

Summary
In the pathological process of acute kidney injury (AKI), innate immune receptors are essential in inflammatory response modulation; however, the precise molecular mechanisms are still unclear. Our study sought to demonstrate the inflammatory response mechanisms in renal tubular epithelial cells via Toll‐like receptor‐4 (TLR‐4) and dendritic cell‐specific intercellular adhesion molecule 3‐grabbing non‐integrin 1 (DC‐SIGN) signalling. We found that DC‐SIGN exhibited strong expression in renal tubular epithelial cells of human acute renal injury tissues. DC‐SIGN protein expression was increased significantly when renal tubular epithelial cells were exposed to lipopolysaccharide (LPS) for a short period. Furthermore, DC‐SIGN was involved in the activation of p65 by TLR‐4, which excluded p38 and c‐Jun N‐terminal kinases (JNK). Interleukin (IL)‐6 and tumour necrosis factor (TNF)‐α expression was decreased after DC‐SIGN knock‐down, and LPS induced endogenous interactions and plasma membrane co‐expression between TLR‐4 and DC‐SIGN. These results show that DC‐SIGN and TLR‐4 interactions regulate inflammatory responses in renal tubular epithelial cells and participate in AKI pathogenesis.
Keywords: acute kidney injury, DC‐SIGN, NF‐κB, renal tubular epithelial cells, TLR‐4
Introduction
Acute kidney injury (AKI) is an acute inflammatory response disease that leads to abrupt loss of kidney function 1. Sepsis is the most common cause of AKI 2, and the combination of sepsis and severe renal dysfunction is a major risk factor for death 3, 4. Endogenous ligands pathogen‐associated molecular patterns (PAMPs) promote tubular epithelial cell (TEC) expression of Toll‐like receptors (TLRs), which participate in inflammatory responses 5. As an important member of TLRs, TLR‐4 has a crucial inflammatory role in AKI 6.
Moreover, lipopolysaccharide (LPS) binds to TLR‐4 and triggers detrimental effects in TECs 7. In our previous study, we have found that renal TECs express dendritic cell‐specific‐ intercellular adhesion molecule 3‐grabbing non‐integrin 1 (DC‐SIGN) in the early stages of nephritis 8. DC‐SIGN, which binds and responds with pathogen‐associated molecular patterns (PAMPs), is a type II transmembrane lectin receptor of the C‐type lectin family. The DC‐SIGN study has become popular due to its central character in mediating DC adhesion, migration, inflammation and activating primary T cells, as well as triggering immune responses 9. LPS up‐regulates the production of proinflammatory mediators via myeloid differentiation primary response 88 (MyD88)‐dependent and ‐independent pathways 10, thereby causing nuclear factor kappa B (NF‐κB) (p65) translocation to the cell nucleus; in turn, NF‐κB regulates cytokine and chemokine expression 11. According to recent studies, yeasts and viruses induce the Raf‐1 proto‐oncogene, serine/threonine kinase (Raf‐1) pathway which, in turn, regulates TLR‐4‐induced p65 activation via DC‐SIGN 12. However, the effect of LPS on the relationship between TLR‐4 and DC‐SIGN in AKI is unclear. Additionally, the mechanism by which DC‐SIGN participates in TLR‐induced signalling is also unknown.
In this study, we observed that the interaction between DC‐SIGN and TLR‐4 affects the MyD88‐independent pathway of TLR‐4‐induced NF‐κB (p65) activation. Using immunohistochemistry, we demonstrated that DC‐SIGN and TLR‐4 co‐localized in TECs of human AKI tissues. Moreover, by immunoprecipitation and immunofluorescence, we found that LPS induced DC‐SIGN binding to TLR‐4 in human kidney 2 (HK‐2) cells. We demonstrated further that DC‐SIGN influences the TLR‐4/NF‐κB pathway in a MyD88‐independent manner. These results reveal a novel pathway, and improve understanding of the inflammatory response of tubular epithelial cells in AKI.
Materials and methods
Reagents and antibodies
LPS (Sigma‐Aldrich, St Louis, MO, USA) was purchased to stimulate HK‐2 cells. Small‐interfering RNA (siRNA) was purchased from Santa Cruz Biotechnology (Dallas, Texas, USA) for knock‐down of DC‐SIGN. Fetal bovine serum (FBS) and Dulbecco's modified Eagle's medium (DMEM)/F12 (1 : 1) complete medium, penicillin and streptomycin were obtained from Gibco BRL (Carlsbad, CA, USA) for culturing HK‐2 cells. Immunohistochemical primary antibodies used to detect the expression of DC‐SIGN, TLR‐4 and cytokeratin 18 (CK18) were obtained from Abcam (Cambridge, MA, USA). Primary antibodies used to detect the phosphorylation and total protein levels of p65, p38, c‐Jun N‐terminal kinase (JNK), IKB kinase (IKKɛ) and TANK binding kinase 1 (TBK1) were purchased from Cell Signaling Technology (Boston, MA, USA). Horseradish peroxidase (HRP)‐conjugated antibodies and Alexa 594‐, Alexa 647‐ and Alexa 488‐conjugated antibodies (Cell Signaling Technology) were used as secondary antibodies. Interleukin (IL)‐6 and tumour necrosis factor (TNF)‐α enzyme‐linked immunosorbent assay (ELISA) kits were used to detected cytokine secretion (Abcam).
