

Summary
T cells from systemic lupus erythematosus (SLE) patients display a wide array of anomalies in peripheral immune tolerance mechanisms. The role of ubiquitin ligases such as Cbl‐b has been described recently in these phenomena. However, its role in resistance to suppression phenotype in SLE has not been characterized, which was the aim of the present study. Thirty SLE patients (20 with active disease and 10 with complete remission) and 30 age‐ and sex‐matched healthy controls were recruited. Effector (CD4+CD25–) and regulatory (CD4+CD25+) T cells (Tregs) were purified from peripheral blood mononuclear cells (PBMCs) by magnetic selection. Suppression assays were performed in autologous and allogeneic co‐cultures and analysed by a flow cytometry assay. Cbl‐b expression and lysine‐63 (K63)‐specific polyubiquitination profile were assessed by Western blotting. We found a defective Cbl‐b expression in Tregs from lupus patients in contrast to healthy controls (1·1 ± 0·9 versus 2·5 ± 1·8, P = 0·003), which was related with resistance to suppression (r = 0·633, P = 0·039). Moreover, this feature was associated with deficient K63 polyubiquitination substrates and enhanced expression of phosphorylated signal transducer and activation of transcription 3 (pSTAT‐3) in Tregs from lupus patients. Our findings support that Cbl‐b modulates resistance to suppression by regulating the K63 polyubiquitination profile in lupus Tregs. In addition, defective K63 polyubiquitination of STAT‐3 is related to increased pSTAT‐3 expression, and might promote the loss of suppressive capacity of Tregs in lupus patients.
Keywords: Cbl‐b, K63‐polyubiquitination, regulatory T cells, resistance to suppression, systemic lupus erythematosus
Introduction
Regulatory T cells (Tregs) in humans are characterized by the expression of diverse molecules such as interleukin 2 receptor α chain (IL‐2Rα, CD25), forkhead box P3 transcription factor (FoxP3) and the absence or low expression of IL‐7 receptor α chain (CD127) 1. They play a crucial role in the regulation of the immune response and maintenance of peripheral tolerance, as they exert a suppressor effect on many cellular subpopulations, such as CD4+ and CD8+ T cells 2, 3, 4, through diverse mechanisms that include the production of suppressor cytokines such as IL‐10 5, IL‐35 6 and transforming growth factor (TGF)‐β 7. In murine models and humans, quantitative and functional alterations in Tregs have been related to the development and maintenance of many organ‐specific and systemic autoimmune diseases 2, 8, 9, 10, 11, 12, 13, 14, 15, 16. In particular, defects in FOXP3 gene, IL‐2Rα signalling pathway and inhibitory markers, such as cytotoxic T lymphocyte‐associated protein‐4 (CTLA‐4) or programmed cell death protein‐1 (PD‐1), as well as over‐expression of T‐bet or signal transducer and activator of transcription‐3 (STAT‐3), appear to be critical in the loss of suppressive function or stability in different T helper type 1 (Th1)‐ and Th17‐dominated autoimmune settings, such as multiple sclerosis, type 1 diabetes, rheumatoid arthritis and systemic lupus erythematosus (SLE) 17.
Diverse post‐translational modifications (PTM) that regulate immunity have been described; among these, ubiquitination has gained recent focus. The conjugation of ubiquitin mainly to a lysine residue from a protein substrate involves three subsequential enzymatic reactions, including the action of activating enzyme (E1), conjugating enzyme (E2) and finally ligase (E3), which is the enzyme that provides specificity to the system. Current evidence supports the role of ubiquitination as a PTM whose outcome can be dependent or independent of proteosomal or lysosomal degradation 18, 19, 20. In particular, in immune response regulation, proteolysis‐independent mechanisms, such as inhibition of phosphorylation, have been described 21, 22. Currently, the role of ubiquitin ligases has been acknowledged as negative modulators of diverse immune responses 23. Our group and others have characterized the role of the E3 ubiquitin ligase Casitas B lineage lymphoma b (Cbl‐b) in the regulation of peripheral tolerance mechanisms through the interplay with multiple substrates associated with the T cell receptor (TCR) signalling pathway, such as protein kinase Cθ (PKCθ), phospholipase Cγ1, Vav‐1, the p85 subunit of phosphatidylinositol 3‐kinase (PI3K‐p85) and phosphatase and tensin homologue deleted on chromosome 10 (Pten). In murine models, Cbl‐b deficiency has been implicated in the development of lupus‐like disease, secondary to impaired peripheral tolerance mechanisms, such as anergy and suppression by Tregs 19, 24, 25, 26, 27, 28, 29, 30.
