

Abstract
The growth of microorganisms involves the conversion of nutrients in the environment into biomass, mostly proteins and other macromolecules. This conversion is accomplished by networks of biochemical reactions cutting across cellular functions, such as metabolism, gene expression, transport and signalling. Mathematical modelling is a powerful tool for gaining an understanding of the functioning of this large and complex system and the role played by individual constituents and mechanisms. This requires models of microbial growth that provide an integrated view of the reaction networks and bridge the scale from individual reactions to the growth of a population. In this review, we derive a general framework for the kinetic modelling of microbial growth from basic hypotheses about the underlying reaction systems. Moreover, we show that several families of approximate models presented in the literature, notably flux balance models and coarse-grained whole-cell models, can be derived with the help of additional simplifying hypotheses. This perspective clearly brings out how apparently quite different modelling approaches are related on a deeper level, and suggests directions for further research.
Keywords: microbial growth, mathematical modelling, metabolic and gene regulatory networks, systems biology
1. Introduction
Bacterial growth curves have exerted much fascination on microbiologists, as eloquently summarized by Frederick Neidhardt in his short commentary ‘Bacterial growth: constant obsession with dN/dt’ published almost 20 years ago [1]. When supplied with a defined mixture of salts, sugar, vitamins and trace elements, a population of bacterial cells contained in liquid medium is capable of growing and replicating at a constant rate in a highly reproducible manner. This observed regularity raises fundamental questions about the organization of the cellular processes converting nutrients into biomass.
Work in microbial physiology has resulted in quantitative measurements of a variety of variables related to the cellular processes underlying growth. These measurements have usually been carried out during steady-state exponential or balanced growth, that is a state in which all cellular components as well as the total volume of the population have the same constant doubling time, implying that the concentrations of the cellular components remain constant [2]. The measurements have enabled the formulation of empirical regularities, also called growth laws [3], relating the macromolecular composition of the cell to the growth rate [4,5]. A classical example is the linear relation between the growth rate and the fraction of ribosomal versus total protein, a proxy for the ribosome concentration, over a large range of growth rates [6–9]. The reported regularities between the growth rate and the macromolecular composition of the cell are empirical correlations and should not be mistaken as representing a causal determination of cellular composition by the growth rate [6,10]. In fact, it has been shown that, for certain combinations of media, the same growth rate of E. coli may correspond to different ribosome concentrations [6].
To unravel causal relations, it is necessary to go beyond correlations and consider the biochemical processes underlying microbial growth. These processes notably include the enzyme-catalysed transformation of substrates into precursor metabolites, the conversion of these precursors into macromolecules by the gene expression machinery, the replication of the cell when its macromolecular content has attained a critical mass and the regulatory mechanisms on different levels controlling these processes [11–14]. Moreover, for identifying causality, a dynamic perspective on microbial growth focusing on transitions between different states of balanced growth, and the time ordering of events during the transitions, is more informative than considering a population at steady state [10]. Whereas most measurements have been obtained under conditions of balanced growth, in which experiments are easier to control and reproduce, data on transitions from one state of balanced growth to another are also available in the literature (reviewed in [4]). One classical example is the measurements of the temporal ordering at which RNA, protein and DNA attain their new steady-state concentrations after a nutrient upshift [5,15]. Recent experimental technologies, allowing gene expression and metabolism to be monitored in real time, have opened new perspectives for studying the dynamics of bacterial growth on the molecular level [16,17].
The large and complex networks of biochemical reactions enabling microbial growth have been mapped in great detail over the past decades and, for some model organisms, much of this information is available in structured and curated databases [18,19]. While a huge amount of knowledge has thus accumulated, a clear understanding of the precise role played by individual constituents and mechanisms in the functioning of the system as a whole has remained elusive. For example, it is well known that in the enterobacterium E. coli the concentration of the second messenger cAMP increases when glycolytic fluxes decrease, leading to the activation of the pleiotropic transcription factor Crp. However, the precise role of this mechanism in the sequential utilization of different carbon sources by E. coli remains controversial [20,21].
Mathematical models have great potential for dissecting the functioning of biochemical reaction networks underlying microbial growth [22–24]. To be useful, they need to satisfy two criteria. First, they should not be restricted to subsystems of the cell, but provide an integrated view of the reaction networks, including transport of nutrients from the environment, metabolism and gene expression. In particular, they should account for the strong coupling between these functions: enzymes are necessary for the functioning of metabolism, while the metabolites thus produced are precursors for enzyme synthesis. In the words of Henrik Kacser, one of the pioneers of metabolic control analysis, ‘to understand the whole, you must look at the whole’ [25]. Second, models of microbial growth should be multilevel in the sense of expressing the growth of a population in terms of the functioning of the biochemical reaction networks inside the cells. Growth amounts to the accumulation of biomass, that is proteins, RNA, DNA, lipids and other cellular components produced in well-defined proportions from nutrients flowing into the cells. The two criteria amount to the requirement that models should capture the autocatalytic nature of microbial growth, the production of daughter cells from growth and division of mother cells.
Precursors of such integrated, multilevel models are the simple autocatalytic models of Hinshelwood, capable of displaying steady-state exponential growth and a variety of responses to perturbations reminiscent of the adaptive behaviour of bacteria [26]. Another early example is the coarse-grained model of a growing and dividing E. coli cell [27], which has evolved over the years into a model of a hypothetical bacterial cell with the minimal number of genes necessary for growing and dividing in an optimal environment [28]. In addition, we mention so-called cybernetic models describing growth of microbial cells on multiple substrates [29–31], and the E-CELL computer environment for whole-cell simulation [32]. In recent years, integrated, multilevel models of the cell have received renewed attention with the landmark achievement of a model describing all individual cellular constituents and reactions of the life cycle of the human pathogens Mycoplasma genitalium [33] and other genome-scale models of bacteria [34]. In addition, several coarse-grained models describing the relation between the macromolecular composition of microorganisms and their growth rate have been published [24,35–39].
At first sight, the above-mentioned models of microbial growth are quite diverse, in the sense that they have a different scope and granularity, make different simplifications, use different approaches to obtain predictions from the model structure and originate in different fields (microbiology, theoretical biology, biophysics and biotechnology). The aim of this review is, first, to show how a general framework for the kinetic modelling of microbial growth, including an analytical expression for the growth rate, can be mathematically derived from few basic hypotheses. Second, we show how additional simplifying assumptions lead to approximate kinetic models that do not require the biochemical reaction networks to be specified in full. The resulting models exemplify two widespread modelling approaches, flux balance analysis (FBA) and coarse-grained whole-cell modelling. The discussion of the different hypotheses and assumptions, including those related to the measurement units employed, which are often not explicit and/or buried in the (older) literature, reveals how the models are related on a deeper level. This will be instrumental for identifying their respective strengths and weaknesses as well as for indicating new directions in the study of the biochemical reaction networks underlying microbial growth.
2. Growth of microbial populations
An obvious view on microbial growth starts by considering the individual cells in a growing population (figure 1a). We denote by n(t) the number of cells at time t (h). Individual cells in a temporal snapshot of the population have different sizes, as they are in different stages between birth and division. Moreover, cell sizes at birth and division are different [40–42]. As a consequence, the size of the cells in a population at time t is best described by a statistical distribution. This distribution may change over time and with the experimental conditions. For instance, in conditions supporting a higher growth rate, the average size of the cell in the population is larger [6,43]. Several models of the cell size distribution and its dependence on the experimental conditions have been proposed, based on different hypotheses about the criterion determining when a cell divides (reviewed in [42,44]). When the size distribution is known at every time t, the number of cells in a growing population can be directly used to estimate the volume of the population.
Figure 1.
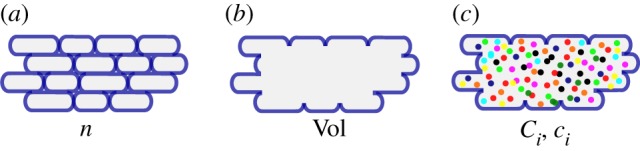
(a) Population of n growing cells with different sizes. (b) Volume Vol of a growing population of cells. (c) Total mass Ci and concentration ci of molecular constituents i in a population with volume Vol (each constituent is indicated by a different colour).
In what follows, however, we will adopt another point of view and ignore the individual cells making up a population. Instead, we directly quantify the growing population in terms of its expanding volume Vol (l) (figure 1b), that is, the sum of the volumes of the cells in the population. This aggregate description is appropriate when one is interested in concentrations of molecular constituents on the population level rather than in individual cells, as in the kinetic models developed below. Moreover, it corresponds to most data available in the experimental literature, obtained by pooling the contents of all cells in a (sample of the) population.
We model the growth of a population of microorganisms by means of a deterministic ordinary differential equation (ODE):
| 2.1 |
that is, the growth rate μ (h−1) of the population is defined as the relative increase of the volume of the population. Both Vol and μ are functions of time t (h). For a constant steady-state growth rate μ = μ*, we obtain the following explicit solution of equation (2.1): Vol(t) = Vol(0) · eμ* · t, where Vol(0) represents the initial population volume. The doubling time of a population with a growth rate μ* is given by t1/2 = ln2/μ*. This is a direct consequence of the solution of equation (2.1), which stipulates that Vol(t1/2) = 2Vol(0) = Vol(0) · eμ* · t1/2, and therefore ln2 = μ* · t1/2.
The growth rate as defined by equation (2.1) is sometimes also called specific growth rate, in order to indicate that it concerns the increase in population volume per unit of population volume (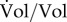 ), instead of the absolute increase in population volume (
), instead of the absolute increase in population volume (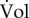 ). In what follows, we will drop the qualifier ‘specific’. The growth rate definition of equation (2.1) should be distinguished from another definition of the growth rate as 1/t1/2, that is, the number of doublings of the population volume per time unit. While the two definitions result in a quantity with the same unit, they do not mean the same thing and differ by a factor of ln2 [4]. Below, we use the growth rate definition of equation (2.1).
). In what follows, we will drop the qualifier ‘specific’. The growth rate definition of equation (2.1) should be distinguished from another definition of the growth rate as 1/t1/2, that is, the number of doublings of the population volume per time unit. While the two definitions result in a quantity with the same unit, they do not mean the same thing and differ by a factor of ln2 [4]. Below, we use the growth rate definition of equation (2.1).
Models that do not distinguish individual cells but lump them into an aggregate volume have been called non-segregated as opposed to segregated models that do make this distinction [45–47]. If the population is composed of cells with the same growth rate, not much is lost by ignoring individual cells and using the population-level description of equation (2.1) (see the electronic supplementary material). There are situations, however, in which this assumption is not appropriate and in which essential features of the growth kinetics are shaped by the heterogeneity of the population [48–51]. For example, it was recently proposed that the lag observed in diauxic growth of E. coli on a glycolytic and gluconeogenic carbon source (e.g. glucose and acetate) is due to the responsive diversification of the population into two subpopulations upon the depletion of the (preferred) glycolytic carbon source and that only one of these subpopulations continues growth on the gluconeogenic carbon source [49]. Non-segregated models are obviously not suitable for describing such phenomena and models describing the dynamics of the distribution of individual cells in a population or of subpopulations need to be used instead.
