

Abstract
Activation of CXC motif chemokine receptor 4 (CXCR4) has been reported to result in lung protective effects in various experimental models. The effects of pharmacological CXCR4 modulation on the development of acute respiratory distress syndrome (ARDS) after lung injury, however, are unknown. Thus, we studied whether blockade and activation of CXCR4 influences development of ARDS in a unilateral lung ischemia-reperfusion injury rat model. Anesthetized, mechanically ventilated animals underwent right lung ischemia (series 1: 30 min; series 2: 60 min) followed by reperfusion for 300 min. In series 1, animals were treated with vehicle or 0.7 μmol/kg of AMD3100 (CXCR4 antagonist) and in series 2 with vehicle, 0.7 or 3.5 μmol/kg ubiquitin (non-cognate CXCR4 agonist) within 5 min of reperfusion. AMD3100 significantly reduced PaO2/FiO2 ratios, converted mild ARDS with vehicle treatment into moderate ARDS (PaO2/FiO2 ratio<200) and increased histological lung injury. Ubiquitin dose-dependently increased PaO2/FiO2 ratios, converted moderate-to-severe into mild-to-moderate ARDS and reduced protein content of bronchoalveolar lavage fluid (BALF). Measurements of cytokine levels (TNFα, IL-6, IL-10) in lung homogenates and BALF showed that AMD3100 reduced IL-10 levels in homogenates from post-ischemic lungs, whereas ubiquitin dose-dependently increased IL-10 levels in BALF from post-ischemic lungs. Our findings establish a cause-effect relationship for the effects of pharmacological CXCR4 modulation on the development of ARDS after lung ischemia-reperfusion injury. These data further suggest CXCR4 as a new drug target to reduce the incidence and attenuate the severity of ARDS after lung injury.
Keywords: AMD3100, ARDS, drug target, lung injury, ubiquitin
Introduction
Despite recent advances in critical care medicine, acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) remain major contributors to morbidity and mortality in critically ill patients (1–7). The American European Consensus Conference definition of ALI/ARDS from 1994 has recently been revised in the 2012 Berlin definition of ARDS, which now defines ALI as mild ARDS (8). Although the etiology of ARDS is multifactorial, it is generally accepted that mild ARDS and its progression into moderate and severe ARDS is caused by local and systemic inflammation in response to the insult, which leads to increased pulmonary capillary permeability, third spacing of fluids into the lung and formation of lung edema, the hallmark of ARDS (9, 10).
While several independent risk factors for development of ARDS have been identified, such as pneumonia, non-pulmonary-sepsis or severe injuries, the molecular events leading to ARDS remain poorly understood (4, 9, 11, 12). Thus, therapeutic options are currently limited to lung-protective ventilation strategies (5, 6, 13). Pharmacological interventions that may attenuate progression of mild ARDS to moderate or severe ARDS are currently not available, but urgently needed.
The G protein-coupled receptor C-X-C motif chemokine receptor 4 (CXCR4) is abundantly expressed in tissues, including lung epithelial and vascular endothelial cells (14, 15). CXCR4 is essential during embryonic development, has pleiotropic roles in the immune system, during tissue repair (16–19) and gained particular attention as a drug target through its role in cancer metastases and HIV infection (20–22). More recently, it has been reported that administration of the cognate CXCR4 agonist CXCL12 (stromal cell-derived factor-1α) and of the non-cognate CXCR4 agonist ubiquitin result in lung protective effects in various experimental models (23–28). While these data suggest CXCR4 as a potential therapeutic target to attenuate lung injury, the effects of pharmacological activation and blockade of CXCR4 on the development of ARDS after lung injury have not been systematically studied. Such data, however, are essential prerequisites to establish CXCR4 as a possible new drug target. Thus, in the present study we tested how blockade of endogenous CXCR4 with the selective CXCR4 antagonist AMD3100 and activation of CXCR4 with the non-cognate CXCR4 agonist ubiquitin modulate development and severity of ARDS in a unilateral lung warm ischemia-reperfusion injury rat model.
Results
Effects of AMD3100 after lung ischemia-reperfusion injury
In the first series of experiments, we tested the effects of the selective CXCR4 antagonist AMD3100 on the development of ARDS after unilateral lung ischemia-reperfusion injury. There were no significant differences for any physiological parameter between animals treated with vehicle and AMD3100 at baseline. Mean arterial blood pressures (MAP, Fig. 1A), blood lactate concentrations (Fig. 1B), creatinine concentrations (Fig. 1B) and hematocrit values (Fig. 1C) were comparable between vehicle- and AMD3100-treated animals throughout the observation period. As compared with vehicle-treated animals, leukocyte counts in AMD3100-treated animals were significantly increased at the end of the experiment (Fig. 1D). While peak inspiratory pressures required to maintain minute volumes were indistinguishable between the groups (Fig. 1E/F), there were significant differences in PaO2/FiO2 (P/F)-ratios between animals with vehicle and AMD3100 treatment (Fig. 1G). In vehicle-treated animals P/F-ratios decreased from 546 ± 18 mmHg at baseline to 299 ± 12 mmHg at t = 300 min. With AMD3100-treatment, P/F-ratios were significantly lower at t = 60 min and remained significantly reduced until the end of the experiment, as compared with vehicle-treated animals. With AMD3100 treatment, animals fulfilled criteria for mild ARDS (P/F ratio < 300 mmHg) at t = 60 min and criteria for moderate ARDS (P/F ration < 200 mmHg) at t = 240 min.
