

Summary
We have demonstrated previously that histone deacetylase (HDAC6) expression is increased in animal models of systemic lupus erythematosus (SLE) and that inhibition of HDAC6 decreased disease. In our current studies, we tested if an orally active selective HDAC6 inhibitor would decrease disease pathogenesis in a lupus mouse model with established early disease. Additionally, we sought to delineate the cellular and molecular mechanism(s) of action of a selective HDAC6 inhibitor in SLE. We treated 20‐week‐old (early‐disease) New Zealand Black (NZB)/White F1 female mice with two different doses of the selective HDAC6 inhibitor (ACY‐738) for 5 weeks. As the mice aged, we determined autoantibody production and cytokine levels by enzyme‐linked immunosorbent assay (ELISA) and renal function by measuring proteinuria. At the termination of the study, we performed a comprehensive analysis on B cells, T cells and innate immune cells using flow cytometry and examined renal tissue for immune‐mediated pathogenesis using immunohistochemistry and immunofluorescence. Our results showed a reduced germinal centre B cell response, decreased T follicular helper cells and diminished interferon (IFN)‐γ production from T helper cells in splenic tissue. Additionally, we found the IFN‐α‐producing ability of plasmacytoid dendritic cells was decreased along with immunoglobulin isotype switching and the generation of pathogenic autoantibodies. Renal tissue showed decreased immunoglobulin deposition and reduced inflammation as judged by glomerular and interstitial inflammation. Taken together, these studies show selective HDAC6 inhibition decreased several parameters of disease pathogenesis in lupus‐prone mice. The decrease was due in part to inhibition of B cell development and response.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease in which the body's immune system produces autoantibodies against normal healthy tissue or cellular components to form immune complexes that are deposited in various tissues, inducing inflammation leading to tissue damage 1. Autoantibodies are produced by activated autoreactive B cells through antibody class‐switching and somatic hypermutation, with help from activated autoreactive T cells 2. In early disease, several studies have shown a critical role for the proinflammatory cytokine interferon (IFN)‐α 3, 4, 5, 6. Additionally, studies using lupus‐prone mice have demonstrated that the ablation of plasmacytoid dendritic cells (pDCs), which are the major source of IFN‐α, prevented lupus nephritis (LN) progression, reduced activation of autoreactive T and B cells and decreased autoantibodies in the circulation and renal deposition 7, 8. These studies highlight the critical involvement of innate immune activation in the initiation of SLE. LN occurs in approximately 50% of SLE patients and is a major cause of morbidity and mortality in this disease 9. Recently, several studies have provided evidence that epigenetic factors play a crucial role in the initiation and progression of the development of SLE, including LN 10.
Epigenetic factors include miRNA, methylation of DNA and acetylation of histones as well as non‐histone proteins. There exists a balance of histone acetylaces (HATs) and histone deacetylases (HDACs) catalyzing the addition or removal of acetyl groups on proteins which alter their stability and function 11. There are 18 mammalian HDACs, which remove acetyl groups from lysine residues on histones and other proteins that can lead to modifications in multiple cellular functions including transcription, cell cycle kinetics, cell signalling and cellular transport processes 12. HDACs are classified based on structure and function homology studied originally in yeast. Currently, HDACs fall into four different classes. Class I HDACs (HDAC1, 2, 3 and 8) reside primarily in the nucleus and are distributed in a wide range of cells and tissues, where they are responsible predominantly for targeting histone proteins to repression gene transcription. Class II HDACs are subdivided into class IIa (HDAC4, 5, 7 and 9) and class IIb (HDAC6 and 10) based on domain organization and exhibit selective tissue expression, and may shuttle between the cytosol and the nucleus 12. Class III comprises the sirtuins, which act through a distinct nicotinamide adenine dinucleotide (NAD+)‐dependent mechanism and are not considered ‘classical’ HDACs 13. HDAC11 is the sole member of class IV, as phylogenetic analysis revealed a very low similarity to HDACs in the other classes 14.
In our previous studies we have demonstrated that inhibition of HDAC6, a class IIb HDAC, resulted in the amelioration of LN in lupus‐prone mice associated with several altered immune responses, with minimal toxicity, suggesting therapeutic potential to treat LN 15, 16, 17. SLE is a chronic inflammatory disease that is divided generally into the early autoimmune initiating stage and the later autoimmune effector stage 18, 19, 20. It is still not well understood whether the effects of HDAC6 inhibition to altered autoimmune responses are at the priming stage or effector stage. Here, using lupus‐prone mice, we demonstrated that HDAC6 inhibition was able to down‐regulate the autoimmune responses in early disease and inhibit the initiation of LN.
Materials and methods
Mice and ACY‐738 treatment
Female New Zealand Black/White F1 (NZB/WF1/J) (NZB/W) and C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained under specific pathogen‐free conditions at Virginia Tech College of Veterinary Medicine. For ACY‐738 treatment, NZB/W mice were given a diet mixed with two different doses of the HDAC6 inhibitor, ACY‐738 (50 mg/kg diet or 200 mg/kg diet), which was purchased from Envigo (Huntingdon, UK). All procedures strictly followed the requirements of the Institutional Animal Care and Use Committee (IACUC) at Virginia Tech.
Leucocyte isolation and flow cytometric analysis
Bone marrow leucocytes. Bones from both hind limbs of each mouse were cracked gently in a mortar containing phosphate‐buffered saline (PBS) using a pestle. Bone marrow was released by gentle stirring after the addition of C10 medium (RPMI‐1640, 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 1% ×100 minimum essential medium (MEM) non‐essential amino acids, 10 mM HEPES, 55 μM 2‐mercaptoethanol, 2 mM L‐glutamine and 100 U/ml penicillin–streptomycin, all from Life Technologies, Grand Island, NY, USA). The suspension was cleared by passing through a 70‐μm sterile cell strainer and layered on top of Ficoll‐Paque Plus (GE Healthcare, Pittsburg, PA, USA). After centrifugation at 1363 g without break for 30 min at room temperature, mononuclear cells in the buffy coat layer were collected. In studies with pure plasmacytoid dendritic cells (pDCs), they were then sorted as 4′,6‐diamidino‐2‐phenylindole (DAPI)–CD11c+CD11b– plasmacytoid dendritic cell antigen 1 (PDCA1)+B220+ cells.
Spleen leucocytes. Spleens were collected, smashed and then washed through 70‐μm cell strainers with C10 medium. Red blood cells were lysed with red blood cell (RBC) lysis buffer (eBioscience, San Diego, CA, USA). Cell suspensions were filtered through 70‐μm cell strainers to remove debris clumps.
Ex‐vivo stimulation of splenocytes
Splenocytes (1 × 106 per well) were stimulated with 50 ng/ml phorbol 12‐myristate 13‐acetate (PMA) (Sigma‐Aldrich, St Louis, MO, USA), 1 μg/ml ionomycin (Sigma‐Aldrich) and ×1 brefeldin A (BFA) (eBioscience) in 200 μl C10 and cultured in a 96‐well flat‐bottomed plate at 37°C with 5% CO2 for 5 h. Splenocytes were then collected for flow cytometry.
