

Abstract
Adult gliomas are aggressive brain tumours associated with low patient survival rates and limited life expectancy. The most important hallmark of this type of tumour is its invasive behaviour, characterized by a markedly phenotypic plasticity, infiltrative tumour morphologies and the ability of malignant progression from low- to high-grade tumour types. Indeed, the widespread infiltration of healthy brain tissue by glioma cells is largely responsible for poor prognosis and the difficulty of finding curative therapies. Meanwhile, mathematical models have been established to analyse potential mechanisms of glioma invasion. In this review, we start with a brief introduction to current biological knowledge about glioma invasion, and then critically review and highlight future challenges for mathematical models of glioma invasion.
Keywords: glioma invasion, cell phenotypic plasticity, malignant progression, infiltrative tumour morphology, mathematical modelling
1. Introduction
Gliomas are the most common primary tumours of the central nervous system (CNS) in adults. They comprise a clinically, histologically and genetically very heterogeneous brain tumour category. Until recently, glioma classification was largely based on microscopic examination of histological sections of tumour specimens by expert pathologists, distinguishing tumours according to their microscopic similarities to different types of glial cells into astrocytomas, oligodendroglioma or ependymomas, the main subtypes of gliomas [1]. The current 2016 World Health Organization (WHO) Classification of Tumours of the CNS for the first time integrates molecular biomarkers together with classic histological features to define distinct glioma entities [1]. This paradigm shift in glioma diagnostics reflects the major progress in our understanding of the molecular biology of brain tumours, which has tremendously increased in the past two decades due to genome-wide molecular-profiling studies that have clarified the genetic basis of gliomas. For example, diffuse gliomas with histologically oligodendroglial features are genetically characterized by mutations in the IDH gene and 1p/19q codeletion, while the diagnosis astrocytoma is usually accompanied by mutations in IDH in combination with ATRX and/or TP53 mutations but intact 1p and 19q. By contrast, classical primary glioblastomas usually do not show mutations in the IDH genes and are therefore referred to as glioblastoma IDH wild-type. In addition, the WHO classification distinguishes four prognostic grades that reflect the degree of malignancy: WHO grade I is assigned to the more circumscribed, benign tumours with low proliferative potential that mainly occur during childhood and in young adults; WHO grade II–IV tumours are diffusely infiltrative with increased cellular abnormalities (diffuse gliomas); WHO grade III tumours also show dedifferentiation and mitotic cell activity; and WHO grade IV tumours exhibit, in addition to the features present in the other grades, pathological proliferation of small vessels and/or necrosis. WHO grade II–IV tumours are characterized by extensive, diffuse infiltration of glioma cells into the host brain tissue, and are therefore referred to as diffuse gliomas. These aggressive brain tumours are typically associated with a poor prognosis, sharp deterioration in the patients' quality of life and markedly low survival rates. In particular, IDH wild-type glioblastoma, the most common (approx. 45% of all gliomas) and malignant primary brain tumour (WHO grade IV), has a 5-year survival of about 5% from the time of diagnosis [2,3]. Even for patients with diffuse low-grade IDH-mutant gliomas (WHO grade II), although survival can be more than 10 years, the prognosis is unfavourable, as these tumours eventually progress to a high-grade malignant lesion (WHO grade III or IV) [4]. Despite significant advances in surgical and medical imaging techniques, as well as in adjuvant radio-, chemo- and immunotherapy [5–10], the inherent tendency of glioma cells to widely disseminate within normal brain parenchyma severely limits treatment responses [11–13]. Therefore, a better understanding of the mechanisms that trigger and govern glioma invasion is of high clinical importance for the development of more effective and less toxic therapeutic strategies.
Although there is a considerable amount of information about the clinical and biological behaviour of gliomas, the high complexity of the invasion mechanisms remains a major challenge in clinical neuro-oncology. Histologically, glioma cells closely resemble glial progenitor cells, which have the ability to proliferate and differentiate into different glial cell types. These cells have a high migratory behaviour in the developing CNS [11,12,14]. This suggests that mechanisms contributing to migration of neuroepithelial cells during embryogenesis are also relevant for glioma invasion [14]. In addition, accumulating evidence indicates that spatial and temporal variations in signalling pathways lead to functional and phenotypic changes in glioma cells, which then affect interactions with neighbouring malignant and non-malignant cells along with other components of the surrounding brain tissue. The exact consequences of the dynamic interplay between heterogeneous cellular entities and their response to alterations in the extracellular microenvironment have not yet been elucidated. Moreover, it remains unclear why metastases outside the CNS are extremely rare in diffuse gliomas [15,16]. From a biological and medical perspective, it is difficult to investigate the connections between clinically observable glioma behaviour and the underlying molecular and cellular processes. The challenge is to integrate the theoretically and empirically acquired knowledge to better understand the mechanisms and factors that contribute to glioma invasion.
In this context, mathematical models provide useful tools towards identifying dependencies and targets of cancer cell migration and invasion. Mathematical models and computational approaches have become increasingly abundant in cancer research to study tumour dynamics and responses to treatment modalities such as chemo- and radiotherapy [17–20]. Mathematical modelling provides a useful theoretical framework to perform in silico experiments, as well as to evaluate assumptions and make predictions that can be experimentally tested [21–32]. In the last two decades, several mathematical models have been developed to investigate key mechanisms governing glioma growth and invasion [23,33–35].
Ten years ago, several of the current co-authors reviewed mathematical models of glioma development, growth and progression [34]. Since then, the field of glioma research has significantly grown. In this review we exclusively focus on mathematical models of glioma invasion. We first introduce current biological knowledge about glioma invasion. Then, we describe biological model systems, in particular, in vitro experiments and in vivo animal models for the analysis of glioma invasion, and medical imaging techniques. We then critically review mathematical models of glioma invasion, and highlight future challenges for mathematical and computational modellers in this research area.
2. Biology of glioma invasion
Infiltration of the brain parenchyma is a prominent feature of diffuse gliomas, making complete surgical resection almost impossible [36]. Diffuse gliomas invade extensively as single cells anywhere within the host brain tissue, with some preference to infiltrate along white matter tracts and the periphery of blood vessel walls [16]. The infiltration of the surrounding brain tissue is determined by complex interactions between glioma cells and the extracellular microenvironment [37]. Here, we review cell intrinsic mechanisms and extrinsic factors that sustain and foster glioma invasion.
2.1. Intrinsic mechanisms: phenotypic plasticity and genetic variability
2.1.1. Epithelial–mesenchymal transition and migration
Glioma cells have the ability to acquire a mesenchymal phenotype in response to microenvironmental cues and migrate through the extracellular matrix (ECM) exhibiting an elongated, often wedge-shaped phenotype [14,38,39]. Migration and invasion of glioma cells are related, multistep processes. Migration is defined as the movement of cells from one site to another, often in response to specific external signals such as chemical gradients or mechanical forces. Epithelial-to-mesenchymal transition (EMT) is an essential process in wound healing, embryonic development and tissue remodelling, consisting in the transdifferentiation of polarized epithelial cells into motile mesenchymal cells (originated from the mesodermal embryonic tissue which develops into connective and skeletal tissues). Accumulating evidence highlights the critical role of EMT during glioma progression and its association with increased glioma cell migration [40]. Individual glioma cells spread by active cell migration rather than by passive movement. Invasion encompasses glioma cell migration, but also involves degradation of the ECM [38]. It is a multifactorial process that consists of interactions between adjacent cancer cells with the ECM coupled with biochemical processes supportive of active cell migration. In general, glioma cell invasion involves four distinct steps [14,38,39]: (1) detachment of invading cells from the primary tumour mass, (2) adhesion to the ECM, (3) degradation of the ECM and (4) cell motility and contractility (active cell migration) (figure 1).
Figure 1.

Glioma cell migration. Schematic of the process of glioma cell invasion into host brain tissue. Invasion of glioma cells involves four distinct steps: (1) detachment of invading cells from the primary tumour mass, a process triggered by downregulation of cell–cell adhesion molecules and microenvironmental changes, (2) integrin-mediated adhesion to the extracellular matrix (ECM), (3) secretion of proteases, which locally degrade ECM components creating routes along which glioma cells invade the brain and (4) migration by extending a prominent leading cytoplasmic protrusion, followed by a burst of forward movement of the cell body. Figure adapted from [39].
At the subcellular level, secretion of proteases, cell adhesion molecules and related signals play an important role in glioma cell migration [37]. Detachment of glioma cells from the primary tumour mass involves several events, including destabilization and disorganization of cell–cell adhesion complexes (cadherin-mediated junctions), loss of expression of neural cell adhesion molecules and cleavage of CD44, a cell-surface protein which anchors the primary tumour mass to the ECM by the metalloproteinase ADAM [16,38]. Integrins are the most common molecules that allow glioma cells to adhere to the ECM, and matrix-metalloproteinases (MMPs) are the most common proteases that degrade the ECM creating migration routes. Several glioma-expressed molecular factors, such as focal adhesion kinase and urokinase-type plasminogen activator (an enzyme participating in ECM degradation), have been found to regulate their expression [38]. Glioma cells migrate similarly as non-transformed neural progenitor cells, with myosin II as the major source for cytoplasmic contractility [41]. Invading glioma cells alter their shape, extending a prominent leading cytoplasmic protrusion followed by a burst of forward movement of the cell body. The complex molecular mechanisms and changes in signalling pathways that occur during glioma invasion are still largely unknown. A major difficulty in understanding the oncogenomics of glioma cell invasion is to determine how and when genetic alterations and signalling cascades interact [42]. Only a few specific pathways have been consistently identified, and there exist multiple possible interactions along with additional unknown factors to be elucidated.
2.1.2. Migration–proliferation dichotomy
At the time of diagnosis, gliomas are already widely disseminated, as they typically grow and invade extensively before the patient experiences any symptoms. This hidden dissemination is a major reason that makes gliomas difficult to treat successfully and a cure almost impossible. Current treatment strategies mainly focus on the highly proliferative tumour mass, but local invasion eventually leads to recurrence of the disease. Enormous efforts have been devoted to identifying the main signalling events that regulate glioma cell motility and invasion. However, therapeutically targeting invasion dynamics is complicated, because it has been observed that migratory and proliferative behaviours of glioma cells are mutually exclusive processes and inversely correlated [11,43,44]. In particular, highly migratory cells have a lower proliferation rate compared to actively proliferating cells that move slowly. This either–or behaviour of proliferative and invasive glioma cells is supported by both in vitro and in vivo experiments [11] and is referred to as the migration–proliferation dichotomy (or ‘Go-or-Grow’ mechanism) [43,44].
The ‘Go-or-Grow’ behaviour has been linked to metabolic stress by several experimental findings. Godlewski et al. [45] identified a glioma-expressed microRNA (small non-coding RNAs that regulate gene expression) that regulates the balance between glioma cell proliferation and migration in response to changes in the available energy. Their experimental data revealed that, in addition to inhibiting glioma cell migration, the microRNA expression also promotes cell proliferation. This suggests that migratory and proliferative events share common signalling pathways, defining a unique intracellular mechanism that regulates both phenomena. More recently, Höring et al. [46] investigated the effects of carboxypeptidase E (CPE), a neuropeptide-processing enzyme, on glioma invasion by means of in vitro and in vivo studies. Their results indicate an oxygen- and nutrient-dependent anti-migratory, but pro-proliferative role of CPE in glioma invasion. Additionally, experiments with glioblastoma-derived neurospheres provided further insight into the ‘Go-or-Grow’ mechanism. EphB2, a receptor of the tyrosine kinase family was found to have both pro-migratory and anti-proliferative effects in vivo [47]. These novel findings suggest that glioma invasion could be attacked by targeting specific cellular and molecular mechanisms associated with the migration–proliferation dichotomy. However, further investigations are required to unravel the underlying signalling pathways regulating glioma cell migration and proliferation.
2.1.3. Cell metabolic plasticity
A common feature of cancer cells is their altered glucose metabolism [48,49]. Unlike non-neoplastic cells that rely on oxidative phosphorylation to generate the energy needed for cellular processes, cancer cells can shift their metabolism from respiration towards glycolysis producing lactic acid. Indeed, cancer cells tend to upregulate aerobic glycolysis even in the presence of sufficient oxygen, a phenomenon known as aerobic glycolysis or the Warburg effect, which is a characteristic metabolic hallmark of tumour development [50–52]. Although the Warburg effect has recently regained attention as a possible therapeutic target [53,54], its biological basis remains elusive. Compared to mitochondrial oxidative metabolism, aerobic glycolysis is an inefficient way to gain energy [55]. However, increased glycolysis creates a hostile acidic environment, in which cancer cells have an evolutionary advantage with respect to normal parenchyma [56]. There is accumulating evidence that acid-induced toxicity is an essential component required for tumour invasion, and therefore a hallmark of invasive cancers [56,57]. Glioma cells are specifically characterized by a high rate of glycolysis and lactate extrusion, with the ability to flourish in a relatively hypoxic environment [58]. In fact, many of the invasive features of gliomas may depend on distorted metabolic functions, which makes the study of metabolic alterations and their effects on invasion processes a growing field in cancer research with potential therapeutic benefits.
2.1.4. Intra- and inter-tumoural heterogeneity
Tumour heterogeneity contributes to disease progression and development of therapy resistance [59,60]. Extensive genetic and phenotypic variations exist among glioma cells within a single tumour (intra-tumoural heterogeneity) and between patients (inter-tumoural heterogeneity) due to extensive molecular diversity and microenvironmental heterogeneity [37,61–68]. It is commonly assumed that tumour heterogeneity arises either from a self-renewing cancer stem cell population or due to clonal competition for common resources driven by the acquisition and expansion of mutations in cancer cells [61,69–71]. Recent clinical and experimental findings have revealed extensive genetic variations in glioma cells due to intra-tumoural evolution [63,64,67,72]. Besides the large genetic heterogeneity, interactions between glioma cells and with the surrounding brain parenchyma lead to functional and phenotypic diversity. Evidence indicates that distinct clones within a tumour may harbor genetic and epigenetic alterations that promote cancer cell migration and invasion. Recently, experiments with mixtures of different cell types to mimic phenotypic heterogeneity have revealed that invasion is driven by the cooperation of multiple tumour-cell subpopulations [73,74]. Patterns of co-invasion were observed with inherently invasive cells acting as the leader and subpopulations of poorly invasive cells as followers. This ‘division of labour’ may facilitate not only tumour invasion, but also malignant progression. Although there is increasing appreciation that intra-tumoural heterogeneity is central to glioma behaviour, little is known about the temporal sequence of genetic and microenvironmental changes or how genomic instabilities and adaptation to microenvironmental conditions contribute to glioma invasiveness.
