

Graphical abstract

Keywords: Benzoxaborole, Trypanosomiasis, Cattle, SAR, Lead optimisation, Protozoan
Abstract
Novel l-valinate amide benzoxaboroles and analogues were designed and synthesized for a structure-activity-relationship (SAR) investigation to optimize the growth inhibitory activity against Trypanosoma congolense (T. congolense) and Trypanosoma vivax (T. vivax) parasites. The study identified 4-fluorobenzyl (1-hydroxy-7-methyl-1,3-dihydrobenzo[c][1,2]oxaborole-6-carbonyl)-l-valinate (5, AN11736), which showed IC50 values of 0.15 nM against T. congolense and 1.3 nM against T. vivax, and demonstrated 100% efficacy with a single dose of 10 mg/kg against both T. congolense and T. vivax in mouse models of infection (IP dosing) and in the target animal, cattle, dosed intramuscularly. AN11736 has been advanced to early development studies.
Animal African Trypanosomiasis (AAT) is a fatal, parasitic wasting disease of livestock and wild animals in Sub-Saharan Africa.1 It is caused primarily by the two protozoan parasites Trypanosoma congolense (T. congolense) and Trypanosoma vivax (T. vivax), which are transmitted by tsetse flies.1 AAT is responsible for 3 million cattle deaths annually and costs African livestock farmers approximately US$ 1–5 billion per year.2 The current standard-of-care drugs, such as diminazene aceturate, isometamidium and homidium chloride, have been in use for several decades and are often ineffective with drug resistance becoming an increasing concern.1 No new trypanocides have been approved for use in cattle for many years. Initial screening of the Anacor Pharmaceuticals library of novel boron-containing compounds identified an active compound (1, Fig. 1), which had an IC50 = 5 nM against T. congolense and 69 nM against T. vivax while its enantiomer was much less active. A quick and simple modification on the amino acid side chain with an isopropyl group generated 2 (Fig. 1) with improved in vitro potency (IC50 = 2 nM against both T. congolense and T. vivax). This encouraging result prompted us to investigate this chemical series further. We designed and synthesized a series of novel benzoxaboroles (3–71, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7, Fig. 8) to optimize anti-parasitic activity, physicochemical and in vitro ADME properties, and the pharmacokinetic profile. Specifically, these molecules were designed to examine the effects of oxaborole 3-substituent variation (3 vs 2, Fig. 1, Fig. 2), oxaborole 7-substituent variation (4 vs 2, Fig. 1, Fig. 2; 5 vs 20–27, Fig. 4), substituent changes on the benzyl group (5–19, Fig. 3), modification of the amino acid (28–32, Fig. 5), heteroaromatic methyl esters (33–48, Fig. 6), introduction of water-solubilizing scaffolds to the benzyl group (49–54, Fig. 7) and aliphatic esters (55–71, Fig. 8). Herein, we report the synthesis and antiparasitic activity against T. congolense and T. vivax of these novel compounds.
Fig. 1.
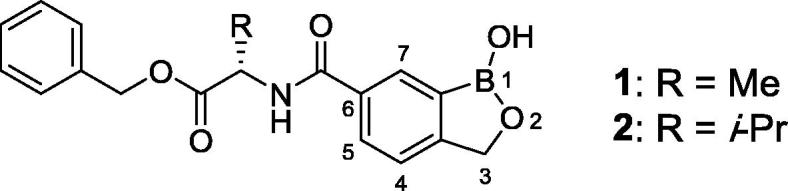
Chemical structures of early hits (1 and 2).
Fig. 2.
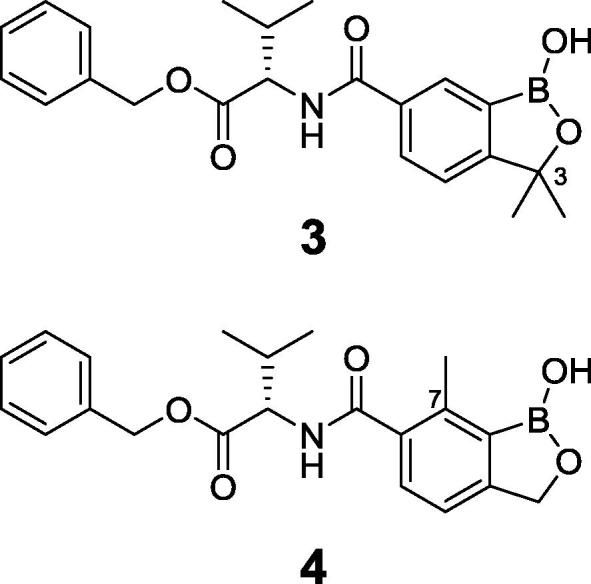
Structures of benzoxaboroles with additional 3,3-Me2 (3) or 7-Me (4) modification as compared to analog 2.
Fig. 3.