3. Volume and macromolecular content of cells
The model of equation (2.1) is unstructured in the sense that it does not take into account the biochemical processes enabling cells to grow. By contrast, so-called structured models [45–47] explicitly describe molecular constituents of the cell and the biochemical reactions in which they are involved. Let Ci (g) be the (dry) mass of molecular constituent i contained in volume Vol (figure 1c). A common assumption supported by experimental data ([52] and references therein) is that the volume of the population is proportional to the biomass, that is, the total mass of the molecular constituents of the cells:
| 3.1 |
with B (g) the biomass. Another way to frame the assumption is to say that the biomass density is constant. In other words,
| 3.2 |
where 1/δ (g l−1) denotes the constant biomass density. For bacterial cells, the cytoplasmic biomass density has a value of about 300 g l−1 [53,54], meaning that 70% of the cell content is water. Macromolecules make up most of the biomass. For E. coli, Bremer & Dennis [6] conclude that the sum of protein, RNA and DNA accounts for between 65% and 73% of the total cellular dry mass, depending on the growth rate, whereas Basan et al. [55] report a stable proportion of approximately 90%. In all of these cases, protein constitutes the largest mass fraction.
Consistent with the decision above to consider the population as a non-segregated volume, we define the concentration ci (g) of each molecular constituent i in a population as
| 3.3 |
If the cells all have the same concentration of constituent i, that is, if molecules are evenly distributed between the cells, then ci also applies to the individual cells (see the electronic supplementary material). While this is a suitable approximation in many cases, there are also situations where variability of enzyme and metabolite concentrations occurs and may lead to a heterogeneous population of cells with different growth phenotypes [50,56].
An immediate consequence of the above definition is the following relation:
| 3.4 |
In words, the assumption of the proportionality of volume and biomass implies that the total concentration of molecular constituents 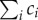 in a growing cell population is constant. While this corresponds to measurements for balanced growth, not many data are available for growth transitions (but see [57]).
in a growing cell population is constant. While this corresponds to measurements for balanced growth, not many data are available for growth transitions (but see [57]).
The dynamics of each molecular constituent i are modelled by means of an ODE, obtained from equations (2.1) and (3.3):
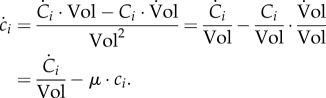 |
3.5 |
Note that a dilution term due to the growth of the population appears in the equation describing the dynamics of ci. As a consequence, if the mass of a specific molecular constituent i remains constant (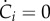 ), but the population continues to grow (μ > 0), its concentration decreases (
), but the population continues to grow (μ > 0), its concentration decreases (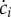 ), as intuitively expected.
), as intuitively expected.
The growth rate itself is directly connected to the concentrations of the molecular constituents, because
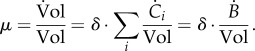 |
3.6 |
Therefore, while it makes sense for a specific constituent i to dilute out when it is not produced, no growth dilution occurs if the mass of all molecular constituents remains constant (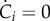 for all i). In the latter case, it follows from equation (3.6) that the growth rate is 0 by definition.
for all i). In the latter case, it follows from equation (3.6) that the growth rate is 0 by definition.
It is increasingly realized that growth dilution may have important physiological consequences [52,58,59] and therefore cannot be neglected in mathematical models of cellular processes. In particular, the interaction of a synthetic circuit with the growth physiology of the cell, and the changes in the growth rate this entails, may have an unexpected nonlinear feedback on the dilution of transcription factors and thus on the functioning of the circuit. This was illustrated by a synthetic circuit in E. coli in which the alternative T7 RNA polymerase regulates itself and a fluorescent protein. Expression of the fluorescent protein causes a metabolic burden, impairing growth and thus growth dilution of T7 RNA polymerase. The resulting positive feedback was shown to lead to two different phenotypes: growth and growth arrest [59].
An important special case of microbial growth occurs when the growth rate and the concentrations of the individual molecular constituents are constant over time, that is, μ = μ* and ci = c*i, for all i. From ci = Ci/Vol = c*i it follows that a doubling of the volume Vol of the population is accompanied by a doubling of the mass Ci of each molecular constituent, which explains why this situation of steady-state exponential growth is also referred to as balanced growth [2,60].
4. Biochemical reactions underlying microbial growth
The molecular constituents of the cell are continually produced and consumed by biochemical reactions. Many of these reactions are enzyme-catalysed, such as the metabolic reactions involved in the conversion of nutrients from the environment into building blocks for macromolecules (amino acids, nucleotides) and energy carriers (ATP, NADH). The building blocks and energy are consumed in large part by the transcription and translation reactions producing macromolecules. The metabolic reactions together form the metabolic network of the cell [14,61].
The term 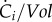 in equation (3.5) represents the net effect of the biochemical reactions on the concentration of molecular constituent i, separate from growth dilution. Usually, for intracellular reactions, the quantities of molecular constituents are expressed in molar rather than mass units. Hence, we introduce Xi = Ci/αi, with Xi (mol) the molar quantity of constituent i and αi (g mol−1) the molar mass of i. The reason for this change in units is that kinetic models of biochemical reactions are based on the physical encounters of molecules in the cell [62,63], which is best expressed in terms of molar quantities. With this unit conversion, and xi = Xi/Vol, equation (3.5) becomes
in equation (3.5) represents the net effect of the biochemical reactions on the concentration of molecular constituent i, separate from growth dilution. Usually, for intracellular reactions, the quantities of molecular constituents are expressed in molar rather than mass units. Hence, we introduce Xi = Ci/αi, with Xi (mol) the molar quantity of constituent i and αi (g mol−1) the molar mass of i. The reason for this change in units is that kinetic models of biochemical reactions are based on the physical encounters of molecules in the cell [62,63], which is best expressed in terms of molar quantities. With this unit conversion, and xi = Xi/Vol, equation (3.5) becomes
| 4.1 |
The term 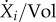 can be further developed by explicitly accounting for the reactions producing and consuming the ith molecular constituent. Consider the jth reaction, in which constituent i participates with stoichiometry Nij, that is, reaction j produces a net change of Nij molecules of constituent i. If the reaction produces constituent i, then Nij > 0, whereas if it consumes constituent i, then Nij < 0 (if constituent i is not altered in the reaction, then Nij = 0). We define Ni as the (row) vector of stoichiometry coefficients of constituent i for all reactions in the system. Moreover, we define the (column) vector of reaction rates v, such that vj is the rate of the jth reaction (mol l−1 h−1).
can be further developed by explicitly accounting for the reactions producing and consuming the ith molecular constituent. Consider the jth reaction, in which constituent i participates with stoichiometry Nij, that is, reaction j produces a net change of Nij molecules of constituent i. If the reaction produces constituent i, then Nij > 0, whereas if it consumes constituent i, then Nij < 0 (if constituent i is not altered in the reaction, then Nij = 0). We define Ni as the (row) vector of stoichiometry coefficients of constituent i for all reactions in the system. Moreover, we define the (column) vector of reaction rates v, such that vj is the rate of the jth reaction (mol l−1 h−1).
With the help of the above concepts, the effect of the biochemical reactions on the concentrations of molecular constituents can be rewritten as
| 4.2 |
or in more compact form, denoting the (column) vector of the concentrations of all molecular constituents by x:
| 4.3 |
This is the classical formulation of stoichiometry models of biochemical reactions, extended with a dilution term [62,64]. Equation (4.3) does not explicitly take into account that the reaction rates v depend on the concentrations of the molecular constituents participating in the reactions. That is, it would be more appropriate to write v in functional form v(x). The model of equation (4.3) describes the biochemical reaction system on the population level. If all cells have the same reaction rates, then the model applies also to the individual cells (see the electronic supplementary material). It should be noted though that reaction rates may differ between cells, even when the concentrations x of cellular constituents are identical, due to the intrinsic stochasticity of biochemical reactions [63].
As a consequence of the conversion of Ci to Xi and the introduction of reaction stoichiometries, the growth rate becomes,
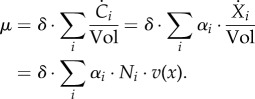 |
4.4 |
The growth rate thus equals the sum of all reaction rates in mass units (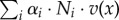 (g l−1 h−1)), that is the net rate of accumulation of intracellular molecular constituents within a unit volume per unit time, relative to the total amount of molecular constituents within a unit volume (1/δ (g l−1)). The latter quantity can equivalently be written as
(g l−1 h−1)), that is the net rate of accumulation of intracellular molecular constituents within a unit volume per unit time, relative to the total amount of molecular constituents within a unit volume (1/δ (g l−1)). The latter quantity can equivalently be written as 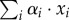 , following equation (3.4) and xi = ci/αi.
, following equation (3.4) and xi = ci/αi.
Combining all of the above, we obtain the following model for a growing microbial population:
| 4.5 |
and
| 4.6 |
We emphasize that the explicit expression for μ in equation (4.6) is not an ad hoc definition, but mechanically follows from the basic modelling assumptions underlying the stoichiometry model of equation (4.5), notably the assumption of constant biomass density. Figure 2a schematically projects the reaction network on a growing microbial population.
Figure 2.
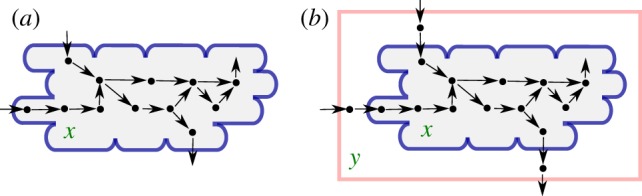
(a) Population of cells with volume Vol growing at a rate μ, described by the model of equations (4.5) and (4.6). The reactions fuelling growth involve intracellular constituents with concentrations x. The dots represent the molecular constituents and the arrows biochemical reactions. (b) Idem, but extended with a bioreactor environment from which the cells take up nutrients and into which they excrete by-products (with concentrations y). This extended system is described by equations (5.4)–(5.7). The boundary of the environment is schematized by the pink rectangle.
Textbooks on the modelling of biochemical reaction systems detail the different rate laws that specify how the reaction rates vj depend on the concentrations x [62,64]. A common choice, relying on first principles, is to assume mass–action kinetics for the reactions, based on the random encounter of molecules in a well-mixed volume [62,63]. In many situations, however, it is more convenient to lump individual reactions into aggregate reactions that are described by approximate rate laws such as (reversible and irreversible) Henri–Michaelis–Menten kinetics, Monod–Wyman–Changeux kinetics, Hill kinetics, etc. [62,64]. The Henri–Michaelis–Menten rate law for an irreversible, enzyme-catalysed reaction with substrate concentration x and enzyme concentration e reads: 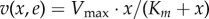 , with
, with 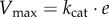 , where kcat (min−1) is the so-called catalytic constant of the enzyme, quantifying the maximum number of substrate molecules converted per enzyme per minute. This expression, and many other approximate kinetic rate laws, can be derived from mass–action kinetics when making appropriate assumptions on the time scale of the rate of the elementary reaction steps. In the case of the Henri–Michaelis–Menten rate law, this concerns the association/dissociation of enzyme and substrate and the formation of the product [65,66].