Flow cytometric analysis
For surface marker staining, cells were blocked with anti‐mouse CD16/32 (eBioscience), stained with fluorochrome‐conjugated antibodies and analysed with BD FACSAria II (BD Biosciences, San Jose, CA, USA) or Attune NxT (ThermoFisher Scientific, Waltham, MA, USA) flow cytometer. For intracellular marker staining, after surface staining cells were fixed and permeabilized with Fix/Perm buffer (eBioscience) before staining intracellular markers. Anti‐mouse antibodies used in this study included: CD11b, CD11c, Siglec‐H, B220, lymphocyte antigen 6 complex, locus A (Ly6a), major histocompatibility complex class II (MHC)‐II, CD19, CD138, immunoglobulin (Ig)D, monoclonal antibody GL7 (GL7), IgM, CD43, CD3, CD4, forkhead box protein 3 (FoxP3), programmed death 1 (PD‐1) (Biolegend), C‐C chemokine receptor type 5 (CCR5), IL‐10 and IFN‐γ (BD Biosciences). Flow cytometry data were analysed with FlowJo software (Tree Star, Ashland, OR, USA.
Ex‐vivo HDAC6 inhibition
For the drug inhibition experiment, bone marrow mononuclear cells (BMMCs) from C57BL/6 mice were isolated and seeded in a 96‐well flat‐bottomed plate at a density of 2 × 106 cells/ml in 200 μl C10 per well. The cells were then incubated with various concentrations of ACY‐738 overnight and stimulated with ODN1585 cytosine–phosphate–guanosine (CpG) (5 μM final) for an additional 6 h. Supernatants were harvested and stored in a freezer at −80°C.
For the small‐interfering RNA (siRNA) interference experiment, pDCs were enriched from BMMCs using negative selection with a magnetic‐activated cell sorting (MACS) kit (MACS catalogue no. 130‐092‐786) and seeded in a 24‐well flat‐bottomed plate at a density of 1·5 × 106 cells/ml in 400 μl C10 per well. siRNA complexes were prepared in HiPerfect reagent (Qiagen, Hilden, Germany) according to the manufacturers' instructions, and added to the cells at 100 μl per well dropwise, followed by 1‐day incubation at 37°C with 5% CO2. pDCs were then stimulated with ODN1585 CpG (5 μM final) for another 6 h. Supernatants were harvested and stored at −80°C.
Enzyme‐linked immunosorbent assay (ELISA)
Blood was collected directly from the heart, transferred into microcentrifuge tubes and incubated for 30 min at room temperature, and then centrifuged at 1500 g for 5 min. Serum was collected and stored at −80°C. Detection of anti‐double‐stranded DNA (dsDNA) IgG was described previously 21. A similar procedure was used to detect anti‐dsDNA IgG2a. Total IgG and IgG2a concentrations were determined using mouse IgG ELISA kits, according to the manufacturer's instructions (Bethyl Laboratories, Montgomery, TX, USA).
Immunofluorescence
Kidneys were embedded in Tissue‐Tek® optimal cutting temperature compound (O.C.T.™) (Sakura Finetek, Torrance, CA, USA) and frozen rapidly in a freezing bath of dry ice and 2‐methylbutane. Frozen OCT samples were cryosectioned and unstained slides were stored at −80°C. Frozen slides were warmed to room temperature and allowed to dry for 30 min, followed by fixation in −20°C cold acetone at room temperature for 10 min. After washing in PBS, slides were blocked with PBS containing 1% bovine serum albumin (BSA) and anti‐mouse CD16/32 for 20 min at room temperature. Slides were then incubated with fluorochrome‐conjugated antibody mixture for 1 h at room temperature in a dark humid box. Slides were mounted with Prolong Gold containing DAPI (Life Technologies, Carlsbad, CA, USA). The following anti‐mouse antibodies were used in immunohistochemical analysis: IgG‐phycoerythrin (PE), IgG 2a‐PE (eBioscience, Santa Clara, CA, USA) and anti‐C3‐fluorescein isothiocyanate (FITC) (Cedarlanelabs, Burlington, Canada). Slides stained with antibodies were read and pictured with EVOS® FL microscope (Advanced Microscopy Group, Grand Island, NY, USA) and a ×40 objective. Six randomly picked glomeruli from each sample were pictured and then analysed by using ImageJ software (National Institutes of Health, Rockville, MD, USA) to calculate the deposition of IgG, IgG2a and C3.
Proteinuria score and kidney histopathology
Proteinuria was measured weekly with Uristix 4 (Siemens, Sacramento, CA, USA). A scale of 0–4 was used that corresponded to negative‐trace, 30, 100, 300 and ≥ 2000 mg/dl total protein, respectively. For renal histopathology, both kidneys were removed at the time of euthanasia. One kidney was fixed in 10% neutral buffered formalin for 24 h, then processed routinely, embedded in paraffin, sectioned at 4–5 μm and stained with periodic acid‐Schiff (PAS). Kidney sections were assigned a glomerular pathology score (0–4) based on a cumulative evaluation of glomerular proliferation, inflammation, crescent formation, necrosis and fibrosis by a board‐certified veterinary anatomic pathologist (M. D. Vieson) in a blinded manner. Briefly, the score ranges from 0 (normal glomeruli) to 4, which includes glomeruli characterized by marked mesangial thickening, hypercellularity, segmental necrosis, crescents and hyalinized end‐stage glomeruli.
Statistical analysis
Unpaired Student's t‐test was used for comparison of two groups. For the comparison of more than two groups, one‐way analysis of variance (anova) and Tukey's post‐test were used. Results were considered statistically significant when P < 0·05. In some experiments, linear regression analysis and Grubbs' test for identification of outliers were used. All analyses were performed with Prism Graphpad (La Jolla, CA, USA) software.
Results
Inhibition of HDAC6 prevents LN progression
We have reported previously on the pharmacokinetics of the selective HDAC6 inhibitor ACY‐783 16. To study the effects of HDAC6 inhibition on the initiation of LN, we treated 20‐week‐old NZB/W F1 female (NZB/W) mice with the selective HDAC6 inhibitor, ACY‐738. Two different concentrations were mixed in the food to evaluate a dose‐dependent effect for this compound. During the treatment, there were no statistically significant changes in food consumption or body weight (data not shown). Weekly monitoring of proteinuria showed that starting from 23 weeks of age, NZB/W mice treated with both doses of ACY‐738 had significantly lower proteinuria scores compared to those without treatment (Fig. 1a). Linear regression analysis showed a significant increase in proteinuria in the untreated group. While proteinuria in the 50 mg/kg ACY‐738‐treated group remained relatively unchanged, there was a statistically significant decrease in proteinuria noted in the 200 mg/kg ACY‐738‐treated group. At termination of the treatment (aged 25 weeks), kidney sections were evaluated for glomerulonephritis (Fig. 1b). Given our scoring parameters, the 50 mg/kg ACY‐738‐treated group was significantly lower than that of the untreated group (Fig. 1b). The 200 mg/kg‐treated group also had a trend of lower score, although it did not reach but was close to statistical significance compared to controls (P = 0·059). Taken together, these results suggest HDAC6 inhibition administered in the early stage of LN can help to prevent the progression of LN.
Figure 1.