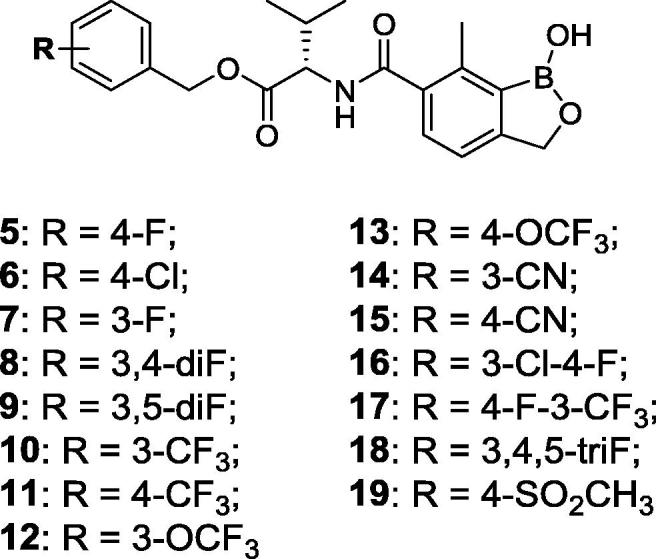
Structures of benzoxaboroles with variation of substituents on the benzyl ring (5–19) as compared to analog 4.
Fig. 4.
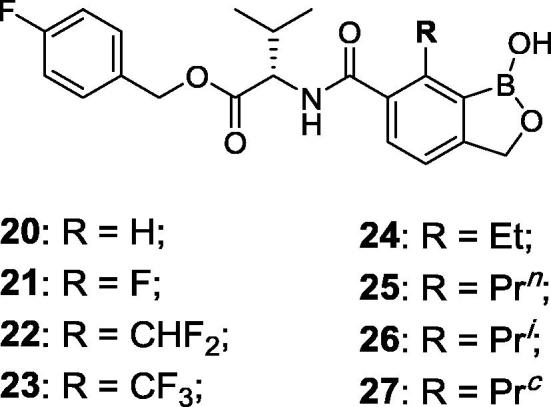
Structures of benzoxaboroles with variation of 7-substituents on the benzene ring (20–27) as compared to analog 5.
Fig. 5.
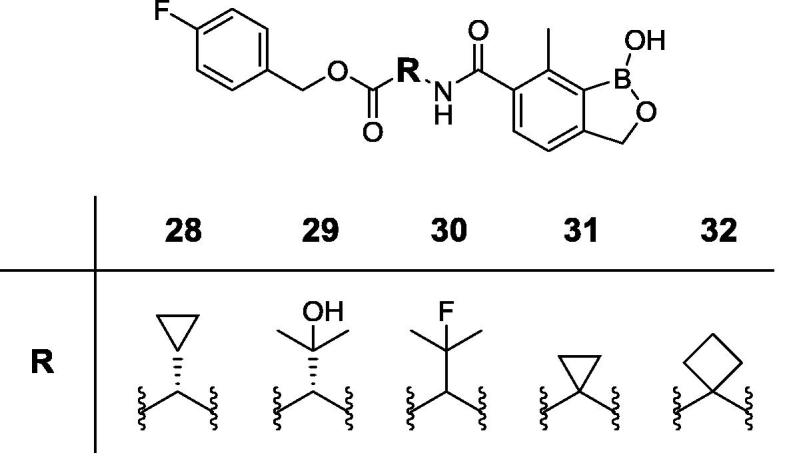
Structures of benzoxaboroles with variation on the amino acid side chain (28–32) as compared to analog 5.
Fig. 6.
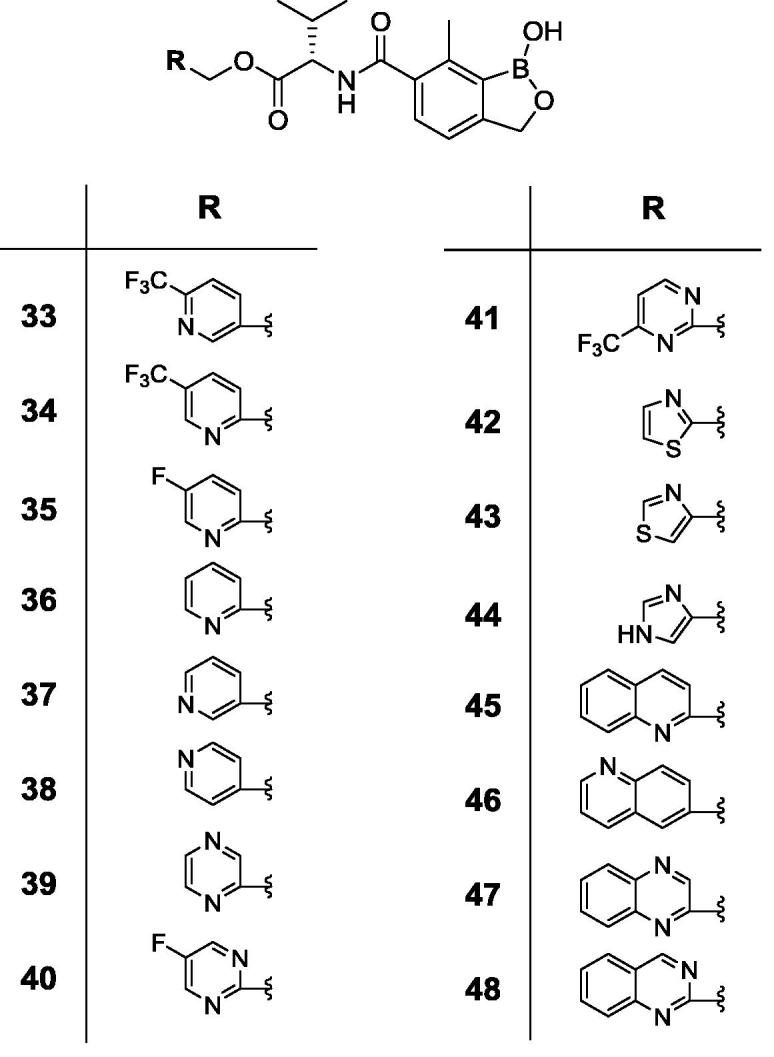
Structures of benzoxaboroles with variation of the left side arylmethyl groups (33–48) as compared to analog 5.
