified genes under OxyR control when organisms were exposed to H2O2, we found that only 8 of the 41 genes listed in Table 2 matched those identified in this study. These genes were betX, ahpB, ahpC, bdlA, katB, nadA, mscL and mvfR, respectively. Among this gene list, phnW was not found by Wei et al., [17] to be under OxyR control and there was also no evidence or prediction by either manual or computer-based programs for any other protein to bind to the upstream sequences of the phnW gene [12].
The phnW gene, encoding 2-aminoethylphosphonate:pyruvate aminotransferase, can be found in the genomes of both pathogenic and non-pathogenic bacteria [25,26], yet its gene product is perceived to be involved in pathogenesis through metabolism of organophosphates. Surprisingly, its phn-based genes are all down-regulated (some several hundred-fold) when PA was grown in blood from burn patients, a phenomenon that is not understood at the mechanistic level [27]. PA PhnW catalyzes the transfer of the amino group of 2-aminoethylphophonate (ciliatine) to pyruvate, resulting in the production of 2-phosphonoacetaldehyde and alanine [28]. In this work, we found that the phnW gene was constitutively expressed, independent of whether OxyR was in its reduced or oxidized states, yet its expression was reduced 80% in an oxyR mutant. In contrast, phnW expression was found to be induced 30% in ahpC mutants relative to wild type bacteria (Fig 2). The significantly lower phnW expression in the oxyR mutant indicates that OxyR is a regulator of the phnW gene (Figs 1 and 2). This was confirmed by the ability of OxyR to bind specifically to phnW upstream sequences using both EMSA and DNase I footprinting analyses (Figs 2 and 3). PA OxyR contains the four putative OxyR binding tetranucleotide sequences with 67% identity to similar sequences in E. coli [29], results that are in agreement with our algorithm predictions. Previous EMSA studies by Wei et al. showed that as little as 84 nM OxyR can bind to the promoter region upstream of the ahpC gene [19]. This is in contrast to the requirement of ~3-fold higher levels of OxyR to bind to the binding domain upstream of the phnW gene. This result was also confirmed by our DNase I footprinting results, indicating that very high concentration of OxyR (500 nM) are needed for binding. A previous study by Ochsner et. al. [6] using EMSA indicated that 100 nM OxyR can bind to the well known OxyR-regulated gene promoters katB, ahpB or ahpC. In addition, DNase I footprinting of E. coli OxyR on the ahpC promoter was studied by Zheng et. al. [30], who showed that as little as 18 nM OxyR was required for promoter binding. This is about 28-fold or 2.5-fold less OxyR than we used in our DNase I footprinting or EMSA experiments, respectively. These two experiments clearly indicated a far lower binding affinity of OxyR to the phnW upstream nucleotides compared to the major OxyR-dependent antioxidant gene, ahpC [6], which is consistent with a very low promoter prediction score (0.49 of a possible perfect score of 1.0). This may result in dramatic differences in the potential for OxyR to control phnW expression in PA. Moreover, it is also supported by the alignment of the putative OxyR binding sequence of phnW with other OxyR-dependent genes base on the results of Wei et al., [17]. Our results indicate that the OxyR binding sequence upstream of phnW is 80% identical to betX (PA3236), 53% to the ahpB, katB and ahpC genes and 46% to bdlA, respectively (S1 Fig).
The ahpB, katB and ahpC are well known OxyR-dependent genes while betX and bdlA were both introduced to the literature by Wei et. al., [6]. Our results show that PhnW has an ability in vivo to partially protect bacteria from t-BOOH toxicity in oxyR and ahpC ohr mutants, two t-BOOH sensitive strains, and also in exponential phase but not stationary phase wild-type bacteria. Complementation of the lower expression of phnW in the oxyR mutant can occur using both oxyR and oxyR C199S, respectively. Interestingly under normal conditions, phnW expression is also lower when the bacteria lack only one of the major proteins involved in organic hydroperoxide detoxification (AhpCF or Ohr), while an absence of both AhpCF and Ohr allow for wild-type phnW expression. This may be a compensatory strategy triggered by PhnW for organisms to protect themselves from t-BOOH toxicity.