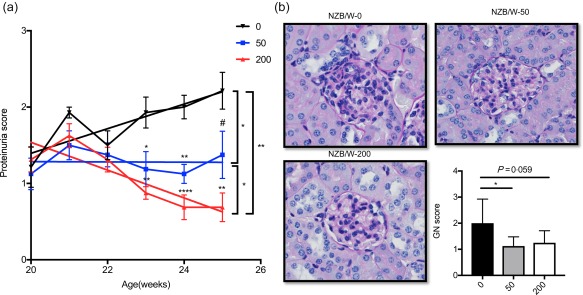
Inhibition of histone deacetylase (HDA)C6 prevents histone deacetylase (LN) progression. (a) New Zealand Black/White (NZB/W) mice showing proteinuria as they age. The tendency of proteinuria changes in three groups are indicated by the slope of lines representatively. 0 = control; 50 (mg/kg ACY‐738); 200 (mg/kg ACY‐738). (b) Representative periodic acid‐Schiff (PAS) image of the glomerulus from NZB/W mice in three different group are shown along with pathology score for glomerular lymphoproliferative mononuclear cell infiltration. Data are shown as mean ± standard error of the mean (s.e.m.), n = 8 mice in each group, #P = 0·057; *P < 0·05; **P < 0·01; ***P < 0·001; one‐way analysis of variance (anova). [Colour figure can be viewed at wileyonlinelibrary.com]
HDAC6 inhibition reduces IFN‐α production from pDCs
To study the mechanism by which selective HDAC6 inhibition prevents the initiation of LN, we focused on the possible changes of the innate immune responses, particularly pDCs, as they have been reported to play a key role in the initiation LN through the production of IFN‐α 7, 8. We found that the administration of ACY‐738 led to a statistically significant dose‐dependent increase of the percentage of MHC‐II+ pDCs (particularly MHC‐II+Ly6a+ pDCs, gated according to the isotype control) and a significant decrease of the percentage of MHC‐II‐Ly6a– pDCs in the BM compared to the untreated group (Supporting information, Fig. S1a, Fig. 2a,b). Furthermore, the absolute number of BM pDCs did not change (Supporting information, Fig. S1b). To evaluate further the effect of HDAC6 inhibition on pDC IFN‐α production, we sorted pDCs from the BM stimulated with CpG, cultured at 37°C for 6 h, and then collected supernatant to detect the IFN‐α production of pDCs. We found that the ex‐vivo IFN‐α‐producing ability of BM pDCs from the 50 mg/kg ACY‐738‐treated group was significantly lower than that of the untreated group (Fig. 2c). The pDCs from the 200 mg/kg ACY‐738‐treated group was also decreased, although it did not reach statistical significance (Fig. 2c). These results suggest that HDAC6 inhibition may inhibit differentiation of BM MHC‐II–Ly6a– pDCs, which are the cells thought to produce the large amounts of IFN‐α at the initial stage of LN 22, 23. Consistent with the reduced IFN‐α production of pDCs in the BM of ACY‐738‐treated groups, through RNA sequencing of total splenocytes we found a set of IFN‐α‐regulated genes significantly different between the 0 mg/kg and 200 mg/kg ACY‐738‐treated groups, as shown in Tables 1 and 2. Compared to the INTERFEROME database 24, we found that the expression of splenic IFN‐α‐down‐regulated (Table 1) and up‐regulated genes (Table 2) were significantly higher and lower in the 200 mg/kg ACY‐738‐treated groups, respectively, suggesting the lower IFN‐α level in the spleens of the HDAC6 inhibition group. To evaluate further the influence of selective HDAC6 inhibition on the function of pDCs, we cultured total BM cells or enriched BM pDCs from C57BL/6 mice in medium containing various concentrations of ACY‐738. In parallel experiments, we used HDAC6 siRNAs to specifically knock down HDAC6. We found that in a concentration‐dependent manner, ACY‐738 at 1 μM suppressed CpG‐induced IFN‐α production significantly from pDCs without significant cytotoxicity (Fig. 3a). Furthermore, using siRNA to knock down HDAC6 directly in pDCs we found a significant decrease of IFN‐α production upon CpG stimulation without affecting the survival of pDCs (Fig. 3b). Taken together, our results suggest that HDAC6 inhibition can suppress IFN‐α production from pDCs which may, in turn, prevent the initiation of LN.
Figure 2.
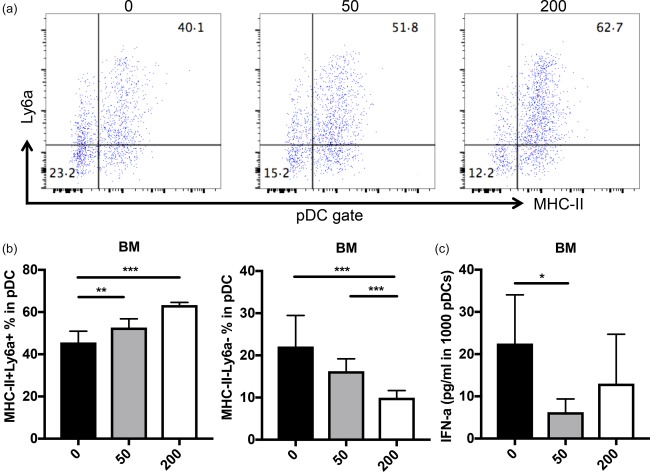
In‐vivo study of histone deacetylase (HDA)C6 inhibition reduces interferon (IFN)‐α production from plasmacytoid dendritic cells (pDCs). (a) Representative flow cytometry plots of subtypes of pDCs in bone marrow are shown. (b) The percentage of pDCs with low ability [major histocompatibility complex (MHC‐II)+ lymphocyte antigen 6 (Ly6a)+ pDCs] and high ability (MHC‐II– Ly6a– pDCs) of secreting IFN‐α. (c) IFN‐α production of sorted pDCs from bone marrow upon cytosine–phosphate–guanosine (CpG) stimulation for 12 h. *P < 0·05; **P < 0·01; ***P < 0·001; one‐way analysis of variance (anova), n ≥ 4. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 1.
Interferon (IFN)‐α down‐regulated genes from RNA sequencing of total splenocytes
| IFN‐α down‐regulated genes | ||
|---|---|---|
| Gene | log2_fold_change (ACY 738: 0 mg/200 mg) | P‐value |
| 1100001G20Rik | −0·79508 | 9·00E‐04 |
| 7SK | −1·05718 | 5·00E‐05 |
| Ankrd13b | −0·743131 | 0·00195 |
| Aqp9 | −0·893753 | 0·00015 |
| Art4 | −0·685621 | 0·0029 |
| Atp7b | −0·951345 | 5·00E‐05 |
| Car3 | −2·26838 | 0·00075 |
| Ccl17 | −1·70116 | 5·00E‐04 |
| Ccl24 | −1·11797 | 5·00E‐05 |
| Ctrb1 | −1·68403 | 5·00E‐05 |
| Ear6 | −1·11203 | 0·00025 |
| Elovl7 | −0·752606 | 0·0033 |
| Epdr1 | −1·09842 | 5·00E‐05 |
| Gstk1 | −1·21378 | 5·00E‐05 |
| Gzma | −0·614498 | 5·00E‐05 |
| Klf1 | −1·41685 | 5·00E‐05 |
| Krt18 | −1·88806 | 5·00E‐05 |
| Lphn1 | −0·714992 | 7·00E‐04 |
| Mgl2 | −1·55538 | 5·00E‐05 |
| Mgst2 | −0·745639 | 0·00265 |
| Myc | −0·468861 | 7·00E‐04 |
| Pdzk1ip1 | −1·06259 | 1·00E‐04 |
| Plekha5 | −0·576601 | 9·00E‐04 |
| Prg2 | −0·713751 | 1·00E‐04 |
| Retnlg | −0·602896 | 0·00085 |
| S100a8 | −0·920403 | 5·00E‐05 |
| S100a9 | −0·942064 | 5·00E‐05 |
| Sbf2 | −0·790673 | 3·00E‐04 |
| Sord | −0·687697 | 0·00015 |
| Sphk1 | −1·4623 | 5·00E‐05 |
| Tcf15 | −2·48992 | 5·00E‐05 |
| Trnp1 | −1·32613 | 0·00015 |
| Wdr60 | −0·824671 | 0·00315 |
Table 2.