Figure 1. AMD3100 treatment promotes development of ARDS after lung ischemia-reperfusion injury.

Animals underwent lung ischemia for 30 min, followed by reperfusion until t = 300 min. Arrows indicate the time point of drug injection. Open symbols: vehicle treatment (n = 5). Grey symbols: AMD3100 (0.7 μmol/kg) treatment (n = 5). Data are mean ± SEM. A. Mean arterial blood pressure (MAP, mmHg). B. Blood lactate (mmol/L, squares) and creatinine (mg/dL, circles) concentrations. C. Hematocrit values (%). D. White blood cell count (WBC, × 109 cells/L). E. Peak inspiratory pressure (PIP, cm H2O). F. Partial pressure of carbon dioxide in arterial blood (PaCO2, mmHg). G. PaO2/FiO2 ratio. *: p<0.05 vs. vehicle (2-way ANOVA with Sidak’s multiple comparisons test).
Fig. 2A shows representative images from H&E stained lung slices of the post-ischemic and contralateral lungs from animals with vehicle and AMD3100 treatment and Fig. 2B the corresponding LIS for all animals. While there were no statistically significant differences between LIS of the contralateral lungs between the groups, LIS of the post-ischemic lungs were significantly higher with AMD3100 treatment (LIS, median (min/max): 7 (4/10)), as compared with vehicle treatment (LIS, median (min/max): 3 (2/6)).
Figure 2. AMD3100 treatment increases histological lung injury in the post-ischemic lung.

A. Representative images from H&E stained lung sections from animals after vehicle (top) and AMD3100 (bottom) treatment, as in Fig. 1. B. Lung injury scores (LIS) from animals in Fig. 1. Boxes extend from the 25th to 75th percentile, the horizontal line shows the median. Error bars show the range of data (min/max). LIS were analyzed with the Mann-Whitney U test. The level of statistical significance is indicated.
There were no significant differences in W/D ratios between the post-ischemic and contra-lateral lung and between animals treated with vehicle or AMD3100 (Fig. 3A). Furthermore, myeloperoxidase activities, malondialdehyde/4-HAE concentrations and TNFα concentrations in lung homogenates were indistinguishable in the post-ischemic and contra-lateral lungs from animals treated with vehicle or AMD3100 (Fig. 3B–D). While IL-6 concentrations were significantly increased in homogenates from the post-ischemic lungs, as compared with homogenates from the contralateral lungs (IL-6, pg/mg (median (interquartile range)): vehicle: - contra-lateral 6 (3/19), -post-ischemic 57 (36/80), p=0.008; AMD3100: - contra-lateral 12 (10/17), - post-ischemic 41 (24/78), p=0.015), there were no differences between animals after vehicle or AMD3100 treatment (Fig. 3E). IL-10 concentrations, however, were significantly lower in homogenates from post-ischemic lungs after AMD3100 treatment, as compared to homogenates from post-ischemic lungs after vehicle treatment (Fig. 3F; IL-10 pg/mg (median (interquartile range): vehicle - 19 (11/27), AMD3100 – 7 (6/10), p = 0.031).
Figure 3. Effects of AMD3100 treatment after lung ischemia-reperfusion injury on lung wet to dry weight ratios and surrogate markers of tissue injury and inflammation.

Data are mean ± SEM. Statistically significant differences between groups are labeled. A. Wet weight to dry weight (W/D) ratios. B. malondialdehyde (MDA, μmol/L/mg). C. Myeloperoxidase (MPO, relative fluorescence units (RFU)/μg). D.–F. TNF-α (pg/mg, D.), IL-6 (pg/mg, E. ) and IL-10 (IL-10, ng/mg F.) concentrations in lung homogenates. Groups were compared with unpaired Student’s t-test. The level of statistical significance is indicated.
Effects of ubiquitin after lung ischemia-reperfusion injury
In the second series of experiments, we tested the effects of the non-cognate CXCR4 agonist ubiquitin. To be able to assess whether activation of CXCR4 with ubiquitin could attenuate ARDS development after lung ischemia-reperfusion injury, we increased the ischemic time period from 30 min to 60 min. There were no significant differences in any physiological parameter between the vehicle-treated and the ubiquitin-treated groups at baseline. Similar to series 1, MAP (Fig. 4A), lactate concentrations, creatinine concentrations (Fig. 4B) and hematocrit values (Fig. 4C) were indistinguishable between the groups throughout the experiment. Furthermore, there were no significant differences in leukocyte counts between groups (Fig. 4D). Peak inspiratory pressures to maintain minute ventilation during the experiment were comparable with vehicle- and ubiquitin-treatment (Fig. 4E/F).
Figure 4. Ubiquitin treatment delays and attenuates development of ARDS after lung ischemia-reperfusion injury.