How could PhnW contribute metabolically to the protection of PA from t-BOOH?
Because PhnW offered protection against t-BOOH in vivo, we sought in earnest to mechanistically define how this might occur. The forward substrate of PhnW, 2-aminoethylphosphonate, is taken into the cell by an ABC transporter system and can be utilized as a sole carbon source for growth [28]. Still, despite the fact that many enzymes can possess dual functions (e.g., catalase/peroxidase), we thought it highly doubtful that PhnW itself possessed any t-BOOH degrading activity and we subsequently proved this experimentally. Rather, we posited that L-alanine, one product of PhnW, could modulate cellular antioxidant levels. L-alanine has been shown to trigger activation of an antioxidant response in eukaryotes [31]. Alternatively, alanine can funnels into pyruvate that subsequently enters the TCA cycle, resulting in the net production of 4 NADH and one FADH2 molecule per pyruvate molecule. The significance of an increase in reducing power (electrons) is that many alkylhydroperoxide reductases such as the E. coli AhpCF require NADH as a bound cofactor [32]. Another possibility is that the reverse reaction of PhnW leads to the production of acetate and acetaldehyde (see panel B inS2 Fig, black arrows on upper left) which can be funneled into the glycolytic pathway, leading to more reducing power starting with the two NADH molecules produced from pyruvate dehydrogenase and the following six NADH and 2 FADH2 molecules from the TCA cycle. The substrate of PhnW protein (aminoethylphosphonic acid) was also investigated its ability to help protect bacteria from t-BOOH toxicity. Our results demonstrated that supplementation of aminoethylphosphonic acid in the media slightly helped to protect bacteria from t-BOOH toxicity at concentrations as low as 1 mM or as high as 10 mM. Surprisingly, the protection afforded by bacteria grown in the presence of aminoethylphosphonic acid was also specific to the ahpC ohr double mutant only. The killing zone with 0.2 M t-BOOH of stationary phase an ahpC ohr double mutant was 42±0.063 mm and 40±0.031 mm for either control bacteria or organisms supplemented with 10 mM aminoethylphosphonic acid, respectively.
Given the frustrating caveat of not knowing the true mechanism by which PhnW influences t-BOOH resistance not directly but possibly indirectly, we propose the following model of phnW expression under different cellular conditions that help protect bacteria from t-BOOH. A cartoon depicting various oxidative conditions in selected strains is shown in Fig 7A–7D. First, there is continuous expression of phnW in wild-type bacteria (Fig 7A).
Fig 7. A model of phnW expression under difference conditions.

(A). In wild-type bacteria, phnW is continuously expressed in both untreated bacteria and during exposure to t-BOOH, while oxidized OxyR triggers activation of ahpC expression as previously shown [6]. Wild-type bacteria used both of these proteins and Ohr to assist in either the direct or indirect degradation of t-BOOH. This results in efficient t-BOOH degradation to a by-product that is not toxic to the bacteria, thereby preventing damage to proteins, DNA and lipid. (B). Expression of the phnW gene is significant lowered in oxyR mutant bacteria (80%) when compared to wild-type bacteria with no induction of ahpC expression when exposed to t-BOOH. This results in greater susceptibility to t-BOOH of this mutant relative to wild-type bacteria since this strain likely only uses Ohr to help detoxify t-BOOH. (C). Lower expression of phnW was also detected in a PAO1 ahpC mutant compared to wild type bacteria (30%). A lack of this major AHP in response to t-BOOH coupled with lower expression of phnW with only Ohr detoxifying power remaining results in less protection of this mutant from t-BOOH toxicity. (D). An ahpC ohr mutant showed a slightly reduced expression of phnW when bacteria were exposed to t-BOOH, but no difference were observed under control conditions when compared to wild-type expression. Thus, a lack of both of the major t-BOOH detoxification proteins and a lower expression of phnW results in this mutant being the most susceptible to t-BOOH. This may aid in bacterial protection from endogenous free radicals that are continuously generated under aerobic