Clinical samples
Human acute renal injury tissues were obtained from three patients who underwent renal puncture. The three patients were diagnosed as acute post‐infectious glomerulonephritis (AGN) complicated with acute kidney injury. Clinical manifestations included increase in serum creatinine (SCr) by ≥ 26·5 μmol/l within 48 h. Kidney histology showed focal segmental glomerular sclerosis and 20% cellular crescents complicated with renal tubular mild atrophy and renal interstitial mild fibrosis. The indication for the biopsy was recorded as post‐infectious diffuse endocapillary proliferative glomerulonephritis, membranoproliferative glomerulonephritis (MPGN) complicated with 20% cellular crescents. The study protocol was approved by the Ethics Committee of RuiJin Hospital, Shanghai Jiaotong University School of Medicine and agreed with the Helsinki Declaration. The study included only adults, and written informed consent was obtained from all patients.
Cell culture
The human proximal tubular epithelial cell line HK‐2 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). HK‐2 cells were cultured in DMEM/F12 (1 : 1) complete medium supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin.
Immunohistochemistry and immunocytochemistry
Human renal puncture tissue (n = 3) samples were fixed in 4% paraformaldehyde overnight and cut into serial cryosections (5 μm thickness) for immunofluorescence analysis. Samples were immunostained with anti‐DC‐SIGN (1 : 50), anti‐TLR‐4 (1 : 50) or anti‐CK18 (1 : 50) antibodies for 12 h at 4°C and then incubated with Alexa 488‐, 594‐ and 647‐conjugated secondary antibodies (1 : 1000), respectively.
HK‐2 cells were treated with LPS to localize DC‐SIGN and TLR‐4. HK‐2 cells (1 × 104/well) were seeded into a four‐well EZ‐SLIDE (Millipore, Billerica, MA, USA). After stimulation with LPS (100 ng/ml) for 12 h, cells were washed twice with phosphate‐buffered saline (PBS) and fixed in 4% paraformaldehyde. Cells were immunostained with anti‐DC‐SIGN (1 : 50) antibody for 12 h at 4°C and incubated with Alexa 594‐conjugated secondary antibody (1 : 1000). The sections were incubated with anti‐TLR‐4 antibody (1 : 50) for 12 h at 4°C and then incubated with Alexa 488‐conjugated secondary antibody (1 : 1000).
After NC and DC‐SIGN siRNA transfection, cells were incubated with LPS (100 ng/ml) for 12 h. Nuclear translocation of p65 was detected by immunofluorescence staining with a primary antibody against NF‐κB p65 (Cell Signaling Technology) and an Alexa Fluor 594‐conjugated secondary antibody (Cell Signaling Technology).
All sections were observed microscopically and photographed (Zeiss Microsystems, Thornwood, NY, USA).
Quantitative real‐time reverse transcription–polymerase chain reaction (RT–PCR)
Total RNA was extracted as described above. Briefly, 5 μg of total RNA was reverse‐transcribed into cDNA using a reverse transcription system (Promega, Madison, WI, USA). PCR amplification was performed with Power SYBR Green PCR Master Mix (Applied BioSystems, Foster City, CA, USA) in a StepOne (Applied BioSystems). The oligonucleotides used in quantitative real‐time RT–PCR analysis are listed in Table 1. Gene expression levels were normalized with beta‐actin, and data were analysed with StepOne software version 2.1 (Applied Biosystems).
Table 1.
The primer has been used for real‐time polymerase chain reaction (PCR)
| Gene | Forward primer | Reverse primer | Products size (bp) |
|---|---|---|---|
| DC‐SIGN | 5′‐TTGGCTGGGCTCCTTGT‐3′ | 5′‐CGTCTTGCCTGGATTGTT‐3′ | 70 |
| α‐tubulin | 5′‐CGTGGACATCCGCAAAG‐3′ | 5′‐TGGAAGGTGGACAGCGA‐3′ | 201 |
DC‐SIGN:dendritic cell‐specific ICAM‐3‐grabbing non‐integrin 1; bp:base pairs.
Western blot analysis
After HK‐2 cells were incubated with different doses of LPS (1, 10 and 100 ng/ml) for 12 h and 100 ng/ml LPS at different time‐points (6, 12 and 24 h), total protein lysates from the cells were prepared in Laemmli buffer. The mixture was electrophoresed on a 10% (w/v) polyacrylamide gel (acrylamide: N′N′‐bis‐methylene acrylamide 30 : 1). Then, the protein was electrotransferred to a polyvinylidene difluoride (PVDF) membrane and immunoblotted overnight with antibodies against DC‐SIGN (1 : 1000) and α‐tubulin (1 : 5000) (Abcam). Finally, the membrane was incubated with horseradish peroxidase‐conjugated secondary antibodies (1 : 5000) for 1 h at room temperature. The bands were exposed with an enhanced chemiluminescent (ECL) detection system (Millipore). Each image was captured, and the intensity of each band was analysed with Quantity One (Bio‐Rad, Hercules, CA, USA).