Fig. 7.
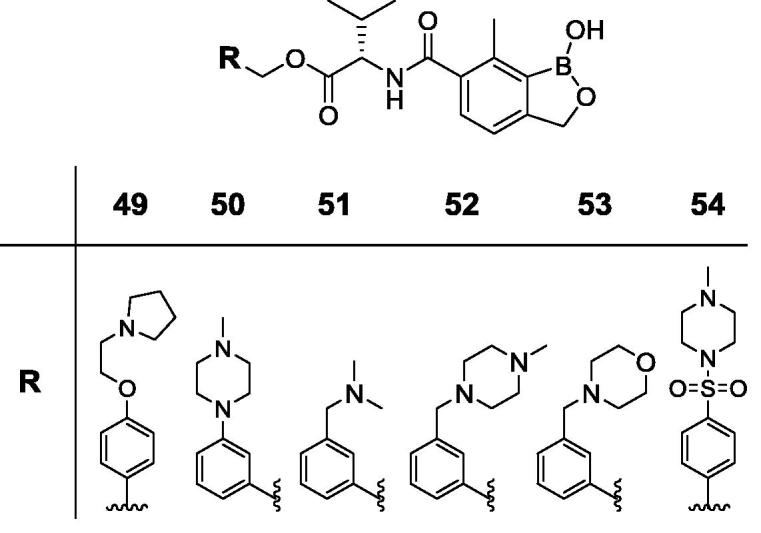
Structures of benzoxaboroles with water-solubilizing scaffolds on the benzyl ring (49–54) as compared to analog 5.
Fig. 8.
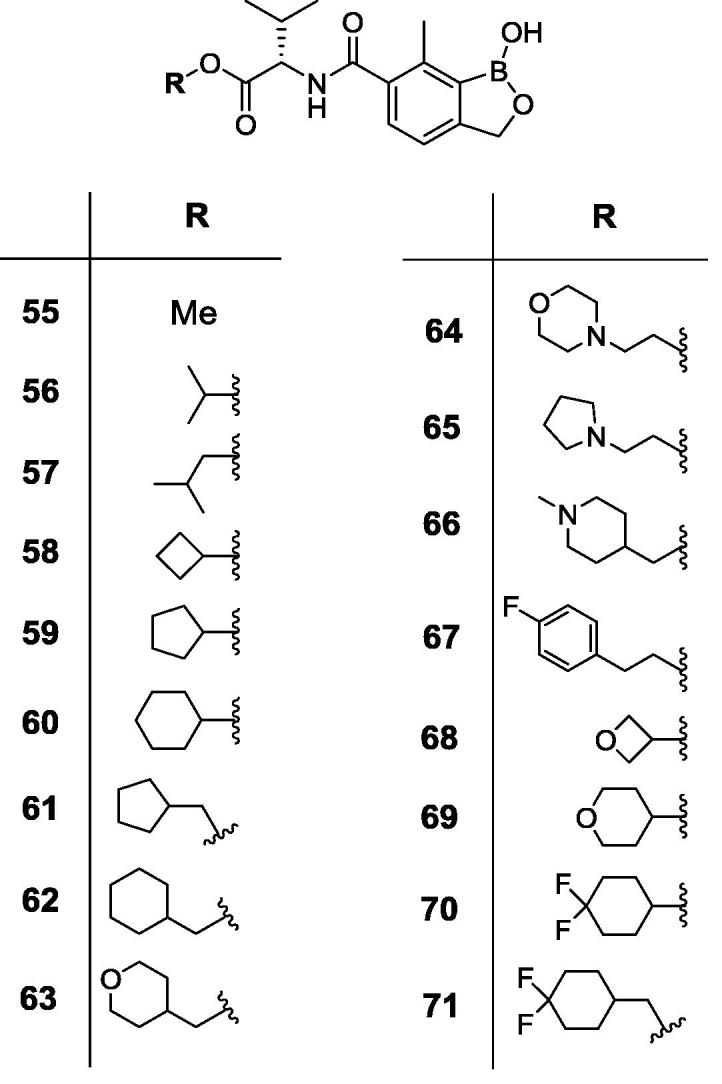
Structures of benzoxaboroles with variation of the left side aliphatic ester groups (55–71) as compared to analog 5.
Compounds 1–71 were convergently synthesized from three building blocks: the left side alcohols (72), amino acid linkers (73) and benzoxaborole 6-carboxylic acids (76).3, 4 The general synthetic route is shown in Scheme 1. Reaction of alcohols 72 with N-Boc protected amino acids 73 gave ester intermediates 74, which were treated with dry hydrogen chloride to generate ester amine salts 75. Condensation of these amine salts 75 with benzoxaborole 6-carboxylic acids 76 provided the final compounds 1–71.