Interferon (IFN)‐α up‐regulated genes from RNA sequencing of total splenocytes
| IFN‐α up‐regulated genes | ||
|---|---|---|
| Gene | log2_fold_change (ACY 738: 0 mg/200 mg) | P‐value |
| Ablim1 | 0·470431 | 0·00075 |
| Apobec1 | 0·431714 | 0·0031 |
| B4galt5 | 0·522463 | 0·00025 |
| Bcl6 | 0·620676 | 5·00E‐05 |
| Cd244 | 0·581184 | 0·0026 |
| Cd83 | 0·722924 | 5·00E‐05 |
| Ceacam1 | 0·833845 | 5·00E‐05 |
| Csrp3 | 1·46752 | 5·00E‐05 |
| Cybb | 0·597421 | 5·00E‐05 |
| Dusp16 | 0·788321 | 5·00E‐05 |
| Ect2 | 0·708839 | 5·00E‐05 |
| Ehd4 | 0·557537 | 5·00E‐05 |
| Gm12250 | 0·650353 | 0·00115 |
| Gm16589 | 0·800247 | 5·00E‐05 |
| Gng11 | 1·22316 | 5·00E‐05 |
| Igkv12–98 | 1·37593 | 5·00E‐05 |
| Igkv3–10 | 0·738 | 5·00E‐05 |
| Igkv5–48 | 0·693764 | 5·00E‐05 |
| Il1b | 0·642912 | 0·00015 |
| Kif11 | 0·530139 | 0·00065 |
| Lag3 | 1·1179 | 5·00E‐05 |
| Lair1 | 1·04921 | 5·00E‐05 |
| Man2a1 | 0·603639 | 5·00E‐05 |
| Manf | 0·689241 | 0·00115 |
| Ms4a6d | 0·818086 | 5·00E‐05 |
| Msr1 | 0·822638 | 0·00335 |
| Nuf2 | 0·544983 | 0·00255 |
| Nupr1 | 1·44185 | 5·00E‐05 |
| Parp14 | 0·609205 | 5·00E‐05 |
| Pdia3 | 0·519802 | 0·00015 |
| Phf6 | 0·532949 | 0·00015 |
| Pik3ap1 | 0·538156 | 5·00E‐05 |
| Serpine1 | 1·33431 | 5·00E‐05 |
| Sh3bp2 | 0·604478 | 1·00E‐04 |
| Tpx2 | 0·498344 | 0·0015 |
Figure 3.
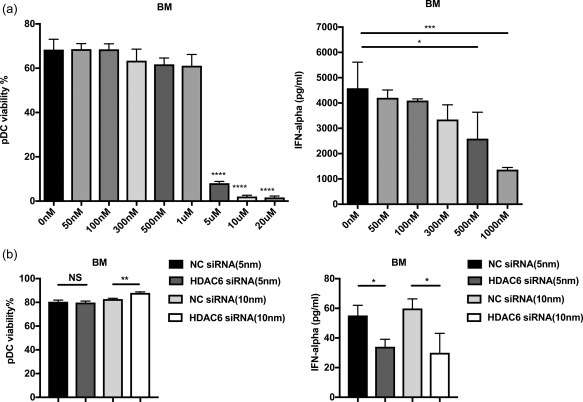
Ex‐vivo study of histone deacetylase (HDA)C6 inhibition reduces interferon (IFN)‐α production from plasmacytoid dendritic cells (pDCs). (a) The viability of sorted pDCs from bone marrow after treatment with different concentrations of the selective HDAC6 inhibitor (ACY‐738) for 24 h. (b) IFN‐α production of treated pDCs with HDAC6 inhibitor from bone marrow upon cytosine–phosphate–guanosine (CpG) stimulation for 24 h. (c) The viability of sorted pDC from bone marrow after treatment with two different concentration of negative control small‐interfering RNA (siRNA) and HDAC6 siRNA, respectively, for 24 h. (d) IFN‐α production of treated pDCs with siRNA from bone marrow upon CpG stimulation for 24 h. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001; one‐way analysis of variance (anova); n = 3.
HDAC6 inhibitor dampens adaptive immunity critical for LN development
As IFN‐α is a broad activator of the immune system 25 and studies have shown the promoting effects of IFN‐α on the activation of B cells and T cells in lupus‐prone mice 26, we sought to determine if changes of B cell and T cell responses occurred following HDAC6 inhibition during the initiation stages of LN in NZB/W mice. Consistent with the reduced IFN‐α responses, the absolute number of plasma cells (CD138+B220–) and both the percentage and absolute numbers of plasmablasts (CD138lowB220+) in the BM of ACY‐738‐treated groups were significantly lower than those of the untreated group (Fig. 4a,c,d). In addition, the spleen weight and the percentage of germinal centre (GC) B cells (GL7+IgD–) in the spleen of the ACY‐738‐treated groups were also decreased significantly (Supporting information, Fig. S2a, Fig. 4b,e). Importantly, the HDAC6 inhibitor‐mediated reduction of spleen weight and B cell responses were dose‐dependent, with more apparent effects in the 200 mg/kg ACY‐738 group compared to the 50 mg/kg ACY‐738 group. GC B cells are responsible for the generation of pathogenic IgGs, particularly the IgG2a isotype 27, with high affinity to self‐antigens in NZB/W mice. This response of GC B cells requires help from corresponding T helper (Th) cells 28. In our studies, we sought to compare Th cell responses in the spleen between ACY‐738‐treated and untreated mice. We found the percentage of T follicular helper cells (PD1+CXCR5+) were significantly lower in the two ACY‐738‐treated groups compared to the untreated group (Fig. 5a). Next, we purified splenic T cells, placed them in culture, then stimulated the cultured cells with phorbol myristate acetate (PMA), ionomycin and brefeldin A (BFA) for 5 h and determined cytokine production using intracellular staining. We found that the percentages of Th cells secreting either IL‐10 or IFN‐γ, both pathogenic in NZB/W mice 29, 30 were reduced significantly in two ACY‐738‐treated groups compared to the untreated group (Fig. 5b,c), while the percentage of IL‐4‐producing Th cells was not changed (Supporting information, Fig. S2b). As CD25+FoxP3+ regulatory T cells (Tregs) did not show a significant difference among the three groups (data not shown), the increased IL‐10‐producing Th cells should be the pathogenic population. In addition, the percentage of IFN‐γ‐producing CD8+ T cells was reduced in the spleen by ACY‐738 treatment (Supporting information, Fig. S2c), indicating a general decrease of the Th1 cytokine response. These results suggest that HDAC6 inhibition down‐regulated B cell responses, as well as the T cell responses that promote B cell responses at the initiation stage of LN in NZB/W mice.