SLE constitutes the prototypical systemic autoimmune disease associated with a breach in peripheral tolerance. It has been well demonstrated that T cells from lupus patients present a wide variety of anomalies in peripheral immune tolerance mechanisms 31, 32, 33. For instance, we have reported that CD4+ T cells from SLE patients display resistance to the anergy phenotype, which is associated with a deficiency in Cbl‐b expression and diverse phosphorylation defects in the TCR signalling pathway 19. Moreover, resistance to suppression has been acknowledged as part of the abnormalities shown by lupus T cells 15, 16, 34.
Therefore, the primary objective of the present study was to assess Cbl‐b protein expression in Tregs from lupus patients in contrast with healthy controls, as well as its relationship with resistance to the suppression phenotype and polyubiquitination profile in SLE.
Materials and methods
Patients and controls
Thirty patients with SLE diagnosis according to the American College of Rheumatology revised classification criteria were included, as well as 30 sex‐ and age‐matched healthy controls 35. Twenty‐five of the patients were female, with a mean age of 34·4 (± 7·8) years. Ten patients were in complete remission, defined as an SLE Disease Activity Index‐2000 (SLEDAI‐2K) of 0 points in the absence of any treatment with corticosteroids or immunosuppressants during 12 months or more 36, and the mean duration of remission was 5·2 (± 8·9) years. The remaining 20 patients had active disease, with a mean SLEDAI‐2K of 18·0 (± 8·9) points without no immunosuppressants, including corticosteroids, during the month prior to study entry (Table 1). The institutional ethics and research committees approved the study (Ref. 441) and all subjects signed the study informed consent prior to inclusion.
Table 1.
Clinical and demographic features of SLE patients
| Female gender, n (%) | 25 (80·0) |
| Age (years), mean (s.d.) | 34·3 (7·8) |
| Time since diagnosis (years), mean (s.d.) | 5·8 (3·7) |
| Time in remission (years), mean (s.d.) | 5·2 (3·4) |
| SLEDAI‐2K (points), mean (s.d.) | 18·0 (8·9) |
SD, standard deviation; SLE, systemic lupus erythematosus; SLEDAI‐2K, Systemic Lupus Erythematosus Disease Activity Index‐2000.
Cell purification
All individuals provided a venous peripheral blood sample. CD4+ T cells were purified from peripheral blood mononuclear cells (PBMCs) by negative selection with depletion column magnetic beads with a purity of > 95%. For the isolation of Tregs we purified CD25+ from this population by positive selection with magnetic beads, with a purity of 90% (MicroBeads CD4+CD25+ Treg isolation kit; Miltenyi Biotec, San Diego, CA, USA). We considered effector T cells to be CD4+CD25– and Tregs to be CD4+CD25+.
Cell cultures and proliferation assays
Before incubation, only the CD25‐depleted cells were stained with carboxyfluorescein diacetate succinimidyl ester (CFSE‐DA; Biochemika‐Fluka, Morris Plains, NJ, USA), according to the manufacturer's protocol. For the suppression assays, CD4+CD25– effector and CD4+CD25+ Tregs were studied in autologous and allogenic co‐cultures (effector : Tregs ratio, 1 : 1) in 24‐well plates and were either left unstimulated (RPMI) or were activated using a combination of plate‐bound anti‐CD3 antibody (5 µg/ml) and soluble anti‐CD28 antibody (2·5 µg/ml) for 48 h. Following this, cells were harvested for proliferation assays. Cell proliferation was evaluated by fluorescence activated cell sorter (FACS) (BD LSRII Fortessa; BD Biosciences, San Jose, CA, USA), according to the CFSE‐DA dilution protocol. The gating strategy was performed according to the FlowJo software proliferation platform (version 10; Tree Star, Inc., Ashland, OR, USA) 15.
Western blotting analysis
Effector and Treg lysates were obtained by using ice‐cold erythrocytes lysis buffer (ELB) and immunoblotted as described previously 19. Approximately 10–30 μg of protein were required for each assay. We used the following antibodies: anti‐Cbl‐b (sc‐8006) and β‐actin (sc‐47778) from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and anti‐K63‐linkage specific polyubiquitin (5621S), total STAT‐3 (12640S) and pSTAT‐3 (2145S) from Cell Signaling (Danvers, MA, USA). Densitometry was performed to determine protein expression using the Chemidoc MP image system (Biorad Laboratories, Berkeley, CA, USA) and ImageLab software. Values were normalized to β‐actin.
Immunoprecipitation assays
Treg lysates (400 μg) were incubated with primary antibody to STAT‐3 (12640S) from Cell Signaling (1 : 1000), and this solution (immunocomplex) was then incubated with Protein G Magnetic DynaBeads® from Invitrogen Thermo Fisher Scientific (Carlsbad, CA, USA), according to the manufacturer's protocol.