DC‐SIGN gene silencing by siRNA
Before transfection, HK‐2 cells were incubated with Opti‐MEM medium for 12 h, when the cell density was at 60–70%. Then, the HK‐2 cells were transfected with DC‐SIGN‐specific or negative siRNA (100 nM/well) with the lipofectamine reagent (Invitrogen, Carlsbad, CA, USA). After 12 h, the cells were incubated with DMEM/F12 (1 : 1) complete culture medium for 48 h, and the transfection efficiency was detected by Western blot analysis. The HK‐2 cells transfected with negative siRNA were used as negative control (NC).
Co‐immunoprecipitation
According to the Thermo Scientific Pierce Co‐Immunoprecipitation Kit (Waltham, MA, USA) protocol, HK‐2 cells were lysed with ice‐cold immunoprecipitation (IP) lysis/wash buffer (1 ml for 107 cells) to extract cellular protein. Then, the total protein level was quantified with the bicinchoninic acid assay (BCA) assay method. Seventy‐five μg of affinity‐purified DC‐SIGN antibody was combined with Amino Link Plus Coupling Resin (Thermo Scientific). The antibody and resin were incubated on a rotator at room temperature for 120 min, and the prey protein mixture and controls were added to the antibody‐coupled resin and incubated with gentle mixing overnight at 4°C. Finally, the antigen–antibody complex was eluted from the resin and analysed with a Western blot assay.
ELISA
After transfection of NC and DC‐SIGN siRNA, HK‐2 cells were stimulated with LPS (100 ng/ml) for 12 h and the supernatant was harvested. Then, the IL‐6 and TNF‐α levels in the supernatants were measured by ELISA, according to the manufacturer's protocol (Abcam).
Statistical analysis
Data are presented as the mean ± standard deviation (s.d.). In‐vitro cell experiments were repeated a minimum of three times. Differences between groups were tested by using one‐way analysis of variance (anova) with the post‐hoc Dunnett's C test. A two‐sided probability level of P < 0·05 was considered statistically significant. All analyses were performed with spss for Windows version 13.0.
Results
Renal tubular epithelial cells during AKI have high DC‐SIGN expression
Renal tissue with AKI were stained with DC‐SIGN, TLR‐4 and CK18. The distribution of DC‐SIGN and TLR‐4 during AKI were investigated by immunofluorescence analysis. Renal TECs (CK18‐positive cells) expressed high levels of DC‐SIGN and TLR‐4 (Fig. 1a,b), and DC‐SIGN and TLR‐4 co‐localized in the renal TECs (Fig. 1a,b). These results indicated that, in AKI patients, renal tubular cells express high levels of DC‐SIGN which co‐localize with TLR‐4.
Figure 1.
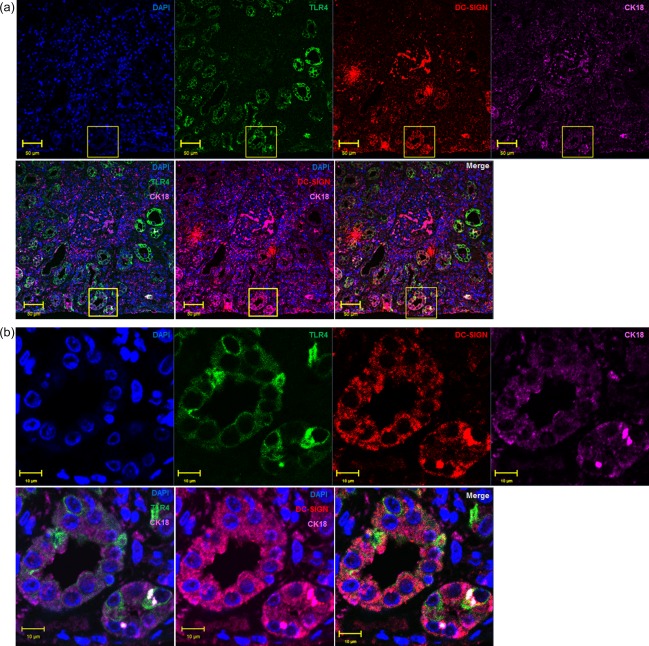