Animals underwent lung ischemia for 60 min, followed by reperfusion until t = 300 min. Arrows indicate the time point of drug injection. Open symbols: vehicle treatment (n = 6). Grey symbols: Ubiquitin (0.7 μmol/kg) treatment (n = 6). Black symbols: Ubiquitin (3.5 μmol/kg) treatment (n = 5). Data are mean ± SEM. A. Mean arterial blood pressure (MAP, mmHg). B. Blood lactate (mmol/L, squares) and creatinine (mg/dL, circles) concentrations. C. Hematocrit values (%). D. White blood cell count (WBC, × 109 cells/L). E. Peak inspiratory pressure (PIP, cm H2O). F. Partial pressure of carbon dioxide in arterial blood (PaCO2, mmHg). G. PaO2/FiO2 ratio. *: p<0.05 vs. vehicle (2-way ANOVA with Sidak’s multiple comparisons test).
The P:F ratios of the animals are shown in Fig. 4G. With vehicle treatment, P:F ratios decreased continuously to 111 ± 10 mmHg at t = 300 min. Vehicle-treated animals fulfilled criteria for mild ARDS (P:F ratio < 300 mmHg) at t=90 min and criteria for moderate ARDS (P:F ratio < 200 mmHg) at t = 180 min. With 0.7 μmol/kg ubiquitin, P:F ratios were significantly higher until t = 120 min (t = 90 min: 378 ± 20 mmHg, t = 120 min: 240 ± 27), as compared with vehicle-treated animals. With this dose of ubiquitin, animals fulfilled criteria for mild ARDS at t = 180 min and criteria for moderate ARDS only at the end of the observation period (t = 300 min). With 3.5 μmol/kg ubiquitin, P:F ratios were significantly higher throughout the entire reperfusion period, as compared with vehicle-treated animals. These animals fulfilled criteria for mild ARDS at t = 180 min and did not progress to moderate ARDS during the observation period.
Histomorphological analyses did not show obvious differences between the experimental groups (Fig. 5A). LIS of the contralateral and post-ischemic lungs were comparable between the groups (Fig. 5B). While W/D ratios of the lungs were also comparable between groups (Fig. 6A), measurements of protein content in BAL fluid revealed significant differences between vehicle- and ubiquitin-treated animals (Fig. 6B/C). With both doses of ubiquitin, protein concentrations in BAL fluids from the contralateral and post-ischemic lungs were significantly lower than in corresponding BAL fluids from vehicle-treated animals. Measurements of IL-10, TNFα and IL-6 concentrations per mg of protein in BAL fluid from contralateral lungs and of TNFα and IL-6 concentrations per mg of protein in in BAL fluid from post-ischemic lungs did not show significant differences among the groups (Fig. 6D–F). IL-10 concentrations per mg of protein in BAL fluids from the post-ischemic lungs increased dose-dependently with ubiquitin treatment (Fig. 6D). There were no significant differences in IL-10, TNFα and IL-6 concentrations in lung homogenates between the groups (Fig. 6G–I).
Figure 5. Lung histology after ischemia reperfusion injury and ubiquitin treatment.
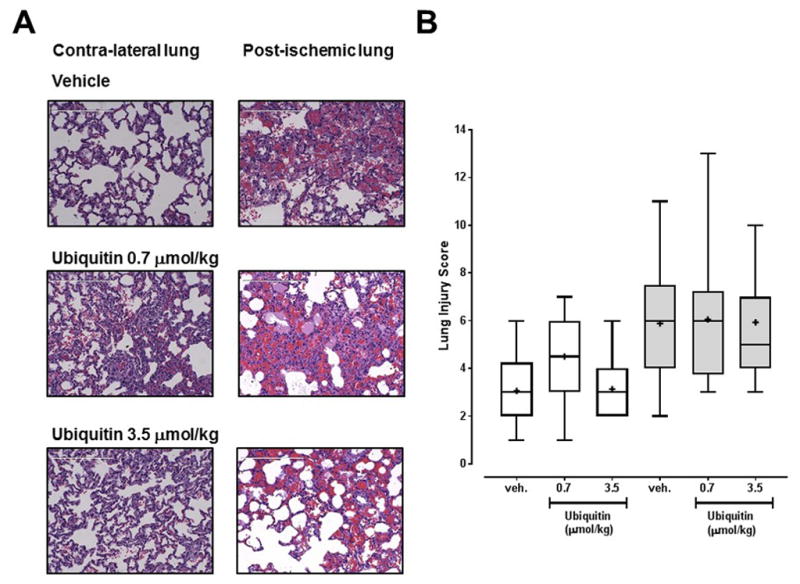
A. Representative images from H&E stained lung sections from animals after vehicle (top), 0.7 μmol/kg ubiquitin (center) and 3.5 μmol/kg ubiquitin (bottom) treatment, as in Fig. 1. B. Lung injury scores (LIS) from animals in Fig. 1. Boxes extend from the 25th to 75th percentile, the horizontal line shows the median. Error bars show the range of data (min/max).
Figure 6. Effects of ubiquitin treatment after lung ischemia-reperfusion injury on lung wet to dry weight ratios and surrogate markers of tissue injury and inflammation.