2.2. Cell-extrinsic factors
2.2.1. Guidance mechanisms
The particular structures of the brain such as blood vessels, white matter tracts and brain parenchyma, and specific tumour cell–ECM adherence mechanisms are crucial factors in glioma invasion [75,76]. At any stage of invasion, glioma cells are confronted with non-neoplastic brain tissue composed of multiple cell types and various ECM components. The ECM in the CNS is different from the ECM in other tissues in that it has low fibrous protein content and high carbohydrate concentrations [77]. In particular, the brain ECM is mainly produced by astrocytes and oligodendrocytes, comprises an estimated 20% of the brain volume in adults and consists primarily of hyaluronic acid, except around blood vessels and at the pial surface (the boundary between grey matter and cerebrospinal fluid) [76]. The invasion of glioma cells into the adjacent brain tissue is guided by a combination of multiple molecular and physical mechanisms along pre-existing tracks of least resistance. The major invasion routes are basement membranes and intercellular tracks provided by myelinated axons and astrocyte processes [76,78]. Glioma cells migrate along blood vessels by using the outward vessel–parenchyma interface and the lumen of the perivascular space [76]. More precisely, blood vessels guide invading glioma cells via laminin- and collagen-IV-mediated integrin engagement (ECM proteins mediated), whereas white matter tracks guide by cell–cell contacts and mechanisms regulating cell–ECM adhesion forces [76]. However, the specific guidance factors and related molecular mechanisms for most dissemination routes remain unclear.
2.2.2. Hypoxia-induced migration
Uncontrolled glioma cell proliferation leads to the development of hypoxic regions. This microregional change produces a local milieu that favours certain glioma cell behaviours such as invasion [60]. A commonly held view is that constitutive upregulation of glycolysis is likely to be an adaptation to the lack of oxygen [57]. According to the WHO classification, detection of vascular proliferation with highly pathological blood vessels and tumour necrosis is essential for the diagnosis of grade IV gliomas [1]. These tumour-induced features are often spatially and temporally related, with sites of pathological neovascularization indicative of the formation of hypoxic and necrotic regions. Quantitative immunohistochemical analysis revealed that while high-grade gliomas may locally show a strong angiogenic activity, many regions of both low- and high-grade gliomas display vascular densities in the range of normal cerebral grey or white matter, indicating limited angiogenesis [79–81]. This supports the observation that in high-grade gliomas different invasive and pro-angiogenic tumour cell phenotypes coexist [39]. Overexpression of pro-angiogenic factors by tumour cells results in local vascular overgrowth with defective blood vessels, which have significantly larger diameters and thicker basement membranes than those in normal brain tissue [82]. It has been observed in vitro that under oxygen-limiting conditions due to vascular abnormalities, glioma cells actively migrate away from hypoxic regions [83].
Different pathological and experimental observations suggest that vaso-occlusion could readily explain the rapid peripheral expansion and diffusely infiltrative growth behaviour of high-grade gliomas [84,85]. Occlusion of vasculature mainly occurs due to increased mechanical pressure by either tumour cells or by intravascular pro-thrombotic mechanisms [83,84,86]. Occluded or collapsed blood vessels induce perivascular tumour hypoxia and necrosis in glioblastoma, which typically form lines with perifocally increased cell density, termed pseudopalisades [83–85,87]. Pseudopalisades around necrotic foci, a common feature of high-grade gliomas, are severely hypoxic and linked to waves of glioma cells actively migrating away from such oxygen-deficient regions [83–85,87]. Experimental studies further suggest that tumour hypoxia results in increased glioma cell migration and invasion, and strongly correlates with tumour malignancy [39,88,89]. The exact pathophysiological factors and mechanisms underlying hypoxia-induced cell migration in gliomas are still not known, and further investigation is required to understand the complex molecular pathways involved in hypoxic responses and metabolic control by glioma cells.
2.2.3. Blood–brain barrier
An important structural component of the brain vasculature is the blood brain barrier (BBB), which is essential for supplying the brain tissue with oxygen and glucose, mediating efflux of waste products, and maintaining a precisely regulated microenvironment for reliable neuronal signalling [90]. The BBB is composed of tightly bound endothelial cells and perivascular astrocytes that restrict the exchange of molecules from the bloodstream much more than capillaries anywhere else in the body. High-grade gliomas have the ability to disrupt the integrity of the BBB, which is associated with increased tumour growth and diffuse invasion into the surrounding brain parenchyma [16,91–93]. Recent studies have shown that the BBB can be heterogeneously disrupted in high-grade gliomas, and the degree of BBB disruption is related to tumour malignancy [92]. However, the precise relationship between glioma-induced BBB dysregulation and cell invasion is still not clear [91]. On the other hand, experimental evidence shows that one of the major obstacles for standard anti-cancer drug delivery in the brain is the BBB, which limits the efficacy of chemotherapy [16,92]. In fact, the presence of an almost intact BBB is one of the main factors that makes the treatment of low-grade gliomas with chemotherapy challenging. Although the BBB may be disrupted at the core of high-grade gliomas, it can be relatively intact at the tumour periphery where invading glioma cells are located [92]. This permits infiltrative glioma cells to escape chemotherapy-induced death, which can result in tumour recurrence.
2.2.4. Immune system engagement
Tumours have long been recognized as wounds that do not heal [94]. Both carcinogenesis and wound healing involve cell proliferation, migration, invasion, angiogenesis, inflammation and astrocyte activation in response to injuries [76,95]. The microenvironment of high-grade gliomas resembles in many ways a chronic wound [95]. In particular, the extensive cell migration and invasion observed in gliomas also accompanies reactive gliosis, a non-specific reaction where astrocytes are activated in response to injuries to the CNS as part of a healing process. The border of gliomas exhibits an increased number of reactive astrocytes, which together with glioma cells secrete various pro-migratory signalling molecules [76]. Recently, a protein (connective tissue growth factor) produced at high levels by reactive astrocytes has been identified to stimulate migration of glioma cells [95]. Indeed, the secreted factor modulates nearly all aspects of the signalling mechanisms that regulate cell invasion, such as modification of growth factor activities, ECM composition, integrin (transmembrane cell-matrix adhesion receptors) and E-cadherin expression (a transmembrane protein that mediates cell–cell adhesion). Thus, the identification of processes involved in glial reactivation may contribute to a better understanding of glioma cell invasion with potential therapeutic implications.
Accumulated histopathological data have established that tissue-resident microglia and macrophages are the predominant infiltrating immune cells in gliomas, accounting for up to 30–50% of the total tumour mass [96]. Monocytes circulate in the bloodstream and are continuously recruited into tumours in response to several tumour-derived chemoattractants. Once inside the tumour, monocytes rapidly differentiate into tumour-associated macrophages (TAMs), and then accumulate in hypoxic/necrotic areas [96]. TAMs produce several factors that not only stimulate the survival and proliferation of tumour cells, but also suppress antitumour immunity [96,97]. There is growing evidence that TAMs are also involved in regulating glioma cell migration and invasion [96,98–100]. Moreover, TAMs contribute to malignant progression of gliomas through secretion of various chemical factors that affect angiogenesis and ECM remodelling, which promotes glioma invasion [96,97,101]. More precisely, TAMs can display two major phenotypes, the classically (M1) and alternatively (M2) activated, which can be viewed as two extreme phenotypes [102,103]. Unlike TAMs displaying an M1-like phenotype with pro-inflammatory and anti-tumour functions, M2 macrophages are immunosuppressive, produce pro-angiogenic factors, contribute to the ECM-remodelling, and thus create a favourable microenvironment for glioma growth and invasion. Although there is growing acceptance that the M1 and M2 states are not dichotomous but rather represent extremes of a continuum of phenotypes, a majority of macrophages in gliomas are macrophages with M2-like phenotype which exhibit pro-invasive properties, particularly in late stages of disease progression [104,105]. Recent evidence indicates that the colony stimulating factor-1 (CSF-1) secreted by glioma cells induces TAMs to promote invasion [104]. It has been observed that CSF-1 levels are elevated in high-grade gliomas, which is associated with the expression of M2 macrophage markers. In addition, the cytokine interleukin-10 (IL-10) is also commonly associated with macrophages of the M2 phenotype and has been found to stimulate glioma cell invasion [104]. Together, chemokines, cytokines and growth factors secreted by TAMs activate different signalling pathways that can switch glioma cells towards more aggressive behaviour.
3. Biological model systems
As it is currently impossible to monitor the entire process of glioma invasion in the human brain, different biological model systems have been introduced. In vitro cell cultures provide the opportunity to generate insights into molecular and cellular pathways related to glioma invasion under controlled conditions. Various in vivo models have been introduced which allow a more realistic representation and monitoring of the complex glioma dynamics.
3.1. In vitro experiments
Experimental protocols in vitro are available to independently observe and control variables of interest at various scales, from single-cell movement to multicellular clonal growth and cancer cell population dynamics [106]. Invasion studies have been performed in both two-dimensional (2D) tumour monolayers and three-dimensional (3D) multicellular spheroids, combining glioma cell migration and proliferation assays, and considering different ECM compositions and substrate rigidities [107]. However, there are significant differences between tumour cell migration on a 2D surface and in a 3D matrix [108], and even more between in vitro and in vivo experiments [106]. Under controlled experimental conditions, glioma cells may exhibit a more fibroblastic shape with a broad lamellipodium and an undistorted nucleus, where the forward movement is continuous and unimpeded [41]. By contrast, glioma cells migrating through the complex heterogeneous brain parenchyma are highly polarized and elongated [38,39]. The mechanical constraints due to small intercellular spaces impede the forward movement of nucleus and cell body until necessary contraction forces are provided [41]. This ability of glioma cells to adapt their motility to the particular microenvironment reinforces the need for assays faithfully representing the environmental conditions of the brain to improve our understanding of invasion mechanisms. Using so-called normal brain cell aggregates derived from fetal rat brains or utilization of rodent or other mammalian brain slices might present a promising compromise to perform 3D in vitro studies that can be more standardized and avoid ethical issues attached to animal studies [109–112].
3.2. In vivo models
Animal models are essential for investigating the interactions between glioma cells and the complex brain microenvironment. The traditional use of animal models involves injecting established tumour cell lines either intravenously or at the target site and then waiting for a tumour to develop before testing a therapy or a given hypothesis. While the complex microenvironments in the animal models mimic the human brain structure much better than in vitro studies, very few established cell lines are able to represent the histopathological characteristics of human gliomas, particularly their invasive nature. Recently, methods have been developed to harvest cells from patients for use in animal models. These are referred to as patient-derived tumour aggregates and are better able to recapitulate patterns of tumour cell invasion observed in patients [113]. Moreover, xenograft models have been extensively employed to assess the efficacy of therapies targeting glioma cells, such as the intra-tumoural administration of IL13-PE toxin (a fusion protein composed of IL-13 and a mutated form of Pseudomonas exotoxin), to monitor glioblastoma angiogenesis and to evaluate anti-angiogenic therapeutical approaches [114]. The main drawback of xenografts is that the histology and genetics of the original tumour are frequently not maintained. Moreover, high-resolution imaging of single glioma cell invasion in xenografts remains laborious, costly and time-consuming.
Tumours can also be induced in animals using retroviruses. Two notable cases use cells infected with retroviruses engineered to overexpress either constitutively activated epidermal growth factor receptor (EGFR) [115] or platelet-derived growth factor (PDGF) [116]. Both retroviruses are able to initiate tumour growth with human glioblastoma characteristics when injected into a rat or mouse brain. In addition to providing a good model system for drug therapy, they also shed light on the tumour initiating process. Another development in animal models is the creation of transgenic mice, where genetic engineering techniques are used to create mice with ubiquitous mutations that are predisposed to developing gliomas [117].
Many in vivo experimental techniques exist, the most common involves sacrificing the animal to allow for staining of the brain tissue, but this only provides a temporal snapshot of tumour composition. Another technique is referred to as ex vivo imaging where brain tissue is harvested and thin slices are placed on nutrient-filled media. This allows to microscopically observe cell movement for a time period up to 24 h [116]. A further option that allows longer observation is the use of bioluminescence (or biofluorescence) at the cell population scale, or multiphoton microscopy at the single-cell level. While any of the aforementioned animal models provides experimental conditions closer to the human brain, the ability of fully monitoring the cancer dynamics is still challenging.
Additionally, the fruit fly Drosophila melanogaster has been considered as an alternative in vivo glioma model because many molecular pathways and cellular functions are fundamentally conserved [118,119]. Advantages include easy handling, a fully sequenced genome, a wide range of available genetic techniques and a well-known anatomical situation [118,120]. Model organisms such as D. melanogaster have been useful not only to visualize tumour cell migration and to investigate the effects of induced metastasis, but also to identify glioma signalling cascades via advanced genetic techniques. However, the D. melanogaster model also has some limitations. Invasion studies in D. melanogaster lack an accurate representation of the human brain parenchyma, including the absence of blood vessels and an adaptive immune system.
4. Medical imaging and histopathology
Conventional computer tomography scanning reveals morphological information of gliomas, but tumours at early stages or small metastatic lesions are often not detected. This technique has been gradually replaced by magnetic resonance imaging (MRI), which is significantly more sensitive to the presence of tumours and has become the standard imaging modality in the evaluation of brain tumours [121,122]. MRI creates non-invasive images by exploiting the magnetic properties of water molecules in the body. By changing the intensity, timing and duration of radiofrequency pulses and directional gradients, myriad non-invasive images with varying contrasts and information can be created [123]. The more common T1- and T2-weighted MRI sequences are mainly used for displaying general anatomic features of gliomas; however, the two types of sequences emphasize features differently (e.g. cerebral spinal fluid (CSF) is bright on T2-weighted images and dark on T1-weighted images). The T1-weighted image can be used to highlight the leaky blood vessels characteristic of glioblastoma by acquiring the image after administration of gadolinium, a contrast agent that appears bright on T1-weighted images. Gadolinium seeps out from the leaky vasculature haphazardly created by the tumour, highlighting what is believed to be the most active/aggressive tumour region on T1-weighted gadolinium-enhanced MRI (T1Gd). The T1Gd and T2 sequences are most commonly used for delineating tumour regions, but it is well known that neither of these sequences is able to provide a precise visualization of tumour abnormality due to the extensive invasion of the tumour cells [124]. In fact, in one study of high-grade gliomas, it was demonstrated that human gliomas grow invasively, with tumour cells demonstrable over 4 cm from the gross tumour [125].