Statistical analysis
Outcomes are expressed as mean ± standard deviation (s.d.), unless noted otherwise. Differences between groups were analysed using independent‐sample Student's t‐tests. The strength of a linear association between percentage of suppression by Tregs in proliferation assays and Cbl‐b protein levels was determined by Pearson's correlation coefficient. P‐values less than 0·05 were considered statistically significant; spss software (version 21.0) was used for statistical analyses.
Results
Effector T cells from SLE patients display a resistance to suppression by Tregs
CD4+CD25+ Tregs and CD4+CD25– effector T cells from SLE patients were tested for their ability to respond and/or suppress after stimulation in vitro with plate‐bound anti‐CD3 antibody and soluble anti‐CD28 antibody, as described in Material and methods. In autologous co‐cultures, CD4+CD25– effector T cells from SLE patients showed a decreased percentage of suppression compared with CD4+CD25– effector T cells from healthy controls in the presence of Tregs (24 ± 3 versus 78 ± 15%, P = 0·002). Conversely, in allogenic co‐cultures CD4+CD25– effector T cells from SLE patients showed an increased percentage of suppression in the presence of CD4+CD25+ Tregs from healthy controls, in contrast with CD4+CD25– effector T cells from SLE patients in the presence of CD4+CD25+ Tregs from SLE patients (45 ± 12 versus 24 ± 3%, P = 0·035). Furthermore, there was no statistically significant difference in the percentage of suppression of CD4+CD25– effector T cells from healthy controls in the presence of CD4+CD25+ Tregs from SLE patients or healthy controls, respectively (Fig. 1). These results suggest a resistance to suppression by Tregs in SLE patients in contrast with healthy controls in proliferation assays.
Figure 1.
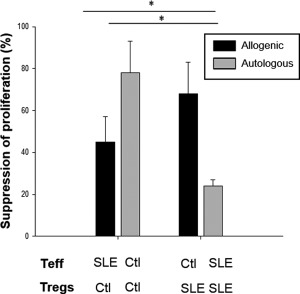
Effector T cells from systemic lupus erythematosus (SLE) patients display a resistance to suppression by regulatory T cells (Tregs). CD4+CD25+ Tregs from SLE patients and healthy controls were tested for their ability to suppress CD4+CD25– effector T cell proliferation in vitro. CD4+CD25– effector and CD4+CD25+ Tregs were studied in autologous and allogenic co‐cultures (effector : Tregs ratio, 1 : 1) stimulated previously with plate‐bound anti‐CD3 antibody (5 µg/ml) and soluble anti‐CD28 antibody (2·5 µg/ml) for 48 h. Before incubation, only the CD25‐depleted effector T cells were stained with carboxyfluorescein diacetate succinimidyl ester (CFSE‐DA; Biochemika‐Fluka). Cells were then harvested for proliferation assays by flow cytometry according to the CFSE‐DA dilution protocol. These are the pooled data from 30 SLE patients and 30 age‐ and sex‐matched healthy control proliferation assays. Ctl = healthy controls; Teff = effector T cells. *P < 0·05.
The E3 ligase Cbl‐b is decreased in Tregs from SLE patients and is associated with resistance to suppression
Cbl‐b protein constitutes an anergy factor and adapter molecule for T cell proliferation and production of IL‐2 37, 38. In previous works, we have reported resistance to anergy phenotype in T cells from SLE patients, which is associated with a deficiency in Cbl‐b expression 19. Moreover, Cbl‐b deficiency has been associated with a TGF‐β‐dependent resistance to suppression by Tregs in murine models 28, 29, 30. Thus, we hypothesized that resistance to suppression would be related to a defect in this E3 ubiquitin ligase expression in SLE patients. We purified CD4+CD25+ Tregs from SLE patients and healthy controls and evaluated Cbl‐b protein expression by Western blotting. As expected, it was decreased in lupus CD4+CD25+ Tregs in comparison to healthy controls (mean ± s.d. fold change: 1·1 ± 0·9 versus 2·5 ± 1·8; P = 0·003). We did not find any significant differences in Cbl‐b expression in CD4+CD25+ Tregs from patients with active SLE compared with those with remitted disease (Fig. 2a,b). Moreover, we found a positive correlation between percentage of suppression by Tregs in proliferation assays and Cbl‐b protein levels (r = 0·633, P = 0·039) (Fig. 2c). Although we also found decreased protein levels of Cbl‐b in lupus effector T cells, it did not correlate with the percentage of suppression by Tregs. Thus, Cbl‐b expression is decreased in CD4+CD25+ Tregs from SLE patients and is associated with resistance to the suppression phenomenon.
Figure 2.
