

Abstract
Purpose
The clinical features, pathogenesis, and outcomes in children with adrenocortical tumors (ACTs) without germline TP53 mutations have not been systematically studied. Herein, we describe these correlates and analyze their association with outcome.
Patients and Methods
Genomic DNA was analyzed for TP53, CTNNB1, CDKN1C, ATRX, and chromosome 11p15 abnormalities. β-catenin expression and Ki-67 labeling index (LI) were evaluated by immunostaining. Primary end points were progression-free (PFS) and overall survival.
Results
Median age of 42 girls and 18 boys was 3.3 years (range, 0.25 to 21.7 years). Complete resection (stages I and II) was achieved in 32 patients, and 28 patients had stage III or IV disease. Constitutional abnormalities of chromosome 11p15 occurred in nine of 40 patients, with six patients not showing phenotype of Beckwith-Wiedemann syndrome. Three-year PFS and overall survival for all patients were 71.4% and 80.5%, respectively. In single-predictor Cox regression analysis, age, disease stage, tumor weight, somatic TP53 mutations, and Ki-67 LI were associated with prognosis. Ki-67 LI and age remained significantly associated with PFS after adjusting for stage and tumor weight. Three-year PFS for 27 patients with Ki-67 LI ≥ 15% was 48.5% compared with 96.2% for 29 patients with Ki-67 LI < 15% (log-rank P = .002), and the rate of relapse increased by 24% with each 1-year increase in age at diagnosis (hazard ratio, 1.24; P = .0057).
Conclusion
Clinicopathologic features and outcomes of children with ACTs without germline TP53 mutations overlapped those reported for children with germline TP53 mutations. Our findings highlight the central role of genetic or epigenetic alterations on chromosome 11p15 in pediatric ACTs. Ki-67 LI is a strong prognostic indicator and should be investigated to improve the histologic classification of pediatric ACTs.
INTRODUCTION
Pediatric adrenocortical tumor (ACT) is an uncommon malignancy frequently associated with Li-Fraumeni syndrome, a familial cancer predisposition disorder caused by germline mutations in the tumor suppressor gene TP53.1,2 ACT is rarely associated with other genetic constitutional disorders such as Beckwith-Wiedemann syndrome (BWS), which results from deregulation of a gene cluster on chromosome 11p153; multiple endocrine neoplasia; neurofibromatosis; familial adenomatous polyposis; or congenital adrenal hyperplasia.4,5
Our recent study6 showed that copy-neutral loss of heterozygosity (LOH) of chromosomes 11 and 17 is the hallmark of pediatric ACTs associated with germline TP53 mutations (mutTP53-ACTs). Combined loss of maternal chromosome 11, duplication of paternal chromosome 11, and loss of chromosome 17 harboring wild-type TP53 occurs early in pediatric adrenocortical tumorigenesis. In general, as the tumor increases in weight, it acquires additional genetic alterations and exhibits increasingly aggressive behavior.6
However, approximately 50% of children with ACTs do not harbor germline TP53 mutations.7 The mechanisms of tumorigenesis and tumor progression as well as prognostic markers of ACT in these patients have not been systematically evaluated. In this retrospective study, we report clinical features, molecular attributes, and outcomes in a relatively large cohort of children with ACTs without germline TP53 mutations (wtTP53-ACTs).
PATIENTS AND METHODS
Patients and Biologic Samples
This was a retrospective study of patients with ACTs carrying germline wild-type TP53 who were age < 22 years and enrolled in the St Jude Children’s Research Hospital International Pediatric Adrenocortical Tumor Registry (ClinicalTrials.gov identifier: NCT00700414; n = 48) between 2003 and 2015 or the Children’s Oncology Group (COG) ARAR0332 study (ClinicalTrials.gov identifier: NCT00304070; n = 12) between 2006 and 2013. Matched blood and tumor DNA was collected from 40 patients. For 20 patients, only tumor DNA (n = 14) or blood DNA (n = 6) was available for molecular studies.
Written informed consent was obtained from parents or legal guardians. The study was approved by the St Jude Institutional Review Board and the COG Rare Tumors Committee. Disease was staged according to reported guidelines.8 Clinical management was uniform for those enrolled in COG ARAR0332 and varied as directed by primary physicians for those enrolled in the International Pediatric Adrenocortical Tumor Registry.
TP53, CDKN1C, ATRX, and CTNNB1 Mutational Status
TP53 and CDKN1C coding regions, including the flanking intronic sequence of each exon, were amplified by polymerase chain reaction and sequenced on a 3730xl DNA Analyzer (Applied Biosystems). Large deletions of TP53 and ATRX were assessed using commercial multiplex ligation-dependent probe amplification (MLPA) kits (PO56 TP53 probemix and P013 ATRX probemix; MRC-Holland, Amsterdam, the Netherlands) as per manufacturer instructions.
Mutational status of CTNNB1 exon 3 was determined as previously described.6 Immunostaining for β-catenin was performed on 4-μm formalin-fixed paraffin-embedded tumor sections. Monoclonal Beta-Catenin (14; Ventana Medical Systems, Tucson, AZ) was used as the primary antibody and visualized using the iVIEW DAB Detection Kit (Ventana Medical Systems).
Chromosome 11p15 Abnormalities
Matched blood and tumor DNA from 40 patients was analyzed for 11p15 chromosomal copy number alterations by using microsatellite analysis.6 Gene dosage and methylation status were evaluated in blood samples by using ME030-C3 BWS/RSS probemix (MRC-Holland) as previously described9 and quantified using Coffalyzer software (MRC-Holland).
Ki-67 Immunostaining
Formalin-fixed paraffin-embedded tumor sections were analyzed for Ki-67 nuclear expression using primary polyclonal rabbit antisera for Ki-67 (dilution, 1:500; Novocastra; Leica Biosystems, Buffalo Grove, IL) and stained using routine protocols. Ten tumor areas were examined under ×400 magnification, and the overall percentage of cells with Ki-67–positive staining among the 10 fields was scored as the Ki-67 labeling index (LI). Ki-67 LI was considered high if ≥ 15% of tumor cell nuclei were stained.