Scheme 1.

General route for syntheses of 1–71. Reagents and conditions: (a) DCC, DMAP, DCM, 0–20 °C, 16 h; (b) HCl/EtOAc, 20 °C, 5 h; (c) HOBt, EDCI, TEA, DCM, 0–20 °C, 15 h.
Scheme 2 illustrates the synthesis of 1-hydroxy-7-methyl-1,3-dihydrobenzo[c][1,2]oxaborole-6-carboxylic acid (83) as an example of key boron intermediates. Esterification of the acid 77 produced the ester 78, which was formylated to yield 79. Treatment of 79 with trifluoromethyl sulfonyl anhydride afforded the triflate compound 80, which was converted to the pinacol boron intermediate 81. Reduction of 81 and subsequent cyclization under aqueous acidic conditions generated the benzoxaborole ester 82. Hydrolysis of the ester group in 82 afforded the acid 83. The experimental procedures for the synthesis of 5 are described in the reference and note section.5
Scheme 2.

Synthetic route for preparation of 83. Reagents and conditions: (a) H2SO4, EtOH, reflux, 24 h; (b) MgCl2, (CH2O)n, TEA, THF, reflux, 14 h; (c) Tf2O, pyridine, DMAP, DCM, 0–15 °C, 1 h; (d) Pin2B2, KOAc, Pd(dppf)Cl2, 1,4-dioxane, N2, 85 °C, 15 h; (e) NaBH4, MeOH, THF, 0–15 °C, 1 h, then HCl, H2O; (f) NaOH,H2O, 40 °C, 3 h, then HCl for acidification.
Activity of compounds 1–71 against T. congolense and T. vivax was determined using the whole cell assays as described 6 and their IC50 values are summarized in Table 1.
Table 1.
Activity of compounds 1–71 against T. congolense (T. c.) and T. vivax (T. v.).a
| Compound | IC50 (nM) |
Compound | IC50 (nM) |
||
|---|---|---|---|---|---|
| T. c. | T. v. | T. c. | T. v. | ||
| 1 | 4.9 | 69 | 37 | 0.78 | 0.50 |
| 2 | 2.0 | 2.0 | 38 | 0.68 | 0.31 |
| 3 | 2580 | 9190 | 39 | 0.57 | 0.24 |
| 4 | 0.46 | 0.79 | 40 | 0.27 | 0.50 |
| 5 | 0.14 | 1.3 | 41 | 0.78 | 0.11 |
| 6 | 0.47 | 2.9 | 42 | 0.20 | 0.19 |
| 7 | 0.59 | 0.10 | 43 | 0.062 | NTb |
| 8 | 0.28 | 0.07 | 44 | 5000 | 980 |
| 9 | 0.18 | 0.10 | 45 | 0.37 | 0.33 |
| 10 | 0.22 | 2.7 | 46 | 0.42 | 0.24 |
| 11 | 0.16 | 24 | 47 | 0.20 | 0.081 |
| 12 | 0.31 | 19 | 48 | <0.005 | 0.71 |
| 13 | 0.23 | 0.04 | 49 | 0.39 | 68 |
| 14 | 0.10 | 0.06 | 50 | 0.32 | 0.48 |
| 15 | 0.15 | 0.05 | 51 | 0.25 | 0.21 |
| 16 | 0.08 | 0.07 | 52 | 1.3 | 0.29 |
| 17 | 0.21 | 0.04 | 53 | 0.51 | 0.35 |
| 18 | 0.20 | 0.06 | 54 | 1.0 | 0.37 |
| 19 | 0.61 | 0.44 | 55 | 4.2 | 3.3 |
| 20 | 3.0 | 1.0 | 56 | 5.2 | 14 |
| 21 | 28 | NTb | 57 | 0.46 | 0.26 |
| 22 | 0.67 | 0.92 | 58 | 0.66 | 0.69 |
| 23 | 37 | 51 | 59 | 0.70 | 0.52 |
| 24 | 0.11 | 0.05 | 60 | <0.005 | 0.78 |
| 25 | 3.0 | 4.7 | 61 | 0.36 | 0.21 |
| 26 | 2.3 | 0.05 | 62 | 0.38 | 0.16 |
| 27 | 3.9 | 0.71 | 63 | 0.39 | 0.09 |
| 28 | 0.12 | 0.38 | 64 | 2.3 | 0.78 |
| 29 | 0.26 | 0.10 | 65 | 9.4 | 1.2 |
| 30 | 0.09 | 0.25 | 66 | 5.9 | 4.1 |
| 31 | 0.13 | 18 | 67 | 0.43 | 0.79 |
| 32 | 0.45 | 2.5 | 68 | 1.8 | 37 |
| 33 | 0.28 | 26 | 69 | 0.47 | 1.6 |
| 34 | 0.14 | 0.07 | 70 | 0.34 | 2.28 |
| 35 | 0.15 | 0.09 | 71 | 0.2 | 0.14 |
| 36 | 0.26 | 0.14 | |||
Experimental procedures are described in the reference and note section.6
NT = Not tested.