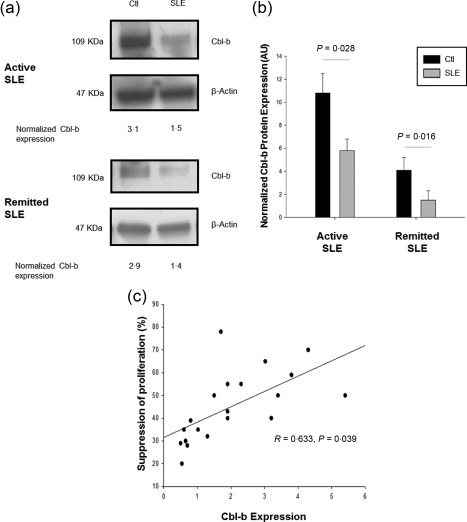
Cbl‐b expression is decreased in regulatory T cells (Tregs) from systemic lupus erythematosus (SLE) patients and is associated with resistance to suppression. Cbl‐b protein expression was assessed in CD4+CD25+ Treg lysates from SLE patients and healthy controls by Western blotting. A representative image of a Western blot for Cbl‐b from an active and a remitted SLE patient versus healthy control is shown (a), as well as the pooled data from 30 SLE patients and 30 age‐ and sex‐matched healthy controls normalized Cbl‐b protein expression (b). There was a positive correlation between percentage of CD4+CD25– effector T cell proliferation suppression and Cbl‐b expression in Tregs from SLE patients (c). Ctl = healthy controls; AU = arbitrary units.
Tregs from SLE patients are deficient in K63 ubiquitinated substrates
Lysine‐63‐specific protein ubiquitination has been implicated in the regulation of signal transduction in immune receptors 39, 40, 41, 42, 43, 44, 45, which is mediated by the E2 ubiquitin‐conjugating enzyme Ubc13, as shown by multiple studies, and directs conjugated proteins to lysosomes 44. As Ubc13 inducible knock‐out mice specific for Tregs display a reminiscent pathological phenotype of the Treg‐deficient mice 46, we hypothesized that Cbl‐b deficiency would modify the ubiquitin profile of Tregs from SLE patients in a K63‐dependent manner, which could contribute to resistance to suppression. We analysed Lys63 (K63) expression by Western blotting in CD4+CD25+ Treg lysates from SLE patients compared to healthy controls. We observed a differential polyubiquitination profile distinguished by a decreased expression of K63 polyubiquitin chains in CD4+CD25+ Tregs from SLE patients related to healthy controls (1·47 ± 0·03 versus 4·31 ± 0·28, P < 0·0001) (Fig. 3a,b). This abnormal K63 polyubiquitination profile was not present in CD4+CD25– effector T cells from SLE patients.
Figure 3.
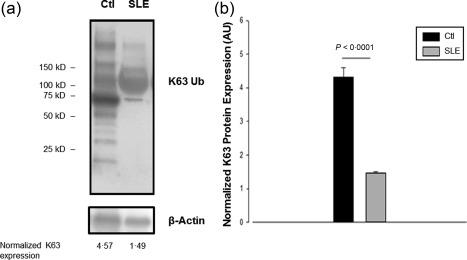
Regulatory T cells (Tregs) from systemic lupus erythematosus (SLE) patients are deficient in K63 ubiquitinated substrates. Lys63 (K63) protein expression was assessed in CD4+CD25+ Treg lysates from SLE patients and healthy controls by Western blotting. A representative image of a Western blot for K63 from an SLE patient versus healthy control is shown (a), as well as the pooled data from 30 SLE patients and 30 age‐ and sex‐matched healthy controls normalized K63 protein expression (b). Ctl = healthy controls; AU = arbitrary units.
Deficient Cbl‐b expression and K63 polyubiquitination are associated with higher levels of activated STAT‐3 in Tregs from SLE patients
We evaluated various signalling molecules known to be prone to K63 regulation (p85, Vav1). Previous studies have proposed that resistance to suppression by Tregs is dependent upon the STAT‐3 signalling pathway, as inhibition of this molecule reverts the resistance to suppression phenotype by Tregs stimulated previously with IL‐6 47. Moreover, the role of different ligases, such as tumour necrosis factor‐associated factor 6 (TRAF‐6), in the ubiquitination of STAT‐3 to promote degradation has been well demonstrated 48. Thus, we hypothesized that deficiency of Cbl‐b and K63 polyubiquitination in Tregs from SLE patients could be associated with an increase in the STAT‐3 signalling pathway. We then immunoprecipitated total STAT‐3 protein from SLE patients and healthy controls CD4+CD25+ Treg lysates and evaluated phosphorylated STAT‐3 (pSTAT‐3) expression by Western blotting. As expected, we observed an increased expression of pSTAT‐3 in lupus Tregs when compared to healthy controls (Fig. 4). Our findings suggest that deficient Cbl‐b expression and K63 polyubiquitination, primarily of STAT‐3, are related to the increased pSTAT‐3 expression in Tregs from SLE patients.