While T1- and T2-weighted MRI sequences remain the dominant images clinically used, there are many other advanced MRI techniques that are being explored such as vascular perfusion imaging, diffusion-weighted imaging (DWI) and proton magnetic resonance spectroscopy (MRS) [126]. Vascular perfusion imaging highlights regions of high vascularity and has been shown to be useful in predicting which patients are responding to anti-angiogenic therapies [127,128]. DWI can be used for various purposes, but for glioblastoma it is most commonly used to quantify the apparent diffusion coefficient, which is thought to be inversely correlated with cell density [129,130]. MRS uses the proton signals to determine relative concentrations of target metabolites rather than an anatomical image. In various studies, MRS has been shown capable of identifying regions of tissue enriched with stem-like cell-enriched foci [131], detecting tumours with mutations in the isocitrate dehydrogenase (IDH) genes [132,133], and assessing response to various therapies such as radiation and PI3K/mTOR inhibitors [134,135]. Further, the use of hyperpolarized (HP) contrast agents can significantly increase the sensitivity of MRS by enhancing the signal-to-noise ratio [136,137]. More generally, MRS has opened up the promising fields of metabolomics (the study of metabolomic signatures in tumours) [138] and radiomics (the use of imaging technology to extrapolate molecular tumour data) [139]. These studies are encouraging, but one must note that the resolution of a standard MRS is much lower than on a standard MRI, voxel sizes being approximately 10 × 10 × 10 mm3 versus 1 × 1 × 1 mm3. Thus, while there are many promising advanced magnetic resonance methods for determining various tumour characteristics, differentiating between normal and pathological tissue on the basis of MRI findings alone is complicated.
Functional imaging techniques like positron emission tomography (PET) scans are useful in providing deeper insights into the biology of gliomas [140,141]. PET imaging is increasingly implemented in neuro-oncology, because it offers unique data about metabolic and physiologic processes such as glucose metabolism, protein/DNA synthesis, cell proliferation and apoptosis, as well as angiogenesis and hypoxia that can reflect the changes in a neoplasm. Assessment of the status of these processes has been shown helpful in delineation of tumour margins, and correlates with clinical metrics such as tumour grade, patient survival and therapy response [140,142,143]. Clinically, this type of information is viewed as complementary to the anatomical MRI, as PET scans generally lack anatomic context, and have a relatively low spatial resolution [144].
Diagnosis and classification of glioma is based on histopathology, referring to the microscopic examination of tissue sections by an experienced pathologist, e.g. neuropathologist. Additionally, histopathology analysis of tissue samples has some potential to provide further information at the single-cell level that is extremely important to quantify and classify intra-tumoural heterogeneity. Histological and immunohistochemical analyses are routinely performed by pathologists for confirmation of the presence or absence of disease, determining glioma grading and assessing disease progression [1,145,146]. However, tissue biopsies can be seen as ‘snapshot’-like frozen scenes of dynamic biological processes providing data severely limited in both space and time. Thus, despite the constant expansion of medical imaging technology, the identification of tumours at an early stage, assessment of intra-tumoural heterogeneity, reduction of radiation exposure and improvement of resolution are challenging for anatomic, functional and metabolic imaging alike.
5. Mathematical modelling of glioma cell migration and invasion
A wide variety of mathematical models have been proposed to investigate the mechanisms of glioma invasion, which is characterized by invasive cell migration, phenotypic plasticity, infiltrative tumour morphologies and the ability of malignant progression. Model types include discrete and continuous approaches such as cellular automaton (CA), lattice-gas cellular automaton (LGCA), cellular Potts model (CPM), partial differential equations (PDE), agent-based models (ABM) and evolutionary game theory models (EGT) [17–20,35,147–153]. We subsequently review mathematical models for invasive cell migration, invasive effects of phenotypic plasticity, infiltrative tumour morphologies and malignant progression. Table 1 provides an overview of the reviewed models.
Table 1.
Overview of reviewed mathematical modelling approaches. Cellular automaton (CA), lattice-gas cellular automaton (LGCA), cellular Potts model (CPM), partial differential equations (PDE), agent-based model (ABM) and evolutionary game theory (EGT).
| mathematical modelling approach |
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Khain et al. [27] | Aubert et al. [154] | Khain et al. [155] | Szabó et al. [156] | Kim et al. [157] | Kim [158] | Tektonidis et al. [25] | Hatzikirou et al. [31] | Böttger et al. [28] | Pham et al. [29] | Gerlee et al. [30] | Alfonso et al. [32] | Sander et al. [159] | Martínez-González et al. [26] | Frieboes et al. [21] | Zhang et al. [160] | Frieboes et al. [161] | Jiao et al. [24] | Basanta et al. [22] | Swanson et al. [23] | ||
| (2D) CA &(1D) PDE | (2D) CA | (2D) CA | (2D) CPM | (2D) PDE | (2D) HYBRID | (2D) LGCA | (2D) LGCA | (2D) LGCA | (2D) PDE | (2D) CA & (1D) PDE | (1D) PDE | (2D) HYBRID | (1D) PDE | (2D) PDE | (3D) ABM | (3D) HYBRID | (2D) STATISTICAL | EGT | (1D) PDE | ||
| main focus of mathematical models | invasive cell migration | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||||||
| phenotypic plasticity | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| tumour morphology | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||||||
| malignant progression | ✓ | ✓ | ✓ | ||||||||||||||||||
| main biological assumptions | cell–cell interactions | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||
| cell–ECM interactions | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||||||||
| hypoxia-induced migration | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||||||
| cell density-dependent invasiveness | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||||||||
| changes in metabolism | ✓ | ✓ | ✓ | ||||||||||||||||||
| clonal heterogeneity | ✓ | ||||||||||||||||||||
| angiogenesis | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
5.1. Invasive cell migration
Based mainly on in vitro experiments, several mathematical models have been developed to investigate the effects of cell–cell adhesion strength under distinct microenvironmental conditions on the invasive behaviour of glioma cells. Khain et al. [27] investigated, both theoretically and experimentally, the effect of cell–cell adhesion on glioma front propagation and the structure of the invasive interface. Migration characteristics of U87-MG cells were measured using a scratch wound-healing assay, and invasion front patterns were simulated by means of both a 2D discrete lattice-based stochastic model and a continuum approach. Simulations of the continuum model show that a small effective cell–cell adhesion change does not influence the propagating front speed, and clusters of glioma cells were not formed (figure 2a). By contrast, the microscopic discrete model shows that a cell–cell adhesion strength exceeding a critical threshold leads to cluster formation in the invasive zone resulting in fingering-like front propagation patterns (figure 2b). The experimental time was characterized as a transient regime, which coincides with the period required for a relatively sharp initial cell density profile to develop into a propagating front. Although simulations successfully reproduced the maximal distance of migration of glioma cells on a plastic substrate, this model underestimated the migration of the main mass of tumour cells, suggesting the presence of chemotactic stimuli.
Figure 2.

Invasive cell migration. Front interface for small (a) and high (b) effective cell–cell adhesion values. Shown are the simulations of a discrete stochastic lattice model; every black dot represents a cell, and every white dot corresponds to an empty site. The system size is 400 × 400 (in units of cell diameter). Figure reproduced with permission from [27].
Glioma cell migration on a substrate of collagen was investigated by Aubert et al. [162]. The proposed 2D CA model indicates that chemotaxis or cell–cell communication through gap junctions (specialized intercellular channels that permit direct cell–cell transfer of ions and molecules) is necessary to reproduce experimental density profiles of glioma cell distributions in tumour spheroids. In a follow-up study, migration patterns of glioma cells in the presence of astrocytes were studied by Aubert et al. [154]. An extended version of the model proposed in [162] was used to analyse the opposite effects of homotypic (between glioma cells) and heterotypic (between glioma cells and surrounding astrocytes) gap junction communication on the invasiveness of gliomas. Lowering glioma cell–cell interactions on a passive substrate of collagen was predicted to enhance the migratory potential, whereas the simultaneous inhibition of glioma cell–cell and glioma cell–normal astrocyte gap junction communication leads to reduced cell migration. This suggests that the interactions between glioma cells and astrocytes play an important role in glioma invasion, due to the effect of heterotypic gap junction inhibition which dominates that of homotypic inhibition. Model simulations are consistent with experimental data of glioma migration patterns in both homotypic and heterotypic situations by only introducing attractive contact between migrating tumour cells. However, the experiments considered in [154,162] only involved relatively small spheroids without central hypoxia or necrosis. Thus, to study migration patters of glioma cells in large spheroids, Aubert et al. [163] assumed a chemorepellent factor produced by cells submitted to stressful conditions in the hypoxic/necrotic microregions. A good agreement between model simulations and experiments allows to conclude the existence of repellent toxic cues that would promote glioma cell detachment and migration, although further work is needed to identify and characterize these chemorepulsive factors.
The role of hypoxia in the regulation of glioma cell–cell adhesion and cell migration was investigated by Khain et al. [155]. A 2D discrete stochastic model was proposed to describe in vitro experiments of U87 glioma cell migration (i) away from tumour spheroids placed on a substrate and (ii) in typical scratch wound-healing assays. The distance migrated (i.e. invasive radius) by both normoxic and hypoxic glioma cells was measured. In the spheroid experiments, the overall migration rate of tumour cells under either normoxic or hypoxic conditions was similar. However, hypoxic glioma cells in the wound-healing assays migrated less than cells under normoxic conditions. This model suggests that lack of oxygen not only suppresses cell motility, but also substantially reduces the strength of cell–cell adhesion. Although oxygen deficiency resulted in reduced cell motility, the decreased cell–cell adhesion allows hypoxic cells to detach from the tumour mass, leading to enhanced glioma invasion. These model predictions are consistent with experimental data showing that hypoxia induces downregulation of E-cadherin, a transmembrane protein that positively regulates extension and strengthening of adhesive contacts, and promotes glioma cell invasion.
To explore the influence of the ECM on glioma cell migration, Szabó et al. [156] considered not only cell–cell adhesion but also cell–ECM interactions. Cell aggregates were prepared from confluent cultures of two different glioblastoma cell lines (GBM1 and U87) placed within a 3D ECM of collagen I gel. The aim was to characterize the collective, large-scale invasion of glioma cells from tumour spheroids into the surrounding ECM. The interplay between haptotaxis, matrix degradation and active cell movement was investigated by means of a 2D CPM. Simulation results suggest that the complex interplay between space-constrained active cell motion, cell–ECM adhesion and degradation of the ECM determines the patterns of migration and increases persistence during cell invasion. In particular, haptotaxis and ECM degradation were observed to destabilize multicellular sprouts as each cell tries to invade the surrounding matrix. By contrast, when both haptotaxis and polarized motion are present, even a homogeneous glioma cell population may be organized into multicellular sprouts within an inhomogeneous ECM environment.
Kim et al. [157] proposed a model that takes into account cell–cell adhesion, haptotaxis and chemotactic effects of a glucose gradient on glioma cell migration in vitro. Model simulations reveal that depending on the chemotactic and haptotactic sensitivities, and the strength of cell–cell adhesion, different migration patterns of glioma cells arise: dispersion, branching, island formation and a mixture of these patterns. In particular, this model reproduced the patterns observed in various invasion assays of in vitro spheroids generated from glioma U87 and mutant U87ΔEGFR cell lines. Moreover, changes of adhesion, haptotactic and chemotactic parameters result in a gradual shift from branching to dispersion as experimentally observed. The main finding was that the front of cell migration can be slowed down by both increasing cell–cell adhesion and blocking the ECM degradation effects of MMPs, a family of enzymes that are capable of breaking down all kinds of proteins, such as collagen, normally found in spaces between tissues (ECM proteins).
In a follow-up study, Kim [158] developed a hybrid multi-scale model in which glioma cell migration and proliferation are regulated by intracellular mechanisms in response to glucose availability and physical constraints in the microenvironment. In particular, a core control system of a single microRNA (miR-451) that regulates AMPK (the 5′-adenosine monophosphate activated protein kinase) signalling [45,164] linked to extracellular glucose was simulated. Recent experimental evidence suggests that, in a glucose-rich environment, miR-451 is up-regulated by tumour cells, leading to AMPK pathway inhibition and in turn cell proliferation. Conversely, sustained AMPK activation under low glucose conditions results in suppression of miR-451, which induces phenotypic changes of glioma cells from a proliferative to a migratory phenotype [165]. Based on the assumption that glucose levels may induce phenotypic changes in glioma cells, fluctuations of the glucose concentration were predicted to trigger migration–proliferation cycles, which in turn increased glioma cell invasion and resulted in faster tumour growth.
5.2. Phenotypic plasticity
The ability of glioma cells to switch their phenotype in response to local cell density and changes in the microenvironment allows adaptation and is believed to have important implications for glioma invasion. In particular, glioma cells can change from a proliferative to a migratory phenotype depending on microenvironmental conditions [43,44]. It is thus crucial to investigate the factors and conditions that drive the transition from the proliferative to the motile phenotype. Several mathematical models have been introduced to analyse implications of this ‘Go-or-Grow’ dichotomy on glioma invasion. Tektonidis et al. [25] proposed a lattice-LGCA model to explain the spatio-temporal evolution of U87 tumour spheroids in vitro reported in [166]. It turns out that the ‘Go-or-Grow’ mechanism combined with self-repulsion and a density-dependent phenotypic switch is required to quantitatively reproduce the experimental observations (figure 3).
Figure 3.

Phenotypic plasticity. Simulations of a lattice-gas cellular automaton (LGCA) model taking into account the ‘Go-or-Grow’ mechanism, cell–cell repulsion and a cell density-dependent switch between a migrating and a proliferative phenotype. Simulation results (in blue) against experimental data (in red). (a) Temporal evolution of the core radius (dotted line) and invasive radius (solid line). (b) Temporal evolution of the ratio of core to invasive radius. (c) Velocity field diagram indicating the direction of glioma cell movement. (d) Visualization of the spatial tumour structure, where the grey level refers to the number of cells per node. Figure reproduced with permission from [25].
Moreover, Hatzikirou et al. [31] demonstrated with a simple population model that a solely mutation-driven phenotypic change is insufficient to explain the fast recurrence of gliomas after extensive resection. However, incorporating the ‘Go-or-Grow’ mechanism in a corresponding LGCA model allows to explain fast glioma recurrence. In addition, a discrete stochastic model was used to characterize and quantify the invasive glioma front width and speed [28]. Model simulations reveal that the ‘Go-or-Grow’ mechanism results in a nonlinear temporal evolution of the invasion front speed and a time-divergent infiltration zone. These findings related to the implications of the ‘Go-or-Grow’ plasticity highlight the important role of intra-tumoural phenotypic heterogeneity in determining the invasive behaviour of gliomas. The ‘Go-or-Grow’ plasticity has been further investigated by Pham et al. [29] using a reaction–diffusion model based on a cell density-dependent phenotypic switch. The model analysis demonstrates that the ‘Go-or-Grow’ mechanism is able to produce complex invasion front patterns characterized by spatio-temporal phenotypic heterogeneity.