conditions and/or at the earliest time period, when exposed to t-BOOH while the responding gene is not yet expressed. When the bacteria are exposed to t-BOOH, oxidized OxyR governs over-expression of AhpCF to help in its detoxification and together with Ohr, an OxyR independent t-BOOH detoxification protein (Fig 7A). A significantly lower expression level of phnW gene (~80%) was revealed in the oxyR mutant under both reduced and oxidized conditions, indicative of OxyR-mediated regulation of the phnW gene (Figs 2 and 1). When the oxyR mutant that had significantly reduced expression of AhpC and PhnW was exposed to t-BOOH, this event triggered an increased susceptibility to this oxidant when compared to wild-type bacteria (Fig 7B). Interestingly, phnW expression levels were also ~30% lower when compared to wild-type levels but are complemented when exposed to t-BOOH in the ahpC but not in the ohr mutant (Fig 2). This could be yet another mechanism by which bacteria used to protect themselves from t-BOOH toxicity when they lack the major t-BOOH detoxifying protein, AhpC (Fig 7C). Both PA oxyR and PA ahpC mutants still have an Ohr (organic hydroperoxide resistance), one of the major proteins that can contribute to t-BOOH detoxification. A genome search for “peroxidase” and “hydroperoxide” revealed 6 and 5 hits, respectively. This indicates that there are likely multiple redundant mechanisms to dispose of hyperoxides such as t-BOOH. Therefore, we expected that phnW expression should be higher in the ohr ahpC double mutant to protect bacteria cell from t-BOOH. Surprisingly, expression was ~20% lower in this strain after exposure to t-BOOH. The lower expression of PhnW after exposure to t-BOOH in the ohr ahpC double mutant may also contribute to the sensitivity of this double mutant to t-BOOH (Fig 7D). AhpC is OxyR-dependenct but Ohr is not. Therefore, it appears that phnW expression levels depend on the present of OxyR in the cell and also the level of AhpC in ahpC or ahpC ohr mutants. Therefore, it is likely that OxyR directly regulated phnW expression level through binding on an upstream sequence of this gene in certain condition to help protect cell from t-BOOH toxicity (Figs 3 and 4). Frustratingly, though the mechanism of PhnW in responding to t-BOOH or regulated by OxyR is still unclear, this study clearly indicates that this protein has the ability to assist in the protection of PA cell from t-BOOH toxicity.
Conclusions
In this study, we revealed a previously unrecognized gene under the control of PA OxyR that plays an as yet unknown role in detoxification of the organic hydroperoxide, t-BOOH. We expose a dynamic that involves a complex interplay between PhnW, that does not appear to have the direct capacity to degrade t-BOOH, but an absence of it in the bacteria shifts the overall expression of other classical AHPs including AhpCF and Ohr. Further studies are required to elucidate the precise detoxification mechanism that PhnW provides to aerobically grown PA.
Supporting information
The OxyR putative binding region of several OxyR-dependent genes based on (18) was used to compare to OxyR binding region of phnW. The MView software from EMBL-EBI was used. The conservation of these sequences in OxyR-dependent genes such as phnW, betX, ahpB, katB, ahpC and bdlA are shown in black. Dark gray represent the nucleotides homologous to the OxyR putative binding region of phnW while lighter gray are not.
(TIFF)
A. Gene orientation of the phnW gene on the PA PAO1 genome. B. KEGG map of a portion of central metabolism of PA with the PhnW protein catalyzing the reaction shown as an arrow. The black arrows on the upper left corner of the diagram indicate the potential flow of acetate and acetaldehyde back in the glycolytic pathway.
(TIFF)
Acknowledgments
The authors wish to thank the Department of Molecular Genetics, Biochemistry and Microbiology at the University of Cincinnati College of Medicine for internal support for this work.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Institutes of Health RO1 GM-69845-01.
References
- 1.WHO. World health statistics. Available at: http://www.who.int/whosis/whostat/EN_WHS08_Full.pdf. Accessed January. World health statistics. Available at: http://www.who.int/whosis/whostat/EN_WHS08_Full.pdf. Accessed January, 2014. 2008.