Dendritic cell‐specific intercellular adhesion molecule 3‐grabbing non‐integrin 1 (DC‐SIGN) expression in the renal tubular epithelial cells of acute kidney injury (AKI) patients. Renal puncture tissues from AKI patients were stained immunofluorescently for DC‐SIGN, Toll‐like receptor (TLR)‐4 and cytokeratin 18 (CK18). (a) The pictures were captured with ×40 magnification, and the yellow square shows the ×1200 magnification (b) in the bottom row. [Colour figure can be viewed at wileyonlinelibrary.com]
LPS promotes DC‐SIGN expression
On the basis of the immunofluorescence findings in AKI patients, we investigated whether DC‐SIGN is involved in LPS‐induced renal TEC activation. HK‐2 cells were stimulated by LPS (for 6, 12 or 24 h and at concentrations of 1, 10 or 100 ng/ml) to assess its effects on mRNA and protein of DC‐SIGN expression (Fig. 2a–d). After 100 ng/ml of LPS stimulation for 12 h, the prolonged incubation times did not induce additional DC‐SIGN expression (mRNA: 6‐h treatment = 1·770‐fold, 12‐h treatment = 2·683‐fold and 24‐h treatment = 2·357‐fold versus con, P < 0·01, Fig. 2a; protein: 6‐h treatment = 2·066‐fold, 12‐h treatment = 3·709‐fold and 24‐h treatment = 2·697‐fold versus con, P < 0·01, Fig. 2c). Different LPS doses increased DC‐SIGN expression in a dose‐dependent manner (mRNA: 1 ng/mL LPS = 1·181‐fold, 10 ng/ml LPS= 1·553‐fold and 100 ng/ml LPS = 2·604‐fold versus con, P < 0·05, Fig. 2b protein: 1 ng/ml LPS = 1·476‐fold, 10 ng/ml LPS = 1·690‐fold and 100 ng/ml LPS= 3·992‐fold versus con, P < 0·05, Fig. 2d). In conclusion, these data indicated that, in renal TECs, the DC‐SIGN levels were regulated rapidly by LPS stimulation.
Figure 2.
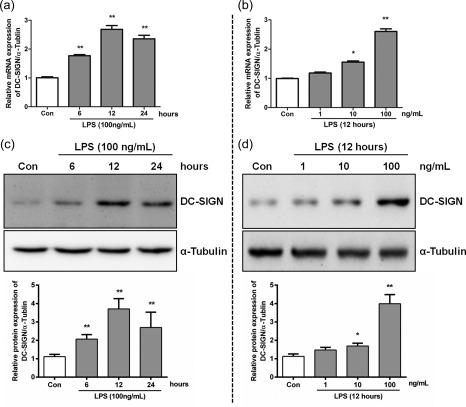
Lipopolysaccharide (LPS)‐induced dendritic cell‐specific intercellular adhesion molecule 3‐grabbing non‐integrin 1 (DC‐SIGN) expression. Human kidney 2 (HK‐2) cells were incubated with 100 ng/ml LPS with increasing time intervals (0, 6, 12 and 24 h) or with increasing doses (0, 1, 10 and 100 ng/ml) for 12 h. DC‐SIGN expression levels were detected by real‐time polymerase chain reaction (PCR) (a,b) and Western blotting (c,d), which were quantified by densitometry in three independent experiments as relative units (DC‐SIGN/α‐tubulin). The data are expressed as the mean ± standard deviation (s.d.) from three independent tests. *P < 0·05; **P < 0·01 compared with LPS untreated HK‐2 cells.
DC‐SIGN mediates p65 activation in a TLR‐4/MyD88‐independent manner
To identify whether DC‐SIGN regulates p65 activation, negative control (NC) or DC‐SIGN siRNAs were transfected into HK‐2 cells (the top panel of Fig. 3a, showing the siRNA efficiency). Compared with the NC, DC‐SIGN knock‐down weakened p65 activation significantly (NC = 3·677 ± 0·567; DC‐SIGN siRNA = 1·133 ± 0·116, versus NC P < 0·01; Fig. 3a). The immunofluorescence results also showed that LPS induced p65 nuclear translocation, which was inhibited by DC‐SIGN knock‐down (Fig. 3b).
Figure 3.