Figure 4.
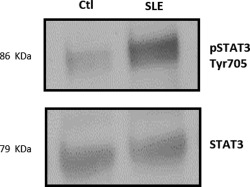
Deficient Cbl‐b expression and K63 polyubiquitination are associated with higher levels of activated signal transducer and activator of transcription 3 (STAT‐3) in regulatory T cells (Tregs) from systemic lupus erythematosus (SLE) patients. The CD4+CD25+ Treg lysates from SLE patients and healthy controls were immunoprecipitated with anti‐STAT‐3 antibody and then subjected to Western blotting with the anti‐STAT‐3 or anti‐pSTAT‐3 antibodies. This is a representative image of a Western blot for pSTAT‐3 from an SLE patient versus healthy control. Ctl = healthy controls.
Discussion
Deficiency in E3 ubiquitin ligases has been related to spontaneous systemic autoimmunity secondary to diverse abnormalities in peripheral tolerance mechanisms. In particular, we have demonstrated that, in patients with SLE, CD4+ T cells display a resistance to the anergy phenotype, which is associated with an abnormal E3 ubiquitin ligase Cbl‐b expression 19. However, the role of ubiquitination in resistance to suppression phenotype in SLE has not been defined completely. Although many studies have suggested that resistance to suppression in SLE is due to an effector T cell resistance and not to an abnormal regulatory function 15, 16, it has been demonstrated that this subpopulation displays some molecular defects which could be associated with the acquisition of an effector phenotype 46, 47.
Consistent with previous works, autologous co‐cultures of effector and Tregs from lupus patients after mitogenic stimulation showed resistance to suppression, as demonstrated by less inhibition of cell proliferation when compared with healthy controls. When effector T cells from SLE patients were studied in the presence of Tregs from healthy controls, the percentage of suppression increased almost twice with statistical significance. Even though we found no difference in the percentage of suppression of effector T cells from healthy controls co‐cultured with Tregs from healthy controls or from SLE patients, our data do not support an intact suppressive capacity of Tregs from SLE patients, as described previously 15, 16.
We have shown for the first time, to our knowledge, that resistance to the suppression phenomenon in CD4+ T cells from patients with SLE is associated with a Cbl‐b deficiency at the protein level. Our results are consistent with those from Wohlfert and colleagues, who demonstrated that Cbl‐b deficiency is associated with TGF‐β‐dependent resistance to suppression by Tregs in murine models 28, even though the loss of Cbl‐b is not as complete in lupus T cells as those from Cbl‐b–/– mice. Cbl‐b seems to be a critical mediator of Tres differentiation and suppressive function through different mechanisms. For instance, Cbl‐b promotes TGF‐β‐mediated induced Tregs (iTregs) conversion by tuning the threshold of T cell activation via an Akt‐2 (protein kinase B)‐dependent mechanism 49. Moreover, mothers against decapentaplegic homologue 7 (SMAD7), a negative regulator of TGF‐β signalling, has been identified as a crucial target of Cbl‐b, thus regulating sensitivity towards TGF‐β effects 50. Thus, iTreg differentiation could be compromised in SLE, as well as CD4+ T cell sensitivity to a TGF‐β suppressive effect. It is important to note that even though we also found a defective Cbl‐b expression in effector lupus T cells, as described previously, it did not correlate with the data from the suppression assays.
As prior studies have illustrated numerous roles of Lys63‐linked (K63) polyubiquitylation in immune responses 39, 40, 41, 42, 43, 44, 45, as well as its relationship with Cbl‐b 27, we determined if the deficiency in this E3 ubiquitin ligase could be associated with an abnormal polyubiquitin profile of Tregs from SLE patients in a K63‐dependent manner. According to our hypothesis, we observed a differential polyubiquitination profile distinguished by a decreased expression of K63 substrates in lupus Tregs, but not in effector T cells. This is in agreement with Chang and colleagues, who stated that E2 ubiquitin‐conjugating enzyme Ube2n, which is the only enzyme that specifically promotes K63‐polyubiquitination 40, seems to be crucial for stability and immunosuppressive in‐vivo function of Tregs in murine models 46. Taken together, there is strong evidence to support that there are several defects in the ubiquitination profile of Tregs from patients with SLE which contribute to resistance to suppression, a phenomenon that is not confined to effector T cell abnormalities. Cbl‐b seems to be necessary for the correct inhibitory function of CTLA‐4 51 and PD‐1 52. Moreover, other E3 ubiquitin ligases have been implicated in resistance to suppression by Tregs, such as stress‐induced phosphoprotein 1 (STIP1) homology and Ubox‐containing protein‐1 (Stub1), which interacts with FoxP3 to promote its K48‐linked (K48) polyubiquitination 53. However, it has not been evaluated in SLE.