Lead compound 2 exhibited an IC50 of 2 nM against both T. congolense and T. vivax. The 3,3-dimethyl analog 3 was essentially inactive (IC50 = 2580 nM against T. c. and 9190 nM against T. v.) but better activity was observed for the 7-methyl analog 4 (IC50 = 0.46 nM against T. c. and 0.79 nM against T. v.). We focused future SAR development on compounds incorporating the 7-methyl group, as in vivo activity of 4 was superior to that observed for 2 (vide infra). A wide range of substituents, such as halogens, trifluoromethyl, trifluoromethoxy, cyano and methylsulfonyl (5–19 in Fig. 3) on the benzyl ring were introduced to examine their effects on the antiparasitic activity. The majority of these fifteen compounds, with exception of 11 and 12, were very potent showing IC50 values around 1 nM (see Table 1). We next explored variation of the substituent at the 7-position of benzoxaborole (20–27 in Fig. 4). The difluoromethyl (22) and ethyl (24) analogs had similar activity to that of 5, but the electron-withdrawing fluoro (21) and trifluoromethyl (23) analogs were of significantly reduced potency. The amino acid linker was also modified (Fig. 5), with the cyclopropyl (28), 2-hydroxyisopropyl (29), 2-fluoroisopropyl (30) and spirocyclobutyl (32) analogs exhibiting potency similar to 5, but the spirocyclopropyl analog (31) exhibited decreased activity against T. v. parasite. Replacement of the 4-fluorophenyl in 5 with various heteroaryl groups (33–48, Fig. 6) resulted in the excellent activity in all cases except the NH-imidazole analog 44. Introduction of basic nitrogen-containing groups on the benzyl ester (49–54, Fig. 7) provided compounds 50–54 that were generally similar to 5. Lastly, aliphatic and heterocyclic esters (55–71, Fig. 8) were synthesized and many of these had IC50 values less than 1 nM as shown in Table 1.
Selected compounds were screened in both mouse and bovine in vitro metabolic stability assays (mouse S9 and bovine S9), as summarized in Table 2. These two species were chosen because the primary in vivo assays were conducted in mice, and the target animal of this research program is cattle. As shown in Table 2, out of 36 compounds tested, 27 compounds had Clint < 10 µL/min/mg protein in both mouse and bovine S9 assays suggesting moderate to excellent in vitro metabolic stability. We evaluated the efficacy of selected compounds in two in vivo mouse models of infection, against T. congolense and T. vivax, respectively. Mice were infected with either 1 × 105 T. c. parasites or 1 × 104 T. v. parasites, and then treated with a test compound via intraperitoneal administration for 1, 2 or 4 consecutive days. The mice were then monitored for the presence of parasitemia for up to 60 days post treatment.7 We tested in T. c. model first, then followed up with T. v. for interesting compounds. As shown in Table 2, the 7-methyl analog 4 was superior to the 7-unsubstituted analog 2 in both T. c. and T. v. mouse models of infection. Of the 38 compounds tested with the in vivo mouse models, seven compounds (5, 8, 33, 34, 49, 62 and 71) demonstrated ≥50% curative efficacy in the T. c.-infected mouse model and 100% curative efficacy in the T. v.-infected mouse model, when tested as a single dose of 10 mg/kg. To select further from these seven compounds, four (5, 8, 33 and 71) had ≥75% curative efficacy in the T. c.-infected mouse model at a single dose of 10 mg/kg, and two (5 and 8) showed 100% curative efficacy. These two compounds were further tested at a single 5 mg/kg dose, but were unable to cure the T. c.-infected mice. We selected compound 5 (AN11736) to progress to exploratory studies to determine the efficacy and safety in a preliminary formulation against induced infections of T. vivax and T. congolense in cattle.8 AN11736 demonstrated 100% curative efficacy with a single intramuscular injection of 10 mg/kg against both T. congolense and T. vivax in cattle.
Table 2.