Gene Expression Analysis
PTTG1, BUB1B, AURKB, HLA-DPA1, and MKI67 mRNA expression data were obtained from the Gene Expression Omnibus (GEO) repository including 63 pediatric ACTs (GEO databases GSE76019 and GSE76021). Experiments were performed as previously described.10
Statistical Analyses
The Kaplan-Meier method was used to estimate progression-free (PFS) and overall survival (OS). PFS was defined as time elapsed from diagnosis to progression or death, with times censored at the date of last follow-up. OS was defined as time elapsed from diagnosis to death, with times for living patients censored at last follow-up. The log-log method was used to calculate the CIs for survival estimates.11,12 The log-rank test was used to compare PFS or OS across groups according to presence or absence of molecular and clinical features. Cox proportional hazards regression models were used to evaluate the association of clinical and molecular features with PFS or OS while accounting for stage (dichotomized as stage ≤ IV). The Wilcoxon rank sum test was used to explore Ki-67 LI status in relation to age and tumor weight. The Wilcoxon rank sum test was also used to determine association between mRNA gene expression and TP53 status, and Cox regression was used to determine association between gene expression and PFS. All tests were two sided, and no multiple-testing adjustments were performed.
RESULTS
Patient Demographics and Clinical Correlates
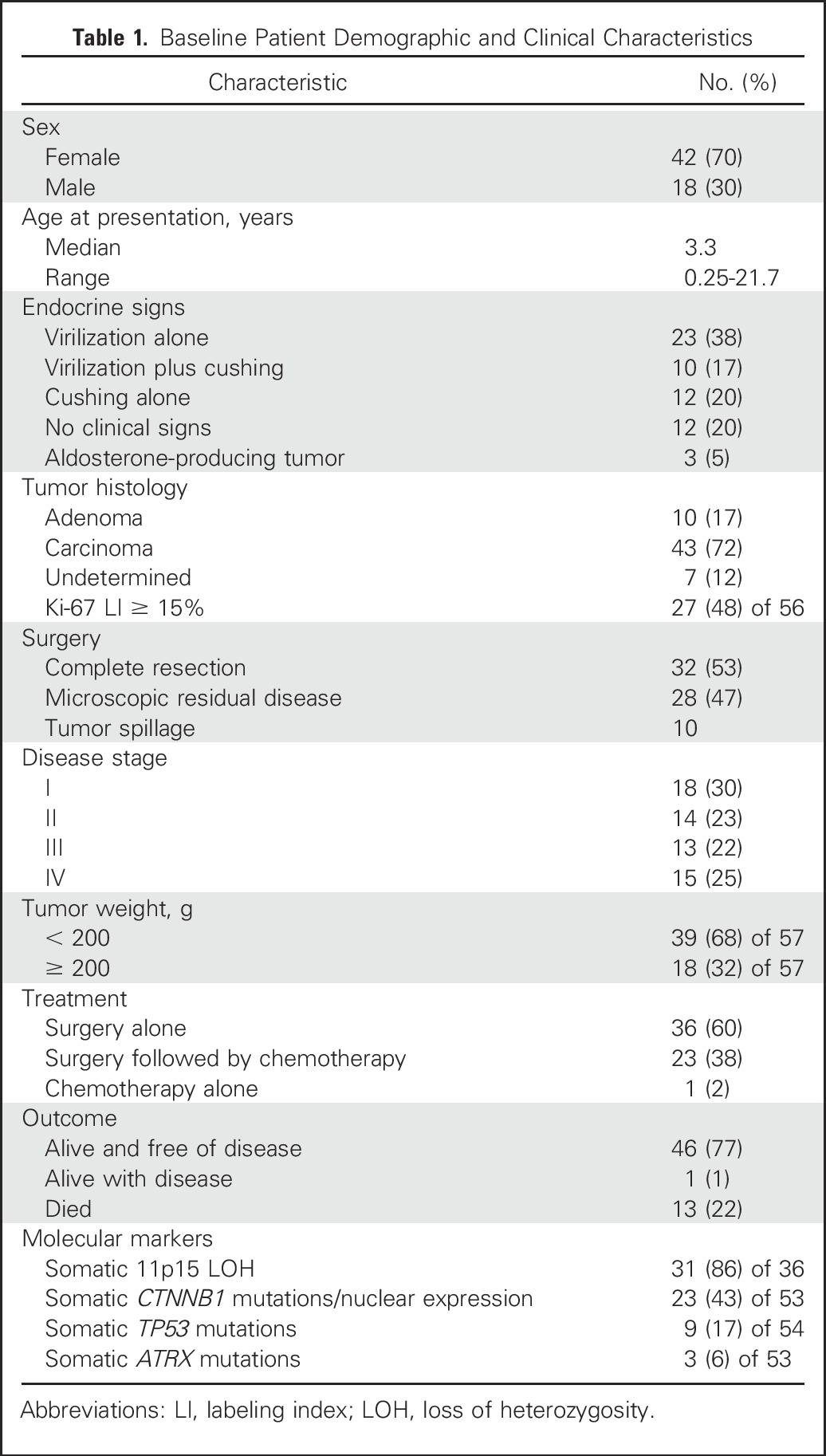
Median age of the 60 eligible patients (42 females and 18 males) was 3.3 years (range, 0.25 to 21.7 years). Signs and symptoms of virilization alone and associated with hypercortisolism (Cushing syndrome) were seen in 23 and 10 patients, respectively. There were no endocrine clinical signs or symptoms in 12 patients. Tumors were histologically characterized as carcinoma (n = 43), adenoma (n = 10), and undetermined (n = 7). Thirty-two patients had localized disease (stage I or II), and 28 had advanced disease (stage III or IV). Clinical data are summarized in Table 1 and detailed in Appendix Table A1 (online only).
Table 1.
Baseline Patient Demographic and Clinical Characteristics
TP53, CTNNB1, and ATRX Mutational Status in Tumor Samples
TP53 was analyzed in 54 of 60 patients, and somatic TP53 alterations were observed in nine samples (Fig 1A), including six missense mutations (R158H, R175H, E180K, M246V, R273C, and R273H), one nonsense mutation in exon 10 (R342*), and one frameshift insertion at exon 5 (c.403_404insT).13 Complete deletion of TP53 occurred in one sample.
Fig 1.

Somatic TP53 mutations in patients with adrenocortical tumors (ACTs) without TP53 mutations. (A) Schematic diagram of acquired TP53 mutations. (B) Kaplan-Meier estimates for progression-free survival (B) in pediatric patients with ACTs with or without acquired TP53 mutations and (C) by TP53 status and disease stage. After discriminating for disease stage and including stage IV disease in the Cox regression model, presence of somatic TP53 mutations was no longer associated with outcome (P = .21). hom, homozygous.