Recent studies support that the key distinctive feature of Treg polarization to an effector phenotype is the STAT‐3 signalling pathway, which favours a Th17‐like effector phenotype through RAR‐related orphan receptor gamma (RORγt) and IL‐23 receptor expression. Specifically, Goodman and colleagues demonstrated that inhibition of STAT‐3 restores immunosuppressive function by Tregs stimulated previously with IL‐6 47. Our results are consistent with this notion, and imply that increased STAT‐3 phosphorylation in Tregs is the hallmark of resistance to suppression in SLE. Moreover, in Ubc13‐deficient Tregs there is a reduced expression of suppressor of cytokine signalling‐1 (SOCS1), which contributes to Treg stability by repressing the output of proinflammatory cytokine receptors, and particularly favours STAT molecule degradation 46. Finally, it is well known that Ubc13 expression is down‐regulated by STAT‐3 operating as a transcriptional repressor of the Ube2n gene and modulating nuclear factor kappa B (NF‐κB) activity, which seems to be crucial for Treg stability and prevention of their conversion into effector‐like T cells 54. Thus, Cbl‐b deficiency could favour STAT‐3 over‐expression by a defect in its degradation in Tregs, and at the same time interferes with the subpopulation stability mediated by the Ubc13‐dependent IKK‐NF‐κB signalling pathway.
In summary, our findings suggest that E3 ubiquitin ligase Cbl‐b modulates the interplay between effector and Tregs, particularly resistance to the suppression phenotype by regulating the K63 polyubiquitination profile in lupus Tregs. Moreover, defective K63 polyubiquitination primarily of STAT‐3 is related to increased pSTAT‐3 expression in this subpopulation and might promote the loss of suppressive capacity of Tregs in SLE, as reported previously for other autoimmune diseases.
Disclosure
The authors have no conflicts of interest to declare.
Acknowledgements
This study was supported by two grants from the Consejo Nacional de Ciencia y Tecnología (CONACYT grants 166033 (SEP‐Ciencia Básica) and 261473 (FOSSIS 2015‐2)). We also would like to thank the Flow Cytometry Unit from the Red de Apoyo a la Investigación CIC‐UNAM for the valuable technical support provided.
References
- 1. Fazekas de St Groth B, Zhu E, Asad S, Lee L. Flow cytometric detection of human regulatory T cells. Methods Mol Biol 2011; 707:263–79. [DOI] [PubMed] [Google Scholar]
- 2. Miyara M, Sakaguchi S. Human FoxP3(+)CD4(+) regulatory T cells: their knowns and unknowns. Immunol Cell Biol 2011; 89:346–51. [DOI] [PubMed] [Google Scholar]
- 3. Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med 2001; 193:1285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levings MK, Sangregorio R, Roncarolo MG. Human CD25(+)CD4(+) T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med 2001; 193:1295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rubtsov YP, Rasmussen JP, Chi EY et al Regulatory T cell‐derived interleukin‐10 limits inflammation at environmental interfaces. Immunity 2008; 28:546–58. [DOI] [PubMed] [Google Scholar]
- 6. Collison LW, Workman CJ, Kuo TT et al The inhibitory cytokine IL‐35 contributes to regulatory T‐cell function. Nature 2007; 450:566–9. [DOI] [PubMed] [Google Scholar]
- 7. Nakamura K, Kitani A, Strober W. Cell contact‐dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface‐bound transforming growth factor beta. J Exp Med 2001; 194:629–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fehervari Z, Sakaguchi S. CD4+ regulatory cells as a potential immunotherapy. Phil Trans R Soc Lond B Biol Sci 2005; 360:1647–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hobeika AC, Morse MA, Osada T, Peplinski S, Lyerly HK, Clay TM. Depletion of human regulatory T cells. Methods Mol Biol 2011; 707:219–31. [DOI] [PubMed] [Google Scholar]
- 10. Kahaly GJ, Shimony O, Gellman YN et al Regulatory T‐cells in Graves’ orbitopathy: baseline findings and immunomodulation by anti‐T lymphocyte globulin. J Clin Endocrinol Metab 2011; 96:422–9. [DOI] [PubMed] [Google Scholar]
- 11. Kollins D, Stoelcker B, Hoffmann U et al FOXP3+ regulatory T‐cells in renal allografts: correlation with long‐term graft function and acute rejection. Clin Nephrol 2011; 75:91–100. [PubMed] [Google Scholar]