, where kcat (min−1) is the so-called catalytic constant of the enzyme, quantifying the maximum number of substrate molecules converted per enzyme per minute. This expression, and many other approximate kinetic rate laws, can be derived from mass–action kinetics when making appropriate assumptions on the time scale of the rate of the elementary reaction steps. In the case of the Henri–Michaelis–Menten rate law, this concerns the association/dissociation of enzyme and substrate and the formation of the product [65,66].
5. Growth in a changing environment
Some of the reactions changing the molecular constituents of the cell correspond to exchanges with the environment, that is the uptake of substrates and the excretion of products. The environment is not explicitly modelled by equations (4.5) and (4.6) and the entries in v corresponding to the rates of these exchange reactions are therefore treated as external inputs. For many purposes, however, it is more appropriate to extend the model and include a (simple) representation of the environment. In what follows, we equate the environment with a bioreactor filled by a liquid medium of fixed volume containing the growing population of microorganisms as well as external substrates and products. The substrate and product concentrations in the medium are denoted by the vector y. Usually, external concentrations are expressed in terms of units g l−1, that is mass in a fixed volume of medium.
The dynamics of the substrate and product concentrations in the medium can be described by the following differential equation:
| 5.1 |
where E is the stoichiometry matrix for the exchange reactions, αy is the diagonal matrix of molar mass coefficients of the external metabolites (g mol−1) and Volmedium is the (constant) volume of the medium (l). Usually, Vol≪Volmedium. The multiplication of αy · E · v(x, y) by Vol expresses the fact that the total rate of consumption of substrates and accumulation of products depends on the volume of the growing microbial population. The division of the resulting product by Volmedium means that we are interested in the concentration of these substrates and products in the medium. Equation (5.1) can be rewritten in a more classical form by explicitly using the biomass variable B (g), introduced in the previous section, and the concentration of biomass in the medium b (g l−1), defined as b = B/Volmedium. It follows with equation (3.2) that
| 5.2 |
and, consequently,
| 5.3 |
The above considerations lead to the following extended model, taking into account the dynamics of exchanges with the environment (figure 2b):
| 5.4 |
| 5.5 |
| 5.6 |
and
| 5.7 |
where we have used the equalities
| 5.8 |
to obtain the biomass differential equation. For some purposes, it is useful to split the reaction rate vector v(x, y) into rates of exchange reactions vex(x, y) and rates of internal reactions vint(x), where obviously the latter do not depend on the concentration of external substrates.
Interestingly, the above model can be used to derive an explicit relation between growth rate and substrate availability. A key insight for the derivation is that due to coupling of the molar mass coefficients and the stoichiometry coefficients, the expressions for the internal reaction rates in the right-hand side of equation (5.6) cancel out. Consider an arbitrary internal reaction, irreversibly converting one molecule of reactant A into nab molecules of reactant B (with molar masses αa and αb, respectively) at a rate vab. Note that the reaction rate vab occurs twice in the sum of equation (5.6): −αavab (for reactant A) and αbnabvab (for reactant B). However, due to mass conservation, we must have αbnab = αa, so that the two terms in the sum cancel out. Extending this argument to every internal reaction gives
| 5.9 |
where Ek denotes the kth row of E, corresponding to external metabolite k, and αy,k the kth diagonal element of αy. In words, the only remaining terms are the rates of the exchange reactions, because they occur only once in the sum of equation (5.6). The minus sign in −E is explained by the fact that, for uptake reactions, the sum of equation (5.6) includes the increase of intracellular biomass components rather than the decrease of extracellular metabolites (the opposite for excretion reactions). Note that it follows from equations (5.3), (5.8) and (5.9) that 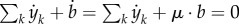 , expressing mass conservation.
, expressing mass conservation.
Furthermore, assume that the exchanges of the cells with the environment can be reduced to the uptake of a single substrate S, used for the production of biomass. The concentration of the substrate in the medium is denoted by s, its molar mass αs and its uptake rate vs. Note that, in this case, y = s, αy = αs and E = − 1, so that we obtain μ = δ · αs · vs(x, s). That is, the growth rate is directly proportional to the substrate uptake rate, a relation sidestepping the biochemical reactions taking place inside the cells. If we further choose a saturating function for the uptake kinetics, 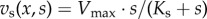 , we obtain the so-called Monod equation [67]
, we obtain the so-called Monod equation [67]
| 5.10 |
with 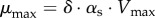 . The Monod equation, which has the same mathematical form as the Henri–Michaelis–Menten rate law, is a well-known phenomenological relation that has been shown to fit quite well data of the steady-state growth rate of bacteria as a function of a single growth-limiting substrate [3,67]. More complex uptake patterns may occur when several substrates are available [68–71]. While in many bacteria the availability of a preferred carbon source represses the utilization of other, secondary carbon sources, a phenomenon known as carbon catabolite repression (CCR) [20], low growth rates or mixtures of secondary carbon sources without the preferred carbon source may disable CCR and lead to the co-utilization of different carbon sources.
. The Monod equation, which has the same mathematical form as the Henri–Michaelis–Menten rate law, is a well-known phenomenological relation that has been shown to fit quite well data of the steady-state growth rate of bacteria as a function of a single growth-limiting substrate [3,67]. More complex uptake patterns may occur when several substrates are available [68–71]. While in many bacteria the availability of a preferred carbon source represses the utilization of other, secondary carbon sources, a phenomenon known as carbon catabolite repression (CCR) [20], low growth rates or mixtures of secondary carbon sources without the preferred carbon source may disable CCR and lead to the co-utilization of different carbon sources.
In equations (5.4)–(5.7) it is implicitly assumed that the only changes in the concentrations of substrates and products in the environment occur through exchanges with the growing microbial population, making it an instance of a batch culture. The model can be easily adapted to other environments, such as continuous culture or fed-batch culture [72,73]. In a continuous culture, a fixed amount of medium per time unit, including microbial cells, is replaced by fresh medium, whereas in a fed-batch culture, nutrients are added over time without removing spent medium (and Volmedium is no longer constant). While these different bioreactor regimes have been mostly used in the context of biotechnological applications, it is interesting to remark that complex natural environments, such as the digestive tracts of vertebrates and insects, can profitably be modelled as coupled series of bioreactors [74,75].
Equations (5.4)–(5.7) form a self-consistent kinetic model of a growing microbial population, taking up nutrients from the environment, converting these into biomass, and excreting by-products. In theory, the model is capable of accomodating all internal reactions and reactions exchanging substrates and products with the environment, from enzymatic reactions to signalling pathways and transcription and translation. Some of the examples of whole-cell models mentioned in the introduction can be seen, to some extent, as instances of this general scheme [28,32]. In practice, such models are not easy to build though. They quickly become very complex to handle, with hundreds of reactions and molecular constituents whose concentrations evolve on very different time scales. Moreover, many of the parameter values will be unknown or known only within an order of magnitude, creating difficult model identification problems [76–78].
6. Connecting metabolism and growth: flux balance analysis
The practical difficulties encountered when dealing with large kinetic models of microbial cells have motivated approximate models that are based on a number of simplifying assumptions. One well-known example are so-called FBA approaches [79–81]. Below we summarize how flux balance models can be obtained from the general modelling framework of equations (5.4)–(5.7), by progressively introducing additional modelling assumptions.
A first simplifying assumption consists in limiting the scope of the models to metabolism alone, disregarding proteins and other macromolecules. It may seem somewhat paradoxical to exclude the major constituents of biomass from a model of microbial growth, but equation (3.6) can be replaced with a new definition of the growth rate, based on the rate of consumption of biomass precursor metabolites. To this end, similar to what was proposed in a recent review of FBA [79], we distinguish between free metabolites and the same metabolites incorporated into proteins and other macromolecules. The former, with concentration vector xM, are included in the model, whereas the latter, with mass vector C′M, are not, although they will be used in the derivation of the model (figure 3). The biomass B (g) is assumed to consist only of the mass of metabolites incorporated into proteins and other macromolecules, that is, 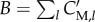 where l runs over the incorporated metabolites. For reasons of consistency, we also restrict δ, the inverse biomass density, to these incorporated metabolites. In agreement with the above, we define a new vector vM, consisting of the rates of the exchange reactions and the reactions that produce metabolites in xM, as well as the corresponding stoichiometry matrix NM.
where l runs over the incorporated metabolites. For reasons of consistency, we also restrict δ, the inverse biomass density, to these incorporated metabolites. In agreement with the above, we define a new vector vM, consisting of the rates of the exchange reactions and the reactions that produce metabolites in xM, as well as the corresponding stoichiometry matrix NM.
Figure 3.
Population of cells growing in a bioreactor with metabolic reactions that involve free metabolites with concentrations xM and metabolites incorporated into biomass with masses C′M. The dots represent the molecular constituents and the arrows biochemical reactions. The dashed reactions represent the incorporation of free metabolites into the biomass. This extended system is described by modifying equations (5.4)–(5.7) with new expressions for the steady-state dynamics of the metabolic network (equation (6.5)) and the growth rate (equation (6.1)).
The coefficients βl = C′M, l/B represent the mass fractions of the incorporated precursor metabolite in the biomass. By definition, βl ≥ 0 and 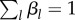 , and we further suppose, as a second simplifying assumption, that these mass fractions are constant. The biomass composition has been empirically determined for several microorganisms, usually for a specific growth condition [82–84]. The incorporation of the precursor metabolites into the biomass, in the proportions βl in which they compose the latter, can be seen as a macroreaction. To unambiguously define this macroreaction, we introduce the reaction rate vector v′M, which describes the rate of incorporation into proteins and other macromolecules of the (free) metabolites. More precisely, v′M, l (mol l−1 h−1) represents the rate of incorporation of the metabolite having concentration xM, l. Many of the rates v′M, l will be 0, because the corresponding metabolites are not included in the biomass (βl = 0). In principle, the degradation of macromolecules back to precursor metabolites would lead to additional reaction rates, but, given that proteins, the main component of biomass, are usually stable on the time scale of interest [85,86], the reverse reactions are ignored here.
, and we further suppose, as a second simplifying assumption, that these mass fractions are constant. The biomass composition has been empirically determined for several microorganisms, usually for a specific growth condition [82–84]. The incorporation of the precursor metabolites into the biomass, in the proportions βl in which they compose the latter, can be seen as a macroreaction. To unambiguously define this macroreaction, we introduce the reaction rate vector v′M, which describes the rate of incorporation into proteins and other macromolecules of the (free) metabolites. More precisely, v′M, l (mol l−1 h−1) represents the rate of incorporation of the metabolite having concentration xM, l. Many of the rates v′M, l will be 0, because the corresponding metabolites are not included in the biomass (βl = 0). In principle, the degradation of macromolecules back to precursor metabolites would lead to additional reaction rates, but, given that proteins, the main component of biomass, are usually stable on the time scale of interest [85,86], the reverse reactions are ignored here.