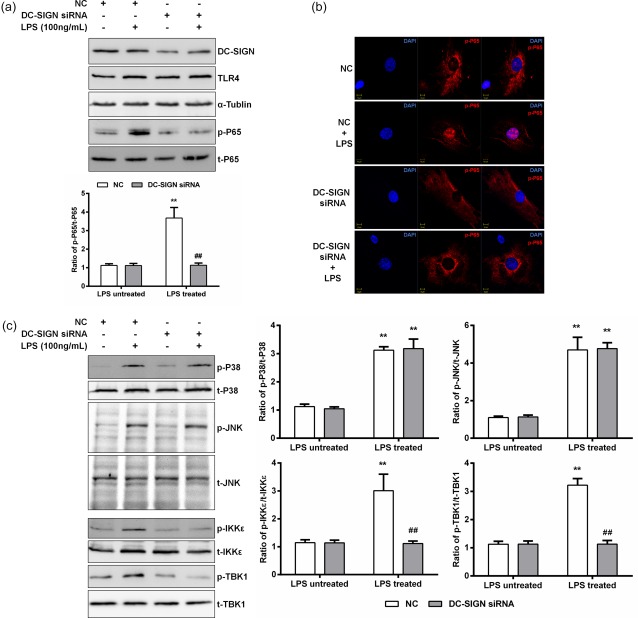
Dendritic cell‐specific intercellular adhesion molecule 3‐grabbing non‐integrin 1 (DC‐SIGN) regulates p65 activation via the myeloid differentiation primary response 88 (MyD88)‐independent pathway. Negative control (NC) or DC‐SIGN siRNAs were transfected into human kidney 2 (HK‐2) cells, and treated or untreated with lipopolysaccharide (LPS) (100 ng/ml) for 60 min. (a) Western blotting was used to detect the DC‐SIGN knock‐down efficiency and p65 phosphorylation, which was quantified by densitometry in three independent experiments as relative units (DC‐SIGN/α‐tubulin, p65 phosphorylated protein/total protein). (b) The immunofluorescence assay used to detect p65 nuclear translocation. (c) Western blotting was used to detect IKB kinase (IKKɛ), TANK binding kinase 1 (TBK1), p38 and c‐Jun N‐terminal kinases (JNK) phosphorylation with DC‐SIGN knock‐down, which was quantified by densitometry in three independent experiments as relative units (IKKɛ, TBK1, p38 and JNK phosphorylated protein/total protein). Data are expressed as the mean ± standard deviation (s.d.) from three independent tests. *P < 0·05; **P < 0·01 compared with LPS untreated; ##P < 0·01 compared with NC in the same group. [Colour figure can be viewed at wileyonlinelibrary.com]
The p65 activation assay results indicated that DC‐SIGN takes part in the TLR‐4 signalling pathway. However, two pathways, MyD88‐dependent (p38 and JNK) and MyD88‐independent (IKKɛ and TBK1), are known to be involved in TLR‐4 signalling and to regulate p65 activation in renal TECs. After LPS (100 ng/ml) stimulation for 60 min, the p65, IKKɛ, TBK1, p38 and JNK phosphorylation levels were increased significantly (P < 0·05). However, DC‐SIGN knock‐down inhibited significantly activation of only the Myd88‐independent pathway (IKKɛ: NC = 3·011 ± 0·587 versus DC‐SIGN siRNA = 1·115 ± 0·090, P < 0·01; TBK1: NC = 3·218 ± 0·235 versus DC‐SIGN siRNA = 1·130 ± 0·127, P < 0·01; Fig. 3c) These results demonstrate that DC‐SIGN reacted with TLR‐4, thereby regulating LPS‐induced inflammatory responses via the MyD88‐independent pathway in renal TECs.
LPS treatment promotes DC‐SIGN interactions with TLR‐4
The immunofluorescence results during AKI demonstrated that DC‐SIGN co‐localized with TLR‐4 in the CK18‐positive regions (Fig. 1). To verify whether LPS induced DC‐SIGN interactions with TLR‐4 in renal TECs, immunoprecipitation and immunofluorescence were performed. The immunoprecipitation assay showed that DC‐SIGN interacted with TLR‐4 after LPS stimulation for 12 h at different doses (1, 10 and 100 ng/ml) (Fig. 4a). Furthermore, DC‐SIGN and TLR‐4 co‐localization was also observed after LPS exposure (Fig. 4b). These results revealed that LPS‐induced DC‐SIGN forms a complex with TLR‐4.
Figure 4.
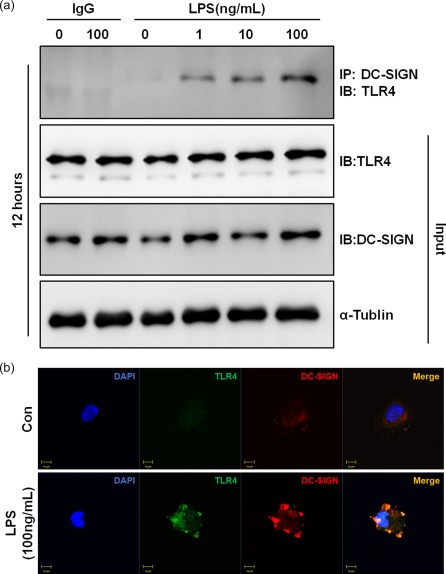
Dendritic cell‐specific intercellular adhesion molecule 3‐grabbing non‐integrin 1 (DC‐SIGN) interacts with Toll‐like receptor (TLR)‐4. (a) human kidney 2 (HK‐2) cells were incubated with lipopolysaccharide (LPS) (0, 1, 10 and 100 ng/ml) for 12 h. Cell lysates were immunoprecipitated with a DC‐SIGN antibody and probed with an antibody against TLR‐4. As loading controls, whole cell lysates were probed with antibodies against total DC‐SIGN, TLR‐4 and α‐tubulin. IB = immunoblotting, IP = immunoprecipitation. (b) Human kidney 2 (HK‐2) stimulated with or without 100 ng/ml LPS and stained with DC‐SIGN (red) and TLR‐4 (green). Images were acquired by confocal microscopy (×1200). Yellow colour indicates co‐localization of the two proteins. [Colour figure can be viewed at wileyonlinelibrary.com]
Knock‐down of DC‐SIGN expression inhibits LPS‐induced IL‐6 and TNF‐α secretion
To test whether DC‐SIGN induced by LPS is essential for expression of inflammatory cytokines in renal TECs, we compared the IL‐6 and TNF‐α secretion responses after LPS treatment in HK‐2 cells transfected with DC‐SIGN siRNA or NC siRNA. The knock‐down efficiency of DC‐SIGN is shown in Fig. 3a. DC‐SIGN or NC siRNA‐transfected HK‐2 cells were incubated with 100 ng/ml LPS for 12 h. In the NC control HK‐2 cells, LPS treatment increased the measured cytokines. However, DC‐SIGN knock‐down HK‐2 cells exhibited a much lower increase in IL‐6 and TNF‐α secretion than did NC‐transfected HK‐2 cells (Fig. 5). This result suggested that DC‐SIGN plays a critical role in LPS‐mediated inflammatory cytokine expression.