- 2.Govan JRW, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;60:539–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hassett DJ, Borchers MT, Panos RJ. Chronic obstructive pulmonary disease (COPD): Evaluation from clinical, immunological and bacterial pathogenesis perspectives. J Microbiol. 2014;52(3):211–26. doi: 10.1007/s12275-014-4068-2 [DOI] [PubMed] [Google Scholar]
- 4.Storz G, Zheng M. Oxidative Stress, In, Bacterial Stress Responses. Storz G. and Hengge-Aronis R. ASM Press, Washington, DC: 2000:47–59. [Google Scholar]
- 5.Heo YJ, Chung IY, Cho WJ, Lee BY, Kim JH, Choi KH, et al. The major catalase gene (katA) of Pseudomonas aeruginosa PA14 is under both positive and negative control of the global transactivator OxyR in response to hydrogen peroxide. Journal of bacteriology. 2010;192(2):381–90. doi: 10.1128/JB.00980-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ. Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB, ahpB, and ahpCF. J Bacteriol. 2000;182:4533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ochsner UA, Hassett DJ, Vasil ML. Genetic and physiological characterization of ohr, encoding a protein involved in organic hydroperoxide resistance in Pseudomonas aeruginosa. Journal of bacteriology. 2001;183(2):773–8. doi: 10.1128/JB.183.2.773-778.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farinha MA, Helinski AM. Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J Bacteriol. 1990;172:3496–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid. 2000;43(1):59–72. doi: 10.1006/plas.1999.1441 [DOI] [PubMed] [Google Scholar]
- 10.West SEH, Schweizer HP, Dall C, Sample AK, Runyen-Janecky LJ. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene. 1994;148:81–6. [DOI] [PubMed] [Google Scholar]
- 11.Schweizer HP, Hoang TT. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene. 1995;158:15–22. [DOI] [PubMed] [Google Scholar]
- 12.Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FS. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic acids research. 2016;44(D1):D646–53. doi: 10.1093/nar/gkv1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hassett DJ, Alsabbagh E, Parvatiyar K, Howell ML, Wilmott RW, Ochsner UA. A protease-resistant catalase, KatA, that is released upon cell lysis during stationary phase, is essential for aerobic survival of a Pseudomonas aeruginosa oxyR mutant at low cell densities. J Bacteriol. 2000;182:4557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ochsner UA, Vasil AI, Johnson Z, Vasil ML. Pseudomonas aeruginosa fur overlaps with a gene encoding a novel outer membrane lipoprotein, OmlA. Journal of bacteriology. 1999;181(4):1099–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller JH. Procedures for working with lac In, A Short Course in Bacterial Genetics Plainview, New York: Cold Spring Harbor Press; 1992:72–4. [Google Scholar]
- 16.Vattanaviboon P, Whangsuk W, Panmanee W, Klomsiri C, Dharmsthiti S, Mongkolsuk S. Evaluation of the roles that alkyl hydroperoxide reductase and Ohr play in organic peroxide-induced gene expression and protection against organic peroxides in Xanthomonas campestris. Biochemical and biophysical research communications. 2002;299(2):177–82. [DOI] [PubMed] [Google Scholar]
- 17.Morgan R, Kohn S, Hwang SH, Hassett DJ, Sauer K. BdlA, a chemotaxis regulator essential for biofilm dispersion in Pseudomonas aeruginosa. Journal of bacteriology. 2006;188(21):7335–43. doi: 10.1128/JB.00599-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei Q, Le Minh PN, Dotsch A, Hildebrand F, Panmanee W, Elfarash A, et al. Global regulation of gene expression by OxyR in an important human opportunistic pathogen. Nucleic acids research. 2012;40(10):4320–33. doi: 10.1093/nar/gks017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei Q, Minh PN, Dotsch A, Hildebrand F, Panmanee W, Elfarash A, et al. Global regulation of gene expression by OxyR in an important human opportunistic pathogen. Nucleic acids research. 2012;40(10):4320–33. doi: 10.1093/nar/gks017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gay C, Collins J, Gebicki JM. Hydroperoxide assay with the ferric-xylenol orange complex. Anal Biochem. 1999;273(2):149–55. doi: 10.1006/abio.1999.4208 [DOI] [PubMed] [Google Scholar]