Activation of the Wnt pathway was determined by Sanger sequencing of CTNNB1 exon 3 (n = 48) and by β-catenin immunostaining (n = 5). Mutations (n = 22) or positive nuclear expression of β-catenin (n = 1) were identified in 23 (43%) of the 53 samples (Figs 2A and 2B). In four of these samples, mutations were acquired in both CTNNB1 and TP53 (WT006, WT007, WT008, and WT050; Appendix Table A1).
Fig 2.

Somatic CTNNB1 mutations in patients with adrenocortical tumors (ACTs) without TP53 mutations. (A) Schematic diagram of activating CTNNB1 mutations. (B) Immunohistochemical analysis of β-catenin and yes-associated protein 1 (YAP1) in pediatric ACTs. YAP serves as a surrogate marker for β-catenin expression. Moderate nuclear immunostaining of (i) β-catenin and (ii) YAP in a tumor with the S45P mutation in CTNNB1. Strong nuclear immunoreactivity for (iii) β-catenin and (iv) YAP in a pediatric patient with unknown CTNNB1 molecular status. These results are indicative of Wnt signaling pathway activation. Size bars represent 50 μm. (C) Kaplan-Meier estimates for progression-free survival by CTNNB1 status (P = .80).
ATRX intragenic deletions were detected in two of 53 ACTs analyzed by MLPA: one sample showed a deletion of exons 11 to 30 in the primary tumor and lung metastasis (WT006), whereas the other showed a deletion in exons 6 to 15 (WT010). A missense ATRX mutation (R2164S) was also identified in one of 12 samples analyzed by whole-exome sequencing (WT009).6 All three tumor samples with an ATRX mutation also harbored a somatic TP53 mutation (Appendix Table A1).
Germline and Somatic 11p15 Genetic and Epigenetic Alterations
Chromosome 11p15 encodes a cluster of imprinted genes that positively and negatively regulate cell proliferation and survival.14-16 These genes are differentially expressed in a parental origin–dependent manner through H19DMR (imprinting control region 1 [IC1]) and KvDMR (IC2). Insulin-like growth factor 2 (IGF2) is selectively expressed from the paternal allele, whereas the cell-cycle inhibitor CDKN1C (p57KIP2) is preferentially expressed from the maternal allele. High expression of IGF2 and low expression of CDKN1C, as seen in most pediatric ACTs,6 likely result from copy-neutral LOH or imprinting defects within the 11p15 locus.6,17
Microsatellite markers and methylation-specific MLPA analysis for chromosome 11p15 in blood DNA of 40 patients revealed a homozygous pattern for all markers and hypermethylation at IC1 and hypomethylation at IC2 (Appendix Fig A1A, online only), indicative of paternal uniparental disomy (UPD), in four patients (WT001, WT031, WT043, and WT049), two of whom did not exhibit clinical signs of BWS (WT0031 and WT043). In another patient (WT002), a mosaic paternal UPD for 11p15 was identified, with partial gain of methylation at IC1 and partial loss of methylation at IC2 (Appendix Fig A1B). An additional four patients (WT003, WT009, WT023, and WT030) exhibited isolated loss of methylation at IC2 (Appendix Fig A1C), suggesting silencing of maternal alleles, as observed in 50% of patients with BWS.18 Of note, patient WT003 had an incomplete BWS phenotype (hemihypertrophy), but no other features of BWS were seen in the remaining three patients. CDKN1C mutations were not detected in the germline DNA from 46 patients. Finally, analysis of corresponding tumors revealed somatic LOH in 31 (86%) of 36 informative patient cases, with selective loss of the maternal chromosome in all patients for whom parental blood DNA was available (n = 11; Appendix Fig A1D). In addition, microsatellite analysis of 14 patients for whom only tumor DNA was available, revealed a homozygous pattern suggestive of LOH in 12 (86%), and UPD was excluded in all patients for whom only blood DNA was available (n = 6).
Ki-67 LI
Immunohistochemical analysis of Ki-67 was performed in tumor samples from 56 patients. Ki-67 LI was positive (≥ 15% tumor-cell nuclei stained) in 27 patients (48%; Fig 3A; Appendix Table A1).
Fig 3.

(A) Hematoxylin and eosin staining and Ki-67 labeling index (LI) in pediatric adrenocortical tumors without TP53 mutations: (i, ii) tumor with a high (≥ 15%) Ki-67 labeling index (LI; red arrows indicate mitotic figures), (iii, iv) tumor with a low (< 5%) LI. Size bars represent 50 μm. (B) Kaplan-Meier estimates for progression-free survival (PFS) according to Ki-67 LI showing that Ki-67 LI < 15% was significantly associated with longer PFS (P = .002). (C) Kaplan-Meier estimates for PFS according to Ki-67 LI and disease stage. Ki-67 LI remained significantly associated with outcome after adjusting for disease stage (P = .0048).
Outcome, Treatment, and Prognostic Factors
Of 60 patients, 32 were considered free of disease after surgery, 13 had microscopic residual disease or tumor spillage during surgery (stage III), and 15 had overt metastatic disease (stage IV). The primary tumor was surgically resected in all cases except in patient WT056. No patients with stage I (n = 18) or II (n = 14) disease (excluding WT017, WT019, and WT027) received adjuvant chemotherapy. Patients with tumor spillage or microscopic residual disease were typically treated with platinum-based chemotherapy and mitotane. At a median follow-up of 3.4 years, 47 patients were alive, and 46 of them were free of disease. Thirteen patients died as a result of progressive disease. The 3-year PFS and OS for all patients were 71.4% (95% CI, 60.3% to 84.4%) and 80.5% (95% CI, 70.2% to 92.3%), respectively.
Consistent with earlier findings from adult ACTs,19 the presence of somatic TP53 mutations was significantly associated with poor prognosis. The 3-year PFS rates for patients with or without somatic TP53 mutations were 38.9% (95% CI, 9.3% to 68.7%) and 79.4% (95% CI, 64.0% to 88.7%; P = .02), respectively (Fig 1B). When stage IV disease was included in the Cox regression model, presence of somatic TP53 mutations was no longer associated with outcome (P = .21; Fig 1C). CTNNB1 status was not significantly associated with PFS (P = .80; Fig 2C).