Figure 5.
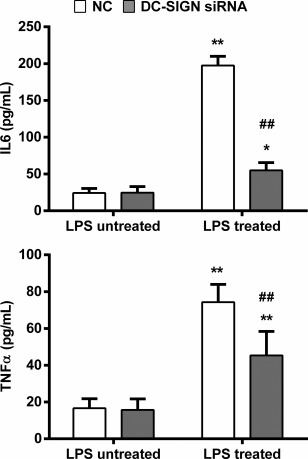
Dendritic cell‐specific intercellular adhesion molecule 3‐grabbing non‐integrin 1 (DC‐SIGN) regulates inflammatory cytokine expression. Negative control (NC) or DC‐SIGN siRNAs were transfected into human kidney 2 (HK‐2) cells and treated with or without lipopolysaccharide (LPS) (100 ng/ml) for 12 h. Enzyme‐linked immunosorbent assay (ELISA) was utilized to measure the interleukin (IL)‐6 and tumour necrosis factor (TNF)‐α secretion. The data are expressed as the mean ± standard deviation (s.d.) from three independent tests. *P < 0·05; **P < 0·01 compared with HK‐2 cells not treated with LPS in the same group, ##P < 0·01 compared with the NC in the same group.
Discussion
Sepsis causes TEC apoptosis and necrosis which leads in turn to AKI 13, and the innate immune response of TECs results in induction of inflammatory cytokine and pathways promoting sepsis‐induced kidney damage 14, 15. Renal TECs are considered to be immune cells, because they exhibit innate and adaptive immune response characteristics throughout the sepsis‐associated AKI process; thus, renal TECs are a critical factor in renal damage 16, 17. Under pathological conditions, adhesion molecules and inflammatory cytokines are up‐regulated in TECs and consequently recruit circulating inflammatory cells to injured renal regions 18; these inflammatory cells then participate in AKI inflammatory responses 19, 20. As a calcium‐dependent carbohydrate‐binding protein, DC‐SIGN is expressed mainly in DCs and macrophage subpopulations. DC‐SIGN induces tethering and transendothelial migration of DCs on endothelial cells by interacting with ICAM‐2 and mediates clustering of DCs with naive T cells through binding of ICAM‐3. As a pathogen recognition receptor, DC‐SIGN induces specific immune responses through interaction with numerous pathogens and distinct carbohydrate structures 9. In our previous studies, we have found that DC‐SIGN is expressed on human renal TECs in renal tubulointerstitial inflammation and fibrosis 8, 21 and on HK‐2 cells stimulated by inflammatory cytokines 22. Additionally, DC‐SIGN is also increased by TNF‐α stimulation in human renal TECs 23. However, these results suggest that DC‐SIGN may participate in immune and inflammatory response processes in chronic renal disease. In the present study, our in‐vivo analyses demonstrated that TECs exhibited strong DC‐SIGN expression during AKI (Fig. 1). In vitro, our results showed that the highest dose and short‐term stimulation with LPS (mimicking the pathophysiological conditions of AKI) caused rapid DC‐SIGN expression (Fig. 2) and DC‐SIGN, in turn, regulated p65 activation and inflammatory cytokine secretion (Figs 3a,b and 5). These results demonstrated that DC‐SIGN not only participates in the pathological processes of chronic renal disease but is also involved in the inflammatory response during acute renal injury.