Recently, Scribner et al. [167] criticized the ‘Go-or-Grow’ hypothesis. Their study suggests that the ‘Go-or-Grow’ mechanism is not necessary to reproduce key features of glioblastoma growth and anti-angiogenic treatment responses such as the formation of the multilayer structure (i.e. necrosis, oedema and contrast-enhancing ring of viable, highly cellular and angiogenic tumour tissue), patterns of progression associated with bevacizumab treatment, and the survival times of glioblastoma patients treated or untreated with bevacizumab. Although the proposed mathematical model replicates clinical and therapeutic features of glioblastoma, the simulation results do not necessarily disprove the ‘Go-or-Grow’ mechanism of glioma cells. Previous ‘Go-or-Grow’ models consider local cell density-dependent diffusive migration of glioma cells [28,31,32,168]. The ‘Go-or-Grow’ model of Scribner et al. [167] assumes a different cell migration mechanism, namely a directed movement of glioma cells (by advection) towards high densities of normal brain cells. In the simulations of this model, advective cell migration dominates the invasive tumour dynamics over the ‘Go-or-Grow’-driven diffusion process. Thus, the ‘Go-or-Grow’ mechanism in the Scribner et al. [167] model is different from the previous ‘Go-or-Grow’ models.
Gerlee et al. [30] investigated the impact of apoptosis on the travelling wave speed and found that tumour progression depends on the apoptosis rate in a discontinuous fashion. In particular, there exists a critical apoptosis rate above which tumours cannot grow (i.e. no travelling wave solutions exist). This model also supported previous findings in suggesting that glioma growth and invasion depend on a non-trivial trade-off between the proliferation and migration switching rates. More recently, the overall effects of vaso-occlusive events and vascular normalization on the glioma front speed and infiltration width were investigated by Alfonso et al. [32]. The existence of critical parameter regimes that distinguish between different glioma invasive patterns with respect to variations in the vaso-occlusion rate was predicted as an emergent consequence of the ‘Go-or-Grow’ plasticity. In particular, a ratio between cell diffusion and proliferation rates separated glioma behaviours into regimes where the front speed and infiltration width are differently affected. While for tumours belonging to one regime, vascular modulations reduce the front speed and increase the infiltration width, for those in the other regime the invasion speed increases and infiltration width decreases. This led to the conclusion that depending on the intrinsic tumour cell features such as glioma cell migration and proliferation, vaso-occlusive events may have opposing effects on glioma invasion. Recently, a new mathematical model based on the migration/proliferation dichotomy reported the existence of a threshold in the glioma cell density that separates tumour growth and extinction dynamics, a phenomenon called the Allee effect in ecology [168].
5.3. Infiltrative tumour morphology
Both, micro- and macroscopically, morphological features are thought to be related to invasiveness and aggressiveness of gliomas. Experimental observations revealed that a morphological instability is correlated with tumours with a higher invasive potential [169]. Several single- and multi-scale mathematical models have been developed to explore the effects of microenvironmental tumour morphology regulators, such as glucose, oxygen and growth factor concentrations, on glioma invasion. Sander et al. [159] formulated a hybrid discrete-continuum model to investigate invasive morphologies of gliomas. Human U87MG EGFR multicellular tumour spheroids in 3D ECM gel were used to calibrate model parameters, as well as to compare the in vitro and model-driven invasive patterns. Invasive glioma growth was assumed to be governed by chemotaxis (heterotype) caused by the gradient of nutrient concentration and homotypic attraction by paracrine production of soluble factors which attracts cells. In the model, the existence of an intrinsic diffusive instability resulted in branch formation of glioma cells within the invasive zone, which is consistent with the in vitro assays. Accordingly, a chemotactic signal that directs cell migration was suggested to be essential for invasive tumour growth as experimentally observed. The model predicted that both strong heterotype chemotaxis and homotypic attraction are required for the formation of branching patterns within invasive tumour regions. While a very strong combination of both chemotaxis and homotypic chemoattraction resulted in well-defined chains of cells that branch and extend around the tumour core, disc-like invasive patterns were obtained by strong heterotypic chemotaxis either with or without homotypic attraction.
Pseudopalisades of cells around hypoxic and necrotic areas have been recognized as a morphologic characteristic that predicts invasive behaviour of gliomas [83–85,87]. Martínez-González et al. [26] investigated the formation of pseudopalisades in glioblastoma by means of a continuous mathematical model that considers the dynamic interplay between normoxic and hypoxic cells competing for space and resources. Based on experimental findings [83], the hypoxic phenotype was assumed to be more migratory and less proliferative than the normoxic phenotype. This model predicted the formation of travelling waves of glioma cells actively migrating away from central hypoxic regions and reproduced the observed histologic patterns of pseudopalisades. A dependency between the characteristic distance from occluded to functional blood vessels, the timescale of palisade formation, lifetime and persistence was also observed. Moreover, simulations showed that under conditions of sufficient oxygen availability palisading waves lead to a faster invasion than pure random motion. This result led to the hypothesis that vaso-occlusion might accelerate glioma invasion, suggesting tumour vasculature normalization as a strategy to reduce cell migration.
Frieboes et al. [21] explored the effects of competition between spatially heterogeneous cell proliferation on tumour morphology. A 2D mathematical model of glioma development describing the diffusion and uptake of nutrients, oxygen and growth factors by tumour cells was developed. This model closely resembles glioma cell spatial arrangements and tumour morphologies from in vitro spheroid cultures of ACBT (human glioblastoma) and BT4C (rat gliomas) cell lines. Simulations led to the hypothesis that unstable invasive glioma morphologies are mainly driven by spatial nutrient variations. In particular, heterogeneous oxygen and nutrient supplies were suggested to drive tumour growth and invasiveness through a diffusional instability mechanism. The formation of subspheroids that break off from the parent tumour was observed to depend more on nutrient gradients than on individual cells with high proliferative potential. This indicates that invasion is a recursive process mediated by the formation of buds at the tumour viable rim and separation from the parent spheroid. Moreover, a spatially uniform distribution of nutrients and homogeneous microenvironmental conditions were demonstrated to be crucial for compact non-invasive tumour morphologies.
To investigate the impact of clonal heterogeneity on glioma growth and invasion dynamics, Zhang et al. [160] developed a 3D multiscale agent-based model that incorporates a simplified tumour progression pathway which leads to the emergence of distinct cancer cell clones. In the model, a heterogeneous glioma consists of five clonal cell populations that emerge sequentially through mutations. The cell phenotype was determined by an EGFR gene–protein interaction network and changes in microenvironmental conditions (i.e. glucose, oxygen and growth factor concentrations). This model predicted that cellular heterogeneity may influence glioma growth patterns and morphology. Higher EGFR expression was demonstrated to promote faster expansion of tumour areas that harbour more aggressive glioma cells due to a temporary competitive advantage. The resulting expansion of glioma cells into areas of nutrient abundance leads to overall growth asymmetries consistent with experimental and clinical observations.
A 3D multiscale approach of functional collective cell migration units (FCCMUs) was proposed by Frieboes et al. [161] to study large-scale morphologies and spatial cell arrangements during glioma growth. The FCCMU model was based on mass and momentum conservation laws and consisted of biologically founded reaction–diffusion equations that govern tumour cell density, nutrient concentration, evolving neo-vasculature, ECM and matrix-degrading enzymes. Angiogenesis involved proliferation and migration of endothelial cells governed by chemotaxis and haptotaxis in response to angiogenic regulators. Simulations reproduced morphologic features such as regions of viable cells, hypoxia and necrosis, as well as the abnormal vasculature of gliomas as observed in vivo (figure 4). Moreover, glioma morphology and invasiveness were predicted to be strongly influenced by diffusion gradients of oxygen and nutrients. Volume loss in the necrotic core moderated the intra-tumoural pressure and led to a morphological instability, resulting in the formation of buds, which, in turn, increased the pressure in the tumour rim. Thus, this model ultimately suggested a cyclic pattern of rapid proliferation and pressure build-up followed by necrosis-induced pressure relief.
Figure 4.

Infiltrative tumour morphology. Multiscale 3D computer model predicts gross morphologic features of a growing glioblastoma. (a) Viable (VT) and necrotic (NT) tissue regions and vasculature (MV, mature blood-conducting vessels in red; NV, new non-conducting vessels in blue) are shown. The time sequence (from left to right, over a period of 3 months) reveals that the morphology is affected by successive cycles of neovascularization, vasculature maturation and vessel cooption (VC). Scale bar, 250 μm. (b) Histology-like section of the last frame of the simulation in (a) (obtained by slicing horizontally through the simulated tumour) reveals viable tumour regions (white) surrounding necrotic tissue (dark). (c) Another view from simulation shown in (a), right. Figure reproduced with permission from [161].
More recently, Jiao et al. [24] developed a statistical approach to identify spatial correlations of normal and abnormal cell nuclei on histological images. Statistics calculated from pair correlation functions, a structure factor and various nearest-neighbour functions were used to compare cell nuclei distributions. This model predicted that tumour cells have a stronger effective cell–cell repulsion and are packed more densely than normal cells. In addition, abnormal cell nuclei were observed to be spatially correlated on intermediate and large length scales, implying that gliomas are organized in a coordinated way rather than randomly. These long-range correlations suggested that some form of glioma cell–cell communication exists, which might lead to collective cell behaviour responsible for invasion. In fact, the existence of non-trivial spatial correlations between glioma cells supports the long-standing view that cancer is a complex and well-organized adaptive system.
5.4. Malignant progression
Glioblastoma can be divided into two different groups: primary and secondary tumours [4]. Primary glioblastomas occur de novo without any clinical or histologic evidence of progression from a less malignant precursor, and represent over 90% of diagnosed gliomas. Secondary glioblastoma develop by malignant transformation of a pre-existent low-grade diffuse astrocytoma (WHO grade II) or anaplastic astrocytoma (WHO grade III), usually in younger patients. An interesting question that arises is how long low-grade gliomas take to reach a clinically detectable size. The answer can be useful to develop screening strategies for early detection. Gerin et al. [170] developed a model to provide estimates on the time of tumour genesis from MRI data of patients diagnosed with low-grade gliomas. Based on the patient's age at time of first MRI examination, two types of tumours were identified: very slowly growing tumours that appear during adolescence and slowly growing tumours that appear later, during early adulthood. Further, the model results suggest that low-grade gliomas become visible on MRI without clinical revelation at a mean patient age of 25–30 years. Although this model provided a description of genesis and growth of low-grade gliomas, predictions on the appearance of malignancy that commonly occur in gliomas were not possible.
An understanding of the factors and mechanisms involved in the malignant progression of gliomas can improve the prognosis and long-term survival rate of patients. This is particularly important given the fact that low-grade gliomas generally progress to higher-grade lesions over time and they grow and invade more quickly [9]. The stepwise process of malignant progression is driven by a series of genetic alterations and microenvironmental changes that lead to phenotypic and molecular heterogeneity. Hatzikirou et al. [31] demonstrated that mutation-based phenotypic changes alone are unable to explain malignant progression of gliomas, and suggested that phenotypic transitions are strongly influenced by the tumour microenvironment. It is thus likely that essential phenotypes for tumour progression might emerge only with the prior presence of other phenotypes.
EGT models have proved successful to analyse various cell interactions by formulating appropriate games [171,172]. Basanta et al. [22] developed an EGT model to investigate the impact of interactions between different glioma cell phenotypes with increasing degree of malignancy on malignant progression. The three phenotypes in the game are autonomous growth (AG), anaerobic glycolysis (GLY) and invasive (INV). It is assumed that tumour cells are initially characterized by AG, and can switch to anaerobic glycolysis for energy production or become increasingly motile/invasive. The tumour dynamics are governed by the fitness costs of cell movement, less efficient glycolytic metabolism and acidification of the microenvironment (table 2). This model predicts that the emergence of the invasive phenotype solely depends on the cost of motility if only glioma cells with a non-glycolytic metabolism are present. On the contrary, the appearance of glycolytic glioma cells supports invasion even for high motility costs. In fact, the presence of glycolytic cells, which increase the acidity of the environment, indirectly reduces the costs of motility. Thus, the invasive phenotype is more likely to evolve after the appearance of glycolytic cells which would explain the ubiquitous presence of invasive growth in high-grade gliomas. This finding suggests that an increase in the fitness cost of switching to anaerobic glycolysis might reduce glioma invasiveness.
Table 2.
Pay-off table of an evolutionary game theory (EGT) approach that represents the change in fitness of a tumour cell with a given phenotype interacting with another tumour cell. The three phenotypes in the game are defined by autonomous growth (AG), invasion (INV) and glycolytic (GLY). The base pay-off in a given interaction is equal to 1 and the cost of moving to another site with respect to the base pay-off is c. The fitness cost of acidity is n, and k is the fitness cost of having a less efficient glycolytic metabolism. The table should be read following the columns, and thus the fitness change for an invasive cell interacting with an AG phenotype is 1 − c. Table reproduced from [22].
| AG | INV | GLY | |
|---|---|---|---|
| AG |  |
1 − c | 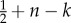 |
| INV | 1 | 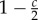 |
1 − k |
| GLY | 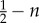 |
1 − c | 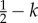 |
A more general proliferation-invasion-hypoxia-necrosis-angiogenesis (PIHNA) model was later proposed by Swanson et al. [23], with the goal of quantifying the role of angiogenesis in malignant progression of gliomas from low- to high-grade analogous to the WHO grading scheme [1]. This approach incorporated microenvironmental factors such as oxygen supply determined by the relative amount of vasculature supplying the tissue and angiogenic factors. Moreover, the proposed mathematical model includes normoxic, hypoxic and necrotic glioma cellular compartments, as well as endothelial cells that form blood vessels. Model simulations quantitatively describe the spectrum of in vivo dynamics of gliomas visualized with medical imaging. Figure 5 shows simulations varying the proliferation rate of tumour cells and reveals stereotypical differences between glioma grades. The observed increase in cellularity, vasculature, hypoxia and necrosis with glioma malignancy is consistent with the currently used histopathologic grading scheme. Furthermore, this model predicts that the accumulation of genetic mutations is not necessarily required for malignant progression. Rather, interactions of tumour cells with fixed net invasion and proliferation rates over the course of the tumour lifespan can present the histopathological characteristics of all four WHO grades of gliomas. This study suggests that dynamic changes in glioma histology (e.g. degrees of cellularity, mitosis, hypoxia-driven neoangiogenesis and necrosis) distinct from changes in cellular phenotype (e.g. proliferation and invasion rates) may be identified. In a later paper, this model's two-dimensional patient-specific predictions of hypoxia were validated in a study using FMISO-PET to image hypoxia [173].