The 3-year PFS for the 27 patients with Ki-67 LI ≥ 15% was 48.5% (95% CI, 27.5% to 66.7%) compared with 96.2% (95% CI, 75.7% to 99.4%) for the 29 patients with Ki-67 LI < 15% (P = .002; Fig 3B). Ki-67 LI remained significantly associated with PFS after adjusting for disease stage (P = .0048; Fig 3C). Cox regression modeling showed that among these 56 patients, rate of progression or death for patients with stage IV disease was 21.9 times that for other patients (95% CI, 5.25 to 91.46; P < .001), and rate of progression or death for patients with Ki-67 LI ≥ 15% was 22.27 times that for patients with Ki-67 LI < 15% (95% CI, 2.57 to 192.53; P = .0048).
Because higher tumor weight and older age are associated with poor prognosis in children with ACTs in general,8,20-22 we analyzed their association with Ki-67 LI. Median tumor weight was significantly higher for patients with Ki-67 LI ≥ 15% than for those with Ki-67 LI < 15% (191 v 93.2 g; P = .02). Ki-67 LI was not significantly associated with age (P = .62), suggesting that Ki-67 LI and age are independently associated with outcome. Thus, we fit a Cox regression model with age, tumor weight, disease stage (stage IV v I to III), and Ki-67 LI (< 15% v ≥ 15%) as predictors of PFS. In this model, tumor weight and disease stage showed potentially meaningful associations with PFS but did not reach statistical significance. However, rate of progression or death for patients with Ki-67 LI ≥ 15% was 41.8 times that for patients with Ki-67 LI < 15% (95% CI, 3.51 to 497.8; P = .003), and rate of relapse increased by 24% with each 1-year increase in age at diagnosis (hazard ratio, 1.24; 95% CI, 1.07 to 1.45; P = .0057; Appendix Table A2, online only).
We analyzed the expression of MKI67, which encodes Ki-67 protein, in two independent gene expression data sets from the GEO repository of pediatric ACTs and found that MKI67 overexpression as a single predictor was associated with worse PFS in both cohorts (GSE76021, P = .001; GSE76019, P = .016) as well as after adjustment for stage IV disease (GSE76021, P = .001; GSE76019, P = .011).
Consistent with these findings, greater mRNA expression of MKI67, PTTG1, BUB1B, and AURKB was found in ACT with adverse prognostic features (Appendix Fig A2, online only). Furthermore, MKI67 overexpression was correlated with low levels of HLA-DPA1 (Appendix Fig A3, online only), which is significantly associated with poor outcome.10,23 Expression of PTTG1, BUB1B, and AURKB was significantly higher in those with mutTP53-ACTs compared with wtTP53-ACTs. However, expression of IGF2 and MKI67 was similar, irrespective of patients’ TP53 status (Appendix Fig A4, online only).
DISCUSSION
Our study reveals significant overlap in clinicopathologic features, molecular attributes, pathogenesis, and outcomes among children with wtTP53-ACTs and mutTP53-ACTs and captures differences between these groups (Appendix Table A3, online only). Approximately 90% of patients with mutTP53-ACTs develop adrenocortical tumors by 5 years of age (peak incidence, 1 to 3 years); thereafter, the risk decreases and remains low throughout life.21,22 In contrast, 37% of our patients with wtTP53-ACTs were diagnosed after the age of 5 years. More than 90% of children with mutTP53-ACTs secrete androgens or androgens plus cortisol,24 whereas this pattern of hormonal secretion is seen in only approximately 55% of patients with wtTP53-ACTs.
The differences in age of onset and hormonal secretion pattern by ACTs may be attributed to adrenal cortex cell types involved in the malignant transformation. Our findings suggest that in children age < 5 years, regardless of genetic background, ACTs arise from the transient embryonic adrenal cortex.25,26 A smaller incidence peak in pubertal patients with wtTP53-ACTs coincides with physiologic changes in the adrenal cortex during adrenarche.27 Strikingly, the female predominance in ACTs at a young age (3:1) was not observed in children age ≥ 10 years (1:1) in this study. Collectively, our findings and previous studies suggest that predisposition to pediatric ACT depends on constitutional factors such as TP53 mutations and chromosome 11p15 abnormalities2,28-31 and physiologic sex- and age-specific developmental changes in the adrenal cortex.32
Remarkable and consistent features of adrenocortical tumors include chromosome 11p15 abnormalities and IGF2 overexpression,6,33-36 which are early events in adrenal cortex tumorigenesis.6 IGF2 overexpression probably occurs as a result of the combination of loss of maternal chromosome 11 and duplication of paternal chromosome 11,6,34 which occurs in virtually all patients, irrespective of TP53 status.6
In this study, dysregulation of imprinted genes in chromosome 11p15 was noted in the germline of nine of 40 patients. Also, insertion of foreign viral DNA (human herpesvirus 6 [HHV-6]) in the telomeric region of the short arm of chromosome 11 occurred in two patients (WT005 and WT030), both of whom inherited integrated HHV-6 from their fathers.6 HHV-6 chromosomal integration is associated with changes in the host methylation machinery to facilitate integration.37 In fact, patient WT030 also had hypomethylation at IC2. These findings suggest that irrespective of chromosome 11p15 status in the germline, adrenocortical tumorigenesis requires IGF2 overexpression and loss of a segment of or the entire maternal chromosome 11p15 (copy-neutral LOH). Our observations are consistent with the observation that in BWS, predisposition to ACT is highest among those with germline paternal UPD.38
Of clinical interest, six (67%) of nine patients in our study harbored germline abnormalities of chromosome 11p15 but did not have clinical signs of BWS. In these patients, IGF2 overexpression (Appendix Fig A1E) might have been restricted to tissues in which chromosome 11 underwent somatic copy-neutral LOH. Consistent with this hypothesis, ACT tissues from all five patients with germline chromosome 11p15 abnormalities had LOH of chromosome 11p15 (Appendix Table A4, online only). Our findings underscore the importance of detailed molecular analysis of chromosome 11p15 status in the germline of all children with embryonic tumors, especially Wilms tumor, hepatoblastoma, rhabdomyosarcoma, ACT, and pancreatoblastoma, all of which are associated with chromosome 11p15 abnormalities.39 Approximately 90% of adult adrenocortical carcinomas also overexpress IGF2.17,30 Attempts to pharmacologically target the IGF2 pathway with the IGF1 receptor inhibitor linsitinib in adult ACT was not associated with improved outcome.40 This strategy has not been used in pediatric ACT.