From the above, and from applying the general growth rate expression of equation (4.6) to the biomass constituents C′M, l, it follows that
| 6.1 |
where vB (g l−1 h−1) is defined as the rate of the biomass macroreaction, that is, the total rate of incorporation of precursor metabolite mass into biomass per unit volume of the cell population. Moreover, the dynamics of the mass of each incorporated metabolite l in the growing microbial population is given by
| 6.2 |
We also obtain from the definition of the biomass composition that
| 6.3 |
so that combining equations (6.2) and (6.3) yields an expression for the rates of the individual incorporation reactions:
| 6.4 |
In words, the rate of incorporation of each individual metabolite is proportional to the rate of the biomass reaction, modulated by the factor βl/αl.
The assumption of a constant biomass composition, leading to equation (6.4), means that the ratio of the time-varying variables C′M, l and B is constant. Hence it follows from equations (3.2) and (3.3) that the concentrations of the pools of incorporated precursor metabolites c′M, l are also constant for all l (i.e. c′M, l = C′M, l/Vol = βl/δ). This can be interpreted as assuming that any changes in a slowly varying environment lead to a rapid adjustment of the rates in the metabolic network, and consistent with this, a rapid adjustment of concentrations of the free metabolites, so as to obtain invariant steady-state concentrations of the incorporated precursor metabolites. In other words, the metabolic system is at quasi-steady state with respect to the environment [62,87]. Indeed, measured in vivo response times of many metabolite pools in E. coli are on the order of seconds to minutes [16,88], whereas the concentrations of external substrates in equation (5.5) vary on a time scale set by the growth rate when they remain well above the half-saturation constant Ks defining the uptake kinetics. As an aside, we note that constant concentrations of incorporated precursor metabolites do not exclude that the concentrations of individual enzymes, not modelled here, may vary over time [89].
When further assuming, third, that growth dilution of metabolite concentrations xM can be ignored, as its effect is negligible with respect to the turn over of metabolite pools by enzyme-catalysed reactions, we obtain the following modification of the stochiometry model of equation (5.4), now restricted to the metabolic network and the consumption of biomass precursor metabolites by the biomass reaction:
| 6.5 |
where NB = ( … , − βl/αl, … )T. The quasi-steady-state value of the metabolite concentrations is indicated by an asterisk (*).
A fourth key simplification underlying FBA, in line with the quasi-steady state of metabolism, is to ignore the kinetics of the reactions and consider only fluxes, that is reaction rates at steady state. As a consequence, the explicit dependence of fluxes on concentrations disappears from the model and the fluxes become the new variables of the system:
| 6.6 |
where we have dropped the steady-state symbol (*) from the fluxes.
Equation (6.6) is a linear system that is usually degenerate, in the sense that the number of rows in the matrix (NM NB) is much smaller than the number of columns. As a consequence, the system does not have a unique, but an infinite number of solutions, given by the kernel of the stoichiometry matrix, ker(NM NB) [90]. Hence, an infinite number of flux distributions satisfy the stoichiometry constraints. The space of solutions can be reduced by taking into account additional inequality constraints on the fluxes, obtained (directly or indirectly) from measurements:
| 6.7 |
where 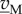 and
and 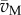 are lower and upper bounds on the fluxes, respectively.
are lower and upper bounds on the fluxes, respectively.
One specific case of interest are measurements of the uptake and excretion fluxes vM, ex. If these measurements are sufficiently precise, then a subset of solutions may be obtained in which the possible values for intracellular, non-measured fluxes remain within tight bounds. This approach, called (stoichiometric) metabolic flux analysis (MFA) [91], underlies, for example, the analysis of the influence of a post-transcriptional regulator, CsrA, on the flux distribution in central carbon metabolism in E. coli [92]. From measurements of the uptake and excretion fluxes of wild-type and mutant strains growing on glucose, estimates of glycolytic fluxes were obtained that, combined with measurements of metabolite pools and gene expression, allowed one to pinpoint the effect of CsrA on the activity of PfkA, a central glycolytic enzyme. If measurements are reduced to exact values, that is if 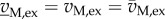 , then the addition of the corresponding equality constraints may under certain conditions lead to a unique solution of equation (6.6) [93].
, then the addition of the corresponding equality constraints may under certain conditions lead to a unique solution of equation (6.6) [93].
While flux measurements can thus be used to reduce the solution space, in many cases this is not enough to obtain sufficiently informative predictions of intracellular fluxes. One way to proceed is to select within the remaining set of solutions those that satisfy some optimization criterion, an approach called FBA [80,94]. The most frequently chosen criterion is the maximization of the growth rate. The choice of this criterion is based on the argument that a higher growth rate provides a selective advantage to microorganisms, because it allows competitors for shared resources to be outgrown. In our case, following equation (6.1), the growth rate is proportional to the rate of the biomass reaction, so that growth-rate maximization results in a linear optimization problem:
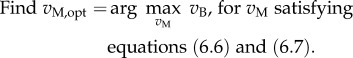 |
6.8 |
FBA has been used in many applications [95], such as predicting growth rates of E. coli on different carbon sources [96] and in different mutants before and after adaptive evolution [97].
Various extensions of classical FBA as summarized by equation (6.8) have been proposed in the literature. For our purpose, a relevant extension is dynamic FBA. In this case, the solution of the FBA problem is embedded in a model of the dynamically changing environment, such that the concentration of external metabolites y provides constraints on the fluxes:
| 6.9 |
In particular, nutrient uptake fluxes depend on the concentration of external metabolites. This dependence may, for example, follow a Henri–Michaelis–Menten rate law, as proposed in the previous section. Following the convention that uptake fluxes are negative, an uptake flux in vM, involving external metabolite k, will typically have an upper bound 0 and a lower bound −ky,k · yk/(Ky,k + yk), where ky,k (mol l−1 h−1) is the maximum uptake rate of external metabolite k, and Ky,k (mol l−1) is its half-saturation constant. In dynamic FBA, in particular the so-called static optimization variant [98], at each time point t with a specific value of y = y(t), the following linear optimization problem is solved:
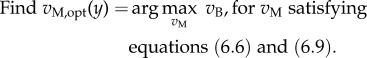 |
6.10 |
The resulting values of the flux distribution vM, opt(y), and the flux of the biomass reaction vB, opt(y) leading to the maximal growth rate δ · vB, opt(y), enter the model of the dynamically changing environment
| 6.11 |
and
| 6.12 |
Notice that, in general, the flux distribution vM, opt(y) is not unique. To make the problem well-posed, additional criteria for selecting optimal solutions need to be specified. To this end, approaches to sample the set of possible flux distributions in a computationally efficient and biologically meaningful manner have been developed [99,100]. Other approaches explore the set of possible solutions by tying its geometry to the structure of the underlying reaction network [101,102].
The main limitation of FBA and dynamic FBA is that these approaches require strong assumptions to be made. To compensate for the absence of kinetic information, cells are hypothesized to optimize a specific objective function, here the growth rate. In many cases the use of growth-rate maximization is debatable [103,104] and it is not straightforward to specify in advance which alternative objective criterion is appropriate. The focus on metabolism excludes proteins and other macromolecules from the model. The absence of these major biomass constituents requires the definition of a new biomass reaction, which comes with additional assumptions on the dynamics of metabolite concentrations. Moreover, FBA models occlude the fundamental autocatalytic nature of the cell, in the sense that the products of metabolism are utilized for synthesizing proteins that in turn control metabolic reactions as well as transcription and translation processes [105]. While a number of extensions of FBA have been proposed in the literature [34,106–112], these do not entirely make up for the above-mentioned limitations.
7. Connecting gene expression, metabolism and growth: coarse-grained whole-cell models
Another way to sidestep the full complexity of the metabolic and gene regulatory networks controlling microbial growth is to preserve the modelling scheme of equations (5.4)–(5.7), but to simplify the equations in a different way. The kinetics of the reactions, and notably the regulatory interactions shaping the kinetics, are no longer ignored, as in the previous section. However, instead of accounting for individual molecular constituents of the cell, these are lumped into a few classes of constituents with their corresponding macroreactions. These approximations result in a model with the same scope, but that provides a more coarse-grained picture of the cell.
An example of this approach are so-called self-replicator models. These models provide a high-level description of the functions involved in the growth of a population, notably the conversion of external substrates into metabolic precursors (metabolism) and the synthesis of macromolecules, notably proteins, from these precursors (gene expression). The self-replicatory nature of the system originates in the catalytic role of the proteins in both metabolism (enzymes) and gene expression (RNA polymerase, ribosome). The principle of self-replicator models of microorganisms can be found in the work of Hinshelwood [26], Gánti [113] and Koch [114], to cite some early examples. More recently, Molenaar et al. [37] used self-replicator models as an analytical tool for explaining the phenomenon of overflow metabolism in various bacteria. They proposed that this wasteful excretion of carbon sources during fast growth arises from a trade-off between what the authors call metabolic efficiency (high production of precursors per unit substrate) and catabolic efficiency (high production of precursors per unit enzyme).
An example of a self-replicator system is shown in figure 4. In this case, following the scheme of equations (5.4)–(5.7), y = s represents the concentration of an external substrate, and x = (p, r, m)T the concentrations of precursor metabolites P, ribosomes and other components of the gene expression machinery R, and enzymes M, respectively. The entries of the reaction rate vector v = (vp, vr, vm)T denote the substrate uptake rate, enzyme production rate and ribosome production rate, respectively. With these substitutions, the general model of equations (5.4)–(5.7) can be rewritten as
 |
7.1 |
| 7.2 |
| 7.3 |
and
| 7.4 |
where np, nr, nm are stoichiometry constants, and αs and αp (g mol−1) are the molar mass coefficient of substrate and precursor molecules, respectively. We also introduce αm and αr, the molar mass coefficients of the components of the metabolic and gene expression machinery, respectively. The expression for the growth rate is obtained from mass conservation, which implies (as explained in §5) that αp · nr = αr and αp · nm = αm.
Figure 4.
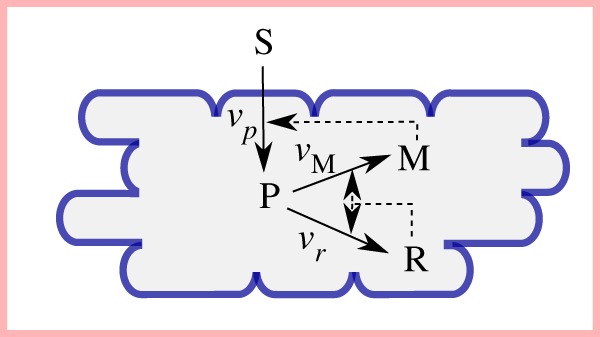
Self-replicator model of bacterial growth, corresponding to the kinetic model of equations (7.1)–(7.4) with three macroreactions describing the conversion of external substrate (S) into metabolic precursors (P) which are used for the synthesis of ribosomes and other components of the gene expression machinery (R) and enzymes making up the metabolic machinery (M) (adapted from [36]). M enables the conversion of external substrates into precursors, while R is responsible for the production of M and R itself. The (auto)catalytic activity of the metabolic machinery and the gene expression machinery thus allows the cell to replicate its protein contents, the major constituent of biomass. Solid arrows represent material flows and dashed arrows regulatory interactions.