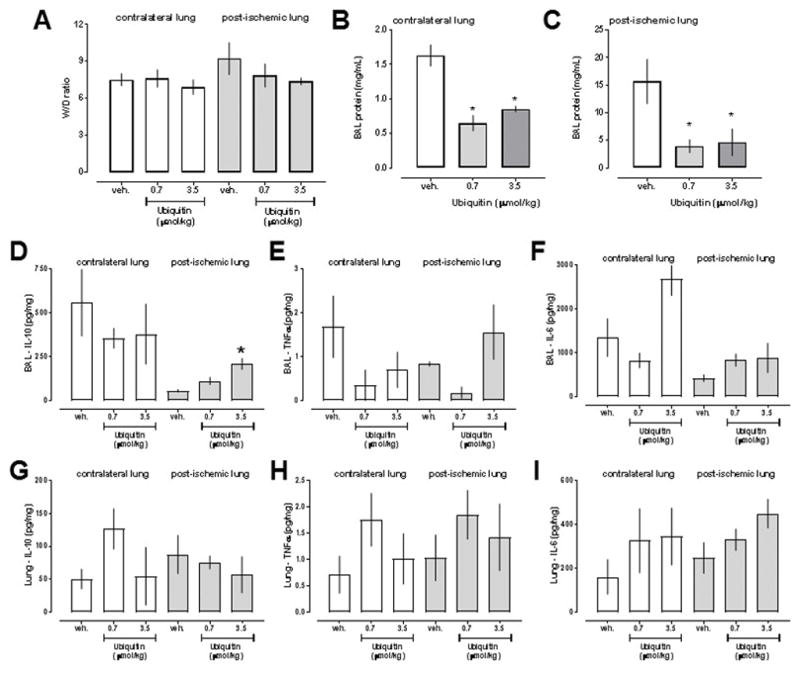
Data are mean ± SEM. A. Wet weight to dry weight (W/D) ratios. B. Protein concentrations (mg/mL) in bronchoalvelar lavage (BAL) fluid from contralateral lungs. C. Protein concentrations (mg/mL) in bronchoalvelar lavage (BAL) fluid from post-ischemic lungs. D.–F. IL-10 (pg/mg,D.), TNFα (pg/mg, E.) and IL-6 (pg/mg, F.) levels in BAL fluid. G.–H. IL-10 (pg/mg,D.), TNFα (pg/mg, E.) and IL-6 (pg/mg, F.) levels in lung homogenates. *: p<0.05 vs. vehicle treatment (1-way ANOVA with Sidak’s multiple comparisons test).
Discussion
In the present study, we tested how pharmacological modulation of CXCR4 influences the development of ARDS after unilateral lung ischemia-reperfusion injury. Our findings suggest that blockade of endogenous CXCR4 after lung ischemia-reperfusion injury promotes development of ARDS, whereas activation of CXCR4 delays development and attenuates severity of ARDS.
While there is no single animal model that is able to resemble all aspects of the human pathophysiology, lung-ischemia reperfusion injury is a well-accepted model that reflects key features of human ARDS (29, 30). The current Berlin definition of ARDS grades the severity of ARDS based on the P/F ratio under PEEP or continuous positive airway pressure of equal or greater than 5 cmH2O (mild – 200 < P/F < 300 mmHg, moderate – 100 < P/F < 200 mmHg, severe – P/F < 100 mmHg) (8). Thus, we ventilated animals with a PEEP of 5 cmH2O during the reperfusion period to be able to monitor the temporal pattern of ARDS development in our model. In series 1, control animals barely reached the criteria for mild ARDS at the end of the observation period. In contrast, AMD3100 treatment shortly after reperfusion accelerated development of ARDS and increased the severity of the reperfusion injury to moderate ARDS. In combination with the observation of increased histological lung injury with AMD3100 treatment, these findings imply that endogenous CXCR4 mediates lung protective effects in the pathophysiology of ARDS. Furthermore, our finding that AMD3100 treatment promotes ARDS development is in agreement with previous reports that CXCR4 blockade increases mortality and tissue injury in models of endotoxemia and polymicrobial sepsis, and that administration of AMD3100 impairs hemodynamic stability in models of traumatic-hemorrhagic shock (31–36). Collectively, these data indicate that endogenous CXCR4 plays important roles in infectious and non-infectious inflammation, and provides tissue/organ protection, including lung protection. The observation that leukocyte counts at the end of the observation period were significantly higher with AMD3100 treatment, as compared with vehicle-treatment, is in line with its known pharmacological properties (32, 37).
In contrast to AMD3100 treatment, ubiquitin treatment dose-dependently increased P/F ratios and converted moderate-to-severe ARDS into mild-to-moderate ARDS. Although we were unable to differentiate the degree of histological lung injury between vehicle- and ubiquitin-treated animals, measurements of BAL fluid protein content showed that protein leakage into the alveolar space was significantly reduced with ubiquitin treatment. As this effect was detectable in BAL fluid from the post-ischemic lung and from the contralateral lung, these findings suggest that CXCR4 activation stabilizes permeability of the alveolar-capillary membrane, through which it reduces direct and remote lung injury, respectively.
Despite significant changes in P/F ratios between vehicle- and AMD3100- or ubiquitin-treated animals, lung W/D ratios were indistinguishable at the end of the observation period. We have shown previously that ubiquitin-treatment significantly reduced W/D ratios of the uninjured lung in a polytrauma (bilateral femur fracture plus unilateral lung contusion) and fluid resuscitation model 7 hours post injury and W/D ratios of the post-ischemic lung after 90 min of ischemia (23, 27). Thus, it appears likely that the combination of a shorter ischemic insult and a shorter observation period in the present study did not provide sufficient time for the development of lung edema that can be differentiated by W/D measurements. This assumption is further supported by the observation that W/D ratios of post-ischemic and contralateral lungs were not significantly different. In addition, the finding that ubiquitin treatment significantly improved P/F ratios while lung histology was not obviously altered indicates that functional and histo-morphological consequences of lung ischemia-reperfusion are not directly correlated with each other.