Figure 5.

Malignant progression. Glioma grade as a function of cellular and microenvironmental compartments. Simulations of glioma grade II–IV varying the net proliferation rate of glioma cells and represented as a plot of density or concentrations of model variables (normoxic cells, hypoxic cells, necrotic tissue, vasculature and angiogenic factor) with respect to the distance from the centre of the in silico tumour. Figure reproduced with permission from [23].
6. Conclusion
Glioma invasion is characterized by invasive cell migration, phenotypic plasticity, infiltrative growth morphologies and the ability of malignant progression from low- to high-grade tumour types. The mathematical models reviewed in this work are targeted at identifying key mechanisms underlying these characteristics. Ultimately, insights gained from the simulation and analysis of the models should allow medical doctors to develop innovative glioma treatments. Accordingly, we have presented the mathematical models in a way useful for both theoreticians and practitioners. In particular, the model descriptions focus on the medical and biological key problem rather than the specific modelling approach chosen. In the following, we summarize the main findings and conclusions of the models described:
— Invasive cell migration: Several models analyse the implications of adhesive cell–cell and cell–ECM interaction on invasive migration under normoxic and hypoxic conditions. The experimental data are mostly taken from in vitro scenarios. In addition, some models analyse haptotactic and chemotactic migration, as well as allow the calculation of the invasive front speed. Several models provide mechanistic hints how hypoxia may trigger glioma cell invasion. It is a challenge to validate modelling results in both in vivo models and human patients.
— Phenotypic plasticity: So far, mathematical models have focused on the analysis of the ‘Go-or-Grow’ plasticity. It turns out that the precise regulation of the ‘Go-or-Grow‘ dichotomy and the metabolic cell state have dramatic implications for glioma growth and invasion. It is a challenge to account for other types of phenotypic plasticity and to analyse implications for invasion in mathematical models in the future. A modelling study on implications of the epithelial–mesenchymal plasticity has already shown that cell dissemination becomes more efficient in the situation of very heterogeneous microenvironments [174].
— Infiltrative morphology: It is known that morphological features are related to glioma invasiveness and aggressiveness. Mathematical models often use data from experiments with multicellular spheroids. With mathematical models, it has been shown why pseudopalisades around hypoxic and necrotic cancer regions allow to predict invasive glioma behaviour. Other models allow to predict mechanisms, e.g. a specific combination of chemotaxis and cell attraction that can lead to infiltrative morphologies. It is a challenge to test these findings in animal models with suitable refined multiscale models that allow to integrate molecular, cell and tissue data.
— Malignant progression: The stepwise process of malignant progression is driven by a series of genetic alterations and microenvironmental changes. Most models so far are formulated in the language of EGT. Model results indicate the important role of the microenvironment, e.g. hypoxic conditions, for malignant progression. Another model shows how progression can result solely from the increase of the cell proliferation rate. So far, these models have not considered genetic mutations. Future models have to analyse the consequences of cancer evolution due to mutation and selection on the progression of adult gliomas. We note that there is already a model for progression of pilocytic astrocytoma, a childhood cancer, which is based on the assumption of neutral evolution, i.e. no fitness gain through mutations [175].
We can conclude that mathematical models have been useful in providing insights into complex dynamic processes in glioma invasion. Several of the mathematical models reviewed here operate at multiple scales to explain existing experimental data, suggest further hypotheses for experimental study and have triggered new lines of research that, hopefully, will improve our understanding of glioma progression and potentially identify novel targets for cancer therapy. Phenomenological models homogenize many of the specific underlying biological processes but can provide insights into glioma growth and invasion [23,33,176]. While their ability to probe into specific outcomes from particular changes in, say, a signalling pathway is limited, phenomenological models are better able to provide insights into long-term, large-scale behaviour than models focused on smaller-scale phenomena. Indeed, phenomenological models have proven useful in characterizing glioma invasion, identifying glioma patients receiving maximal benefit from therapeutic interventions [177] and defining a more prognostic response metric for patients than is currently available [178,179].
However, several problems related to glioma invasion have not yet been addressed by mathematical approaches. In particular, the impact of molecular, cell and microenvironmental heterogeneity remains to be determined. Reher et al. [180] have shown with a mathematical model that cell-adhesion heterogeneity reinforces tumour cell dissemination. Recent experimental findings support this model prediction [181,182]. In particular, disseminated tumour cells isolated from breast cancer patients show an extensive variability in the expression of epithelial cell adhesion molecules. It is an open question if this cell-adhesion heterogeneity is also found in gliomas. However, it is already known that expression levels of the EGFR which has also effects on adhesion can be highly heterogeneous within glioblastomas. Moreover, with the help of a mathematical model it has been shown that heterogeneous microenvironments have a positive impact on cancer cell dissemination [174]. Thus, mathematical models offer an excellent opportunity to analyse implications of specific types of heterogeneity on tumour dynamics. However, the precise implications on glioma invasion and progression are still an open research topic.
Another problem concerns the discovery of quantitative biomarkers for the early detection and diagnosis of different types of tumours. In addition, several statistical and computer-based approaches have been developed to analyse gene and protein networks in gliomas that can be linked to cell motility and invasion. Reverse-engineering of a glioma-specific regulatory network revealed a transcriptional module that activates the expression of mesenchymal genes in malignant gliomas [183]. Furthermore, network models derived from gene expression and gene copy number data of glioblastoma patients were used to predict the effects of gene copy number mutations on global gene expression, revealing potential disease-relevant driver genes and predictors associated with patient survival [184]. In addition, different omics layers were integrated to predict key micro-RNAs and transcription factors that drive common or subtype-specific gene expression programs in glioblastomas [185]. A transcriptional regulatory network associated with expression differences between pilocytic and diffuse astrocytoma enabled the identification of differentially expressed major regulators involved in brain development, chromatin remodelling and cell-cycle control [186]. The observed strong overexpression of fractalkine and its receptor in pilocytic astrocytoma may contribute to the absence of invasive growth [186]. Moreover, recently a transcriptional regulatory network extracted from cancer cell lines allowed to classify glioblastoma patients into short- and long-lived groups [187].
A further challenge concerns the improvement of glioma treatments. While the vast majority of cancer therapies are cytotoxic, the aggressive invasive nature of glioma cells has prompted interest in administering therapies that focus on halting the cells’ capability to move. These therapies would not directly kill the glioma cells, but, if successful, they could potentially enhance the effectiveness of other localized cytotoxic therapies such as radiation and surgery. A few drugs under investigation target signalling pathways directing cell movement, such as those involving PDGF (i.e. Gleevac [188]) and EGF (i.e. gefitinib [189] and erlotinib [190]). Unfortunately, due to the redundancy in signalling pathways, the therapeutic effect of these drugs remains limited. Another such drug under investigation, blebbistatin, is a direct inhibitor of myosin II [191]. Myosin II is a key protein required for elongating the cells and allowing them to move through tight spaces. Recently, it was demonstrated that blebbistatin could significantly reduce glioma cell migration in in vitro experiments as well as in ex vivo brain slices even in the presence of multiple motogens such as the growth factors EGF and/or PDGF [192]. These results are promising, but further studies need to be done to demonstrate effectiveness in humans. Mathematical modelling has been demonstrated useful to predict patient-specific responses to treatment (such as anti-angiogenic therapy, chemotherapy, radiotherapy and immunotherapy) and suggest novel therapeutic avenues against cancer [176,193–209]. In the future, appropriate mathematical models should be considered to optimize treatment plans and help to identify causes underlying glioma treatment resistance.
With the wealth of biomedical data now becoming available at numerous scales ranging from population averages to images of individual tumours to genome variants within single cells, there is a large push for personalized medicine. However, across all fields of oncology research there is a fundamental gap that is currently hampering progress in individualized cancer treatment and that is the lack of meaningful methods to integrate the data across these scales. This is particularly true for glioma treatment. While the mathematical models presented here represent approaches at tackling this problem, there are still several challenges not directly handled that need to be addressed to push personalized medicine forwards. Some of them include grappling with the uncertainty in the data, integrating data of many types and extrapolating single cell/single time point genomic data to the gross tumour behaviour. We believe that the framework provided by mathematical modelling is powerful and general enough to handle these challenges, but much work remains to be done. The state of the art in mathematical modelling of glioma invasion has come a long way in 10 years. We hope that in another 10 years we will be able to write another review describing how the field of glioma modelling has bridged this gap.
Data accessibility
This article has no additional data.
Author's contributions
J.C.L.A., K.T., H.H. and A.D. designed the plan and structure of the paper. K.R.S., H.H. and A.D. supervised the study. J.C.L.A., K.T., M.S., B.K., A.D., K.R.S., H.H. and A.D. wrote and revised the manuscript. All the authors read and approved the final manuscript.
Competing interests
We declare we have no competing interests.
Funding
This work was partially supported by the Free State of Saxony and European Social Fund of the European Union (ESF, grant GlioMath-Dresden). J.C.L.A. and H.H. gratefully acknowledge the funding support of the German Federal Ministry of Education and Research (BMBF) for the eMED project SYSIMIT (01ZX1308D). A.D. and B.K. acknowledge the support by Deutsche Krebshilfe. A.D. also acknowledges support by the German Research Foundation (Deutsche Forschungsgemeinschaft) within the projects SFB-TR 79 ‘Materials for tissue regeneration within systemically altered bones’ and Research Cluster of Excellence ‘Center for Advancing Electronics Dresden’. K.R.S. and A.H.-D. gratefully acknowledge the funding support of the James S. McDonnell Foundation and the National Institutes of Health (R01 NS060752, R01 CA16437, U54 R01 CA193489, U54 CA210180)
References
- 1.Louis DN, et al. 2016. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820. ( 10.1007/s00401-016-1545-1) [DOI] [PubMed] [Google Scholar]
- 2.Ohgaki H, Kleihues P. 2005. Epidemiology and etiology of gliomas. Acta Neuropathol. 109, 93–108. ( 10.1007/s00401-005-0991-y) [DOI] [PubMed] [Google Scholar]
- 3.Ostrom QT, et al. 2014. The epidemiology of glioma in adults: a ‘state of the science’ review. Neuro Oncol. 16, 896–913. ( 10.1093/neuonc/nou087) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohgaki H, Kleihues P. 2013. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 19, 764–772. ( 10.1158/1078-0432.CCR-12-3002) [DOI] [PubMed] [Google Scholar]
- 5.Stupp R, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996. ( 10.1056/NEJMoa043330) [DOI] [PubMed] [Google Scholar]
- 6.Weller M, Stupp R, Reifenberger G, Brandes A, van den Bent M, Wick W, Hegi M. 2010. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat. Rev. Neurol. 6, 39–51. ( 10.1038/nrneurol.2009.197) [DOI] [PubMed] [Google Scholar]
- 7.Bovenberg MSS, Degeling MH, Tannous BA. 2013. Cell-based immunotherapy against gliomas: from bench to bedside. Mol. Ther. 21, 1297–1305. ( 10.1038/mt.2013.80) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlsson SK, Brothers SP, Wahlestedt C. 2014. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 6, 1359–1370. ( 10.15252/emmm.201302627) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen PY, Reardon DA. 2016. Neuro-oncology in 2015: progress in glioma diagnosis, classification and treatment. Nat. Rev. Neurol. 12, 69–70. ( 10.1038/nrneurol.2015.242) [DOI] [PubMed] [Google Scholar]
- 10.Batich KA, et al. 2017. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin. Cancer Res. 23, 1898–1909. ( 10.1158/1078-0432.CCR-16-2057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giese A, Bjerkvig R, Berens ME, Westphal M. 2003. Cost of migration: invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 21, 1624–1636. ( 10.1200/JCO.2003.05.063) [DOI] [PubMed] [Google Scholar]
- 12.Westphal M, Lamszus K. 2011. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat. Rev. Neurosci. 12, 495–508. ( 10.1038/nrn3060) [DOI] [PubMed] [Google Scholar]
- 13.Campos B, Olsen L, Urup T, Poulsen H. 2016. A comprehensive profile of recurrent glioblastoma. Oncogene 35, 5819–5825. ( 10.1038/onc.2016.85) [DOI] [PubMed] [Google Scholar]