Activation of the Wnt signaling pathway is common in adult adrenocortical adenomas and carcinomas,41-44 suggesting its occurrence early in tumor formation.6,43 In our patients with wtTP53-ACTs, activating mutations in CTNNB1 were frequent and also observed in both adenomas and carcinomas. In contrast, activating mutations in CTNNB1 are rare in patients with germline TP53 mutations and seem to be mutually exclusive.6
Histologic classification of pediatric ACT has been controversial because of the lack of prognostic specificity.20,45 Because clinical presentation (age of onset and tumor pattern of hormonal secretion) and molecular (germline TP53 mutations, chromosome 11p15 abnormalities, and overexpression of IGF2) findings for pediatric ACT are similar to those for adenoma, carcinoma, and undetermined histology, we believe that they all represent a spectrum of the same disease.
A strong association between Ki-67 LI and prognosis in adult ACT has recently been established.46 Ki-67 is a cell proliferation marker that is critical for chromosome segregation during cell division.47 Given these findings, we investigated the prognostic implications of Ki-67 LI in our pediatric cohort. Ki-67 LI ≥ 15% and age were each independently associated with poor prognosis. To further explore Ki-67 prognostic correlates, we reanalyzed public data from our recent studies showing that expression of cell-cycle regulators and HLA class II genes was associated with outcome in pediatric ACT.6,10 When MKI67 mRNA expression was included in the analysis, it was positively associated with expression of cell-cycle regulatory genes and strongly associated with outcome. Therefore, Ki-67 LI may serve as an additional prognostic marker that should be considered in the current histopathologic classification of pediatric ACT.
In summary, our study reveals that clinical features, prognostic factors, and outcomes of pediatric ACT with or without germline TP53 mutations are overlapping. However, some genetic alterations contributing to tumorigenesis are likely diverse between both groups, with CTNNB1 mutations occurring almost exclusively in patients without germline TP53 mutations. On the basis of this study and other previous findings, constitutional 11p15 abnormalities should be considered in all children with ACTs irrespective of the absence of features of BWS or other growth disorders. We also recommend that Ki-67 LI be included in histologic characterization to improve the pathologic classification of pediatric ACT.
ACKNOWLEDGMENT