- 12. Koyabu M, Uchida K, Miyoshi H et al Analysis of regulatory T cells and IgG4‐positive plasma cells among patients of IgG4‐related sclerosing cholangitis and autoimmune liver diseases. J Gastroenterol 2010; 45:732–41. [DOI] [PubMed] [Google Scholar]
- 13. La Scaleia R, Morrone S, Stoppacciaro A et al Peripheral and intestinal CD4+ T cells with a regulatory phenotype in pediatric patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 2010; 51:563–72. [DOI] [PubMed] [Google Scholar]
- 14. Lourenco EV, La Cava A. Natural regulatory T cells in autoimmunity. Autoimmunity 2011; 44:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vargas‐Rojas MI, Crispin JC, Richaud‐Patin Y, Alcocer‐Varela J. Quantitative and qualitative normal regulatory T cells are not capable of inducing suppression in SLE patients due to T‐cell resistance. Lupus 2008; 17:289–94. [DOI] [PubMed] [Google Scholar]
- 16. Venigalla RK, Tretter T, Krienke S et al Reduced CD4+, CD25– T cell sensitivity to the suppressive function of CD4+, CD25high,CD127–/low regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum 2008; 58:2120–30. [DOI] [PubMed] [Google Scholar]
- 17. Rakebrandt N, Littringer K, Joller N. Regulatory T cells: balancing protection versus pathology. Swiss Med Wkly 2016; 146:w14343. [DOI] [PubMed] [Google Scholar]
- 18. Liu YC, Penninger J, Karin M. Immunity by ubiquitylation: a reversible process of modification. Nat Rev Immunol 2005; 5:941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gomez‐Martin D, Ibarra‐Sanchez M, Romo‐Tena J et al Casitas B lineage lymphoma B is a key regulator of peripheral tolerance in systemic lupus erythematosus. Arthritis Rheum 2013; 65:1032–42. [DOI] [PubMed] [Google Scholar]
- 20. Gomez‐Martin D, Diaz‐Zamudio M, Alcocer‐Varela J. Ubiquitination system and autoimmunity: the bridge towards the modulation of the immune response. Autoimmun Rev 2008; 7:284–90. [DOI] [PubMed] [Google Scholar]
- 21. Ciechanover A, Orian A, Schwartz AL. Ubiquitin‐mediated proteolysis: biological regulation via destruction. Bioessays 2000; 22:442–51. [DOI] [PubMed] [Google Scholar]
- 22. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem 2001; 70:503–33. [DOI] [PubMed] [Google Scholar]
- 23. MacKenzie DA, Schartner J, Lin J et al GRAIL is up‐regulated in CD4+ CD25+ T regulatory cells and is sufficient for conversion of T cells to a regulatory phenotype. J Biol Chem 2007; 282:9696–702. [DOI] [PubMed] [Google Scholar]
- 24. Lutz‐Nicoladoni C, Wolf D, Sopper S. Modulation of immune cell functions by the E3 ligase Cbl‐b. Front Oncol 2015; 5:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heissmeyer V, Macian F, Im SH et al Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol 2004; 5:255–65. [DOI] [PubMed] [Google Scholar]
- 26. Macian F, Garcia‐Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 2002; 109:719–31. [DOI] [PubMed] [Google Scholar]
- 27. Guo H, Qiao G, Ying H et al E3 ubiquitin ligase Cbl‐b regulates Pten via Nedd4 in T cells independently of its ubiquitin ligase activity. Cell Rep 2012; 1:472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wohlfert EA, Callahan MK, Clark RB. Resistance to CD4+CD25+ regulatory T cells and TGF‐beta in Cbl‐b–/– mice. J Immunol 2004; 173:1059–65. [DOI] [PubMed] [Google Scholar]
- 29. Wohlfert EA, Gorelik L, Mittler R, Flavell RA, Clark RB. Cutting edge: deficiency in the E3 ubiquitin ligase Cbl‐b results in a multifunctional defect in T cell TGF‐beta sensitivity in vitro and in vivo . J Immunol 2006; 176:1316–20. [DOI] [PubMed] [Google Scholar]
- 30. Adams CO, Housley WJ, Bhowmick S et al Cbl‐b(‐/‐) T cells demonstrate in vivo resistance to regulatory T cells but a context‐dependent resistance to TGF‐beta. J Immunol 2010; 185:2051–8. [DOI] [PubMed] [Google Scholar]
- 31. Vratsanos GS, Jung S, Park YM, Craft J. CD4(+) T cells from lupus‐prone mice are hyperresponsive to T cell receptor engagement with low and high affinity peptide antigens: a model to explain spontaneous T cell activation in lupus. J Exp Med 2001; 193:329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsokos GC, Liossis SN. Immune cell signaling defects in lupus: activation, anergy and death. Immunol Today 1999; 20:119–24. [DOI] [PubMed] [Google Scholar]