- 14.Nakada M, Kita D, Watanabe T, Hayashi Y, Teng L, Pyko IV, Hamada JI. 2011. Aberrant signaling pathways in glioma. Cancers 3, 3242–3278. ( 10.3390/cancers3033242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beauchesne P. 2011. Extra-neural metastases of malignant gliomas: myth or reality? Cancers 3, 461–477. ( 10.3390/cancers3010461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuddapah VA, Robel S, Watkins S, Sontheimer H. 2014. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 15, 455–465. ( 10.1038/nrn3765) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrne H, Alarcon T, Owen M, Webb S, Maini P. 2006. Modelling aspects of cancer dynamics: a review. Phil. Trans. R. Soc. A 364, 1563–1578. ( 10.1098/rsta.2006.1786) [DOI] [PubMed] [Google Scholar]
- 18.Anderson AR, Quaranta V. 2008. Integrative mathematical oncology. Nat. Rev. Cancer 8, 227–234. ( 10.1038/nrc2329) [DOI] [PubMed] [Google Scholar]
- 19.Byrne HM. 2010. Dissecting cancer through mathematics: from the cell to the animal model. Nat. Rev. Cancer 10, 221–230. ( 10.1038/nrc2808) [DOI] [PubMed] [Google Scholar]
- 20.Altrock PM, Liu LL, Michor F. 2015. The mathematics of cancer: integrating quantitative models. Nat. Rev. Cancer 15, 730–745. ( 10.1038/nrc4029) [DOI] [PubMed] [Google Scholar]
- 21.Frieboes HB, Zheng X, Sun C-H, Tromberg B, Gatenby R, Cristini V. 2006. An integrated computational/experimental model of tumor invasion. Cancer Res. 66, 1597–1604. ( 10.1158/0008-5472.CAN-05-3166) [DOI] [PubMed] [Google Scholar]
- 22.Basanta D, Simon M, Hatzikirou H, Deutsch A. 2008. Evolutionary game theory elucidates the role of glycolysis in glioma progression and invasion. Cell Prolif. 41, 980–987. ( 10.1111/j.1365-2184.2008.00563.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swanson KR, Rockne RC, Claridge J, Chaplain MA, Alvord EC, Anderson AR. 2011. Quantifying the role of angiogenesis in malignant progression of gliomas: in silico modeling integrates imaging and histology. Cancer Res. 71, 7366–7375. ( 10.1158/0008-5472.CAN-11-1399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiao Y, Berman H, Kiehl TR, Torquato S. 2011. Spatial organization and correlations of cell nuclei in brain tumors. PLoS ONE 6, e27323 ( 10.1371/journal.pone.0027323) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tektonidis M, Hatzikirou H, Chauvière A, Simon M, Schaller K, Deutsch A. 2011. Identification of intrinsic in vitro cellular mechanisms for glioma invasion. J. Theor. Biol. 287, 131–147. ( 10.1016/j.jtbi.2011.07.012) [DOI] [PubMed] [Google Scholar]
- 26.Martínez-González A, Calvo GF, Perez Romasanta LA, Perez-Garcia VM. 2012. Hypoxic cell waves around necrotic cores in glioblastoma: a biomathematical model and its therapeutic implications. Bull. Math. Biol. 74 ( 10.1007/s11538-012-9786-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khain E, Katkowski M, Charteris N, Jiang F, Chopp M. 2012. Migration of adhesive glioma cells: front propagation and fingering. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 86, 011904 ( 10.1103/PhysRevE.86.011904) [DOI] [PubMed] [Google Scholar]
- 28.Böttger K, Hatzikirou H, Chauviere A, Deutsch A. 2012. Investigation of the migration/proliferation dichotomy and its impact on avascular glioma invasion. Math. Model Nat. Phenom. 7, 105–135. ( 10.1051/mmnp/20127106) [DOI] [Google Scholar]
- 29.Pham K, Chauviere A, Hatzikirou H, Li X, Byrne HM, Cristini V, Lowengrub J. 2012. Density-dependent quiescence in glioma invasion: instability in a simple reaction–diffusion model for the migration/proliferation dichotomy. J. Biol. Dyn. 6, 54–71. ( 10.1080/17513758.2011.590610) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerlee P, Nelander S. 2012. The impact of phenotypic switching on glioblastoma growth and invasion. PLoS Comput. Biol. 8, e1002556 ( 10.1371/journal.pcbi.1002556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatzikirou H, Basanta D, Simon M, Schaller K, Deutsch A. 2012. 'Go or Grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 29, 49–65. ( 10.1093/imammb/dqq011) [DOI] [PubMed] [Google Scholar]
- 32.Alfonso JCL, Köhn-Luque A, Stylianopoulos T, Feuerhake F, Deutsch A, Hatzikirou H. 2016. Why one-size-fits-all vaso-modulatory interventions fail to control glioma invasion: in silico insights. Sci. Rep. 6, 37283 ( 10.1038/srep37283) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harpold HL, Alvord EC, Swanson KR. 2007. The evolution of mathematical modeling of glioma proliferation and invasion. J. Neuropathol. Exp. Neurol. 66, 1–9. ( 10.1097/nen.0b013e31802d9000) [DOI] [PubMed] [Google Scholar]
- 34.Hatzikirou H, Deutsch A, Schaller C, Simon M, Swanson K. 2005. Mathematical modelling of glioblastoma tumour development: a review. Math. Models Methods Appl. Sci. 15, 1779–1794. ( 10.1142/S0218202505000960) [DOI] [Google Scholar]
- 35.Martirosyan NL, Rutter EM, Ramey WL, Kostelich EJ, Kuang Y, Preul MC. 2015. Mathematically modeling the biological properties of gliomas: a review. Math. Biosci. Eng. 12, 879–905. ( 10.3934/mbe.2015.12.879) [DOI] [PubMed] [Google Scholar]
- 36.Eyüpoglu IY, Buchfelder M, Savaskan NE. 2013. Surgical resection of malignant gliomas-role in optimizing patient outcome. Nat. Rev. Neurol. 9, 141–151. ( 10.1038/nrneurol.2012.279) [DOI] [PubMed] [Google Scholar]
- 37.Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG. 2004. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 36, 1046–1069. ( 10.1016/j.biocel.2004.01.013) [DOI] [PubMed] [Google Scholar]
- 38.Tate MC, Aghi MK. 2009. Biology of angiogenesis and invasion in glioma. Neurotherapeutics 6, 447–457. ( 10.1016/j.nurt.2009.04.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onishi M, Ichikawa T, Kurozumi K, Date I. 2011. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 28, 13–24. ( 10.1007/s10014-010-0007-z) [DOI] [PubMed] [Google Scholar]
- 40.Iwadate Y. 2016. Epithelial-mesenchymal transition in glioblastoma progression (review). Oncol. Lett. 11, 1615–1620. ( 10.3892/ol.2016.4113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beadle C, Assanah MC, Monzo P, Vallee R, Rosenfeld SS, Canoll P. 2008. The role of myosin II in glioma invasion of the brain. Mol. Biol. Cell 19, 3357–3368. ( 10.1091/mbc.E08-03-0319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanu OO, Hughes B, Di C, Lin N, Fu J, Bigner H, Yan DD, Adamson C. 2009. Glioblastoma multiforme oncogenomics and signaling pathways. Clin. Med. Oncol. 3, 39–52. ( 10.4137/CMO.S1008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giese A, Loo M, Tran N, Haskett D, Coons S, Berens M. 1996. Dichotomy of astrocytoma migration and proliferation. Int. J. Cancer 67, 275–282. ( 10.1002/(SICI)1097-0215(19960717)67:2%3C275::AID-IJC20%3E3.0.CO;2-9) [DOI] [PubMed] [Google Scholar]
- 44.Giese A, Kluwe L, Laube B, Meissner H, Berens ME, Westphal M. 1996. Migration of human glioma cells on myelin. Neurosurgery 38, 755–764. ( 10.1227/00006123-199604000-00026) [DOI] [PubMed] [Google Scholar]
- 45.Godlewski J, Bronisz A, Nowicki MO, Chiocca EA, Lawler S. 2010. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle 9, 2742–2748. ( 10.4161/cc.9.14.12248) [DOI] [PubMed] [Google Scholar]
- 46.Höring E, et al. 2012. The ‘go or grow’ potential of gliomas is linked to the neuropeptide processing enzyme carboxypeptidase e and mediated by metabolic stress. Acta Neuropathol. 1, 83–97. ( 10.1007/s00401-011-0940-x) [DOI] [PubMed] [Google Scholar]
- 47.Wang SD, Rath P, Lal B, Richard J-P, Li Y, Goodwin CR, Laterra J, Xia S. 2012. EphB2 receptor controls proliferation/migration dichotomy of glioblastoma by interacting with focal adhesion kinase. Oncogene 31, 5132–5143. ( 10.1038/onc.2012.16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gatenby R. 1996. Altered glucose metabolism and the invasive tumor phenotype. Int. J. Oncol. 8, 597–601. ( 10.3892/ijo.8.3.597) [DOI] [PubMed] [Google Scholar]
- 49.Griguer CE, Oliva CR, Gillespie GY. 2005. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J. Neurooncol. 74, 123–133. ( 10.1007/s11060-004-6404-6) [DOI] [PubMed] [Google Scholar]
- 50.Warburg O. 1956. On the origin of cancer cells. Science 123, 309–314. ( 10.1126/science.123.3191.309) [DOI] [PubMed] [Google Scholar]
- 51.Koppenol WH, Bounds PL, Dang CV. 2011. Otto warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337. ( 10.1038/nrc3038) [DOI] [PubMed] [Google Scholar]
- 52.Jang M, Kim SS, Lee J. 2013. Cancer cell metabolism: implications for therapeutic targets. Exp. Mol. Med. 45, e45 ( 10.1038/emm.2013.85) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang CV, Hamaker M, Sun P, Le A, Gao P. 2011. Therapeutic targeting of cancer cell metabolism. J. Mol. Med. 89, 205–212. ( 10.1007/s00109-011-0730-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao Y, Butler EB, Tan M. 2013. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 4, e532 ( 10.1038/cddis.2013.60) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. ( 10.1126/science.1160809) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gillies RJ, Robey I, Gatenby RA. 2008. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 49(Suppl. 2), 24S–42S. ( 10.2967/jnumed.107.047258) [DOI] [PubMed] [Google Scholar]
- 57.Gatenby RA, Gillies RJ. 2004. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899. ( 10.1038/nrc1478) [DOI] [PubMed] [Google Scholar]
- 58.Turner DA, Adamson DC. 2011. Neuronal-astrocyte metabolic interactions: understanding the transition into abnormal astrocytoma metabolism. J. Neuropathol. Exp. Neurol. 70, 167–176. ( 10.1097/NEN.0b013e31820e1152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marusyk A, Polyak K. 2010. Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta 1805, 105–117. ( 10.1016/j.bbcan.2009.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marusyk A, Almendro V, Polyak K. 2012. Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer 12, 323–334. ( 10.1038/nrc3261) [DOI] [PubMed] [Google Scholar]
- 61.Bonavia R, Inda MM, Cavenee WK, Furnari FB. 2011. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 71, 4055–4060. ( 10.1158/0008-5472.CAN-11-0153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klink B, Schlingelhof B, Klink M, Stout-Weider K, Patt S, Schrock E. 2011. Glioblastomas with oligodendroglial component-common origin of the different histological parts and genetic subclassification. Cell. Oncol. (Dordr) 34, 261–275. ( 10.1007/s13402-011-0034-8) [DOI] [PubMed] [Google Scholar]
- 63.Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavaré S. 2013. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl Acad. Sci. USA 110, 4009–4014. ( 10.1073/pnas.1219747110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel AP, et al. 2014. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. ( 10.1126/science.1254257) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyer M, et al. 2015. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl Acad. Sci. USA 112, 851–856. ( 10.1073/pnas.1320611111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Welch DR. 2016. Tumor heterogeneity–a ‘contemporary concept’ founded on historical insights and predictions. Cancer Res. 76, 4–6. ( 10.1158/0008-5472.CAN-15-3024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abou-El-Ardat K, et al. 2017. Comprehensive molecular characterization of multifocal glioblastoma proves its monoclonal origin and reveals novel insights into clonal evolution and heterogeneity of glioblastomas. Neuro. Oncol. 19, 546–557. ( 10.1093/neuonc/now231) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venteicher AS, et al. 2017. Decoupling genetics, lineages, and microenvironment in idh-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 ( 10.1126/science.aai8478) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shackleton M, Quintana E, Fearon ER, Morrison SJ. 2009. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 138, 822–829. ( 10.1016/j.cell.2009.08.017) [DOI] [PubMed] [Google Scholar]
- 70.Michor F, Polyak K. 2010. The origins and implications of intratumor heterogeneity. Cancer Prev. Res. 3, 1361–1364. ( 10.1158/1940-6207.CAPR-10-0234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meacham CE, Morrison SJ. 2013. Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337. ( 10.1038/nature12624) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gerlinger M, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892. ( 10.1056/NEJMoa1113205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shin Y, Han S, Chung E, Chung S. 2014. Intratumoral phenotypic heterogeneity as an encourager of cancer invasion. Integr. Biol. 6, 654–661. ( 10.1039/C4IB00022F) [DOI] [PubMed] [Google Scholar]
- 74.Chapman A, del Ama LF, Ferguson J, Kamarashev J, Wellbrock C, Hurlstone A. 2014. Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep. 8, 688–695. ( 10.1016/j.celrep.2014.06.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Claes A, Idema AJ, Wesseling P. 2007. Diffuse glioma growth: a guerilla war. Acta Neuropathol. 114, 443–458. ( 10.1007/s00401-007-0293-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gritsenko PG, Ilina O, Friedl P. 2012. Interstitial guidance of cancer invasion. J. Pathol. 226, 185–199. ( 10.1002/path.3031) [DOI] [PubMed] [Google Scholar]
- 77.Kwok J, Yang S, Fawcett JW. 2013. Neural ECM in regeneration and rehabilitation. Prog. Brain Res. 214, 179–192. ( 10.1016/B978-0-444-63486-3.00008-6) [DOI] [PubMed] [Google Scholar]
- 78.Hoelzinger DB, Demuth T, Berens ME. 2007. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J. Natl. Cancer Inst. 99, 1583–1593. ( 10.1093/jnci/djm187) [DOI] [PubMed] [Google Scholar]
- 79.Wesseling P, van der Laak JA, de Leeuw H, Ruiter DJ, Burger PC. 1994. Quantitative immunohistological analysis of the microvasculature in untreated human glioblastoma multiforme computer-assisted image analysis of whole-tumor sections. J. Neurosurg. 81, 902–909. ( 10.3171/jns.1994.81.6.0902) [DOI] [PubMed] [Google Scholar]
- 80.Wesseling P, van der Laak JA, Link M, Teepen HL, Ruiter DJ. 1998. Quantitative analysis of microvascular changes in diffuse astrocytic neoplasms with increasing grade of malignancy. Hum. Pathol. 29, 352–358. ( 10.1016/S0046-8177(98)90115-0) [DOI] [PubMed] [Google Scholar]
- 81.Bernsen H, van der Laak J, Küsters B, van der Ven A, Wesseling P. 2005. Gliomatosis cerebri: quantitative proof of vessel recruitment by cooptation instead of angiogenesis. J. Neurosurg. 103, 702–706. ( 10.3171/jns.2005.103.4.0702) [DOI] [PubMed] [Google Scholar]