Protective and therapeutically relevant effects of natural and synthetic CXCR4 agonists have been reported in multiple previous pre-clinical studies, including animal models of endotoxemia, sepsis, ischemia-reperfusion injury, trauma and hemorrhagic shock (23–27, 32–34, 38–44). The findings of the present study confirm that CXCR4 agonists have therapeutic potential, demonstrate that administration of the non-cognate CXCR4 agonist ubiquitin delays and attenuates development of ARDS after lung ischemia-reperfusion injury and provide initial information on its dose-effect profile.
While our observations on the effects of pharmacological blockade and activation of CXCR4 are consistent with the assumption that CXCR4 fulfills protective roles during inflammation and development of tissue injury, the molecular mechanisms underlying the observed effects in the present and in previous studies are not well understood. Although single time point measurements of surrogate markers of inflammation and tissue injury in BAL fluid or lung extracts are inherently difficult to interpret, such measurements may provide initial insights into possible mechanisms. Similar to our observations in previous animal models, we did not detect differences in lipid peroxidation products and MPO activities in lung extracts between groups (27). The finding that there were no differences in TNFα or IL-6 levels in lung homogenates and BAL fluids was not surprising due to their large inter-individual variability, their dynamic nature and the small sample size in each group. Thus, we cannot exclude that larger sample sizes or detailed time course measurements would demonstrate differences between vehicle-treated and AMD3100- or ubiquitin-treated animals. Nevertheless, our observations that AMD3100-treatment reduced IL-10 concentrations per mg of protein in homogenates from post-ischemic lung, whereas ubiquitin-treatment dose-dependently increased IL-10 concentrations per mg of protein in BAL fluid from post-ischemic lungs, could point towards anti-inflammatory and immune modulatory effects of CXCR4 as a contributing mechanism. This assumption is supported by previous observations suggesting enhanced pro-inflammatory responses after pharmacological CXCR4 blockade and enhanced anti-inflammatory responses following CXCR4 activation (23, 31, 32, 41). As CXCR4 has been reported to regulate vascular endothelial barrier function, direct effects of pharmacological CXCR4 modulators on pulmonary vascular permeability, independent of the cytokine network, may also account for the observed effects (45). To delineate the exact underlying mechanisms and target cell populations, detailed mechanistic studies will be required. Such studies, however, are beyond the scope of the present study.
In conclusion, the present study confirms previous reports suggesting therapeutic potential of CXCR4 agonists to attenuate lung injury in various animal models, establishes cause-effect relationships for the effects of pharmacological CXCR4 modulation on the development of ARDS after lung ischemia-reperfusion injury and provides initial pre-clinical evidence that CXCR4 agonists could be used to attenuate progression of mild ARDS to moderate and severe ARDS. Our findings point towards CXCR4 as a drug target to reduce the incidence and attenuate the severity of ARDS. Future studies delineating the molecular mechanisms underlying the protective effects of CXCR4 activation as well as pre-clinical efficacy studies in other ARDS models will be required to characterize and better understand the translational potential of this new therapeutic approach.
Methods
Lung ischemia-reperfusion injury model
All procedures were performed according to National Institutes of Health Guidelines for Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Loyola University Chicago and the Animal Care and Use Review Office of the U.S. Army Medical Research and Materiel Command. Male Sprague-Dawley rats (300 – 350 g) were purchased from Harlan (Indianapolis, IN, USA). Anesthetized (isoflurane inhalation) animals were oro-tracheally intubated with a 16-gauge EXEL disposable safelet angiocatheter (EXELINT International, Los Angeles, CA, USA) and mechanically ventilated with a SomnoSuite small animal anesthesia system (Kent Scientific Corporation, Torrington, CT, USA). Animals were ventilated with an initial positive end expiratory pressure (PEEP) of 2 mmHg, a fraction of inspired oxygen (FiO2) of 1.0 and anesthetized with isoflurane at 2.5%. Tidal volumes were titrated to maintain normal PaCO2 (35–45 mmHg). The femoral artery was then cannulated with a 24-gauge BD angiocath shielded IV catheter (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) to allow for monitoring of arterial blood pressure, blood withdrawal, and drug administration. Animals underwent a right lateral thoracotomy and a suture was placed around the hilum of the right lung. Five units of heparin in 500 μL normal saline (NS) solution was administered and allowed to circulate for 5 min. The suture was then tied around the hilum of the right lung, occluding the pulmonary artery, vein, and right main stem bronchus. After 30 min (AMD3100 treatment) or 60 min (ubiquitin treatment) of ischemia, the suture was removed and animals were ventilated with FiO2 1.0, PEEP 5 mmHg. A sigh breath was administered every 15 breaths for the first 10 min following reperfusion and then every 90 breaths until completion of the experiment. Hemodynamics were continuously monitored with the surgivet invasive blood pressure monitor (Med-Electronics, Beltsville, MD, USA) and blood pressures values were recorded every 5 min throughout the experiment. Arterial blood gases and routine laboratory parameters were determined in regular intervals throughout the experiment. At t=300 min, animals were euthanized (5% isoflurane, bilateral pneumothorax) and both lungs harvested for histology and measurements of inflammatory markers in whole lung homogenates. All experiments were performed randomized and blinded. Animals received vehicle (0.5 mL NS, n=5) or AMD3100 (700 nmol/kg, n=5) in 0.5 mL NS intra-arterially within 5 min of reperfusion after a 30 min period of ischemia. Animals received vehicle (0.5 mL NS, n=6), 0.7 μmol/kg ubiquitin (n=6) or 3.5 μmol/kg ubiquitin (n=5) intra-arterially within 5 min of reperfusion following a 60 min period of ischemia. Dosing of AMD3100 and ubiquitin was selected based on our previous studies in other animal models (27, 33, 34, 46).