Note that, like in the previous section, protein degradation is ignored in the model, motivated by the observations that the half-lives of proteins are usually sufficiently long to be ignored on the time scale of interest. Moreover, the only macromolecules we consider are proteins, thus excluding RNA and DNA. This is motivated by the fact that the mass fraction of RNA and DNA is limited, maximally approximately 20% in E. coli [6], but it should be remarked that the gene expression machinery includes ribosomal RNA in addition to ribosomal proteins.
Equation 7.3, the expression for the growth rate, can be further analysed by making some additional assumptions beyond the fundamental hypothesis of constant biomass density [36]. Neglecting the contribution of the metabolic precursors to the biomass, we obtain from equation (3.2) that
| 7.5 |
where R + M is the total amount of protein (in units g). As R = αr · r · Vol and M = αm · m · Vol, it follows from equation (7.5) that αr · r + αm · m = 1/δ and therefore 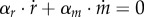 . The equations describing the dynamics of r and m are therefore not independent, and one of them may be dropped from the system of equation (7.1). Moreover, substituting the expressions for
. The equations describing the dynamics of r and m are therefore not independent, and one of them may be dropped from the system of equation (7.1). Moreover, substituting the expressions for 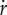 and
and 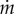 into
into 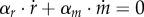 , and using the equalities between the stoichiometry constants and the molar mass coefficients due to mass conservation, allows us to obtain an insightful approximate expression for the growth rate:
, and using the equalities between the stoichiometry constants and the molar mass coefficients due to mass conservation, allows us to obtain an insightful approximate expression for the growth rate:
| 7.6 |
| 7.7 |
That is, the growth rate equals the total mass of protein synthesized per unit time and unit volume, or equivalently the total mass of precursors consumed for protein synthesis per unit time and unit volume (αp · (nr · vr + nm · vm) (g l−1 h−1)), normalized by the total mass of protein per unit volume (1/δ (g l−1)).
In what follows, we will write vps = nr · vr + nm · vm for the total protein synthesis rate (mol l−1 h−1). Furthermore, we introduce the following kinetic expressions for vps and vp:
| 7.8 |
and
| 7.9 |
where kr, km are catalytic constants (min−1) and Kr, Km half-saturation constants (mol l−1). Note that m, while not explicitly included in the model, is given by the conservation equation 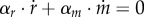 .
.
Giordano et al. [36] set nr · vr = λ · vps and nm · vm = (1 − λ) · vps, for 0 ≤ λ ≤ 1, and by means of the above expressions for vps and vp, the value of λ resulting in the maximum growth rate during steady-state exponential growth was determined. The empirical regularities relating the growth rate to the ribomal protein mass fraction [24] could thus be reproduced. The analysis can be generalized to the situation where the system is not in steady state, but makes a transition from one state of balanced growth to another following a nutrient upshift. In this case, λ is not constant, but time-varying. Using concepts from optimal control theory [115], it can be shown that the λ leading to optimal biomass accumulation has a bang-bang profile, alternating periods of exclusive synthesis of R with periods of exclusive synthesis of M, until the new steady state is reached. A regulatory strategy defining λ in terms of p and r was proposed that approximates this optimal solution. Interestingly, this strategy has structural similarities with the action of the ppGpp system in E. coli, known to play an important role in growth control [116]. Several other coarse-grained models based on assumptions similar to the ones developed above can be found in the literature, all describing aggregated autocatalytic processes converting nutrients into proteins [24,35,37–39,117,118]. Some of the models are analysed from an optimization perspective, whereas others detail regulatory mechanisms controlling the growth rate in response to changes in the environment.
In the example above, coarse-graining of the microbial cell was carried out a priori, based on our understanding of the major cellular functions involved in microbial growth. An alternative to this top-down approach would be to start from an extensive characterization of the individual molecular constituents and the biochemical reactions in which they are involved and to group these together into functional modules. This bottom-up approach relies on appropriate criteria for defining modules, based on the structure or the dynamics of the network. A discussion of the wide variety of criteria proposed is beyond the scope of this review (see [119] instead). Once a modular structure of the network has been determined, however, the dynamics of each module can be described by formulating a macroreaction and defining a kinetic rate law for the macroreaction. Such an approach has been used, for example, for modelling the accumulation of lipids and carbohydrates in unicellular microalgae [120]. The modules in this study were defined by a time scale decomposition, grouping together molecular constituents that are at quasi-steady state on a given time scale (see also [121]).
The use of an abstract representation of cellular components and processes is a strength of self-replicators and other coarse-grained models, but also their limitation. It notably makes it more difficult to quantitatively account for data on the molecular level, for example perturbations of specific reactions or the addition of specific components to the growth medium. By contrast, the representation of individual biochemical reactions is a strength of FBA models discussed in the previous section. However, these models lack the dynamic feedback from gene expression and growth to metabolism that distinguishes self-replicator models. Can one imagine hybrid FBA–self-replicator models that combine the strengths of both? Given that the model simplifications underlying the two approaches are quite different, this may not be easy to achieve, although some interesting variants of FBA, including additional flux constraints derived from the catalytic activity and molecular weight of proteins, should be mentioned here [34,106,107,109,110]. An alternative strategy would be to embed a detailed kinetic model of some module of interest within a coarse-grained model of the entire cell. The latter strategy of localized fine-graining in a global coarse-grained model may strike an adequate compromise between the simultaneous needs of molecular detail, model tractability and adequacy with the experimental data.
8. Concluding remarks
The growth of microorganisms arises from the conversion of nutrients in the environment into biomass, mostly proteins and other macromolecules, by intracellular networks of biochemical reactions. The aim of this paper has been to review the literature in the context of a general modelling framework derived from basic assumptions about microbial growth and biochemical reaction networks. In particular, we have considered the cells in a population as a non-segregrated aggregate, characterized by their combined volume rather than by a distribution of individual cells. Concentrations of molecular constituents were correspondingly defined over the entire population volume and, at all times, the total mass of molecular constituents was assumed proportional to the population volume (constant biomass density). The dynamics of this system was described by a deterministic ODE model. Figure 5 summarizes some of the fundamental modelling choices underlying the modelling framework developed here [45–47].
Figure 5.
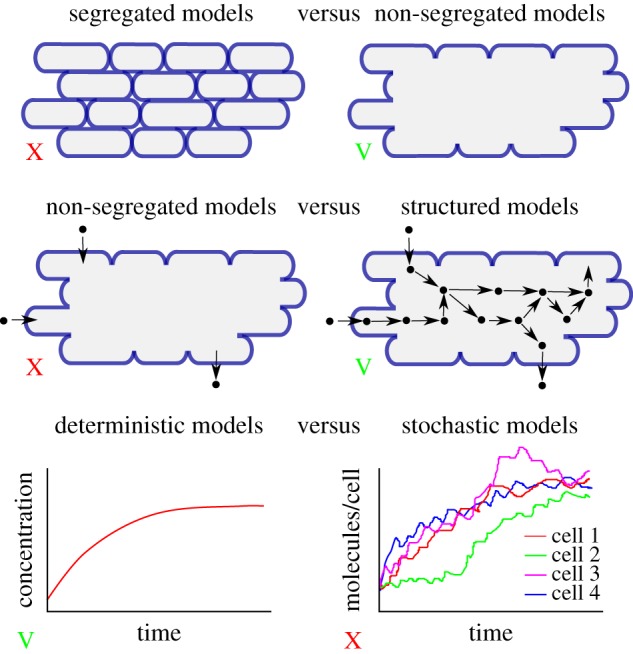
Overview of some of the fundamental modelling choices made in the development of the modelling framework of equations (5.4)–(5.7), within the range of choices proposed in [45–47]. On each dimension, the choice is indicated by a green V or red X.
The modelling framework has allowed the discussion of a broad variety of models integrating growth of microbial populations with the dynamics of the underlying reaction networks. The contribution of this paper does not so much lie in the derivation of the modelling framework, because most of the assumptions made and arguments advanced can be found in the (older) literature. Rather, the interest lies in bringing these insights together and making explicit modelling assumptions that are often forgotten or whose consequences may not always be recognized, including the careful consideration of the units of the different quantities. For example, this has brought to the fore that the first-order growth dilution term appearing in many models originates from the proportionality of the biomass and aggregate population volume. Moreover, the definition of biomass as the mass sum of the molecular consituents in the cell population was seen to lead to an explicit, analytic expression for the growth rate (instead of a heuristic definition added a posteriori). Finally, the fact that the total concentration of molecular constituents is constant contributes a constraint that can be usefully exploited for model calibration [37,38]. In general, making explicit the assumptions that underlie a model is critical for its use as a ‘logical machine’ converting assumptions about biological processes into testable predictions [122].
The focus on non-segregated, deterministic models entails a bias in that it ignores such important phenomena as transport, cell division and population heterogeneity. The existence of a lipid membrane containing proteins that allow the uptake and secretion of metabolites is one of the defining characteristics of microbial cells. A specific class of self-replicator models, sometimes referred to as protocells, addresses this issue by coupling biochemical processes inside the cell to the growth of the cell membrane, in some cases explicitly accounting for the three-dimensional cell shape and cell division [37,123,124]. The engineering of actual protocells is an interesting branch of ongoing work at the frontier of biological chemistry and biophysics [125,126], with applications in biotechnology [127]. Biomass synthesis and cell division are precisely coordinated during microbial growth [128], but the underlying mechanisms involved are still not well understood. Some variants of the above-mentioned protocell models, describing biomass accumulation and cell division in yeast on the global level, have integrated a simplified representation of the network controlling the cell cycle to provide a mechanistic basis for the synchronization of growth and division [124,129].
Population heterogeneity plays a key role in such diverse phenomena as resistance to antibiotics and biofilm formation. Heterogeneity often arises from the stochasticity of biochemical reactions, amplified by the small numbers of the cellular constituents involved in the reactions, especially in gene expression [63,130]. Stochastic models are necessary to explore bistability, the mathematical property that lies at the heart of the above-mentioned forms of population heterogeneity, but that cannot be analysed with the deterministic models discussed here. While full-scale stochastic models of the biochemical networks underlying cellular growth and division are rare, some models do introduce stochastic variables for mRNA and protein constituents [33,129]. For instance, one of the interesting aspects of the whole-cell model of M. genitalium [33] is that it combines a variety of different modelling formalisms for different cellular functions, including deterministic (FBA) models of metabolism, deterministic (ODE) models for cell division, and stochastic models for transcription, translation, and degradation of mRNA and proteins.
To a first approximation, current modelling efforts push in two directions. The first strategy attempts to construct whole-cell models that are as complete as possible, including a maximum of knowledge of cellular components and their interactions on the molecular level. The resulting models provide a detailed executable map of the cell with a variety of uses, for example the in silico screening of the effects of drug candidates, the design of genetically-modified organisms or the identification of gaps in our knowledge [131]. Owing to their size and complexity, the models are difficult to build, maintain, and revise however, requiring a sustained community effort for all but the simplest cells. Moreover, the level of detail included in the models may not make them most suitable for apprehending global principles of growth control shared between different microorganisms.