We thank study participants, clinicians who referred children with adrenocortical tumors to the International Pediatric Adrenocortical Tumor Registry study, and research staff at participating institutions. We also thank the Hartwell Center for Bioinformatics and Biotechnology and the Anatomic Pathology staff for their technical assistance and Vani Shanker for scientific editing. We thank the Speer Charitable Trust for its continued support of St Jude research.
Appendix
Table A1.
Characteristics of Patients With ACTs

Table A2.
Cox Regression Modeling Showing Association of PFS With Disease Stage, Tumor Weight, Age, and Ki-67 LI Status
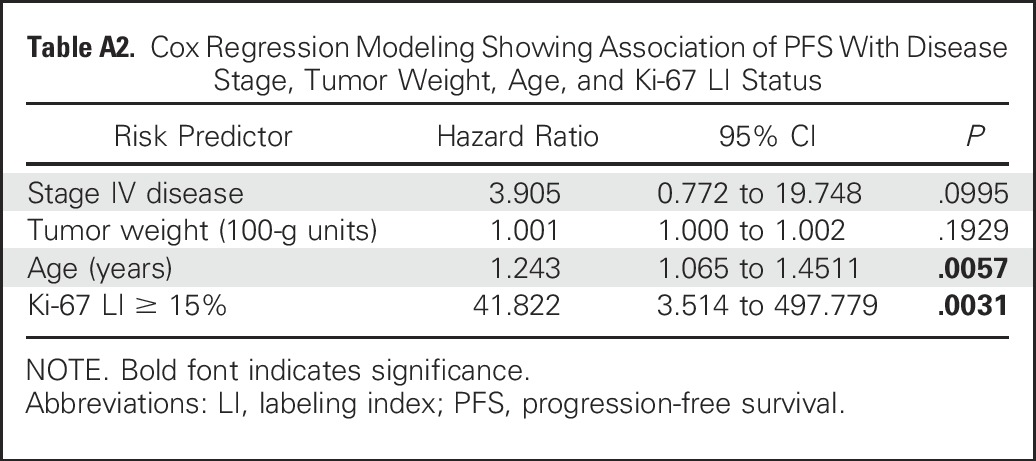
Table A3.
Characteristics of Patients With ACTs Harboring Mutations or No Mutations in TP53
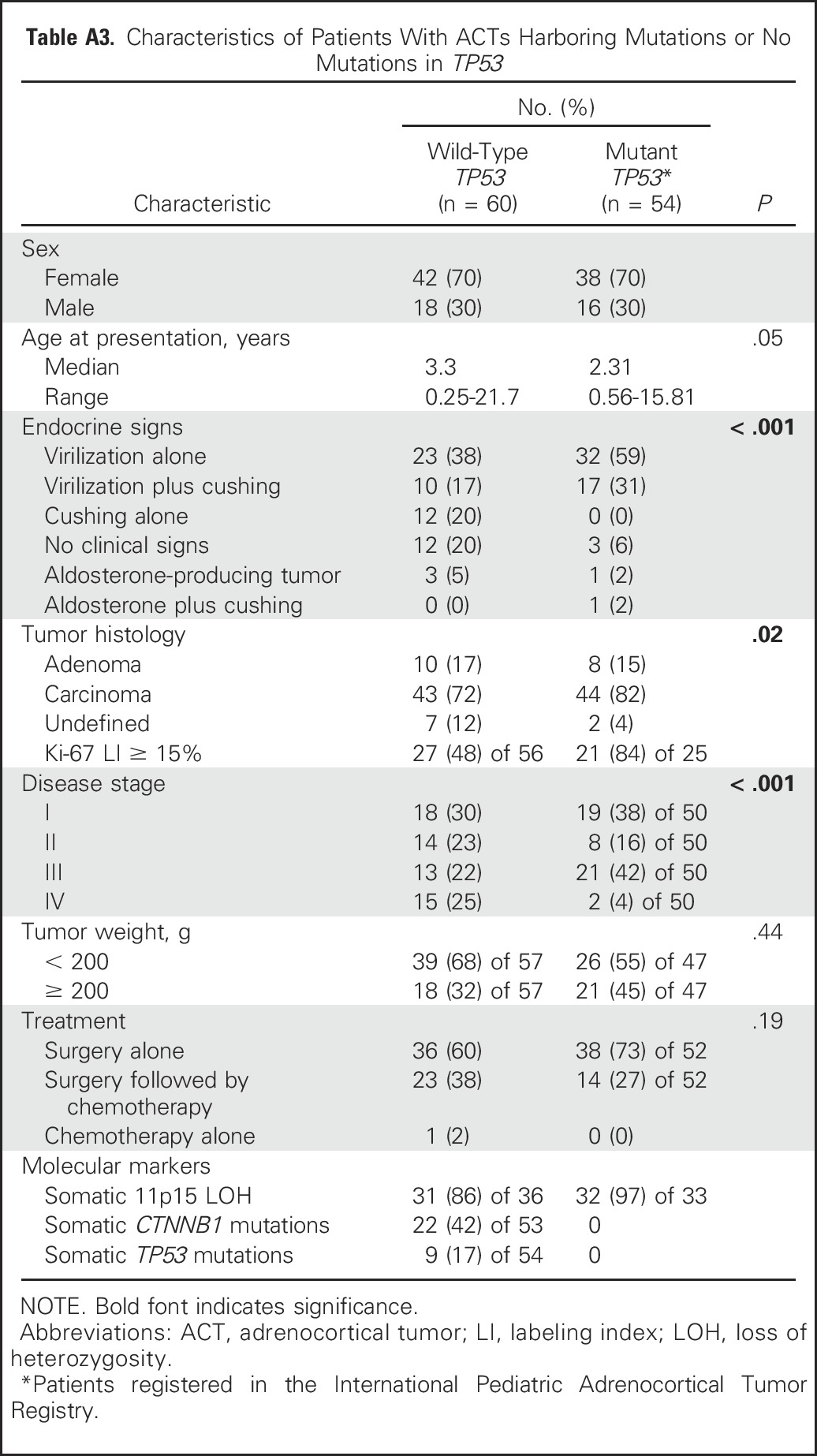
Table A4.
Characteristics of Patients With ACTs and Chromosome 11 Abnormalities
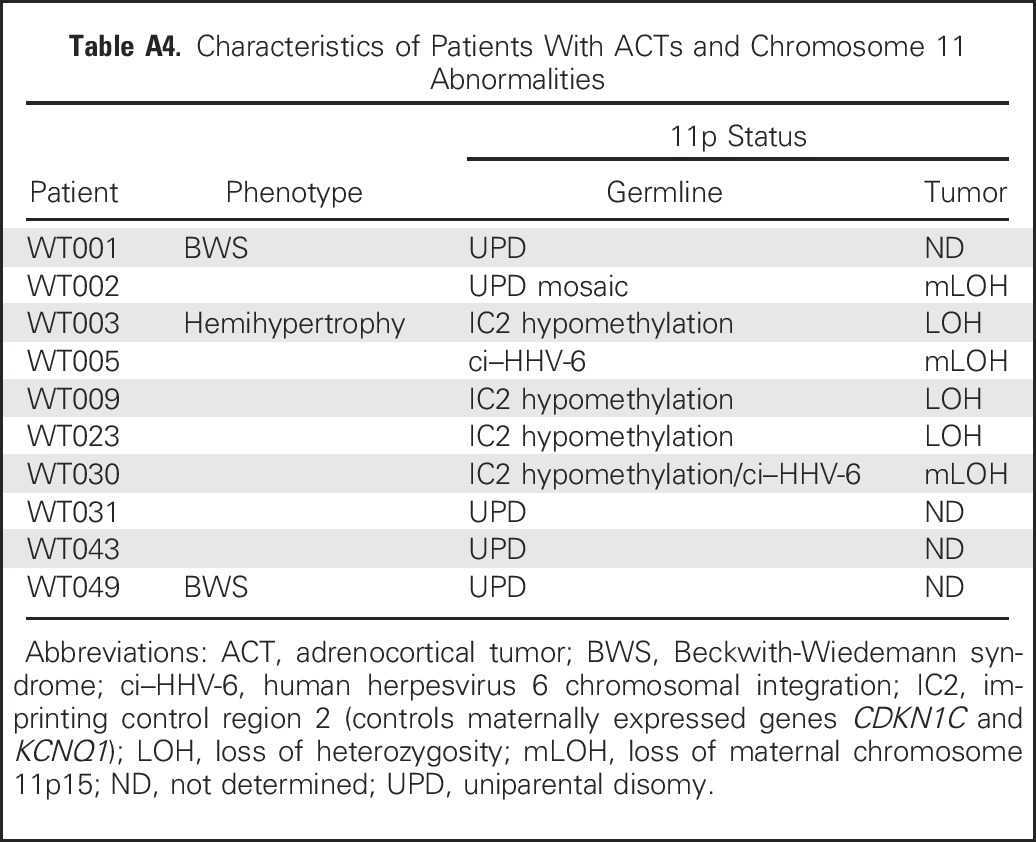
Fig A1.

Chromosome 11p15 abnormalities as visualized by methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) and microsatellite analysis in DNA from pediatric patients with adrenocortical tumors (ACTs) without TP53 mutations. (A) MS-MLPA analysis of WT031 blood DNA showing (upper panel) normal copy number of chromosome 11p15 and (lower panel) gain of methylation at imprinting control region 1 (IC1) with loss of methylation at imprinting control region 2 (IC2) indicating uniparental disomy. (B) WT002 blood DNA showing partial gain of methylation at IC1 and partial loss of methylation at IC2. (C) MS-MLPA analysis of WT023 blood DNA showing loss of methylation at IC2. (D) Representative microsatellite marker analysis of parents (blood) and patient (blood and tumor) DNA. The father (F) is homozygous (185/185) and the mother (M) heterozygous (192/194) for the D11S4046 marker. Patient blood DNA shows inheritance of alleles 185 from father and 194 from mother; (bottom panel) allele 194 (maternal origin) is selected against in the tumor. (E) Western blot analysis performed with 50 µg of protein with goat polyclonal antihuman antibody directed against insulin-like growth factor 2 (IGF2; 1:500 dilution; Sigma-Aldrich, St Louis, MO) as previously reported.23 β-actin (1:2,000; Sigma-Aldrich) was used as the loading control. This analysis included patients with 11p15 abnormalities on germline (n = 6) and with germline-mutated TP53, as indicated. Levels of IGF2 were higher in samples than in control (two normal adrenocortical tissues obtained during nephrectomy for Wilms tumor) and were not affected by genotype (wild-type or mutated TP53) or histology (carcinoma, adenoma, or undetermined [Und]). ACA, adrenocortical adenoma; ACC, adrenocortical carcinoma; CNV, copy number variation; al, allele; ht, height.