Figure 4.
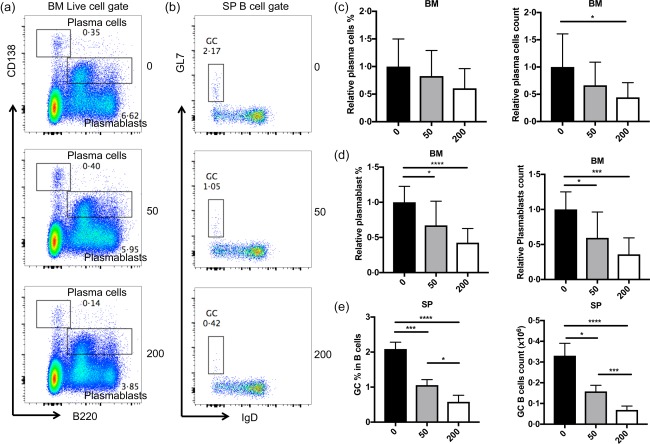
Histone deacetylase (HDA)C6 inhibition decreases B cell responses. (a) Representative flow cytometry plots for plasma cells and plasmablasts in the bone marrow. (c,d) Graphical representation of the relative percentage and absolute cell count of plasma cells and plasmablasts in the bone marrow. (b) Representative flow cytometry plots of B cells of germinal centre in the spleen. (e) Graphical representation of relative percentage and absolute cell numbers of B cells of germinal centre in the spleen. Representative flow cytometry plots are shown. *P < 0·05; ***P < 0·001; one‐way analysis of variance (anova); n ≥ 4. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 5.
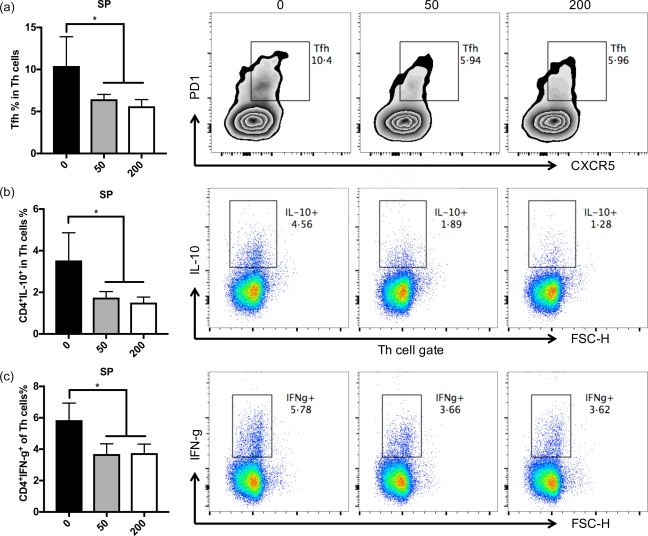
Histone deacetylase (HDA)C6 inhibition reduces pathogenic T cell responses. (a) Graphical representation and histogram of the percentage of follicular T cells in total T helper cells in the spleen in New Zealand Black/White (NZB/W) mice treated with different doses of ACY‐783. (b) Graphical representation and histogram of the percentage of interleukin (IL)‐10‐secreting cells in total CD4+ T helper cells in the spleen of NZB/W mice treated with different doses of ACY‐783. (c) Graphical representation and histogram showing percentage of interferon (IFN)‐γ‐secreting cells in total CD4+ T helper cells in the spleen of NZB/W mice treated with different doses of ACY‐783. *P < 0·05; **P < 0·01; one‐way analysis of variance (anova). Representative images are shown; n = 4. [Colour figure can be viewed at wileyonlinelibrary.com]
Pathogenic autoantibodies decrease with HDAC6 inhibition
A direct inducer of LN is the generation and deposition of pathogenic autoantibodies in the glomerular regions of the kidney. In LN, the activation of pDCs, B cells and T cells all result in the generation of pathogenic autoantibodies. To determine if HDAC6 inhibition would decrease pathogenic Ig production we measured IgG antibody levels, including autoreactive IgGs with or without HDAC6 inhibition. The results showed that the levels of total IgG and IgG2a in the serum of the 50 mg/kg ACY‐738‐treated group were significantly lower than those of the untreated group (Supporting information, Fig. S3a). The levels of anti‐dsDNA IgG and IgG2a in the serum of both the 50 and 200 mg/kg ACY‐738‐treated groups had a trend to decrease compared to the untreated group, but did not reach statistical significance (Supporting information, Fig. S3b). Conversely, the deposition of both IgG and IgG2a was reduced significantly in two ACY‐738‐treated groups compared to the untreated group (Fig. 6a,b). Moreover, C3 deposition was also reduced significantly in the 200 mg/kg ACY‐738 group (Fig. 6c), suggesting down‐regulation of the complement system upon HDAC6 inhibition. Together, these results suggest that the pathogenic autoantibody responses in the kidney of NZB/W mice were suppressed by HDAC6 inhibition.
Figure 6.