Bronchoalveolar lavage
At the completion of the 60 min ischemia groups (t = 300 min) bronchoalveolar lavages (BAL) were performed from both lungs. After a midline sternotomy, the left main stem bronchus was occluded with a vascular clamp, 1.5 mL of normal saline were injected via the endotracheal tube and the BAL fluid was aspirated. The vascular clamp was then placed on the right main stem bronchus and the BAL procedure repeated. Recovery of BAL fluid was 70–83%. The BAL fluid was then centrifuged (0.5 × g for 5 min), the supernatant aspirated, snap frozen in liquid nitrogen and stored at −80°C until further analysis.
Arterial blood gases and routine laboratory parameters
Arterial blood gases, electrolytes, creatinine, lactate, hematocrit and hemoglobin were analyzed using the Element point of care veterinary blood gas, electrolyte and critical care analyzer (Cuattro Veterinary USA, Loveland, CO, USA). Complete blood counts were analyzed using the Hematrue hematology analyzer (Cuattro Veterinary USA, Loveland, CO, USA).
Histopathological analysis of lung tissue
For histomorphological examination, lung specimens were placed in formalin fixative solution and sent to AML Labs (Saint Augustine FL), where they were embedded in paraffin wax, sliced into 5 μm sections and stained with hematoxylin and eosin (H&E). From each lung specimen, 6 slides were prepared. The slides were examined under a light microscope by 3 investigators who were blinded as to the identity of the specimens. Histopathology was assessed using a previously described lung injury score (LIS), with slight modifications (47). In brief, each investigator rendered a score of 0 (no damage) to 4 (maximal damage) based on the following criteria: 1.) alveolar congestion, 2.) presence of hemorrhage, 3.) interstitial edema, and 4.) alveolar wall thickness.
Lung Wet Weight to Dry Weight Ratios
The ratio of the tissue wet weight to dry weight (W/D) was determined gravimetrically, as previously described (23, 48).
Lung Extract Preparation
Snap frozen tissues were homogenized in 1/10 phosphate buffered saline, pH 7.4 (1:5 weight/volume), centrifuged (20,000 × g, 4°C, 30 min) and supernatants (= extracts) aliquoted, as described (48, 49).
Enzyme Linked Immunosorbent Assays (ELISA)
Interleukin 6 (IL-6), IL-10 and tumor necrosis factor α (TNF-α) were measured in lung extracts and BAL samples with commercially available enzyme-linked immunosorbent assay kits (DuoSet ELISA, all from R&D Systems, Minneapolis, MN) according to the manufacturer’s protocols.
Myeloperoxidase (MPO)
MPO was measured in lung extracts using a commercially available assay kit (Invitrogen, Carlsbad CA) according to the manufacturer’s protocol.
Lipid peroxidation assay
Malondialdehyde (MDA) in combination with 4-hydroxyalkenals (4-HAE) were measured as an indicator of lipid peroxidation in the lung extracts using a commercially available colorimetric microplate assay (No. FR 22, Oxford Biomedical Research, Oxford, MI), as described (23).
Data Analyses and Statistics
Data are presented as mean ± standard error or median with interquartile range (25th/75th percentile). Parametric data were analyzed with unpaired Student’s t-test, 1-way ANOVA or 2-way ANOVA with Sidak’s multiple comparisons test, as appropriate. Non-parametric data were analyzed with the Mann-Whitney U test. Data analyses were calculated with the GraphPad Prism program (GraphPad Software). A two-tailed p<0.05 was considered significant.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (Awards R01GM107495 and T32GM008750) and by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award No. W81XWH-15-1-0262. The content is solely the responsibility of the authors.
Footnotes
The authors have no conflicts of interest.