Fig A2.

Association of PTTG1, BUB1B, AURKB, HLA-DPA1, and MKI67 mRNA expression with progression-free survival (PFS). mRNA expression was obtained from Gene Expression Omnibus databases GSE76019 and GSE76021. Plots represent mRNA expression according to each probe set for those selected genes. Higher expression of AURKB, BUB1B, PTTG1, and MKI67 was significantly associated with worse PFS.
Fig A3.

Correlations of mRNA expression of selected cell-cycle genes within the (A) Children’s Oncology Group (GSE76019) and International Pediatric Adrenocortical Tumor Registry cohorts (GSE76021). The panels provide the Spearman correlation and P value for each pair of cell-cycle gene probe sets in both cohorts. Each entry in the upper triangle gives the Spearman correlation of the expression of the pair of genes indicated by the row and column labels. Each entry in the lower triangle gives the P value for the association of the pair of genes indicated by the row and column labels. NA, not applicable.
Fig A4.

Representative plots of mRNA expression of MKI67, AURKB, BUB1B, and PTTG1 according to TP53 status from adrenocortical tumor (ACT) samples in available public databases (Gene Expression Omnibus databases GSE76019 and GSE7602). Expression levels of PTTG1, BUB1B, and AURKB were significantly higher in ACTs with TP53 mutations than in those without. Expression of IGF2 and MKI67 was not significantly different,according to TP53 status. Expression levels are represented on the positive quantile transformation (PQT) scale.
Footnotes
Supported by National Cancer Institute, National Institutes of Health, Cancer Center Support Grant No. CA21765, by Centocor, and by the American Lebanese Syrian Associated Charities.
AUTHOR CONTRIBUTIONS
Conception and design: Emilia Modolo Pinto, Gerard P. Zambetti, Raul C. Ribeiro
Provision of study materials or patients: Emilia Modolo Pinto, Carlos Rodriguez-Galindo, Catherine G. Lam, Carolyn Fein Levy, Alberto S. Pappo, Gerard P. Zambetti, Raul C. Ribeiro
Collection and assembly of data: Emilia Modolo Pinto, Stanley B. Pounds, Lei Wang, Michael R. Clay, Geoffrey Neale, Elizabeth A.R. Garfinkle, Raul C. Ribeiro
Data analysis and interpretation: Emilia Modolo Pinto, Carlos Rodriguez-Galindo, Stanley B. Pounds, Lei Wang, Michael R. Clay, Geoffrey Neale, Catherine G. Lam, Carolyn Fein Levy, Alberto S. Pappo, Raul C. Ribeiro
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Identification of Clinical and Biologic Correlates Associated With Outcome in Children With Adrenocortical Tumors Without Germline TP53 Mutations: A St Jude Adrenocortical Tumor Registry and Children’s Oncology Group Study
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Emilia Modolo Pinto
No relationship to disclose
Carlos Rodriguez-Galindo
Honoraria: Novimmune
Stanley B. Pounds
No relationship to disclose
Lei Wang
No relationship to disclose
Michael R. Clay
No relationship to disclose
Geoffrey Neale
No relationship to disclose
Elizabeth A.R. Garfinkle
No relationship to disclose
Catherine G. Lam
Stock or Other Ownership: PharmaJet (I)
Consulting or Advisory Role: Baxalta (I), Sanofi (I), Sigma-Tau (I), Jazz Pharmaceuticals (I)
Carolyn Fein Levy
Stock or Other Ownership: Pfizer
Alberto S. Pappo
No relationship to disclose
Gerard P. Zambetti
Research Funding: Centocor
Raul C. Ribeiro
No relationship to disclose
REFERENCES
- 1.Li FP, Fraumeni JF, Jr, Mulvihill JJ, et al. : A cancer family syndrome in twenty-four kindreds. Cancer Res 48:5358-5362, 1988 [PubMed] [Google Scholar]
- 2.Malkin D, Li FP, Strong LC, et al. : Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233-1238, 1990 [DOI] [PubMed] [Google Scholar]
- 3.Weksberg R, Shuman C, Beckwith JB: Beckwith-Wiedemann syndrome. Eur J Hum Genet 18:8-14, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribeiro RC, Pinto EM, Zambetti GP: Familial predisposition to adrenocortical tumors: Clinical and biological features and management strategies. Best Pract Res Clin Endocrinol Metab 24:477-490, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Else T: Association of adrenocortical carcinoma with familial cancer susceptibility syndromes. Mol Cell Endocrinol 351:66-70, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinto EM, Chen X, Easton J, et al. : Genomic landscape of paediatric adrenocortical tumours. Nat Commun 6:6302, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wasserman JD, Novokmet A, Eichler-Jonsson C, et al. : Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: A Children’s Oncology Group study. J Clin Oncol 33:602-609, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ribeiro RC, Pinto EM, Zambetti GP, et al. : The International Pediatric Adrenocortical Tumor Registry initiative: Contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol Cell Endocrinol 351:37-43, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Priolo M, Sparago A, Mammì C, et al. : MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur J Hum Genet 16:565-571, 2008 [DOI] [PubMed] [Google Scholar]
- 10.Pinto EM, Rodriguez-Galindo C, Choi JK, et al. : Prognostic significance of major histocompatibility complex class II expression in pediatric adrenocortical tumors: A St Jude and Children’s Oncology Group study. Clin Cancer Res 22:6247-6255, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collett D: Modeling Survival Data in Medical Research. London, United Kingdom, Chapman and Hall/CRC, 1994 [Google Scholar]
- 12.Lachin JM: Biostatistical Methods: The Assessment of Relative Risks. New York, NY, John Wiley & Sons, 2000 [Google Scholar]
- 13.Bouaoun L, Sonkin D, Ardin M, et al. : TP53 variations in human cancers: New lessons from the IARC TP53 database and genomics data. Hum Mutat 37:865-876, 2016 [DOI] [PubMed] [Google Scholar]
- 14.Kaneda A, Wang CJ, Cheong R, et al. : Enhanced sensitivity to IGF-II signaling links loss of imprinting of IGF2 to increased cell proliferation and tumor risk. Proc Natl Acad Sci USA 104:20926-20931, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuoka S, Edwards MC, Bai C, et al. : p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev 9:650-662, 1995 [DOI] [PubMed] [Google Scholar]