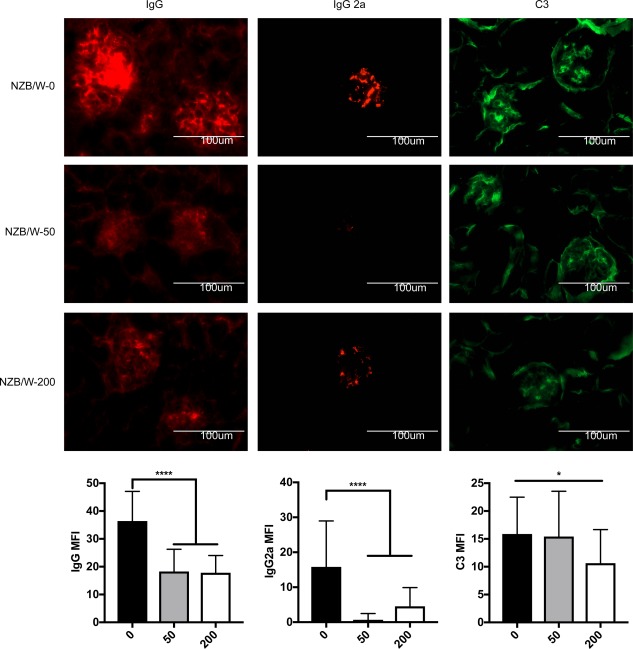
Pathogenic autoantibodies decrease with histone deacetylase (HDA)C6 inhibition. (Top) Representative images of fluorescent staining for immunoglobulin (Ig)G, IgG2a and C3 in the glomerulus of New Zealand Black/White (NZB/W) mice treated with various doses of ACY‐738. (Bottom) Graphical representation of mean fluorescent intensity (MFI) of IgG, IgG2a and C3, *P < 0·05; **P < 0·01; one‐way analysis of variance (anova) n = 24. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
HDAC inhibitors are epigenetic regulators that have been used effectively to treat haematological malignancies 31. Despite the different classes of HDAC inhibitors that have been used successfully as anti‐cancer agents, their use in immunology has not been reported widely. As published recently by Grammer and Lipsky, HDAC inhibition in lupus will effect pathways and not work solely through a signal enzyme or gene 32. They reported that the QUADrATIC study showed 447 significant positive connections to the HDAC signature in SLE and predicted that the HDAC inhibitors vorinostat and valproic acid would have positive effects in altering the lupus gene signature. As both vorinostat and valproic acid are considered non‐selective HDAC inhibitors affecting HDAC1, 2, 3 and 6, our studies were aimed to determine the efficacy of selectively inhibiting HDAC6. Our previous work using NZB/W mice demonstrated that HDAC6 inhibition has beneficial effects in lupus nephritis 15, 16. However, there are two major differences between our current study and previous studies of HDAC6 inhibition in NZB/W mice. First, in our previous studies we examined the changes of established lupus nephritis symptoms at the late stage influenced by continuous HDAC6 inhibition starting from the early pre‐disease stage until the end‐point of the late stage. Although in these studies we demonstrated the beneficial effects of the HDAC6 inhibitor on lupus nephritis, it was not known whether HDAC6 inhibition disrupted the priming of autoreactive B cell responses at the early stage or simply suppressed the induced inflammation in the kidney at the later effector stage. To answer this question further, our current study focused on the effects of HDAC6 inhibition to the priming of autoimmune responses at the early stage. We found that the beneficial effects of HDAC6 inhibition started early before the onset of lupus nephritis symptoms. Secondly, the immune mechanisms we investigated behind HDAC6 inhibition are different between our previous studies and the current study. In previous studies, we focused mainly on the changes of B cell and T cell development from precursors to immature naive cells to mature naive cells and the changes of different subpopulations of B cells in the spleen. However, in the current study, we moved forward to examine mainly not only the adaptive immune factors, but also the innate immune factors critical for the priming of autoimmune responses. In our current studies, we show that an orally active selective HDAC6 inhibitor can decrease disease in a murine model of SLE with early established disease. Inhibition of HDAC6 in early disease led to a reduction of BM plasmablasts as well as plasma cells, which are considered to be the producers of pathogenic antibodies in SLE. Additionally, we saw a dose‐dependent decrease in splenic GC B cells which was not observed in previous studies. Also, a decrease in IgG deposition and glomerular pathology scores in the kidney was consistent with the effect of long‐term HDAC6 inhibition until late stage of LN in previous studies. These results suggest that HDAC6 inhibition to decrease LN starts early at the priming stage of humoral autoimmune responses. Moreover, the short‐term HDAC6 inhibition ended at the early stage of LN, resulting in reduced IFN‐α‐producing Th1 cells and T follicular helper cells. However, there was no change of Th17 cells, which have been shown in previous studies to reduce with long‐term HDAC6 inhibition until the late stage of LN. These different influences of HDAC6 inhibition on different effector T cell subtypes at different stages of LN may reflect the stepwise pathogenic functions of various T cells with Th1 and T follicular helper cells critical at the early stage to induce pathogenic autoIgG2 antibodies and Th17 cells important at the late stage to promote the inflammation in the nephritic kidney.
There have been other studies published on the use of non‐selective (pan‐HDAC) inhibitors, including the compound romidepsin, which was found to reduce B cell survival, leaving proliferation and differentiation intact. Interestingly, different pan‐HDAC inhibitors have shown various results with regard to targeting B cells and/or T cells. This was evident in the studies comparing the pan‐HDAC inhibitor panobinostat and vorinostat, which altered B cell function in multiple myeloma and T cell function in cutaneous T cell lymphoma, respectively 33, 34. Also of interest, the action of panobinostat on B cells occurred at low concentrations (in the nanomolar range), whereas at elevated concentrations effects on T cell function were observed 35. In our prior studies we showed the pan‐HDAC inhibitor vorinostat (also known as suberanilohydroxamic acid) could decrease disease in lupus mice through targeting T cells, although it had some toxicity at elevated concentrations when administered long‐term to lupus mice 36, 37.
There have been many different ways to approach treating disease pathogenesis in lupus, including targeting B cells, T cells, plasma cells or cells of the innate immune system. In support of specifically target B cells in SLE, the B cell‐depleting antibody rituximab (anti‐CD20) has been used therapeutically to dampen the antibody response and reduce autoimmunity 38. However, clinical trials were somewhat disappointing, possibly because rituximab was not very effective at targeting plasma cells that are CD20‐negative. The studies using panobinostat showed efficacy in targeting all B cells, including plasma cells 35, which may be a better therapeutic target. Although these results are promising, the rationale for more selective HDAC inhibition remains paramount as we seek to understand their precise mechanism. In our studies, we saw a decrease in plasma cell differentiation along with a decrease in IFN‐α production, suggesting that HDAC6i inhibition may be an effective treatment in the early stages of SLE. We did not observe any alterations in weight with the treatment between the treated and untreated groups. As we continue to investigate the therapeutic efficacy of long‐term HDAC6 inhibition in SLE it will be imperative that, by decreasing renal disease, we increase the life span of animals with this therapy. Future studies are currently being planned. In addition, whether HDAC6 inhibition will be a first‐line defence to treat lupus nephritis still needs further animal models, preclinical and even clinical studies. For current clinical relevance, it is indeed more important to investigate the effects of the later intervention by HDAC6 inhibition starting after disease onset, which we have not yet performed, but is worth conducting in future studies. However, with the improvement of early disease biomarker studies, one of the future directions in lupus treatment should be early diagnosis and early prevention of symptom onset. From our previous and current studies, HDAC6 inhibition has the ability to suppress autoimmune responses early and sustains this ability to ameliorate the symptoms at later stages of lupus nephritis, which provides a potential early intervention strategy for lupus nephritis along with the development of early diagnosis in the future.
As non‐selective blockade of classes I and II HDACs does not allow elucidation of their mechanism in renal damage, more studies using selective HDACs are warranted. HDAC6 is a class IIb HDAC that localizes within the cytoplasm due to its inclusion of both a nuclear export signal and Ser‐Glu‐containing tetrapeptide domain 39, 40. HDAC6 affects multiple cellular functions in the cytoplasm, including cell signalling, activation, survival, motility and protein degradation 41, all of which can contribute to inflammation and immunity. One rationale for targeting HDAC6 is that knock‐out mice exhibit a viable phenotype, develop normally and have no life‐limiting defects. This is not necessarily true for all HDACs, as several other HDAC knock‐out murine models show either embryonic lethality or severe developmental defects. Interestingly, lymphocyte development and numbers in these mice are normal, although there is a mild decrease in the immune response after antigenic stimulation 42. With regard to SLE, we have previously observed increased expression and activity of HDAC6 within B cells, T cells and glomerular cells of diseased lupus‐prone mice 17.