- 82.Jain RK, Di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. 2007. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 8, 610–622. ( 10.1038/nrn2175) [DOI] [PubMed] [Google Scholar]
- 83.Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B, Van Meir EG. 2004. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 64, 920–927. ( 10.1158/0008-5472.CAN-03-2073) [DOI] [PubMed] [Google Scholar]
- 84.Brat D, Van Meir E. 2004. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Invest. 84, 397–405. ( 10.1038/labinvest.3700070) [DOI] [PubMed] [Google Scholar]
- 85.Rong Y, Brat D. 2009. Vaso-occlusive mechanisms that intiate hypoxia and necrosis in glioblastoma: the role of thrombosis and tissue factor. In: CNS Cancer: models, markers, prognostic factors, targets, and therapeutic approaches (cancer drug discovery and development) (ed. EG Meir), pp. 507–528. Berlin, Germany: Springer. [Google Scholar]
- 86.Padera T, Stoll B, Tooredman J, Capen D, di Tomaso E, Jain R. 2004. Pathology: cancer cells compress intratumour vessels. Nature 427, 695 ( 10.1038/427695a) [DOI] [PubMed] [Google Scholar]
- 87.Rong Y, Durden DL, Van Meir EG, Brat DJ. 2006. Pseudopalisading necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 65, 529 ( 10.1097/00005072-200606000-00001) [DOI] [PubMed] [Google Scholar]
- 88.Evans SM, et al. 2004. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin. Cancer Res. 10, 8177–8184. ( 10.1158/1078-0432.CCR-04-1081) [DOI] [PubMed] [Google Scholar]
- 89.Elstner A, Holtkamp N, von Deimling A. 2007. Involvement of HIF-1 in desferrioxamine-induced invasion of glioblastoma cells. Clin. Exp. Metastasis 24, 57–66. ( 10.1007/s10585-007-9057-y) [DOI] [PubMed] [Google Scholar]
- 90.Abbott NJ, Rönnbäck L, Hansson E. 2006. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 7, 41–53. ( 10.1038/nrn1824) [DOI] [PubMed] [Google Scholar]
- 91.Lee J, Lund-Smith C, Borboa A, Gonzalez AM, Baird A, Eliceiri BP. 2009. Glioma-induced remodeling of the neurovascular unit. Brain Res. 1288, 125–134. ( 10.1016/j.brainres.2009.06.095) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Agarwal S, Manchanda P, Vogelbaum MA, Ohlfest JR, Elmquist WF. 2013. Function of the blood-brain barrier and restriction of drug delivery to invasive glioma cells: findings in an orthotopic rat xenograft model of glioma. Drug Metab. Dispos. 41, 33–39. ( 10.1124/dmd.112.048322) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dubois LG, Campanati L, Righy C, D'Andrea-Meira I, Ximenes-da Silva A, Lopes MC, Faveret E, Gasparetto EL, Moura-Neto V. 2014. Gliomas and the vascular fragility of the blood brain barrier. Front. Integr. Neurosci. 8, 418 ( 10.3389/fncel.2014.00418) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dvorak HF. 1986. Tumors: wounds that do not heal: similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659. ( 10.1056/NEJM198612253152606) [DOI] [PubMed] [Google Scholar]
- 95.Halliday JJ, Holland EC. 2011. Connective tissue growth factor and the parallels between brain injury and brain tumors. J. Natl. Cancer. Inst. 103, 1141–1143. ( 10.1093/jnci/djr261) [DOI] [PubMed] [Google Scholar]
- 96.Hambardzumyan D, Gutmann DH, Kettenmann H. 2016. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19, 20–27. ( 10.1038/nn.4185) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Garris C, Pittet MJ. 2013. Therapeutically reeducating macrophages to treat GBM. Nat. Med. 19, 1207–1208. ( 10.1038/nm.3355) [DOI] [PubMed] [Google Scholar]
- 98.Bettinger I, Thanos S, Paulus W. 2002. Microglia promote glioma migration. Acta Neuropathol. 103, 351–355. ( 10.1007/s00401-001-0472-x) [DOI] [PubMed] [Google Scholar]
- 99.Solinas G, et al. 2010. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J. Immunol. 185, 642–652. ( 10.4049/jimmunol.1000413) [DOI] [PubMed] [Google Scholar]
- 100.Quail DF, Joyce JA. 2013. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. ( 10.1038/nm.3394) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. 2011. The brain tumor microenvironment. Glia 59, 1169–1180. ( 10.1002/glia.21136) [DOI] [PubMed] [Google Scholar]
- 102.Li W, Graeber MB. 2012. The molecular profile of microglia under the influence of glioma. Neuro. Oncol. 8, 958–978. ( 10.1093/neuonc/nos116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. 2008. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 66, 1–9. ( 10.1016/j.critrevonc.2007.07.004) [DOI] [PubMed] [Google Scholar]
- 104.Coniglio SJ, Segall JE. 2013. Review: molecular mechanism of microglia stimulated glioblastoma invasion. Matrix Biol. 32, 372–380. ( 10.1016/j.matbio.2013.07.008) [DOI] [PubMed] [Google Scholar]
- 105.Powell Perng ML. 2015. Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front. Oncol. 5, 153 ( 10.3389/fonc.2015.00153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kam Y, Rejniak KA, Anderson AR. 2012. Cellular modeling of cancer invasion: integration of in silico and in vitro approaches. J. Cell. Physiol. 227, 431–438. ( 10.1002/jcp.22766) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kunz-schughart LA, Kreutz M, Knuechel R. 1998. Multicellular spheroids: a three-dimensional in vitro culture system to study tumour biology. Int. J. Exp. Pathol. 79, 1–23. ( 10.1046/j.1365-2613.1998.00051.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zaman MH, Trapani LM, Sieminski AL, MacKellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. 2006. Migration of tumor cells in 3d matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl Acad. Sci. USA 103, 10 889–10 894. ( 10.1073/pnas.0604460103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bjerkvig R, Laerum OD, Mella O. 1986. Glioma cell interactions with fetal rat brain aggregates in vitro and with brain tissue in vivo. Cancer Res. 46, 4071–4079. [PubMed] [Google Scholar]
- 110.Terzis AJA, Arnold H, Fiskerstrand T, Refsum H, Ueland PM, Bjerkvig R. 1993. Proliferation, migration and invasion of human glioma cells exposed to antifolate drugs. Int. J. Cancer 54, 112–118. ( 10.1002/ijc.2910540118) [DOI] [PubMed] [Google Scholar]
- 111.Murakami M, Goto S, Yoshikawa M, Goto T, Hamasaki T, Rutka JT, Kuratsu JI, Ushio Y. 2001. The invasive features of glial and non-central nervous system tumor cells are different on organotypic brain slices from newborn rats. Int. J. Oncol. 18, 721–728. ( 10.3892/ijo.18.4.721) [DOI] [PubMed] [Google Scholar]
- 112.Schichor C, Kerkau S, Visted T, Martini R, Bjerkvig R, Tonn JC, Goldbrunner R. 2005. The brain slice chamber, a novel variation of the boyden chamber assay, allows time-dependent quantification of glioma invasion into mammalian brain in vitro. J. Neurooncol. 73, 9–18. ( 10.1007/s11060-004-3341-3) [DOI] [PubMed] [Google Scholar]
- 113.Wang J, et al. 2009. A reproducible brain tumour model established from human glioblastoma biopsies. BMC Cancer 9, 465 ( 10.1186/1471-2407-9-465) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Candolfi M, et al. 2007. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J. Neurooncol. 85, 133–148. ( 10.1007/s11060-007-9400-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Holland EC, Hively WP, DePinho RA, Varmus HE. 1998. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 12, 3675–3685. ( 10.1101/gad.12.23.3675) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, Canoll P. 2006. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J. Neurosci. 26, 6781–6790. ( 10.1523/JNEUROSCI.0514-06.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ding H, et al. 2001. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 61, 3826–3836. [PubMed] [Google Scholar]
- 118.Witte HT, Jeibmann A, Klämbt C, Paulus W. 2009. Modeling glioma growth and invasion in drosophila melanogaster. Neoplasia 11, 882–888. ( 10.1593/neo.09576) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gonzalez C. 2013. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 13, 172–183. ( 10.1038/nrc3461) [DOI] [PubMed] [Google Scholar]
- 120.Adams MD, et al. 2000. The genome sequence of Drosophila melanogaster. Science 287, 2185–2195. ( 10.1126/science.287.5461.2185) [DOI] [PubMed] [Google Scholar]
- 121.Upadhyay N, Waldman A. 2011. Conventional MRI evaluation of gliomas. Br. J. Radiol. 84, 107–111. ( 10.1259/bjr/65711810) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Langen K-J, Galldiks N, Hattingen E, Shah NJ. 2017. Advances in neuro-oncology imaging. Nat. Rev. Neurol. 13, 279–289. ( 10.1038/nrneurol.2017.44) [DOI] [PubMed] [Google Scholar]
- 123.Brown RW, Cheng Y-CN, Haacke EM, Thompson MR, Venkatesan R (eds). 2014. Magnetic resonance imaging: physical principles and sequence design, 2nd edn New York, NY: John Wiley & Sons, Inc. [Google Scholar]
- 124.Sattur MG, Vogelbaum MA. 2016. Chapter 37—{RANO} criteria: application to response assessment in clinical trials. In Handbook of Neuro-Oncology Neuroimaging (Second Edition) (ed. Newton HB.), 2nd edn, pp. 409–418. San Diego, CA: Academic Press. [Google Scholar]
- 125.Silbergeld DL, Chicoine MR. 1997. Isolation and characterization of human malignant glioma cells from histologically normal brain. J. Neurosurg. 86, 525–531. ( 10.3171/jns.1997.86.3.0525) [DOI] [PubMed] [Google Scholar]
- 126.Lee E, Ahn K, Lee E, Lee Y, Kim D. 2013. Potential role of advanced MRI techniques for the peritumoural region in differentiating glioblastoma multiforme and solitary metastatic lesions. Clin. Radiol. 68, e689–e697. ( 10.1016/j.crad.2013.06.021) [DOI] [PubMed] [Google Scholar]
- 127.Sorensen AG, et al. 2012. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 72, 402–407. ( 10.1158/0008-5472.CAN-11-2464) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Batchelor TT, et al. 2013. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc. Natl Acad. Sci. USA 110, 19 059–19 064. ( 10.1073/pnas.1318022110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen L, Liu M, Bao J, Xia Y, Zhang J, Zhang L, Huang X, Wang J. 2013. The correlation between apparent diffusion coefficient and tumor cellularity in patients: a meta-analysis. PLoS ONE 8, e79008 ( 10.1371/journal.pone.0079008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Eidel O, et al. 2016. Automatic analysis of cellularity in glioblastoma and correlation with ADC using trajectory analysis and automatic nuclei counting. PLoS ONE 11, e0160250 ( 10.1371/journal.pone.0160250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.He T, et al. 2017. Multivoxel magnetic resonance spectroscopy identifies enriched foci of cancer stem-like cells in high-grade gliomas. Onco. Targets Ther. 10, 195–203. ( 10.2147/OTT.S118834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Leather T, Jenkinson MD, Das K, Poptani H. 2017. Magnetic resonance spectroscopy for detection of 2-hydroxyglutarate as a biomarker for IDH mutation in gliomas. Metabolites 7, E29 ( 10.3390/metabo7020029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tietze A. 2017. Noninvasive assessment of isocitrate dehydrogenase mutation status in cerebral gliomas by magnetic resonance spectroscopy in a clinical setting. J. Neurosurg. 3, 1–8. ( 10.3171/2016.10.JNS161793) [DOI] [PubMed] [Google Scholar]
- 134.Agliano A, Balarajah G, Ciobota DM, Sidhu J, Clarke PA, Jones C, Workman P, Leach MO, Al-Suffar NM. 2017. Pediatric and adult glioblastoma radiosensitization induced by PI3K/mTOR inhibition causes early metabolic alterations detected by nuclear magnetic resonance spectroscopy. Oncotarget 8, 47 969–47 983. ( 10.18632/oncotarget.18206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hyare H, Thust S, Rees J. 2017. Advanced MRI techniques in the monitoring of treatment of gliomas. Curr. Treat. Options Neurol. 19, 11 ( 10.1039/C3MB70602H) [DOI] [PubMed] [Google Scholar]
- 136.Adamson E, Ludwig K, Mummy D, Fain SB. 2017. Magnetic resonance imaging with hyperpolarized agents: methods and applications. Phys. Med. Biol. 62, R81 ( 10.1088/1361-6560/aa6be8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.James ML, Gambhir SS. 2012. A molecular imaging primer: modalities, imaging agents, and applications. Physiol. Rev. 92, 897–965. ( 10.1152/physrev.00049.2010) [DOI] [PubMed] [Google Scholar]
- 138.Pandey R, Caflisch L, Lodi A, Brenner AJ, Tiziani S. 2017. Metabolomic signature of brain cancer. Mol. Carcinog. 56, 2355–2371. ( 10.1002/mc.22694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lopez CJ, Nagornaya N, Parra NA, Kwon D, Ishkanian F, Markoe AM, Maudsley A, Stoyanova R. 2017. Association of radiomics and metabolic tumor volumes in radiation treatment of glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 97, 586–595. ( 10.1016/j.ijrobp.2016.11.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.la Fougére C, Suchorska B, Bartenstein P, Kreth F, Tonn J. 2011. Molecular imaging of gliomas with pet: opportunities and limitations. Neuro Oncol. 13, 806–819. ( 10.1093/neuonc/nor054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Herholz K. 2017. Brain tumors: an update on clinical pet research in gliomas. Semin. Nucl. Med. 47, 5–17. ( 10.1053/j.semnuclmed.2016.09.004) [DOI] [PubMed] [Google Scholar]
- 142.Leiva-Salinas C, Schiff D, Flors L, Patrie JT, Rehm PK. 2016. FDG PET/MR imaging coregistration helps predict survival in patients with glioblastoma and radiologic progression after standard of care treatment. Radiology 283, 508–514. ( 10.1148/radiol.2016161172) [DOI] [PubMed] [Google Scholar]
- 143.Laukamp KR, et al. 2017. Multimodal imaging of patients with gliomas confirms 11c-MET PET as a complementary marker to MRI for noninvasive tumor grading and intraindividual follow-up after therapy. Mol. Imaging 16, 1536012116687651 ( 10.1177/1536012116687651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Götz I, Grosu AL. 2013. [18f] fet-pet imaging for treatment and response monitoring of radiation therapy in malignant glioma patients–a review. Front. Oncol. 3, 104 ( 10.3389/fonc.2013.00104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brat DJ, Prayson RA, Ryken TC, Olson JJ. 2008. Diagnosis of malignant glioma: role of neuropathology. J. Neurooncol. 89, 287–311. ( 10.1007/s11060-008-9618-1) [DOI] [PubMed] [Google Scholar]
- 146.Dunbar E, Yachnis AT. 2010. Glioma diagnosis: immunohistochemistry and beyond. Adv. Anat. Pathol. 17, 187–201. ( 10.1097/PAP.0b013e3181d98cd9) [DOI] [PubMed] [Google Scholar]
- 147.Moreira J, Deutsch A. 2002. Cellular automaton models of tumor development: a critical review. Advs. Complex Syst. 5, 247–267. ( 10.1142/S0219525902000572) [DOI] [Google Scholar]
- 148.Deutsch A, Dormann S. 2005. Cellular automaton modeling of biological pattern formation: characterization, applications, and analysis. Boston, MA: Birkhäuser. [Google Scholar]