A second strategy consists in increasing the coarseness of the models while preserving their scope, notably by coupling growth to intracellular biochemical processes. The resulting models are much more tractable from a mathematical and computational point of view, and they are particularly suited for exploring the consequences of hypotheses on the global architecture of growth control. On the other hand, by stripping away molecular details and focusing on a few explanatory principles, such coarse-grained models run the risk of losing key features of microbial cells. In particular, the complexity of regulatory mechanisms may lead to unexpected cross-talk between cellular functions not accounted for in abstract models but possibly critical for their predictive success. Moreover, in addition to contributing to the beauty of living systems [132], the molecular details of regulatory mechanisms may also be important for matching the model with quantitative data and for understanding evolutionary trajectories of microorganisms. As an illustration of the latter point, a recent study attributed the increased growth of an E. coli strain in minimal media observed in adaptive laboratory evolution experiments to specific point mutations in the β subunit of RNA polymerase [133].
In our view, one of the most promising directions for further work lies in finding original combinations of the above-mentioned strategies. In particular, local fine-graining of functions of interest in a coarse-grained model of the cellular machinery responsible for growth and division may yield models that are at the same time robust over a range of growth conditions and that can be related to specific regulatory mechanisms on the molecular level. From the point of view of experimental validation, such models would have the advantage that predictions of the behaviour of modules developed in molecular detail can be directly tested against experimental data, as they will correspond to measurable concentrations of molecular constituents. At the same time, the embedding of detailed modules in a global model of cellular physiology will widen its applicability to experimental scenarios in which growth or other major aspects of the physiological state are perturbed. The approach also exemplifies the well-known adage that models are not universal but developed for a specific question. Indeed, combining local fine-graining with a coarse-grained view of cellular physiology does not yield a single model, but rather a family of models each developing in detail a specific function or mechanism, depending on the question at hand.
Supplementary Material
Data accessibility
Electronic supplementary material is available from the journal web site.
Author's contributions
All authors contributed to the development of the ideas summarized in the manuscript. H.d.J. drafted the manuscript and S.C., N.G., E.C., D.R., J.G. and J.-L.G. helped draft the manuscript. All the authors gave their final approval for publication.
Competing interests
We declare we have no competing interests.
Funding
This work was funded by the Programme Investissements d'Avenir, Bio-informatique, RESET (ANR-11-BINF-0005) and the Inria Project Lab AlgaeInSilico. We thank the research program Labex SIGNALIFE (ANR-11-LABX-0028-01) and Conseil Régional PACA for partial funding of the PhD thesis of S.C.
References
- 1.Neidhardt FC. 1999. Bacterial growth: constant obsession with dn/dt. J. Bacteriol. 181, 7405–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fishov I, Zaritsky A, Grover NB. 1995. On microbial states of growth. Mol. Microbiol. 15, 789–794. ( 10.1111/j.1365-2958.1995.tb02349.x) [DOI] [PubMed] [Google Scholar]
- 3.Hwa T, Scott M. 2011. Bacterial growth laws and their applications. Curr. Opin. Biotechnol. 22, 559–565. ( 10.1016/j.copbio.2011.04.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ehrenberg M, Bremer H, Dennis PP. 2013. Medium-dependent control of the bacterial growth rate. Biochimie 95, 643–658. ( 10.1016/j.biochi.2012.11.012) [DOI] [PubMed] [Google Scholar]
- 5.Maaløe O, Kjeldgaard NO. 1966. Control of macromolecular synthesis: a study of DNA, RNA, and protein synthesis in bacteria. New York, NY: Benjamin. [Google Scholar]
- 6.Bremer H, Dennis PP. 2013. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. In Ecosal plus: cellular and molecular biology of E. coli, Salmonella, and the Enterobacteriaceae (ed. Slauch JM.). Washington, DC: ASM Press. [DOI] [PubMed] [Google Scholar]
- 7.Forchhammer J, Lindahl L. 1971. Growth rate of polypeptide chains as a function of the cell growth rate in a mutant of Escherichia coli 15. J. Mol. Biol. 55, 563–568. ( 10.1016/0022-2836(71)90337-8) [DOI] [PubMed] [Google Scholar]
- 8.Gausing K. 1977. Regulation of ribosome production in Escherichia coli: synthesis and stability of ribosomal RNA and of ribosomal protein messenger RNA at different growth rates. J. Mol. Biol. 115, 335–354. ( 10.1016/0022-2836(77)90158-9) [DOI] [PubMed] [Google Scholar]
- 9.Scott M, Gunderson CW, Mateescu EM, Zhang Z, Hwa T. 2010. Interdependence of cell growth and gene expression: origins and consequences. Science 330, 1099–1103. ( 10.1126/science.1192588) [DOI] [PubMed] [Google Scholar]
- 10.Levy S, Barkai N. 2009. Coordination of gene expression with growth rate: a feedback or a feed-forward strategy? FEBS Lett. 583, 3974–3978. ( 10.1016/j.febslet.2009.10.071) [DOI] [PubMed] [Google Scholar]
- 11.Chubukov V, Gerosa L, Kochanowski K, Sauer U. 2014. Coordination of microbial metabolism. Nat. Rev. Microbiol. 12, 327–340. ( 10.1038/nrmicro3238) [DOI] [PubMed] [Google Scholar]
- 12.Marr AG. 1991. Growth rate of Escherichia coli. Microbiol. Rev. 55, 316–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nierlich DP. 1974. Regulation of bacterial growth. Science 184, 1043–1050. ( 10.1126/science.184.4141.1043) [DOI] [PubMed] [Google Scholar]
- 14.Schachter M, Ingraham JL, Neidhardt FC. 2006. Microbe. Washington, DC: ASM Press. [Google Scholar]
- 15.Brunschede H, Dove TL, Bremer H. 1977. Establishment of exponential growth after a nutritional shift-up in Escherichia coli B/r: accumulation of deoxyribonucleic acid, ribonucleic acid, and protein. J. Bacteriol. 129, 1020–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Link H, Fuhrer T, Gerosa L, Zamboni N, Sauer U. 2015. Real-time metabolome profiling of the metabolic switch between starvation and growth. Nat. Methods 12, 1091–1097. ( 10.1038/nmeth.3584) [DOI] [PubMed] [Google Scholar]
- 17.Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, Liebermeister W, Surette MG, Alon U. 2006. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 3, 623–628. ( 10.1038/nmeth895) [DOI] [PubMed] [Google Scholar]
- 18.Gama-Castro S, et al. 2016. RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 44, D133–D143. ( 10.1093/nar/gkv1156) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karp PD, et al. 2014. The EcoCyc database. EcoSal. Plus. 6, ESP–0009–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Görke B, Stülke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624. ( 10.1038/nrmicro1932) [DOI] [PubMed] [Google Scholar]
- 21.You C, et al. 2013. Coordination of bacterial proteome with metabolism by cyclic AMP signalling. Nature 500, 301–306. ( 10.1038/nature12446) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kafri M, Metzl-Raz E, Jonas F, Barkai N. 2016. Rethinking cell growth models. FEMS Yeast Res. 16, 1–6. ( 10.1093/femsyr/fow081) [DOI] [PubMed] [Google Scholar]
- 23.Kremling A, Geiselmann J, Ropers D, de Jong H. 2015. Understanding carbon catabolite repression in Escherichia coli using quantitative models. Trends Microbiol. 23, 99–109. ( 10.1016/j.tim.2014.11.002) [DOI] [PubMed] [Google Scholar]
- 24.Scott M, Klumpp S, Mateescu EM, Hwa T. 2014. Emergence of robust growth laws from optimal regulation of ribosome synthesis. Mol. Syst. Biol. 10, 747 ( 10.15252/msb.20145379) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kacser H. 1986. On parts and wholes in metabolism. In The Organization of Cell Metabolism (eds Welch GR, Clegg JS). NATO ASI Series, vol. 127, pp. 327–37. New York, NY: Plenum Press. [Google Scholar]
- 26.Hinshelwood CN. 1952. On the chemical kinetics of autosynthetic systems. J. Chem. Soc. (Res.) 745–755. ( 10.1039/jr9520000745) [DOI] [Google Scholar]
- 27.Shuler ML, Leung S, Dick CC. 1979. A mathematical model for the growth of a single cell. Ann. N. Y. Acad. Sci. 326, 35–52. ( 10.1111/j.1749-6632.1979.tb14150.x) [DOI] [Google Scholar]
- 28.Shuler ML, Foley P, Atlas J. 2012. Modeling a minimal cell. Methods Mol. Biol. 881, 573–610. ( 10.1007/978-1-61779-827-6_20) [DOI] [PubMed] [Google Scholar]
- 29.Kompala DS, Ramkrishna D, Jansen NB, Tsao GT. 1986. Investigation of bacterial growth on mixed substrates: experimental evaluation of cybernetic models. Biotechnol. Bioeng. 28, 1044–1055. ( 10.1002/bit.260280715) [DOI] [PubMed] [Google Scholar]
- 30.Kompala DS, Ramkrishna D, Tsao GT. 1984. Cybernetic modeling of microbial growth on multiple substrates. Biotechnol. Bioeng. 26, 1272–1281. ( 10.1002/bit.260261103) [DOI] [PubMed] [Google Scholar]
- 31.Ramkrishna D, Song HS. 2012. Dynamic models of metabolism: review of the cybernetic approach. AIChE J. 58, 986–997. ( 10.1002/aic.13734) [DOI] [Google Scholar]
- 32.Tomita M, et al. 1999. E-CELL: software environment for whole-cell simulation. Bioinformatics 15, 72–84. ( 10.1093/bioinformatics/15.1.72) [DOI] [PubMed] [Google Scholar]