We have found previously that ACY‐783 decreased the activation of nuclear factor kappa B (NF‐κB) in kidney mesangial cells although there may be additional targets in the renal tissue that are affected by HDAC6 inhibition 15. In our current studies, we found that that the selective HDAC6 inhibitor ACY‐738 decreased the percentage of BM pDCs dose dependently. It did not affect the viability of the cultured BM pDCs up to 1 μM, but did decrease significantly the production of IFN‐α in the nanomolar range. This indicates that ACY‐738 could exert therapeutic effects with no apparent toxicity. The ability of the HDAC6 inhibitor to inhibit IFN‐α production was confirmed using siRNA to HDAC6. These results are significant, as several studies have shown that IFN‐α facilitates the maturation of myeloid DC and contributes to T cell activation and follicular Th cell differentiation 43, 44, 45. IFN‐α also has the ability to stimulate CD4 T cells directly and enhance antigen‐specific B cell activation 46, 47. Of note, in our studies IFN‐α has been shown to increase Toll‐like receptor (TLR)‐7 expression in B cells, and promotes B cell proliferation and differentiation into early plasmablasts which we were able to abrogate through HDAC6 inhibition. It has also been reported by Mathian and co‐workers 48 that IFN‐α is critical for GC formation in NZB/W mice and for inducing pathogenic IgG2a autoantibodies. In our studies we noted that, with ACY‐738, both GC formation as well as IgG2a antibody formation were decreased. We are not sure in this study whether HDAC6 inhibition has direct effects on autoreactive B and T cell responses or only indirect effects through down‐regulating IFN‐α. As one of our previous studies showed that B cells and T cells in another lupus model, Murphy‐Roths large lupus‐prone mice (MRL/lpr) mice, expressed HDAC6, even at significantly higher levels compared to those in non‐lupus mice 17, we speculate that HDAC6 inhibition should have both direct and indirect effects on the alteration of B and T cell responses. To demonstrate this hypothesis, we will create NZB/W mice with pDC‐, T cell‐ and B cell‐specific HDAC6 depletion, respectively, in future studies.
With regard to the decrease in glomerulonenephritis seen in our studies, there is emerging evidence that HDACs may play a role in renal fibrogenesis 49. The mechanisms by which HDACs mediate renal damage remain elusive, but may be associated with regulating the expression of inflammatory and profibrotic genes and activation of cell signalling pathways that mediate renal fibrosis 50. In another model of renal damage, Pang et al. 51 demonstrated that treatment with trichostatin A (TSA), a pan‐HDAC inhibitor that can block both class I and class II HDACs, can attenuate renal fibrosis in a murine model of unilateral ureteral obstruction. In our prior studies we found TSA to exert protective effects in both the NZB/W and MRL/lpr animal models 36, 52. In a recent paper by Manson et al. 53, they demonstrated that TSA treatment induced BM P‐7 expression, which is important as an anti‐fibrotic molecule. Furthermore, Marumo et al. 54 and Liu et al. 50 showed that HDAC inhibition alleviates renal fibrosis through suppression of inflammatory responses in the injured kidney.
In summary, we found that we could inhibit HDAC6 using an orally active compound, and that this leads to decreased IFN‐α production and the inhibition of BM plasmablasts as well as plasma cells. Additionally, the decrease in IFN‐α resulted in reduced GC formation and decreased pathogenic IgG2a antibody production. These coupled with improved renal scores and decreased proteinuria suggest that HDAC6 inhibition may be therapeutically beneficial for the treatment of SLE.
Disclosure
The authors declare no conflicts of interest.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. (a) The percentage of activated plasmacytoid dendritic cells (pDCs) [major histocompatibility complex class II (MHC‐II)]+ pDCs in the bone marrow. (b) The absolute cell counts of pDCs in the bone marrow. *P < 0·05; **P < 0·01; one‐way analysis of variance (anova); n = 8.
Fig. S2. (a) Spleen weight. (b) The percentage of interleukin (IL)‐4 secreting cells in total CD4+ T helper cells in the spleen. (c) The percentage of interferon (IFN)‐γ‐secreting cells in total CD8+ T helper cells in the spleen. *P < 0·05, **P < 0·01; one‐way analysis of variance (anova); n ≥ 4.
Fig. S3. (a) The amount of total immunoglobulin (Ig)G and IgG2a in the serum. (b) The optical density (OD) (450 nm) value of anti‐dsDNA IgG and anti‐dsDNA IgG2a in the serum. *P < 0·05; **P < 0·01; one‐way analysis of variance (anova); n = 4.
Acknowledgements
We thank Melissa Makris for the use of flow cytometry core facility at Virginia Tech. This work was supported by NIH R15AR062883 and a grant from Acetylon Inc.
References
- 1. Apostolidis SA, Crispin JC, Tsokos GC. IL‐17‐producing T cells in lupus nephritis. Lupus 2011; 20:120–4. [DOI] [PubMed] [Google Scholar]
- 2. Nacionales DC, Weinstein JS, Yan XJ et al B cell proliferation, somatic hypermutation, class switch recombination, and autoantibody production in ectopic lymphoid tissue in murine lupus. J Immunol 2009; 182:4226–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.[No authors listed]. Belimumab: anti‐BLyS human monoclonal antibody, anti‐BLyS monoclonal antibody, BmAb, human monoclonal antibody to B‐lymphocyte stimulator. Drugs R&D 2008; 9:197–202. [DOI] [PubMed] [Google Scholar]
- 4. Asokan R, Hua J, Young KA et al Characterization of human complement receptor type 2 (CR2/CD21) as a receptor for IFN‐alpha: a potential role in systemic lupus erythematosus. J Immunol 2006; 177:383–94. [DOI] [PubMed] [Google Scholar]
- 5. Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN‐alpha induces early lethal lupus in preautoimmune (New Zealand Black × New Zealand White) F1 but not in BALB/c mice. J Immunol 2005; 174:2499–506. [DOI] [PubMed] [Google Scholar]
- 6. Santiago‐Raber ML, Baccala R, Haraldsson KM et al Type‐I interferon receptor deficiency reduces lupus‐like disease in NZB mice. J Exp Med 2003; 197:777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sisirak V, Ganguly D, Lewis KL et al Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med 2014; 211:1969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rowland SL, Riggs JM, Gilfillan S et al Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med 2014; 211:1977–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol 2016; 12:825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xiao G, Zuo X. Epigenetics in systemic lupus erythematosus. Biomed Rep 2016; 4:135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009; 10:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol 2011; 32:335–43. [DOI] [PubMed] [Google Scholar]
- 14. Alchi B, Jayne D, Labopin M et al Autologous haematopoietic stem cell transplantation for systemic lupus erythematosus: data from the European Group for Blood and Marrow Transplantation registry. Lupus 2012; 22:245–53. [DOI] [PubMed] [Google Scholar]
- 15. Vieson MD, Gojmerac AM, Khan D et al Treatment with a selective histone deacetylase 6 inhibitor decreases lupus nephritis in NZB/W mice. Histol Histopathol 2017; 11885. [DOI] [PubMed] [Google Scholar]
- 16. Regna NL, Vieson MD, Luo XM et al Specific HDAC6 inhibition by ACY‐738 reduces SLE pathogenesis in NZB/W mice. Clin Immunol 2016; 162:58–73. [DOI] [PubMed] [Google Scholar]