- 149.Bellomo N, Li NK, Maini PK. 2008. On the foundations of cancer modelling: selected topics, speculations, and perspectives. Math. Models Methods Appl. Sci. 18, 593–646. ( 10.1142/S0218202508002796) [DOI] [Google Scholar]
- 150.Edelman LB, Eddy JA, Price ND. 2010. In silico models of cancer. Wiley Interdiscip. Rev. Syst. Biol. Med. 2, 438–459. ( 10.1002/wsbm.75) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rejniak KA, McCawley LJ. 2010. Current trends in mathematical modeling of tumor–microenvironment interactions: a survey of tools and applications. Exp. Biol. Med. 235, 411–423. ( 10.1258/ebm.2009.009230) [DOI] [PubMed] [Google Scholar]
- 152.Deisboeck TS, Wang Z, Macklin P, Cristini V. 2011. Multiscale cancer modeling. Annu. Rev. Biomed. Eng. 13, 127–155. ( 10.1146/annurev-bioeng-071910-124729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hatzikirou H, Chauviere A, Bauer AL, Leier A, Lewis MT, Macklin P, Marquez-Lago TT, Bearer EL, Cristini V. 2011. Integrative physical oncology. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 1–14. ( 10.1002/wsbm.158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Aubert M, Badoual M, Christov C, Grammaticos B. 2008. A model for glioma cell migration on collagen and astrocytes. J. R. Soc. Interface 5, 75–83. ( 10.1098/rsif.2007.1070) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Khain E, Katakowski M, Hopkins S, Szalad A, Zheng Z, Jiang F, Chopp M. 2011. Collective behavior of brain tumor cells: the role of hypoxia. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 83, 031920 ( 10.1103/PhysRevE.83.031920) [DOI] [PubMed] [Google Scholar]
- 156.Szabó A, Varga K, Garay T, Hegedus B, Czirók A. 2012. Invasion from a cell aggregate–the roles of active cell motion and mechanical equilibrium. Phys. Biol. 9, 016010 ( 10.1088/1478-3975/9/1/016010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim Y, Lawler S, Nowicki MO, Chiocca EA, Friedman A. 2009. A mathematical model for pattern formation of glioma cells outside the tumor spheroid core. J. Theor. Biol. 260, 359–371. ( 10.1016/j.jtbi.2009.06.025) [DOI] [PubMed] [Google Scholar]
- 158.Kim Y. 2013. Regulation of cell proliferation and migration in glioblastoma: new therapeutic approach. Front. Oncol. 3, 359–371. ( 10.3389/fonc.2013.00053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sander LM, Deisboeck TS. 2002. Growth patterns of microscopic brain tumors. Phys. Rev. E 66, 051901 ( 10.1103/PhysRevE.66.051901) [DOI] [PubMed] [Google Scholar]
- 160.Zhang L, Strouthos CG, Wang Z, Deisboeck TS. 2009. Simulating brain tumor heterogeneity with a multiscale agent-based model: linking molecular signatures, phenotypes and expansion rate. Math. Comput. Model. 49, 307–319. ( 10.1016/j.mcm.2008.05.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Frieboes HB, Lowengrub JS, Wise S, Zheng X, Macklin P, Bearer EL, Cristini V. 2007. Computer simulation of glioma growth and morphology. Neuroimage 37, S59–S70. ( 10.1016/j.neuroimage.2007.03.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Aubert M, Badoual M, Fereol S, Christov C, Grammaticos B. 2006. A cellular automaton model for the migration of glioma cells. Phys. Biol. 3, 93–100. ( 10.1088/1478-3975/3/2/001) [DOI] [PubMed] [Google Scholar]
- 163.Aubert M, Badoual M, Grammaticos B. 2008. A model for short-and long-range interactions of migrating tumour cell. Acta Biotheor. 56, 297–314. ( 10.1007/s10441-008-9061-x) [DOI] [PubMed] [Google Scholar]
- 164.Kim Y, Roh S, Lawler S, Friedman A. 2011. miR451 and AMPK mutual antagonism in glioma cell migration and proliferation: a mathematical model. PLoS ONE 6, e28293 ( 10.1371/journal.pone.0028293) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ansari KI, Ogawa D, Rooj AK, Lawler SE, Krichevsky AM, Johnson MD, Chiocca EA, Bronisz A, Godlewski J. 2015. Glucose-based regulation of miR-451/AMPK signaling depends on the OCT1 transcription factor. Cell Rep. 11, 902–909. ( 10.1016/j.celrep.2015.04.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stein AM, Demuth T, Mobley D, Berens M, Sander LM. 2007. A mathematical model of glioblastoma tumor spheroid invasion in a three-dimensional in vitro experiment. Biophys. J. 92, 356–365. ( 10.1529/biophysj.106.093468) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Scribner E, Fathallah-Shaykh HM. 2017. Single cell mathematical model successfully replicates key features of GBM: go-or-grow is not necessary. PLoS ONE 12, e0169434 ( 10.1371/journal.pone.0169434) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Böttger K, Hatzikirou H, Voss-Böhme A, Cavalcanti-Adam EA, Herrero MA, Deutsch A. 2015. An emerging Allee effect is critical for tumor initiation and persistence. PLoS Comput. Biol. 11, e1004366 ( 10.1371/journal.pcbi.1004366) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sanga S, Frieboes HB, Zheng X, Gatenby R, Bearer EL, Cristini V. 2007. Predictive oncology: a review of multidisciplinary, multiscale in silico modeling linking phenotype, morphology and growth. Neuroimage 37, 120–134. ( 10.1016/j.neuroimage.2007.05.043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gerin C, et al. 2012. Improving the time-machine: estimating date of birth of grade II gliomas. Cell Prolif. 45, 76–90. ( 10.1111/j.1365-2184.2011.00790.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bohl K, Hummert S, Werner S, Basanta D, Deutsch A, Schuster S, Theissen G, Schroeter A. 2014. Evolutionary game theory: molecules as players. Mol. Biosyst. 10, 3066–3074. ( 10.1039/C3MB70601J) [DOI] [PubMed] [Google Scholar]
- 172.Hummert S, Bohl K, Basanta D, Deutsch A, Werner S, Theißen G, Schroeterc A, Schuster S. 2014. Evolutionary game theory: cells as players. Mol. Biosyst. 10, 3044–3065. ( 10.1039/C3MB70602H) [DOI] [PubMed] [Google Scholar]
- 173.Gu S, et al. 2012. Applying a patient-specific bio-mathematical model of glioma growth to develop virtual [18f]-fmiso-pet images. Math. Med. Biol. 29, 31–48. ( 10.1093/imammb/dqr002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Talkenberger K, Cavalcanti-Adam EA, Voss-Böhme A, Deutsch A. 2017. Amoeboid-mesenchymal migration plasticity promotes invasion only in complex heterogeneous microenvironments. Sci. Rep. 7, 9237 ( 10.1038/s41598-017-09300-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Buder T, Deutsch A, Klink B, Voss-Böhme A. 2015. Model-based evaluation of spontaneous tumor regression in pilocytic astrocytoma. PLoS Comput. Biol. 11, e1004662 ( 10.1371/journal.pcbi.1004662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Alfonso JCL, Buttazzo G, García-Archilla B, Herrero MA, Núñez L. 2012. A class of optimization problems in radiotherapy dosimetry planning. Discr. Cont. Dyn. Syst. B 17, 1651–1672. ( 10.3934/dcdsb.2012.17.1651) [DOI] [Google Scholar]
- 177.Baldock AL, et al. 2014. Patient-specific metrics of invasiveness reveal significant prognostic benefit of resection in a predictable subset of gliomas. PLoS ONE 9, e99057 ( 10.1371/journal.pone.0099057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Neal ML, et al. 2013. Discriminating survival outcomes in patients with glioblastoma using a simulation-based, patient-specific response metric. PLoS. ONE. 8, e51951 ( 10.1371/journal.pone.0051951) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Neal ML, et al. 2013. Response classification based on a minimal model of glioblastoma growth is prognostic for clinical outcomes and distinguishes progression from pseudoprogression. Cancer Res. 73, 2976–2986. ( 10.1158/0008-5472.CAN-12-3588) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Reher D, Klink B, Deutsch A, Voss-Böhme A. 2017. Cell adhesion heterogeneity reinforces tumour cell dissemination: novel insights from a mathematical model. Biol. Direct. 12, 18 ( 10.1186/s13062-017-0188-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yu M, et al. 2013. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584. ( 10.1126/science.1228522) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ellsworth RE, Blackburn HL, Shriver CD, Soon-Shiong P, Ellsworth DL. 2017. Molecular heterogeneity in breast cancer: state of the science and implications for patient care. Semin. Cell Dev. Biol. 64, 65–72. ( 10.1016/j.semcdb.2016.08.025) [DOI] [PubMed] [Google Scholar]
- 183.Carro MS, et al. 2010. The transcriptional network for mesenchymal transformation of brain tumors. Nature 463, 318–325. ( 10.1038/nature08712) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jörnsten R, et al. 2011. Network modeling of the transcriptional effects of copy number aberrations in glioblastoma. Mol. Syst. Biol. 7, 486 ( 10.1038/msb.2011.17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Setty M, et al. 2012. Inferring transcriptional and microRNA-mediated regulatory programs in glioblastoma. Mol. Syst. Biol. 8, 605 ( 10.1038/msb.2012.37) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Seifert M, Garbe M, Friedrich B, Mittelbronn M, Klink B. 2015. Comparative transcriptomics reveals similarities and differences between astrocytoma grades. BMC Cancer 15, 952 ( 10.1186/s12885-015-1939-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Seifert M, Friedrich B, Beyer A. 2016. Importance of rare gene copy number alterations for personalized tumor characterization and survival analysis. Genome Biol. 17, 204 ( 10.1186/s13059-016-1058-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Holdhoff M, et al. 2005. Imatinib mesylate radiosensitizes human glioblastoma cells through inhibition of platelet-derived growth factor receptor. Blood Cells Mol. Dis. 34, 181–185. ( 10.1016/j.bcmd.2004.11.006) [DOI] [PubMed] [Google Scholar]
- 189.Rich JN, et al. 2004. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 22, 133–142. ( 10.1200/JCO.2004.08.110) [DOI] [PubMed] [Google Scholar]
- 190.Raizer JJ. 2010. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy? Neuro. Oncol. 12, 95–103. ( 10.1093/neuonc/nop015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kovács M, Tóth J, Hetényi C, Málnási-Csizmadia A, Sellers JR. 2004. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 279, 35 557–35 563. ( 10.1074/jbc.M405319200) [DOI] [PubMed] [Google Scholar]
- 192.Ivkovic S, Beadle C, Noticewala S, Massey SC, Swanson KR, Toro LN, Bresnick AR, Canoll P, Rosenfeld SS. 2012. Direct inhibition of myosin II effectively blocks glioma invasion in the presence of multiple motogens. Mol. Biol. Cell 23, 533–542. ( 10.1091/mbc.E11-01-0039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tracqui P, Cruywagen G, Woodward D, Bartoo G, Murray J, Alvord E. 1995. A mathematical model of glioma growth: the effect of chemotherapy on spatio-temporal growth. Cell Prolif. 28, 17–31. ( 10.1111/j.1365-2184.1995.tb00036.x) [DOI] [PubMed] [Google Scholar]
- 194.d'Onofrio A. 2006. Tumour-immune system interaction: modeling the tumour-stimulated proliferation of effectors and immunotherapy. Math. Models Methods Appl. Sci. 16, 1375–1401. ( 10.1142/S0218202506001571) [DOI] [Google Scholar]
- 195.de Pillis L, Gu W, Radunskaya A. 2006. Mixed immunotherapy and chemotherapy of tumors: modeling, applications and biological interpretations. J. Theor. Biol. 238, 841–862. ( 10.1016/j.jtbi.2005.06.037) [DOI] [PubMed] [Google Scholar]
- 196.Powathil G, Kohandel M, Sivaloganathan S, Oza A, Milosevic M. 2007. Mathematical modeling of brain tumors: effects of radiotherapy and chemotherapy. Phys. Med. Biol. 52, 3291–3306. ( 10.1088/0031-9155/52/11/023) [DOI] [PubMed] [Google Scholar]
- 197.Rockne R, Alvord EC, Rockhill JK, Swanson KR. 2009. A mathematical model for brain tumor response to radiation therapy. J. Math. Biol. 58, 561–578. ( 10.1007/s00285-008-0219-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rockne R. 2010. Predicting the efficacy of radiotherapy in individual glioblastoma patients in vivo: a mathematical modeling approach. Phys. Med. Biol. 55, 3271–3285. ( 10.1088/0031-9155/55/12/001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Baker GJ, et al. 2014. Mechanisms of glioma formation: iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 16, 543–561. ( 10.1016/j.neo.2014.06.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Borasi G, Nahum A. 2016. Modelling the radiotherapy effect in the reaction-diffusion equation. Phys. Medica. 32, 1175–1179. ( 10.1016/j.ejmp.2016.08.020) [DOI] [PubMed] [Google Scholar]
- 201.Gao X, McDonald JT, Hlatky L, Enderling H. 2013. Acute and fractionated irradiation differentially modulate glioma stem cell division kinetics. Cancer Res. 73, 1481–1490. ( 10.1158/0008-5472.CAN-12-3429) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Alfonso J, Jagiella N, Núñez L, Herrero M, Drasdo D. 2014. Estimating dose painting effects in radiotherapy: a mathematical model. PLoS. ONE. 9, e89380 ( 10.1371/journal.pone.0089380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Badoual M, Gerin C, Deroulers C, Grammaticos B, Llitjos J-F, Oppenheim C, Varlet P, Pallud J. 2014. Oedema-based model for diffuse low-grade gliomas: application to clinical cases under radiotherapy. Cell Prolif. 47, 369–380. ( 10.1111/cpr.12114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Alfonso J, Buttazzo G, García-Archilla B, Herrero M, Núñez L. 2014. Selecting radiotherapy dose distributions by means of constrained optimization problems. Bull. Math. Biol. 76, 1017–1044. ( 10.1007/s11538-014-9945-7) [DOI] [PubMed] [Google Scholar]
- 205.Enderling H, Chaplain MA. 2014. Mathematical modeling of tumor growth and treatment. Curr. Pharm. Des. 20, 4934–4940. ( 10.2174/1381612819666131125150434) [DOI] [PubMed] [Google Scholar]
- 206.Hatzikirou H, Alfonso JCL, Mühle S, Stern C, Weiss S, Meyer-Hermann M. 2015. Cancer therapeutic potential of combinatorial immuno-and vasomodulatory interventions. J. R. Soc. Interface 12, 20150439 ( 10.1098/rsif.2015.0439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Michor F, Beal K. 2015. Improving cancer treatment via mathematical modeling: surmounting the challenges is worth the effort. Cell 163, 1059–1063. ( 10.1016/j.cell.2015.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Reppas AI, Alfonso JCL, Hatzikirou H. 2016. In silico tumor control induced via alternating immunostimulating and immunosuppressive phases. Virulence 7, 174–186. ( 10.1080/21505594.2015.1076614) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Walker R, Enderling H. 2016. From concept to clinic: mathematically informed immunotherapy. Curr. Probl. Cancer 40, 68–83. ( 10.1016/j.currproblcancer.2015.10.004) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.