References
- 1.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 2.Gattinoni L, Quintel M. Fifty Years of Research in ARDS Why Is Acute Respiratory Distress Syndrome So Important for Critical Care? Am J Respir Crit Care Med. 2016;194(9):1051–2. doi: 10.1164/rccm.201604-0662ED. [DOI] [PubMed] [Google Scholar]
- 3.Laffey JG, Kavanagh BP. Am J Respir Crit Care Med. 2017. Feb 1, Fifty Years of Research in ARDS. Insight into ARDS - From Models to Patients. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 4.Pham T, Rubenfeld GD. Fifty Years of Research in ARDS. The Epidemiology of ARDS: A Fiftieth Birthday Review. Am J Respir Crit Care Med. 2017;195(7):860–870. doi: 10.1164/rccm.201609-1773CP. [DOI] [PubMed] [Google Scholar]
- 5.Sahetya SK, Goligher EC, Brower RG. Fifty Years of Research in ARDS. Setting Positive End-expiratory Pressure in the Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med. 2017 Feb 1; doi: 10.1164/rccm.201610-2035CI. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yadav H, Thompson BT, Gajic O. Fifty Years of Research in ARDS. Is Acute Respiratory Distress Syndrome a Preventable Disease? Am J Respir Crit Care Med. 2017;195(6):725–36. doi: 10.1164/rccm.201609-1767CI. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida T, Fujino Y, Amato MB, Kavanagh BP. Fifty Years of Research in ARDS. Spontaneous Breathing During Mechanical Ventilation - Risks, Mechanisms & Management. Am J Respir Crit Care Med. 2017;195(8):985–92. doi: 10.1164/rccm.201604-0748CP. [DOI] [PubMed] [Google Scholar]
- 8.Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–33. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 9.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27(4):337–49. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 10.Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. Journal of aerosol medicine and pulmonary drug delivery. 2010;23(4):243–52. doi: 10.1089/jamp.2009.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller PR, Croce MA, Kilgo PD, Scott J, Fabian TC. Acute respiratory distress syndrome in blunt trauma: identification of independent risk factors. Am Surg. 2002;68(10):845–50. discussion 50–1. [PubMed] [Google Scholar]
- 12.Pierrakos C, Karanikolas M, Scolletta S, Karamouzos V, Velissaris D. Acute respiratory distress syndrome: pathophysiology and therapeutic options. J Clin Med Res. 2012;4(1):7–16. doi: 10.4021/jocmr761w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levitt JE, Matthay MA. Clinical review: Early treatment of acute lung injury - paradigm shift toward prevention and treatment prior to respiratory failure. Crit Care. 2012;16(3):223. doi: 10.1186/cc11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh MC, Makena PS, Gorantla V, Sinclair SE, Waters CM. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L846–56. doi: 10.1152/ajplung.00321.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135(3):561–71. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. 2007;1768(4):952–63. doi: 10.1016/j.bbamem.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karin N. The multiple faces of CXCL12 (SDF-1alpha) in the regulation of immunity during health and disease. J Leukoc Biol. 2010;88(3):463–73. doi: 10.1189/jlb.0909602. [DOI] [PubMed] [Google Scholar]
- 18.Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382(6592):635–8. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- 19.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393(6685):591–4. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 20.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16(11):2927–31. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 21.Veldkamp CT, Ziarek JJ, Peterson FC, Chen Y, Volkman BF. Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J Am Chem Soc. 2010;132(21):7242–3. doi: 10.1021/ja1002263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagasawa T, Tachibana K, Kishimoto T. A novel CXC chemokine PBSF/SDF-1 and its receptor CXCR4: their functions in development, hematopoiesis and HIV infection. Semin Immunol. 1998;10(3):179–85. doi: 10.1006/smim.1998.0128. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Covarrubias L, Manning EW, 3rd, Sorell LT, Pham SM, Majetschak M. Ubiquitin enhances the Th2 cytokine response and attenuates ischemia-reperfusion injury in the lung. Crit Care Med. 2008;36(3):979–82. doi: 10.1097/CCM.0B013E318164E417. [DOI] [PubMed] [Google Scholar]
- 24.Majetschak M, Cohn SM, Nelson JA, Burton EH, Obertacke U, Proctor KG. Effects of exogenous ubiquitin in lethal endotoxemia. Surgery. 2004;135(5):536–43. doi: 10.1016/j.surg.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Earle SA, Proctor KG, Patel MB, Majetschak M. Ubiquitin reduces fluid shifts after traumatic brain injury. Surgery. 2005;138(3):431–8. doi: 10.1016/j.surg.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Guo W, Li Z, Xie X, Tan T, Wang S, Xie N, et al. Stromal cell-derived factor-1alpha attenuates oleate-induced acute lung injury in rabbits. Biochem Biophys Res Commun. 2014;52(1):191–6. doi: 10.1016/j.bbrc.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 27.Baker TA, Romero J, Bach HHt, Strom JA, Gamelli RL, Majetschak M. Effects of exogenous ubiquitin in a polytrauma model with blunt chest trauma. Crit Care Med. 2012;40(8):2376–84. doi: 10.1097/CCM.0b013e3182514ed9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saini V, Marchese A, Majetschak M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J Biol Chem. 2010;285(20):15566–76. doi: 10.1074/jbc.M110.103408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44(5):725–38. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rocco PR, Nieman GF. ARDS: what experimental models have taught us. Intensive Care Med. 2016;42(5):806–10. doi: 10.1007/s00134-016-4268-9. [DOI] [PubMed] [Google Scholar]