- 17. Regna NL, Vieson MD, Gojmerac AM, Luo XM, Caudell DL, Reilly CM. HDAC expression and activity is upregulated in diseased lupus‐prone mice. Int Immunopharmacol 2015; 29:494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Z, Davidson A. Taming lupus‐a new understanding of pathogenesis is leading to clinical advances. Nat Med 2012; 18:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eriksson C, Kokkonen H, Johansson M, Hallmans G, Wadell G, Rantapaa‐Dahlqvist S. Autoantibodies predate the onset of systemic lupus erythematosus in northern Sweden. Arthritis Res Ther 2011; 13:R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arbuckle MR, James JA, Dennis GJ et al Rapid clinical progression to diagnosis among African‐American men with systemic lupus erythematosus. Lupus 2003; 12:99–106. [DOI] [PubMed] [Google Scholar]
- 21. Liao X, Ren J, Wei CH et al Paradoxical effects of all‐trans‐retinoic acid on lupus‐like disease in the MRL/lpr mouse model. PLOS ONE 2015; 10:e0118176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 2015; 15:471–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niederquell M, Kurig S, Fischer JA et al Sca‐1 expression defines developmental stages of mouse pDCs that show functional heterogeneity in the endosomal but not lysosomal TLR9 response. Eur J Immunol 2013; 43:2993–3005. [DOI] [PubMed] [Google Scholar]
- 24. Rusinova I, Forster S, Yu S et al Interferome v2.0: an updated database of annotated interferon‐regulated genes. Nucleic Acids Res 2013; 41:D1040–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity 2006; 25:373–81. [DOI] [PubMed] [Google Scholar]
- 26. Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol 2014; 192:5459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 2006; 203:553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014; 41:529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ishida H, Muchamuel T, Sakaguchi S, Andrade S, Menon S, Howard M. Continuous administration of anti‐interleukin 10 antibodies delays onset of autoimmunity in NZB/W F1 mice. J Exp Med 1994; 179:305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ozmen L, Roman D, Fountoulakis M, Schmid G, Ryffel B, Garotta G. Experimental therapy of systemic lupus erythematosus: the treatment of NZB/W mice with mouse soluble interferon‐gamma receptor inhibits the onset of glomerulonephritis. Eur J Immunol 1995; 25:6–12. [DOI] [PubMed] [Google Scholar]
- 31. Popovic R, Shah MY, Licht JD. Epigenetic therapy of hematological malignancies: where are we now? Ther Adv Hematol 2013; 4:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grammer AC, Lipsky PE. Drug repositioning strategies for the identification of novel therapies for rheumatic autoimmune inflammatory diseases. Rheum Dis Clin North Am 2017; 43:467–80. [DOI] [PubMed] [Google Scholar]
- 33. Pratt G. Histone deacetylase inhibitors in multiple myeloma. Lancet Oncol 2013; 14:1038–9. [DOI] [PubMed] [Google Scholar]
- 34. Duvic M. Histone deacetylase inhibitors for cutaneous T‐cell lymphoma. Dermatol Clin 2015; 33:757–64. [DOI] [PubMed] [Google Scholar]
- 35. Waibel M, Christiansen AJ, Hibbs ML et al Manipulation of B‐cell responses with histone deacetylase inhibitors. Nat Commun 2015; 6:6838. [DOI] [PubMed] [Google Scholar]
- 36. Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL‐lpr/lpr mouse. J Clin Invest 2003; 111:539–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reilly CM, Mishra N, Miller JM et al Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J Immunol 2004; 173:4171–8. [DOI] [PubMed] [Google Scholar]
- 38. Beckwith H, Lightstone L. Rituximab in systemic lupus erythematosus and lupus nephritis. Nephron Clin Pract 2014; 128:250–4. [DOI] [PubMed] [Google Scholar]
- 39. Bertos NR, Gilquin B, Chan GK, Yen TJ, Khochbin S, Yang XJ. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J Biol Chem 2004; 279:48246–54. [DOI] [PubMed] [Google Scholar]
- 40. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003; 370:737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y, Shin D, Kwon SH. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J 2013; 280:775–93. [DOI] [PubMed] [Google Scholar]
- 42. Albert D, Dunham J, Khan S et al Variability in the biological response to anti‐CD20 B cell depletion in systemic lupus erythaematosus. Ann Rheum Dis 2008; 67:1724–31. [DOI] [PubMed] [Google Scholar]
- 43. Korthals M, Safaian N, Kronenwett R et al Monocyte derived dendritic cells generated by IFN‐alpha acquire mature dendritic and natural killer cell properties as shown by gene expression analysis. J Trans Med 2007; 5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tough DF. Modulation of T‐cell function by type I interferon. Immunol Cell Biol 2012; 90:492–7. [DOI] [PubMed] [Google Scholar]
- 45. Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson‐Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph‐node‐resident T follicular helper cells. Immunity 2009; 31:491–501. [DOI] [PubMed] [Google Scholar]
- 46. Gallagher KM, Lauder S, Rees IW, Gallimore AM, Godkin AJ. Type I interferon (IFN alpha) acts directly on human memory CD4+ T cells altering their response to antigen. J Immunol 2009; 183:2915–20. [DOI] [PubMed] [Google Scholar]
- 47. Kiefer K, Oropallo MA, Cancro MP, Marshak‐Rothstein A. Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol 2012; 90:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mathian A, Gallegos M, Pascual V, Banchereau J, Koutouzov S. Interferon‐alpha induces unabated production of short‐lived plasma cells in pre‐autoimmune lupus‐prone (NZB×NZW)F1 mice but not in BALB/c mice. Eur J Immunol 2011; 41:863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Markopoulou A, Kyttaris VC. Small molecules in the treatment of systemic lupus erythematosus. Clin Immunol 2013; 148:359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu N, He S, Ma L et al Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF‐beta and EGFR signaling. PLOS ONE 2013; 8:e54001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pang M, Kothapally J, Mao H et al Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 2009; 297:F996–F1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reilly CM, Thomas M, Gogal R Jr et al The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/W F1 mice. J Autoimmun 2008; 31:123–30. [DOI] [PubMed] [Google Scholar]
- 53. Manson SR, Song JB, Hruska KA, Austin PF. HDAC dependent transcriptional repression of Bmp‐7 potentiates TGF‐beta mediated renal fibrosis in obstructive uropathy. J Urol 2014; 191:242–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marumo T, Hishikawa K, Yoshikawa M, Fujita T. Epigenetic regulation of BMP7 in the regenerative response to ischemia. J Am Soc Nephrol 2008; 19:1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. (a) The percentage of activated plasmacytoid dendritic cells (pDCs) [major histocompatibility complex class II (MHC‐II)]+ pDCs in the bone marrow. (b) The absolute cell counts of pDCs in the bone marrow. *P < 0·05; **P < 0·01; one‐way analysis of variance (anova); n = 8.
Fig. S2. (a) Spleen weight. (b) The percentage of interleukin (IL)‐4 secreting cells in total CD4+ T helper cells in the spleen. (c) The percentage of interferon (IFN)‐γ‐secreting cells in total CD8+ T helper cells in the spleen. *P < 0·05, **P < 0·01; one‐way analysis of variance (anova); n ≥ 4.
Fig. S3. (a) The amount of total immunoglobulin (Ig)G and IgG2a in the serum. (b) The optical density (OD) (450 nm) value of anti‐dsDNA IgG and anti‐dsDNA IgG2a in the serum. *P < 0·05; **P < 0·01; one‐way analysis of variance (anova); n = 4.