- 16.Smith AC, Choufani S, Ferreira JC, et al. : Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatr Res 61:43R-47R, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Guillaud-Bataille M, Ragazzon B, de Reyniès A, et al. : IGF2 promotes growth of adrenocortical carcinoma cells, but its overexpression does not modify phenotypic and molecular features of adrenocortical carcinoma. PLoS One 9:e103744, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper WN, Luharia A, Evans GA, et al. : Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur J Hum Genet 13:1025-1032, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Libè R, Groussin L, Tissier F, et al. : Somatic TP53 mutations are relatively rare among adrenocortical cancers with the frequent 17p13 loss of heterozygosity. Clin Cancer Res 13:844-850, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Wieneke JA, Thompson LD, Heffess CS: Adrenal cortical neoplasms in the pediatric population: A clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol 27:867-881, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Michalkiewicz E, Sandrini R, Figueiredo B, et al. : Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol 22:838-845, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Varley JM, McGown G, Thorncroft M, et al. : Are there low-penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet 65:995-1006, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West AN, Neale GA, Pounds S, et al. : Gene expression profiling of childhood adrenocortical tumors. Cancer Res 67:600-608, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Marques-Pereira R, Delacerda L, Lacerda HM, et al. : Childhood adrenocortical tumours: A review. Hered Cancer Clin Pract 4:81-89, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spencer SJ, Mesiano S, Lee JY, et al. : Proliferation and apoptosis in the human adrenal cortex during the fetal and perinatal periods: Implications for growth and remodeling. J Clin Endocrinol Metab 84:1110-1115, 1999 [DOI] [PubMed] [Google Scholar]
- 26.Ishimoto H, Jaffe RB: Development and function of the human fetal adrenal cortex: A key component in the feto-placental unit. Endocr Rev 32:317-355, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baquedano MS, Saraco N, Berensztein E, et al. : Identification and developmental changes of aromatase and estrogen receptor expression in prepubertal and pubertal human adrenal tissues. J Clin Endocrinol Metab 92:2215-2222, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Birch JM, Alston RD, McNally RJ, et al. : Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene 20:4621-4628, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Henry I, Jeanpierre M, Couillin P, et al. : Molecular definition of the 11p15.5 region involved in Beckwith-Wiedemann syndrome and probably in predisposition to adrenocortical carcinoma. Hum Genet 81:273-277, 1989 [DOI] [PubMed] [Google Scholar]
- 30.Gicquel C, Bertagna X, Schneid H, et al. : Rearrangements at the 11p15 locus and overexpression of insulin-like growth factor-II gene in sporadic adrenocortical tumors. J Clin Endocrinol Metab 78:1444-1453, 1994 [DOI] [PubMed] [Google Scholar]
- 31.Gicquel C, Le Bouc Y: Molecular markers for malignancy in adrenocortical tumors. Horm Res 47:269-272, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Hui XG, Akahira J, Suzuki T, et al. : Development of the human adrenal zona reticularis: Morphometric and immunohistochemical studies from birth to adolescence. J Endocrinol 203:241-252, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ilvesmäki V, Kahri AI, Miettinen PJ, et al. : Insulin-like growth factors (IGFs) and their receptors in adrenal tumors: High IGF-II expression in functional adrenocortical carcinomas. J Clin Endocrinol Metab 77:852-858, 1993 [DOI] [PubMed] [Google Scholar]
- 34.Gicquel C, Raffin-Sanson ML, Gaston V, et al. : Structural and functional abnormalities at 11p15 are associated with the malignant phenotype in sporadic adrenocortical tumors: Study on a series of 82 tumors. J Clin Endocrinol Metab 82:2559-2565, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Wilkin F, Gagné N, Paquette J, et al. : Pediatric adrenocortical tumors: Molecular events leading to insulin-like growth factor II gene overexpression. J Clin Endocrinol Metab 85:2048-2056, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Gicquel C, Bertagna X, Gaston V, et al. : Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors. Cancer Res 61:6762-6767, 2001 [PubMed] [Google Scholar]
- 37.Engdahl E, Dunn N, Niehusmann P, et al. : Human herpesvirus 6B induces hypomethylation on chromosome 17p13.3 correlating with increased gene expression and virus integration. J Virol 91:e02105-e02116, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maas SM, Vansenne F, Kadouch DJ, et al. : Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A 170:2248-2260, 2016 [DOI] [PubMed] [Google Scholar]
- 39.Chung WY, Yuan L, Feng L, et al. : Chromosome 11p15.5 regional imprinting: Comparative analysis of KIP2 and H19 in human tissues and Wilms’ tumors. Hum Mol Genet 5:1101-1108, 1996 [DOI] [PubMed] [Google Scholar]
- 40.Fassnacht M, Berruti A, Baudin E, et al. : Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: A double-blind, randomised, phase 3 study. Lancet Oncol 16:426-435, 2015 [DOI] [PubMed] [Google Scholar]
- 41.Penny MK, Finco I, Hammer GD: Cell signaling pathways in the adrenal cortex: Links to stem/progenitor biology and neoplasia. Mol Cell Endocrinol 445:42-54, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Assié G, Letouzé E, Fassnacht M, et al. : Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46:607-612, 2014 [DOI] [PubMed] [Google Scholar]
- 43. doi: 10.1016/j.ccell.2016.04.002. Zheng S, Cherniack AD, Dewal N, et al: Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 29:723-736, 2016 [Erratum: Cancer Cell 30:363, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tissier F, Cavard C, Groussin L, et al. : Mutations of beta-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res 65:7622-7627, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Dehner LP, Hill DA: Adrenal cortical neoplasms in children: Why so many carcinomas and yet so many survivors? Pediatr Dev Pathol 12:284-291, 2009 [DOI] [PubMed] [Google Scholar]
- 46.Beuschlein F, Weigel J, Saeger W, et al. : Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab 100:841-849, 2015 [DOI] [PubMed] [Google Scholar]
- 47.Cuylen S, Blaukopf C, Politi AZ, et al. : Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 535:308-312, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]