- 33.Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Assad-Garcia N, Glass JI, Covert MW. 2012. A whole-cell computational model predicts phenotype from genotype. Cell 150, 389–401. ( 10.1016/j.cell.2012.05.044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Brien EJ, Lerman JA, Chang RL, Hyduke DR, Palsson BO. 2013. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 9, 693 ( 10.1038/msb.2013.52) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bosdriesz E, Molenaar D, Teusink B, Bruggeman FJ. 2015. How fast-growing bacteria robustly tune their ribosome concentration to approximate growth-rate maximization. FEBS J. 282, 2029–2044. ( 10.1111/febs.13258) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giordano N, Mairet F, Gouzé JL, Geiselmann J, de Jong H. 2016. Dynamical allocation of cellular resources as an optimal control problem: novel insights into microbial growth strategies. PLoS Comput. Biol. 12, e1004802 ( 10.1371/journal.pcbi.1004802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molenaar D, van Berlo R, de Ridder D, Teusink B. 2009. Shifts in growth strategies reflect tradeoffs in cellular economics. Mol. Syst. Biol. 5, 323 ( 10.1038/msb.2009.82) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiße AY, Oyarzún DA, Danos V, Swain PS. 2015. Mechanistic links between cellular trade-offs, gene expression, and growth. Proc. Natl Acad. Sci. USA 112, E1038–E1047. ( 10.1073/pnas.1416533112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaslaver A, Kaplan S, Bren A, Jinich A, Mayo A, Dekel E, Alon U, Itzkovitz S. 2009. Invariant distribution of promoter activities in Escherichia coli. PLoS Comput. Biol. 5, e1000545 ( 10.1371/journal.pcbi.1000545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campos M, Surovtsev IV, Kato S, Paintdakhi A, Beltran B, Ebmeier SE, Jacobs-Wagner C. 2014. A constant size extension drives bacterial cell size homeostasis. Cell 159, 1433–1446. ( 10.1016/j.cell.2014.11.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osella M, Nugent E, CosentinoLagomarsino M. 2014. Concerted control of Escherichia coli cell division. Proc. Natl Acad. Sci. USA 111, 3431–3435. ( 10.1073/pnas.1313715111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taheri-Araghi S, Bradde S, Sauls JT, Hill NS, Levin PA, Paulsson J, Vergassola M, Jun S. 2015. Cell-size control and homeostasis in bacteria. Curr. Biol. 25, 385–391. ( 10.1016/j.cub.2014.12.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Volkmer B, Heinemann M. 2011. Condition-dependent cell volume and concentration of Escherichia coli to facilitate data conversion for systems biology modeling. PLoS ONE 6, e23126 ( 10.1371/journal.pone.0023126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robert L, Hoffmann M, Krell N, Aymerich S, Robert J, Doumic M. 2014. Division in Escherichia coli is triggered by a size-sensing rather than a timing mechanism. BMC Biol. 12, 17 ( 10.1186/1741-7007-12-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey JE. 1998. Mathematical modeling and analysis in biochemical engineering: Past accomplishments and future opportunities. Biotechnol. Prog. 14, 8–20. ( 10.1021/bp9701269) [DOI] [PubMed] [Google Scholar]
- 46.Blanch HW. 1981. Microbial growth kinetics. Chem. Eng. Commun. 8, 181–211. ( 10.1080/00986448108912580) [DOI] [Google Scholar]
- 47.Fredrickson AG, Megee RD, Tsuchiya HM. 1970. Mathematical models for fermentation processes. Adv. Appl. Microbiol. 13, 419–465. ( 10.1016/S0065-2164(08)70413-1) [DOI] [Google Scholar]
- 48.Kiviet DJ, Nghe P, Walker N, Boulineau S, Sunderlikova V, Tans SJ. 2014. Stochasticity of metabolism and growth at the single-cell level. Nature 514, 376–379. ( 10.1038/nature13582) [DOI] [PubMed] [Google Scholar]
- 49.Kotte O, Volkmer B, Radzikowski JL, Heinemann M. 2014. Phenotypic bistability in Escherichia coli's central carbon metabolism. Mol. Syst. Biol. 10, 736 ( 10.15252/msb.20135022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Heerden JH, et al. 2014. Lost in transition: Start-up of glycolysis yields subpopulations of nongrowing cells. Science 343, 1245114 ( 10.1126/science.1245114) [DOI] [PubMed] [Google Scholar]
- 51.Westermayer SA, Frit G, Gutiérrez J, Megerle JA, Weißl MPS, Schnetz K, Gerland U, Rädler JO. 2016. Single-cell characterization of metabolic switching in the sugar phosphotransferase system of Escherichia coli. Mol. Microbiol. 100, 472–485. ( 10.1111/mmi.13329) [DOI] [PubMed] [Google Scholar]
- 52.Klumpp S, Zhang Z, Hwa T. 2009. Growth rate-dependent global effects on gene expression in bacteria. Cell 139, 1366–1375. ( 10.1016/j.cell.2009.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGuffee SR, Elcock AH. 2010. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput. Biol. 6, e1000694 ( 10.1371/journal.pcbi.1000694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zimmerman SB, Trach SO. 1991. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 222, 599–620. ( 10.1016/0022-2836(91)90499-V) [DOI] [PubMed] [Google Scholar]
- 55.Basan M, Zhu M, Dai X, Warren M, Sévin D, Wang Y-P, Hwa T. 2015. Inflating bacterial cells by increased protein synthesis. Mol. Syst. Biol. 11, 836 ( 10.15252/msb.20156178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Afroz T, Biliouris K, Kaznessis Y, Beisel CL. 2014. Bacterial sugar utilization gives rise to distinct single-cell behaviours. Mol. Microbiol. 93, 1093–1103. ( 10.1111/mmi.12695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Y, Vazquez A, Wise A, Warita T, Warita K, Bar-Joseph Z, Oltvai ZN. 2013. Carbon catabolite repression correlates with the maintenance of near invariant molecular crowding in proliferating E. coli cells. BMC Syst. Biol. 7, 138 ( 10.1186/1752-0509-7-138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Narang A, Pilyugin SS. 2007. Bacterial gene regulation in diauxic and non-diauxic growth. J. Theor. Biol. 244, 326–348. ( 10.1016/j.jtbi.2006.08.007) [DOI] [PubMed] [Google Scholar]
- 59.Tan C, Marguet P, You L. 2009. Emergent bistability by a growth-modulating positive feedback circuit. Nat. Chem. Biol. 5, 842–848. ( 10.1038/nchembio.218) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell A. 1957. Synchronization of cell division. Bacteriol. Rev. 21, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gottschalk G. 1986. Bacterial metabolism, 2nd edn New York, NY: Springer. [Google Scholar]
- 62.Heinrich R, Schuster S. 1996. The regulation of cellular systems. New York, NY: Chapman & Hall. [Google Scholar]
- 63.Gillespie DT. 2007. Stochastic simulation of chemical kinetics. Annu. Rev. Phys. Chem. 58, 35–55. ( 10.1146/annurev.physchem.58.032806.104637) [DOI] [PubMed] [Google Scholar]
- 64.Kremling A. 2013. Systems biology: mathematical modeling and model analysis. Boca Raton, FL: Chapman & Hall/CRC. [Google Scholar]
- 65.Chen WW, Niepel M, Sorger PK. 2010. Classic and contemporary approaches to modeling biochemical reactions. Genes Dev. 24, 1861–1876. ( 10.1101/gad.1945410) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Segel LA, Slemrod M. 1989. The quasi-steady-state assumption: a case-study in perturbation. SIAM Rev. 31, 446–477. ( 10.1137/1031091) [DOI] [Google Scholar]
- 67.Monod J. 1949. The growth of bacterial cultures. Ann. Rev. Microbiol. 3, 371–394. ( 10.1146/annurev.mi.03.100149.002103) [DOI] [Google Scholar]
- 68.Aidelberg G, Towbin BD, Rothschild D, Dekel E, Bren A, Alon U. 2014. Hierarchy of non-glucose sugars in Escherichia coli. BMC Syst. Biol. 8, 1 ( 10.1186/s12918-014-0133-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harder W, Dijkhuizen L. 1982. Strategies of mixed substrate utilization in microorganisms. Phil. Trans. R. Soc. Lond. B. 297, 459–480. ( 10.1098/rstb.1982.0055) [DOI] [PubMed] [Google Scholar]
- 70.Hermsen R, Okano H, You C, Werner N, Hwa T. 2015. A growth-rate composition formula for the growth of E. coli on co-utilized carbon substrates. Mol. Syst. Biol. 11, 801 ( 10.15252/msb.20145537) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramakrishna R, Ramkrishna D, Konopka AE. 1996. Cybernetic modeling of growth in mixed, substitutable substrate environments: preferential and simultaneous utilization. Biotechnol. Bioeng. 52, 141–51. ( 10.1002/(SICI)1097-0290(19961005)52:1%3C141::AID-BIT14%3E3.0.CO;2-R) [DOI] [PubMed] [Google Scholar]
- 72.Bastin G, Dochain D. 1990. On-line estimation and adaptive control of bioreactors. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 73.Stephanopoulos GN, Aristidou AA, Nielsen J. 1998. Metabolic engineering: principles and methodologies. San Diego, CA: Academic Press. [Google Scholar]
- 74.Godon J-J, Arcemisbéhère L, Escudié R, Harmand J, Miambi E, Steyer J-P. 2013. Overview of the oldest existing set of substrate-optimized anaerobic processes: digestive tracts. BioEnergy Res. 6, 1063–1081. ( 10.1007/s12155-013-9339-y) [DOI] [Google Scholar]
- 75.Muñoz-Tamayo R, Laroche B, Walter E, Doré J, Leclerc M. 2010. Mathematical modelling of carbohydrate degradation by human colonic microbiota. J. Theor. Biol. 266, 189–202. ( 10.1016/j.jtbi.2010.05.040) [DOI] [PubMed] [Google Scholar]
- 76.Ashyraliyev M, Fomekong Nanfack Y, Kaandorp JA, Blom JG. 2009. Systems biology: parameter estimation for biochemical models. FEBS J. 276, 886–902. ( 10.1111/j.1742-4658.2008.06844.x) [DOI] [PubMed] [Google Scholar]
- 77.Berthoumieux S, Brilli M, Kahn D, de Jong H, Cinquemani E. 2013. On the identifiability of metabolic network models. J. Math. Biol. 67, 1795–1832. ( 10.1007/s00285-012-0614-x) [DOI] [PubMed] [Google Scholar]
- 78.Villaverde AF, Banga JR. 2013. Reverse engineering and identification in systems biology: strategies, perspectives and challenges. J. R. Soc. Interface 11, 20130505 ( 10.1098/rsif.2013.0505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gottstein W, Olivier BG, Bruggeman FJ, Teusink B. 2016. Top-down analysis of temporal hierarchy in biochemical reaction networks. J. R. Soc. Interface 13, 20160627 ( 10.1098/rsif.2016.0627) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Palsson BO. 2015. Systems biology: constraint-based reconstruction and analysis, 2nd edn Cambridge, UK: Cambridge University Press. [Google Scholar]
- 81.Price ND, Reed JL, Palsson BO. 2004. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev Microbiol. 2, 886–897. ( 10.1038/nrmicro1023) [DOI] [PubMed] [Google Scholar]
- 82.Feist AM, Palsson BO. 2010. The biomass objective function. Curr. Opin. Microbiol. 13, 344–349. ( 10.1016/j.mib.2010.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nookaew I, Jewett MC, Meechai A, Thammarongtham C, Laoteng K, Cheevadhanarak S, Nielsen J, Bhumiratana S. 2008. The genome-scale metabolic model iIN800 of Saccharomyces cerevisiae and its validation: a scaffold to query lipid metabolism. BMC Syst. Biol. 2, 71 ( 10.1186/1752-0509-2-71) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Teusink B, Wiersma A, Molenaar D, Francke C, de Vos WM, Siezen RJ, Smid EJ. 2006. Analysis of growth of Lactobacillus plantarum WCFS1 on a complex medium using a genome-scale metabolic model. J. Biol. Chem. 281, 40 041–40 048. ( 10.1074/jbc.M606263200) [DOI] [PubMed] [Google Scholar]