LPS induces a ‘cytokine storm’ during the early stage of infection and triggers an acute inflammatory response during AKI 24. TLR‐4, a main sensor of Gram‐negative bacteria, triggers the ‘cytokine storm’ which causes renal injury and dysfunction 11. However, the exact regulatory pathways of LPS‐induced TLR‐4‐activation remain unknown in sepsis‐induced AKI; hence, TLR‐4 activation probably recruits other molecules. To verify this hypothesis, we observed the novel finding that DC‐SIGN combines with TLR‐4 in renal TECs and subsequently regulates the inflammatory response. In Fig. 1, DC‐SIGN co‐expression with TLR‐4 is shown in human renal TECs during AKI. Additionally, LPS promoted DC‐SIGN and TLR‐4 endogenous interactions (Fig. 4a) and co‐localization of these two proteins on renal TEC membranes in vitro (Fig. 4b). Furthermore, DC‐SIGN knock‐down down‐regulated p65 activation and inflammatory cytokine expression (Figs 3a,b and 5). SIGN‐R1 (one type of DC‐SIGN in mice) knock‐out mice have been found to have lower susceptibility to LPS‐induced shock compared with wild‐type mice, but double‐knock‐out SIGN‐R1 and TLR‐4 exhibit the lowest susceptibility 25. SIGNR1 associates with TLR‐4 and then captures LPS and induces signalling pathway activation, thereby evoking innate macrophage responses 23. These results suggest that DC‐SIGN in complex with TLR‐4 mediates LPS‐induced inflammatory responses during AKI.
Even though the results of the LPS‐induced DC‐SIGN interactions with TLR‐4 in renal TECs were partly similar to those in macrophages, the AKI infection factor effect elicited by LPS on DC‐SIGN and TLR‐4 has not, to our knowledge, been reported previously. First, we found that LPS increased DC‐SIGN expression at 12 h and promoted the maximum amount of DC‐SIGN/TLR‐4 complex formation, which was observed during the same period (Figs 2a and 4a). This result indicated that DC‐SIGN, after its rapid increase, complexes with TLR‐4 and participates in the inflammatory response acute phase. Secondly, DC‐SIGN knock‐down suppressed LPS‐enhanced p65 phosphorylation (Fig. 3a,b). This result demonstrated that DC‐SIGN participates in the LPS‐induced inflammatory response via the TLR‐4–NF‐κB axis. Thirdly, TLR‐4 recruited adaptor proteins in response to LPS, which then activate downstream pathways, including Myd88‐dependent and ‐independent pathways. These two pathways activate the NF‐kB pathway and induce proinflammatory genes 26, 27, 28 p38 and JNK, which act downstream of the Myd88‐dependent pathway, and IKKɛ and TBK1, which act downstream of the Myd88‐independent pathway and participate in the NF‐kB pathway. In this study, we found that DC‐SIGN knock‐down inhibited phosphorylation of only IKKɛ and TBK1, and not p38 and JNK (Fig. 3c). These results demonstrated that LPS regulates DC‐SIGN binding with TLR‐4 and participates in the inflammatory response via the Myd88‐independent pathway.
However, in the LPS‐induced TLR‐4 signalling pathway, LBP (LPS binding protein) transfers LPS to CD14 29, which presents LPS to the TLR‐4–MD‐2 complex and triggers multiple signalling components, including NF‐κB and IFN transcription factor 3 (IRF3) 30, 31, 32. Thus, our data indicated that LPS promotes DC‐SIGN binding to TLR‐4 and consequently triggers the inflammatory response. These results suggest that DC‐SIGN has a similar function to CD14. It is likely that mechanisms other than those elucidated in this study also regulate DC‐SIGN binding to TLR‐4, which will be clarified in our future studies.
In summary, we have demonstrated that DC‐SIGN is expressed in renal TECs during AKI and co‐localizes with TLR‐4. Our endogenous interaction assay showed that DC‐SIGN directly binds TLR‐4. DC‐SIGN participates in the MyD88‐independent pathway of TLR‐4, thereby regulating NF‐kB activation. The clinical implications of this study are that weakened DC‐SIGN or DC‐SIGN/TLR‐4 complex activation may be beneficial in AKI.
Disclosure
None to declare.
Acknowledgement
This work was supported by grants from the National Natural Science Foundation of China (81200204, 81470547, 81770384, 81170363, 81270801 and 81470941) and a grant from Natural Science Foundation of Shanghai (15ZR1426100).