- 31.Seemann S, Lupp A. Administration of AMD3100 in endotoxemia is associated with pro-inflammatory, pro-oxidative, and pro-apoptotic effects in vivo. J Biomed Sci. 2016;23(1):68. doi: 10.1186/s12929-016-0286-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bach HH, Saini V, Baker TA, Tripathi A, Gamelli RL, Majetschak M. Initial assessment of the role of CXC chemokine receptor 4 after polytrauma. Mol Med. 2012;18:1056–66. doi: 10.2119/molmed.2011.00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bach HHt, Wong YM, LaPorte HM, Gamelli RL, Majetschak M. Pharmacological targeting of chemokine (C-X-C motif) receptor 4 in porcine polytrauma and hemorrhage models. J Trauma Acute Care Surg. 2016;80(1):102–10. doi: 10.1097/TA.0000000000000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bach HH, Wong YM, Tripathi A, Nevins AM, Gamelli RL, Volkman BF, et al. Chemokine (C-X-C motif) receptor 4 and atypical chemokine receptor 3 regulate vascular alpha(1)-adrenergic receptor function. Mol Med. 2014;20:435–47. doi: 10.2119/molmed.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Novoa B, Bowman TV, Zon L, Figueras A. LPS response and tolerance in the zebrafish (Danio rerio) Fish Shellfish Immunol. 2009;26(2):326–31. doi: 10.1016/j.fsi.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delano MJ, Kelly-Scumpia KM, Thayer TC, Winfield RD, Scumpia PO, Cuenca AG, et al. Neutrophil mobilization from the bone marrow during polymicrobial sepsis is dependent on CXCL12 signaling. J Immunol. 2011;187(2):911–8. doi: 10.4049/jimmunol.1100588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liles WC, Broxmeyer HE, Rodger E, Wood B, Hubel K, Cooper S, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102(8):2728–30. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]
- 38.Majetschak M. Extracellular ubiquitin: immune modulator and endogenous opponent of damage-associated molecular pattern molecules. J Leukoc Biol. 2011;89(2):205–19. doi: 10.1189/jlb.0510316. [DOI] [PubMed] [Google Scholar]
- 39.Majetschak M, Cohn SM, Obertacke U, Proctor KG. Therapeutic potential of exogenous ubiquitin during resuscitation from severe trauma. J Trauma. 2004;56(5):991–9. doi: 10.1097/01.ta.0000127770.29009.5a. [DOI] [PubMed] [Google Scholar]
- 40.Ahn HC, Yoo KY, Hwang IK, Cho JH, Lee CH, Choi JH, et al. Ischemia-related changes in naive and mutant forms of ubiquitin and neuroprotective effects of ubiquitin in the hippocampus following experimental transient ischemic damage. Exp Neurol. 2009;220(1):120–32. doi: 10.1016/j.expneurol.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 41.Fan H, Wong D, Ashton SH, Borg KT, Halushka PV, Cook JA. Beneficial Effect of a CXCR4 Agonist in Murine Models of Systemic Inflammation. Inflammation. 2012;35(1):130–7. doi: 10.1007/s10753-011-9297-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guan S, Guo C, Zingarelli B, Wang L, Halushka PV, Cook JA, et al. Combined treatment with a CXCL12 analogue and antibiotics improves survival and neutrophil recruitment and function in murine sepsis. Immunology. 2014 Sep 8; doi: 10.1111/imm.12382. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seemann S, Lupp A. Administration of a CXCL12 Analog in Endotoxemia Is Associated with Anti-Inflammatory, Anti-Oxidative and Cytoprotective Effects In Vivo. PLoS One. 2015;10(9):e0138389. doi: 10.1371/journal.pone.0138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griebenow M, Casalis P, Woiciechowsky C, Majetschak M, Thomale UW. Ubiquitin reduces contusion volume after controlled cortical impact injury in rats. J Neurotrauma. 2007;24(9):1529–35. doi: 10.1089/neu.2007.0306. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi K, Sato K, Kida T, Omori K, Hori M, Ozaki H, et al. Stromal cell-derived factor-1alpha/C-X-C chemokine receptor type 4 axis promotes endothelial cell barrier integrity via phosphoinositide 3-kinase and Rac1 activation. Arterioscler Thromb Vasc Biol. 2014;34(8):1716–22. doi: 10.1161/ATVBAHA.114.303890. [DOI] [PubMed] [Google Scholar]
- 46.Tripathi A, Vana PG, Chavan TS, Brueggemann LI, Byron KL, Tarasova NI, et al. Heteromerization of chemokine (C-X-C motif) receptor 4 with alpha1A/B-adrenergic receptors controls alpha1-adrenergic receptor function. Proc Natl Acad Sci U S A. 2015;112(13):E1659–68. doi: 10.1073/pnas.1417564112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manning EW, 3rd, Patel MB, Garcia-Covarrubias L, Rahnemai-Azar AA, Pham SM, Majetschak M. Proteasome peptidase activities parallel histomorphological and functional consequences of ischemia-reperfusion injury in the lung. Exp Lung Res. 2009;35(4):284–95. doi: 10.1080/01902140802668823. [DOI] [PubMed] [Google Scholar]
- 48.Geng Q, Romero J, Saini V, Baker TA, Picken MM, Gamelli RL, et al. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem Biophys Res Commun. 2009;390(4):1136–41. doi: 10.1016/j.bbrc.2009.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Majetschak M, Patel MB, Sorell LT, Liotta C, Li S, Pham SM. Cardiac proteasome dysfunction during cold ischemic storage and reperfusion in a murine heart transplantation model. Biochem Biophys Res Commun. 2008;365(4):882–8. doi: 10.1016/j.bbrc.2007.11.092. [DOI] [PubMed] [Google Scholar]