- 21.Christman MF, Morgan R, Jacobson FS, Ames BN. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell. 1985;41:753–62. [DOI] [PubMed] [Google Scholar]
- 22.Winsor GL, Lo R, Ho Sui SJ, Ung KS, Huang S, Cheng D, et al. Pseudomonas aeruginosa Genome Database and PseudoCAP: facilitating community-based, continually updated, genome annotation. Nucleic acids research. 2005;33(Database issue):D338–43. doi: 10.1093/nar/gki047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christman MF, Storz G, Ames BN. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc Natl Acad Sci. 1989;86:3484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seth D, Hausladen A, Wang YJ, Stamler JS. Endogenous protein S-Nitrosylation in E. coli: regulation by OxyR. Science. 2012;336(6080):470–3. doi: 10.1126/science.1215643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altuvia S, Weinstein-Fischer D, Zhang A, Postow L, Storz G. A small, stable RNA induced by oxidative stress: role as a pleiotropic regulator and antimutator. Cell. 1997;90(1):43–53. [DOI] [PubMed] [Google Scholar]
- 26.Kim AD, Baker AS, Dunaway-Mariano D, Metcalf WW, Wanner BL, Martin BM. The 2-aminoethylphosphonate-specific transaminase of the 2-aminoethylphosphonate degradation pathway. Journal of bacteriology. 2002;184(15):4134–40. doi: 10.1128/JB.184.15.4134-4140.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borisova SA, Christman HD, Metcalf ME, Zulkepli NA, Zhang JK, van der Donk WA, et al. Genetic and biochemical characterization of a pathway for the degradation of 2-aminoethylphosphonate in Sinorhizobium meliloti 1021. The Journal of biological chemistry. 2011;286(25):22283–90. doi: 10.1074/jbc.M111.237735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruczek C, Kottapalli KR, Dissanaike S, Dzvova N, Griswold JA, Colmer-Hamood JA, et al. Major Transcriptome Changes Accompany the Growth of Pseudomonas aeruginosa in Blood from Patients with Severe Thermal Injuries. PloS one. 2016;11(3):e0149229 doi: 10.1371/journal.pone.0149229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dumora C, Lacoste AM, Cassaigne A. Purification and properties of 2-aminoethylphosphonate:pyruvate aminotransferase from Pseudomonas aeruginosa. European journal of biochemistry. 1983;133(1):119–25. [DOI] [PubMed] [Google Scholar]
- 30.Toledano MB, Kullik I, Trinh F, Baird PT, Schneider TD, Storz G. Redox-dependent shift of OxyR-DNA contacts along an extended DNA-binding site: a mechanism for differential promoter selection. Cell. 1994;78:897–909. [DOI] [PubMed] [Google Scholar]
- 31.Zheng M, Wang X, Doan B, Lewis KA, Schneider TD, Storz G. Computation-directed identification of OxyR DNA binding sites in Escherichia coli. Journal of bacteriology. 2001;183(15):4571–9. doi: 10.1128/JB.183.15.4571-4579.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grosser N, Oberle S, Berndt G, Erdmann K, Hemmerle A, Schroder H. Antioxidant action of L-alanine: heme oxygenase-1 and ferritin as possible mediators. Biochemical and biophysical research communications. 2004;314(2):351–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The OxyR putative binding region of several OxyR-dependent genes based on (18) was used to compare to OxyR binding region of phnW. The MView software from EMBL-EBI was used. The conservation of these sequences in OxyR-dependent genes such as phnW, betX, ahpB, katB, ahpC and bdlA are shown in black. Dark gray represent the nucleotides homologous to the OxyR putative binding region of phnW while lighter gray are not.
(TIFF)
A. Gene orientation of the phnW gene on the PA PAO1 genome. B. KEGG map of a portion of central metabolism of PA with the PhnW protein catalyzing the reaction shown as an arrow. The black arrows on the upper left corner of the diagram indicate the potential flow of acetate and acetaldehyde back in the glycolytic pathway.
(TIFF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.