- 33. Tsokos GC, Mitchell JP, Juang YT. T cell abnormalities in human and mouse lupus: intrinsic and extrinsic. Curr Opin Rheumatol 2003; 15:542–7. [DOI] [PubMed] [Google Scholar]
- 34. Banica LM, Besliu AN, Pistol GC et al Dysregulation of anergy‐related factors involved in regulatory T cells defects in systemic lupus erythematosus patients: rapamycin and vitamin D efficacy in restoring regulatory T cells. Int J Rheum Dis 2016; 19:1294–303. [DOI] [PubMed] [Google Scholar]
- 35. Tan EM, Cohen AS, Fries JF et al The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–7. [DOI] [PubMed] [Google Scholar]
- 36. Urowitz MB, Feletar M, Bruce IN, Ibanez D, Gladman DD. Prolonged remission in systemic lupus erythematosus. J Rheumatol 2005; 32:1467–72. [PubMed] [Google Scholar]
- 37. Chiang YJ, Kole HK, Brown K et al Cbl‐b regulates the CD28 dependence of T‐cell activation. Nature 2000; 403:216–20. [DOI] [PubMed] [Google Scholar]
- 38. Bachmaier K, Krawczyk C, Kozieradzki I et al Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl‐b. Nature 2000; 403:211–6. [DOI] [PubMed] [Google Scholar]
- 39. Branigan E, Plechanovova A, Jaffray EG, Naismith JH, Hay RT. Structural basis for the RING‐catalyzed synthesis of K63‐linked ubiquitin chains. Nat Struct Mol Biol 2015; 22:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Erpapazoglou Z, Walker O, Haguenauer‐Tsapis R. Versatile roles of k63‐linked ubiquitin chains in trafficking. Cells 2014; 3:1027–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. French ME, Klosowiak JL, Aslanian A, Reed SI, Yates JR III, Hunter T. Mechanism of ubiquitin chain synthesis employed by a HECT ubiquitin ligase. J Biol Chem 2017; 292:10398–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martinez‐Forero I, Azpilikueta A, Bolanos‐Mateo E et al T cell costimulation with anti‐CD137 monoclonal antibodies is mediated by K63‐polyubiquitin‐dependent signals from endosomes. J Immunol 2013; 190:6694–706. [DOI] [PubMed] [Google Scholar]
- 43. Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K. The K48–K63 branched ubiquitin chain regulates NF‐kappaB signaling. Mol Cell 2016; 64:251–66. [DOI] [PubMed] [Google Scholar]
- 44. Wu X, Karin M. Emerging roles of Lys63‐linked polyubiquitylation in immune responses. Immunol Rev 2015; 266:161–74. [DOI] [PubMed] [Google Scholar]
- 45. Tenekeci U, Poppe M, Beuerlein K et al K63‐ubiquitylation and TRAF6 pathways regulate mammalian P‐body formation and mRNA decapping. Mol Cell 2016; 63:540. [DOI] [PubMed] [Google Scholar]
- 46. Chang JH, Xiao Y, Hu H et al Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector‐like T cells. Nat Immunol 2012; 13:481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goodman WA, Young AB, McCormick TS, Cooper KD, Levine AD. Stat3 phosphorylation mediates resistance of primary human T cells to regulatory T cell suppression. J Immunol 2011; 186:3336–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wei J, Yuan Y, Jin C et al The ubiquitin ligase TRAF6 negatively regulates the JAK‐STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLOS ONE 2012; 7:e49567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qiao G, Zhao Y, Li Z et al T cell activation threshold regulated by E3 ubiquitin ligase Cbl‐b determines fate of inducible regulatory T cells. J Immunol 2013; 191:632–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gruber T, Hinterleitner R, Hermann‐Kleiter N et al Cbl‐b mediates TGFbeta sensitivity by downregulating inhibitory SMAD7 in primary T cells. J Mol Cell Biol 2013; 5:358–68. [DOI] [PubMed] [Google Scholar]
- 51. Li D, Gal I, Vermes C et al Cutting edge: Cbl‐b: one of the key molecules tuning CD28‐ and CTLA‐4‐mediated T cell costimulation. J Immunol 2004; 173:7135–9. [DOI] [PubMed] [Google Scholar]
- 52. Fujiwara M, Anstadt EJ, Clark RB. Cbl‐b deficiency mediates resistance to programmed death‐ligand 1/programmed death‐1 regulation. Front Immunol 2017; 8:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Z, Barbi J, Bu S et al The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 2013; 39:272–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang H, Hu H, Greeley N et al STAT3 restrains RANK‐ and TLR4‐mediated signalling by suppressing expression of the E2 ubiquitin‐conjugating enzyme Ubc13. Nat Commun 2014; 5:5798. [DOI] [PMC free article] [PubMed] [Google Scholar]