In vitro metabolic stability and in vivo mouse efficacy of selected compounds.a
| Compound | Clint (µL/min/mg protein) |
Efficacy in mouse modelb |
||
|---|---|---|---|---|
| Mouse S9 | Bovine S9 | T. c. | T. v. | |
| 2 | NTc | NTc | 0/5 (2 × 10) | 5/5 (4 × 10) 1/5 (1 × 10) |
| 4 | NTc | NTc | 4/5 (2 × 10) | 5/5 (2 × 10) 4/4 (1 × 10) |
| 5 | 5.4 | 9.3 | 5/5 (2 × 10) 4/4 (1 × 10) 0/4 (1 × 5) |
5/5 (2 × 10) 5/5 (1 × 10) 4/4 (1 × 10) |
| 6 | 17 | 9.1 | 0/4 (1 × 10) | NTc |
| 8 | 7.9 | 5.6 | 4/4 (1 × 10) 0/4 (1 × 5) |
4/4 (1 × 10) |
| 14 | 9.1 | 2.7 | 0/4 (1 × 10) | NTc |
| 16 | 15 | 8.9 | 4/4 (2 × 10) 1/4 (1 × 10) |
NTc |
| 17 | 14 | 10 | 0/4 (1 × 10) | NTc |
| 18 | 9.7 | 3.1 | 1/4 (1 × 10) | NTc |
| 19 | 2.6 | 1.3 | 0/4 (1 × 10) | NTc |
| 20 | 1.5 | 1.3 | 0/5 (4 × 10) | NTc |
| 21 | <1 | <1 | 1/4 (2 × 10) | 4/4 (2 × 10) |
| 22 | 1.4 | 0.9 | 0/4 (1 × 10) | NTc |
| 24 | 21 | 23 | 1/4 (2 × 10) | NTc |
| 26 | 32 | 32 | 0/4 (1 × 10) | NTc |
| 29 | <1 | <1 | 0/4 (1 × 10) | NTc |
| 30 | 3.7 | 3.1 | 0/4 (1 × 10) | NTc |
| 31 | <1 | <1 | 0/4 (1 × 10) | NTc |
| 33 | 6.3 | 2.0 | 3/4 (1 × 10) | 4/4 (1 × 10) |
| 34 | 11.8 | 4.8 | 2/4 (1 × 10) | 4/4 (1 × 10) |
| 35 | 5.2 | 7.8 | 0/4 (1 × 10) | NTc |
| 39 | 1.6 | 3.9 | 0/4 (1 × 10) | NTc |
| 41 | 1.3 | 1.8 | 0/4 (1 × 10) | NTc |
| 46 | 7.4 | 7.0 | 1/4 (1 × 10) | NTc |
| 47 | 4.8 | 10 | 0/4 (1 × 10) | NTc |
| 48 | 4.0 | 23 | 1/4 (1 × 10) | NTc |
| 49 | 1.5 | 4.0 | 2/4 (1 × 10) | 4/4 (1 × 10) |
| 50 | 8.6 | 12.7 | 1/4 (1 × 10) | 4/4 (1 × 10) |
| 51 | 1.8 | 5.0 | 0/4 (1 × 10) | 4/4 (1 × 10) |
| 52 | 1.8 | 6.0 | 0/4 (1 × 10) | NTc |
| 53 | 5.4 | 6.8 | 0/4 (1 × 10) | NTc |
| 54 | 5.5 | 3.8 | 0/4 (1 × 10) | NTc |
| 55 | <1 | 3.1 | 0/4 (1 × 10) | NTc |
| 62 | 10 | 20 | 2/4 (1 × 10) | 4/4 (1 × 10) |
| 63 | 0.9 | 1.5 | 1/4 (1 × 10) | 4/4 (1 × 10) |
| 69 | 0.7 | 0.6 | 1/4 (1 × 10) | 1/4 (1 × 10) |
| 70 | 1.5 | 0.3 | 1/4 (1 × 10) | 4/4 (1 × 10) |
| 71 | 6.4 | 8.1 | 3/4 (1 × 10) | 4/4 (1 × 10) |
Methods for testing compound efficacy in mouse models are described in the reference and note section.7
The efficacy data in the table is presented as n/m (q × 10), where n = number of mice survived, m = total number of mice in the study group, q = how many time dosed, and 10 = 10 mg/kg.
NT = Not tested.
In summary, a novel series of l-valinate amide benzoxaboroles was discovered to be active against T. congolense and T. vivax, which are the main causative agents of Animal African Trypanosomiasis (AAT) in cattle. Two compounds (5 and 8) showed 100% curative efficacy in both T. c.- and T. v.-infected mice with a single dose of 10 mg/kg. Compound 5 (AN11736) demonstrated 100% curative efficacy with a single IM dose of 10 mg/kg against both T. congolense and T. vivax in cattle for a duration of 100 days. AN11736, as a novel chemical entity, was selected as a potential developmental candidate for the treatment of AAT.
Acknowledgement
The research was conducted with a grant from the Global Alliance for Livestock Veterinary Medicines (GALVmed) and funded by the Bill & Melinda Gates Foundation [OPP1093639] and the UK Government. The findings and conclusions contained are those of the authors and do not necessarily reflect positions or policies of the Bill & Melinda Gates Foundation or the UK Government.