Contributor Information
K. Yang, Email: ykk_ykkk@126.com
T. Zhou, Email: zhoutong_cn@hotmail.com.
References
- 1. Ronco C, Kellum JA, Bellomo R, House AA. Potential interventions in sepsis‐related acute kidney injury. Clin J Am Soc Nephrol 2008; 3:531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol 2011; 22:999–1006. [DOI] [PubMed] [Google Scholar]
- 3. Bagshaw SM, Uchino S, Bellomo R et al Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol 2007; 2:431–9. [DOI] [PubMed] [Google Scholar]
- 4. Cruz DN, Ferrer‐Nadal A, Piccinni P et al Utilization of small changes in serum creatinine with clinical risk factors to assess the risk of AKI in critically ill adults. Clin J Am Soc Nephrol 2014; 9:663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Allam R, Scherbaum CR, Darisipudi MN et al Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 2012; 23:1375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol 2008; 19:923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frei R, Steinle J, Birchler T et al MHC class II molecules enhance Toll‐like receptor mediated innate immune responses. PLOS ONE 2010; 5:e8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou T, Li X, Zou J et al Effects of DC‐SIGN expression on renal tubulointerstitial fibrosis in nephritis. Front Biosci (Landmark edn) 2009; 14:3814–24. [DOI] [PubMed] [Google Scholar]
- 9. den Dunnen J, Gringhuis SI, Geijtenbeek TB. Innate signaling by the C‐type lectin DC‐SIGN dictates immune responses. Cancer Immunol Immunother 2009; 58:1149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll‐like receptor 4 to the induction of interferon‐beta. Nat Immunol 2008; 9:361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderberg SB, Luther T, Frithiof R. Physiological aspects of Toll‐like receptor 4 activation in sepsis‐induced acute kidney injury. Acta Physiol (Oxf) 2017; 219:573–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TB. C‐type lectin DC‐SIGN modulates Toll‐like receptor signaling via Raf‐1 kinase‐dependent acetylation of transcription factor NF‐kappaB. Immunity 2007; 26:605–16. [DOI] [PubMed] [Google Scholar]
- 13. Emlet DR, Shaw AD, Kellum JA. Sepsis‐associated AKI: epithelial cell dysfunction. Semin Nephrol 2015; 35:85–95. [DOI] [PubMed] [Google Scholar]
- 14. Nakhoul N, Batuman V. Role of proximal tubules in the pathogenesis of kidney disease. Contrib Nephrol 2011; 169:37–50. [DOI] [PubMed] [Google Scholar]
- 15. Gentle ME, Shi S, Daehn I et al Epithelial cell TGFbeta signaling induces acute tubular injury and interstitial inflammation. J Am Soc Nephrol 2013; 24:787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duann P, Lianos EA, Ma J, Lin PH. Autophagy, innate immunity and tissue repair in acute kidney injury. Int J Mol Sci 2016; 17:E662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med 2016; 67:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leemans JC, Butter LM, Teske GJ, Stroo I, Pulskens WP, Florquin S. The Toll interleukin‐1 receptor (IL‐1R) 8/single Ig domain IL‐1R‐related molecule modulates the renal response to bacterial infection. Infect Immun 2012; 80:3812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ho AW, Wong CK, Lam CW. Tumor necrosis factor‐alpha up‐regulates the expression of CCL2 and adhesion molecules of human proximal tubular epithelial cells through MAPK signaling pathways. Immunobiology 2008; 213:533–44. [DOI] [PubMed] [Google Scholar]
- 20. Meng XM, Nikolic‐Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol 2014; 10:493–503. [DOI] [PubMed] [Google Scholar]
- 21. Cai M, Zhou T, Li X et al DC‐SIGN modulates DC maturation and function in rat renal tubulointerstitial lesions. Front Biosci (Landmark edn) 2012; 17:1795–803. [DOI] [PubMed] [Google Scholar]
- 22. Zhou T, Zhang Y, Sun G et al PsL‐EGFmAb inhibits the stimulatory functions of human dendritic cells via DC‐SIGN. Front Biosci 2008; 13:7269–76. [DOI] [PubMed] [Google Scholar]
- 23. Nagaoka K, Takahara K, Tanaka K et al Association of SIGNR1 with TLR4‐MD‐2 enhances signal transduction by recognition of LPS in gram‐negative bacteria. Int Immunol 2005; 17:827–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suh SH, Kim CS, Choi JS, Bae EH, Ma SK, Kim SW. Acute kidney injury in patients with sepsis and septic shock: risk factors and clinical outcomes. Yonsei Med J 2013; 54:965–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saunders SP, Barlow JL, Walsh CM et al C‐type lectin SIGN‐R1 has a role in experimental colitis and responsiveness to lipopolysaccharide. J Immunol 2010; 184:2627–37. [DOI] [PubMed] [Google Scholar]
- 26. Murillo LS, Morre SA, Pena AS. Toll‐like receptors and NOD/CARD proteins: pattern recognition receptors are key elements in the regulation of immune response. Drugs Today (Barc) 2003; 39:415–38. [PubMed] [Google Scholar]
- 27. Jonuleit H, Schmitt E, Steinbrink K, Enk AH. Dendritic cells as a tool to induce anergic and regulatory T cells. Trends Immunol 2001; 22:394–400. [DOI] [PubMed] [Google Scholar]
- 28. Cohen PJ, Cohen PA, Rosenberg SA, Katz SI, Mule JJ. Murine epidermal Langerhans cells and splenic dendritic cells present tumor‐associated antigens to primed T cells. Eur J Immunol 1994; 24:315–9. [DOI] [PubMed] [Google Scholar]
- 29. Vremec D, Zorbas M, Scollay R et al The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J Exp Med 1992; 176:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murugan R, Kellum JA. Acute kidney injury: what's the prognosis? Nat Rev Nephrol 2011; 7:209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim DH, Jung YJ, Lee AS et al COMP‐angiopoietin‐1 decreases lipopolysaccharide‐induced acute kidney injury. Kidney Int 2009; 76:1180–91. [DOI] [PubMed] [Google Scholar]
- 32. Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med 2004; 351:159–69. [DOI] [PubMed] [Google Scholar]