- 85.Larrabee KL, Phillips JO, Williams GJ, Larrabee AR. 1980. The relative rates of protein synthesis and degradation in a growing culture of Escherichia coli. J. Biol. Chem. 255, 4125–4130. [PubMed] [Google Scholar]
- 86.Mosteller RD, Goldstein RV, Nishimoto KR. 1980. Metabolism of individual proteins in exponentially growing Escherichia coli. J. Biol. Chem. 255, 2524–2532. [PubMed] [Google Scholar]
- 87.Khalil HK. 2001. Nonlinear systems, 3rd edn Upper Saddle River, NJ: Prentice Hall. [Google Scholar]
- 88.Taymaz-Nikerel H, De Mey M, Baart G, Maertens J, Heijnen JJ, van Gulik W. 2013. Changes in substrate availability in Escherichia coli lead to rapid metabolite, flux and growth rate responses. Metab. Eng. 16, 115–129. ( 10.1016/j.ymben.2013.01.004) [DOI] [PubMed] [Google Scholar]
- 89.Baldazzi V, Ropers D, Markowicz Y, Kahn D, Geiselmann J, de Jong H. 2010. The carbon assimilation network in Escherichia coli is densely connected and largely sign-determined by directions of metabolic fluxes. PloS Comput. Biol. 6, e1000812 ( 10.1371/journal.pcbi.1000812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schilling CH, Schuster S, Palsson BO, Heinrich R. 1999. Metabolic pathway analysis: basic concepts and scientific applications in the post-genomic era. Biotechnol. Prog. 15, 296–303. ( 10.1021/bp990048k) [DOI] [PubMed] [Google Scholar]
- 91.Antoniewicz MR. 2015. Methods and advances in metabolic flux analysis: a mini-review. J. Ind. Microbiol. Biotechnol. 42, 317–325. ( 10.1007/s10295-015-1585-x) [DOI] [PubMed] [Google Scholar]
- 92.Morin M, Ropers D, Letisse F, Laguerre S, Portais JC, Cocaign-Bousquet M, Enjalbert B. 2016. The post-transcriptional regulatory system CSR controls the balance of metabolic pools in upper glycolysis of Escherichia coli. Mol. Microbiol. 100, 686–700. ( 10.1111/mmi.13343) [DOI] [PubMed] [Google Scholar]
- 93.Provost A, Bastin G. 2004. Dynamic metabolic modelling under the balanced growth condition. J. Process Control 14, 717–728. ( 10.1016/j.jprocont.2003.12.004) [DOI] [Google Scholar]
- 94.Orth JD, Thiele I, Palsson BO. 2010. What is flux balance analysis? Nat. Biotechnol. 28, 245–248. ( 10.1038/nbt.1614) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bordbar A, Monk JM, King ZA, Palsson BO. 2014. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120. ( 10.1038/nrg3643) [DOI] [PubMed] [Google Scholar]
- 96.Edwards JS, Ibarra RU, Palsson BO. 2001. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19, 125–130. ( 10.1038/84379) [DOI] [PubMed] [Google Scholar]
- 97.Ibarra RU, Edwards JS, Palsson BO. 2002. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420, 186–189. ( 10.1038/nature01149) [DOI] [PubMed] [Google Scholar]
- 98.Mahadevan R, Edwards JS, Doyle FJ. 2002. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 83, 1331–1340. ( 10.1016/S0006-3495(02)73903-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Megchelenbrink W, Huynen M, Marchiori E. 2014. optGpSampler: an improved tool for uniformly sampling the solution-space of genome-scale metabolic networks. PLoS ONE 9, e86587 ( 10.1371/journal.pone.0086587) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Price ND, Schellenberger J, Palsson BO. 2004. Uniform sampling of steady-state flux spaces: means to design experiments and to interpret enzymopathies. Biophys. J. 87, 2172–2186. ( 10.1529/biophysj.104.043000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schuster S, Fell DA, Dandekar T. 2000. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 18, 326–332. ( 10.1038/73786) [DOI] [PubMed] [Google Scholar]
- 102.Kelk SM, Olivier BG, Stougie L, Bruggeman FJ. 2012. Optimal flux spaces of genome-scale stoichiometric models are determined by a few subnetworks. Sci. Rep. 2, 580 ( 10.1038/srep00580) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schuetz R, Kuepfer L, Sauer U. 2007. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 3, 119 ( 10.1038/msb4100162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schuster S, Pfeiffer T, Fell DA. 2008. Is maximization of molar yield in metabolic networks favoured by evolution? J. Theor. Biol. 252, 497–504. ( 10.1016/j.jtbi.2007.12.008) [DOI] [PubMed] [Google Scholar]
- 105.de Lorenzo V. 2014. From the selfish gene to selfish metabolism: revisiting the central dogma. Bioessays 36, 226–235. ( 10.1002/bies.201300153) [DOI] [PubMed] [Google Scholar]
- 106.Adadi R, Volkmer B, Milo R, Heinemann M, Shlomi T. 2012. Prediction of microbial growth rate versus biomass yield by a metabolic network with kinetic parameters. PLoS Comput. Biol. 8, e1002575 ( 10.1371/journal.pcbi.1002575) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Beg QK, Vazquez A, Ernst J, de Menezes MA, Bar-Joseph Z, Barabási A-L, Oltvai ZN. 2007. Intracellular crowding defines the mode and sequence of substrate uptake by Escherichia coli and constrains its metabolic activity. Proc. Natl Acad. Sci. USA 104, 12 663–12 668. ( 10.1073/pnas.0609845104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Covert MW, Xiao N, Chen TJ, Karr J. 2008. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 24, 2044–2050. ( 10.1093/bioinformatics/btn352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Goelzer A, Fromion V. 2011. Bacterial growth rate reflects a bottleneck in resource allocation. Biochim. Biophys. Acta 1810, 978–988. ( 10.1016/j.bbagen.2011.05.014) [DOI] [PubMed] [Google Scholar]
- 110.Mori M, Hwa T, Martin OC, De Martino A, Marinari E. 2016. Constrained allocation flux balance analysis. PLoS Comput. Biol. 12, e1004913 ( 10.1371/journal.pcbi.1004913) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Waldherr S, Oyarzún DA, Bockmayr A. 2015. Dynamic optimization of metabolic networks coupled with gene expression. J. Theor. Biol. 365, 469–485. ( 10.1016/j.jtbi.2014.10.035) [DOI] [PubMed] [Google Scholar]
- 112.Zhuang K, Vemuri GN, Mahadevan R. 2011. Economics of membrane occupancy and respiro-fermentation. Mol. Syst. Biol. 7, 500 ( 10.1038/msb.2011.34) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gánti T. 1975. Organization of chemical reactions into dividing and metabolizing units: the chemotons. Biosystems 7, 15–21. ( 10.1016/0303-2647(75)90038-6) [DOI] [PubMed] [Google Scholar]
- 114.Koch AL. 1988. Why can't a cell grow infinitely fast? Can. J. Microbiol. 34, 421–426. ( 10.1139/m88-074) [DOI] [PubMed] [Google Scholar]
- 115.Stengel RF. 1994. Optimal control and estimation. Mineola, NY: Dover Publications. [Google Scholar]
- 116.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu. Rev. Microbiol. 62, 35–51. ( 10.1146/annurev.micro.62.081307.162903) [DOI] [PubMed] [Google Scholar]
- 117.Bollenbach T, Quan S, Chait R, Kishony R. 2009. Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell 139, 707–718. ( 10.1016/j.cell.2009.10.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van den Berg HA, Kiselev YN, Orlov MV. 2002. Optimal allocation of building blocks between nutrient uptake systems in a microbe. J. Math. Biol. 44, 276–296. ( 10.1007/s002850100123) [DOI] [PubMed] [Google Scholar]
- 119.Kaltenbach HM, Stelling J. 2012. Modular analysis of biological networks. Adv. Exp. Med. Biol. 736, 3–17. ( 10.1007/978-1-4419-7210-1_1) [DOI] [PubMed] [Google Scholar]
- 120.Baroukh C, Muñoz-Tamayo R, Steyer JP, Bernard O. 2014. DRUM: a new framework for metabolic modeling under non-balanced growth. application to the carbon metabolism of unicellular microalgae. PLoS ONE 9, e104499 ( 10.1371/journal.pone.0104499) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jamshidi N, Palsson BO. 2008. Top-down analysis of temporal hierarchy in biochemical reaction networks. PLoS Comput. Biol. 4, e1000177 ( 10.1371/journal.pcbi.1000177) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gunawardena J. 2014. Models in biology: ‘accurate descriptions of our pathetic thinking’. BMC Biol. 12, 29 ( 10.1186/1741-7007-12-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bigan E, Paulevé L, Steyaert JM, Douady S. 2016. Necessary and sufficient conditions for protocell growth. J. Math. Biol. 73, 1627–1664. ( 10.1007/s00285-016-0998-0) [DOI] [PubMed] [Google Scholar]
- 124.Surovstev IV, Morgan JJ, Lindahl PA. 2007. Whole-cell modeling framework in which biochemical dynamics impact aspects of cellular geometry. J. Theor. Biol. 244, 154–166. ( 10.1016/j.jtbi.2006.07.020) [DOI] [PubMed] [Google Scholar]
- 125.Noireaux V, Maeda YT, Libchaber A. 2011. Development of an artificial cell, from self-organization to computation and self-reproduction. Proc. Natl Acad. Sci. USA 108, 3473–3480. ( 10.1073/pnas.1017075108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rasmussen S, Bedau MA, Chen L, Deamer D, Krakauer DC, Packard NH, Stadler PF. 2009. Protocells: bridging nonliving and living matter. Cambridge, MA: MIT Press. [DOI] [PubMed] [Google Scholar]
- 127.de Jong H, Geiselmann J, Ropers D. 2017. Resource reallocation in bacteria by reengineering the gene expression machinery. Trends Microbiol. 25, 480–493. ( 10.1016/j.tim.2016.12.009) [DOI] [PubMed] [Google Scholar]
- 128.Si F, et al. 2017. Invariance of initiation mass and predictability of cell size in Escherichia coli. Curr. Biol. 27, 1278–1287. ( 10.1016/j.cub.2017.03.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Spiesser W, Müller C, Schreiber G, Krantz M, Klipp E. 2012. Size homeostasis can be intrinsic to growing cell populations and explained without size sensing or signalling. FEBS J. 279, 4111–4145. ( 10.1111/febs.12014) [DOI] [PubMed] [Google Scholar]
- 130.Kaern M, Elston TC, Blake WJ, Collins JJ. 2005. Stochasticity in gene expression From theories to phenotypes. Nat. Rev. Genet. 6, 451–464. ( 10.1038/nrg1615) [DOI] [PubMed] [Google Scholar]
- 131.Carrera J, Covert MW. 2015. Why build whole-cell models? Trends Cell Biol. 25, 719–722. ( 10.1016/j.tcb.2015.09.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Goodsell DS. 2009. The machinery of life. New York, NY: Copernicus Books. [Google Scholar]
- 133.Utrilla J, O'Brien EJ, Chen K, McCloskey D, Cheung J, Wang H, Armenta-Medina D, Feist AM, Palsson BO. 2016. Global rebalancing of cellular resources by pleiotropic point mutations illustrates a multi-scale mechanism of adaptive evolution. Cell Syst. 2, 260–271. ( 10.1016/j.cels.2016.04.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Electronic supplementary material is available from the journal web site.