References
- 1.Giordani F., Morrison L.J., Rowan T.G., de Koning H.P., Barrett M.P. Parasitology. 2016;143:1862. doi: 10.1017/S0031182016001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw A.P., Cecchi G., Wint G.R., Mattioli R.C., Robinson T.P. Prev Vet Med. 2014;113:197. doi: 10.1016/j.prevetmed.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 3.Li X., Zhang S., Zhang Y.-K. Bioorg Med Chem Lett. 2011;21:2048. [Google Scholar]
- 4.Ding C.Z., Zhang Y.-K., Li X. Bioorg Med Chem Lett. 2010;20:7317. doi: 10.1016/j.bmcl.2010.10.071. [DOI] [PubMed] [Google Scholar]
- 5.Experimental procedures for the synthesis of 4-fluorobenzyl (1-hydroxy-7-methyl-1,3-dihydrobenzo[c][1,2]oxaborole-6-carbonyl)-L-valinate (5, AN11736): To a solution of 77 (1.65 kg, 10.8 mol) in EtOH (6.50 L) was added conc. H2SO4 (326 g, 3.25 mol). The reaction mixture was stirred at 105 °C for 24 h. TLC (thin layer chromatography) showed the starting material was consumed completely. The mixture was cooled to 15 °C and concentrated. The residue was poured into aqueous 2 M NaHCO3 (3 L) and the solid was filtered off. The filtrate was concentrated to give 78 (1.75 kg, yield 90%) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 7.41 (d, J = 7.9 Hz, 1H), 7.11 (t, J = 7.9 Hz, 1H), 6.94 (d, J = 7.9 Hz, 1H), 4.58 (br s, 1H), 4.37 (q, J = 7.4 Hz, 2H), 2.46 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H). To a solution of 78 (800 g, 4.44 mol) in THF (6.50 L) were added MgCl2 (634 g, 6.66 mol), TEA (1.80 kg, 17.8 mol) and (HCHO)n (600 g, 6.66 mol). The mixture was immediately heated to 90 °C and was stirred for 14 h. The reaction mixture was cooled to 15 °C. To it were added ice H2O (3 L) and then 12 N HCl (1.5 L) slowly. The mixture was stirred for 0.5 h and then extracted with EtOAc (2 L). The combined organic layer was washed with sat. NaHCO3 to neutral, dried over Na2SO4, filtered and concentrated under reduced pressure to give crude 79 (880 g) as a brown oil. 1H NMR (400 MHz, CDCl3): δ 11.40 (s, 1H), 9.93 (s, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 4.40 (q, J = 7.4 Hz, 2H), 2.44 (s, 3H), 1.41 (t, J = 7.1 Hz, 3H). To a solution of 79 (900 g, 4.32 mol) in DCM (7.56 L) were added pyridine (1.02 kg, 12.9 mol) and DMAP (27 g, 221 mmol). The mixture was cooled to 0 °C and Tf2O (1.60 kg, 5.66 mol) was added dropwise. The reaction mixture was warmed to 15 °C and stirred for 1 h. The mixture was quenched with water (7.65 L) and then extracted with DCM (2 × 7.65 L). The combined organic layer was washed with water (2 L), dried over Na2SO4, filtered and concentrated under reduced pressure to give 80 (685 g, 47%) as a light yellow oil. 1H NMR (400 MHz, CDCl3): δ 10.27 (s, 1H), 7.99 (d, J = 8.0 Hz, 1H), 7.91–7.87 (m, 1H), 4.43 (q, J = 7.0 Hz, 2H), 2.64 (s, 3H), 1.43 (t, J = 7.3 Hz, 3H). To a solution of 80 (1.00 kg, 2.94 mol), bis(pinacolato)diboron (1.12 kg, 4.41 mol) and KOAc (573 g, 5.84 mol) in 1,4-dioxane (6.50 L) was added Pd(dppf)Cl2·CH2Cl2 (150 g, 184 mmol). The mixture was stirred at 85 °C for 15 h under N2. The mixture was cooled to 15 °C, filtered and concentrated to give the crude product. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 40/1 to 4:1) to give crude 81 (942 g) as a yellow oil. To a solution of 81 (1.20 kg, 3.77 mol) in MeOH (300 mL) and THF (6 L) was added NaBH4 (80 g, 2.11 mol) in portions at 0 °C. Then the reaction mixture was stirred at 15 °C for 1 h. HPLC showed 81 was consumed completely. The reaction solution was adjusted to pH = 4 with 2 M HCl. The organic layer was removed in vacuum and the mixture was filtered. The cake was washed with petroleum ether (5 L) and dried in vacuum to give 82 (665 g, 80%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 9.18 (s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 5.00 (s, 2H), 4.30 (q, J = 7.0 Hz, 2H), 2.68 (s, 3H), 1.33 (t, J = 7.0 Hz, 3H). To a mixture of 82 (867 g, 3.94 mol) in H2O (5 L) was added NaOH (394 g, 9.85 mol) in one portion. The solution was heated at 40 °C for 3 h. HPLC showed 82 was consumed completely. This batch was worked-up together with the other batches and acidified with 2 N HCl to pH = 2. The solid was filtered and washed with H2O (10 L). The cake was dried to give 83 (2.00 kg, yield 87%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 12.75 (s, 1H), 9.13 (s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 4.98 (s, 2H), 2.68 (s, 3H); HPLC purity: 100% at both 220 nm and 254 nm; MS (ESI+): m/z = 193 (M+1). To a solution of 4-fluorobenzylalcohol (72, R1 = 4-fluorobenzyl, 290 g, 2.30 mol, 248.10 mL) and N-Boc-(S)-valine (73, R2 = isopropyl, R3 = H, 500 g, 2.30 mol) in dry DCM (6.0 L) were added DCC (854 g, 4.14 mol, 838 mL) and DMAP (39.36 g, 322.19 mmol). The reaction mixture was stirred at 25 °C for 15 h. The mixture was filtered and washed with DCM (2 L) and concentrated to give the crude product. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 50:1 to 10:1) to give 4-fluorobenzyl (tert-butoxycarbonyl)-l-valinate (74, R1 = 4-fluorobenzyl, R2 = isopropyl, R3 = H, 708 g, 95% yield) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.35 (dd, J = 8.2 & 5.5 Hz, 2H), 7.05 (t, J = 8.6 Hz, 2H), 5.19–5.08 (m, 2H), 5.01 (d, J = 8.4 Hz, 1H), 4.25 (dd, J = 8.4 & 4.4 Hz, 1H), 2.13 (dd, J = 11.9 & 6.2 Hz, 1H), 1.44 (s, 9H), 0.93 (d, J = 7.1 Hz, 3H), 0.84 (d, J = 7.1 Hz, 3H). The mixture of 4-fluorobenzyl (tert-butoxycarbonyl)-l-valinate (74, R1 = 4-fluorobenzyl, R2 = isopropyl, R3 = H, 1.06 kg, 3.26 mol) in HCl/EtOAc (6.0 L) was stirred at 25 °C for 14 h. The solvent was removed under reduced pressure to give 4-fluorobenzyl l-valinate hydrochloride (75, R1 = 4-fluorobenzyl, R2 = isopropyl, R3 = H, 780 g, 91% yield) was obtained as a white solid. 1H NMR (400 MHz, CDCl3): δ 8.90 (br. s, 3H), 7.37 (dd, J = 8.2 & 5.5 Hz, 2H), 7.03 (t, J = 8.4 Hz, 2H), 5.29–5.10 (m, 2H), 3.95 (br, s, 1H), 2.44 (dd, J = 11.0 & 6.6 Hz, 1H), 1.08 (dd, J = 10.1 & 7.1 Hz, 6H). To the mixture of 83 (150 mg, 0.77 mmol), 75 (203 mg, 0.77 mmol) and DIEA (0.4 mL, 2.33 mmol) in DMF was added HATU (325 mg, 0.86 mmol). The mixture was stirred at rt for 3 h. The crude product was purified by preparative TLC and preparative HPLC to get the final product 5 (AN11736, 125 mg, 40% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.03 (s, 1H), 8.57 (d, J = 7.2 Hz, 1H), 7.47–7.43 (m, 2H), 7.33 (d, J = 7.6 Hz, 1H), 7.23–7.18 (m, 3H), 5.21 (d, J = 12.4 Hz, 1H), 5.11 (d, J = 12.4 Hz, 1H), 4.96 (s, 2H), 4.33 (t, J = 7.2 Hz, 1H), 2.42 (s, 3H), 2.19-2.10 (m, 1H), 0.94 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H); HPLC purity: 100% at both 220 nm and 254 nm; MS (ESI+): m/z = 400 (M+1).
- 6.Assay methods for determination of T. c. and T. v. IC50 values: Bloodstream-form Trypanosoma congolense (T. congolense IL-3000 strain) were cultured in 24-well plates at 34 °C and 5% CO2. Culture media consisted of complete HMI-9 (IMDM) with 20% bovine serum. To ensure log growth phase, trypanosomes were sub-cultured at appropriate dilutions every 2–3 days. Bloodstream form Trypanosoma vivax (T. vivax) cannot be cultured axenically, therefore trypanosomes (T. vivax STIB719/ILRAD 560 strain) were harvested from a highly parasitemia mouse via cardiac puncture and used directly in the ex vivo drug sensitivity assay on the same day. IC50 determination was carried out using the Alamar Blue assay for T. congolense (72 h in vitro assay) and modified slightly for T. vivax (48 h ex vivo assay). Parasite starting concentrations of 2 × 105 (T. c.) and 4 × 105 (T. v.) were calculated respectively, using a cell analyzer system or haemo-cytometer, followed by quantification at 536 nm excitation and 588 nm emission wavelengths using a flow cytometer reader. Test compounds were prepared as 10 mg/mL DMSO stocks for each assay run. Compounds were assayed in at least three separate, independent test runs and an 11-point dilution curve was used to determine the IC50 values. Data points were averaged to generate sigmoidal dose-response curves and IC50 values were determined using Softmax Pro 5.2 software.
- 7.Methods for testing compound efficacy in mouse models: In vivo mouse efficacy studies were performed at the Swiss TPH, using established mouse models of infection for T. c. and T. v. NMRI female mice were independently infected either with 1 × 105T. c. parasites/mouse (STIB736/IL1180 strain) or 1 × 104T. v. parasites/mouse (STIB719/ILRAD560 strain) using an intraperitoneal route. Parasitemia was allowed to develop over 7 days (T. c.) or 3 days (T. v.), respectively, before treatment was administered. Compounds were administered via intraperitoneal injection in 10% DMSO/water. Four mice were used per treatment group. Mice were monitored for the presence of trypanosomes, via tail blood examination microscopically with twice-per-week parasitemia checks for 60 days post treatment. Parasitemia was graded on a scale of 0–3, with 0 indicating no trypanosomes seen in 20 fields of view, 1 indicating the presence of 1–5 trypanosomes per field, 2 indicating the presence of 6–20 trypanosomes per field and 3 indicating greater than 20 trypanosomes per field. Mice scoring 2 or 3 were immediately euthanized. After 60 days, aparasitemic mice were considered cured. Untreated control mice survive on average for 10 and 6 days post infection for T. c. and T. v., respectively. All in vivo mouse experiments were conducted in accordance with the strict guidelines set out by the Swiss Federal Veterinary Office, under the ethical approval of license number #2813.
- 8.Method for testing compound efficacy in cattle: The cattle studies were conducted in accordance with the method described for cattle by Eisler et al. (2001) in studies conducted in fly-proof facilities and using T. congolense and T. vivax isolates that had previously been confirmed resistant in cattle to diminazine (7 mg/kg live weight) and/or isometamidium (1 mg/kg live weight). Cattle studies included negative (saline) controls, all assessments were made for 100 days post treatment (unless animals relapsed sooner) and were conducted by staff blinded (masked) to allocation of animals to treatment groups and in accordance with the principles of veterinary good clinical practice (VICH, 2000).