

Summary
Cathelicidin peptide LL‐37 plays an important role in the early host response against invading pathogens via its broad‐spectrum anti‐microbial activity. In this study, we investigated LL‐37 expression in the inflamed mucosa of inflammatory bowel disease (IBD) patients. Furthermore, the regulatory mechanism of LL‐37 induction was investigated in human colonic subepithelial myofibroblasts (SEMFs). LL‐37 mRNA expression and protein secretion were analysed using real‐time polymerase chain reaction and enzyme‐linked immunosorbent assay, respectively. Intracellular signalling pathways were analysed using immunoblotting and specific small interference RNA (siRNA). The expression of LL‐37 mRNA was increased significantly in the inflamed mucosa of ulcerative colitis and Crohn's disease. The Toll‐like receptor (TLR)‐3 ligand, polyinosinic‐polycytidylic acid (poly(I:C), induced LL‐37 mRNA expression and stimulated LL‐37 secretion in colonic SEMFs. The transfection of siRNAs specific for intracellular signalling proteins [Toll/IL‐1R domain‐containing adaptor‐inducing interferon (IFN) (TRIF), tumour necrosis factor receptor‐associated factor (TRAF)6, transforming growth factor β‐activated kinase (TAK)1] suppressed the poly(I:C)‐induced LL‐37 mRNA expression significantly. Poly(I:C)‐induced phosphorylation of mitogen‐activated protein kinases (MAPKs) and activated nuclear factor kappa B (NF‐κB) and activating factor protein (AP)‐1. siRNAs specific for NF‐κB and c‐Jun inhibited poly(I:C)‐induced LL‐37 mRNA expression. LL‐37 suppressed lipopolysaccharide (LPS)‐induced interleukin (IL)‐6 and IL‐8 expression significantly in colonic SEMFs. The expression of LL‐37 was up‐regulated in the inflamed mucosa of IBD patients. LL‐37 was induced by TLR‐3 stimulation and exhibited an anti‐microbial effect via interaction with lipopolysaccharide (LPS).
Keywords: anti‐microbial peptides, LL‐37, myofibroblasts
Introduction
Inflammatory bowel disease (IBD), principally Crohn's disease (CD) and ulcerative colitis (UC), is a chronic inflammation of the intestine. Although the precise pathogenesis of IBD remains poorly understood, it is clear that dysregulated host–microbial interactions are involved in initiating and perpetuating IBD 1, 2.
The mucosal surface of the intestinal tract is exposed continuously to a large number of microorganisms such as bacteria, fungi, viruses and parasites that could act as pathogens 3, 4. Epithelial cells separate these microorganisms from internal tissues over an enormous surface area. To cope with the microbial exposure, epithelial cells produce anti‐microbial peptides (AMPs) which inhibit the growth and the invasion of pathogens 5. Human AMP cathelicidin LL‐37 is a cationic peptide and it plays an important role in the early host response against invading pathogens. LL‐37 is generated by extracellular cleavage of the C‐terminal end of the proform hCAP18 by the kallikreins 6. Recently, LL‐37 has been reported to be expressed mainly by the epithelial cells of various tissues 7 and to exhibit broad‐spectrum anti‐microbial activity against Gram‐positive and ‐negative bacteria, protozoa, fungi and viruses 8.
Recent studies have reported that LL‐37 is involved in the pathophysiology of diverse inflammatory disorders, such as gingivitis, bronchitis, dermatitis, keratitis, pneumonitis and enteritis 9, 10, 11, 12. However, there are few reports of LL‐37 expression in IBD. Initial study showed that mucosal mRNA expression of LL‐37 is enhanced in UC patients but not in CD patients 12. However, the authors did not perform precise characterization of LL‐37 expressing cells and the regulatory mechanism of LL‐37 induction 12. Another study focused on circulating neutrophils and demonstrated that LL‐37 expression was up‐regulated in peripheral neutrophils in patients with CD 13. Butyrate has been reported to induce LL‐37mRNA in colon cancer cell lines HT‐29 14, but extensive study of the regulatory mechanisms of LL‐37 induction has not been performed in any other cell types.
Colonic subepithelial myofibroblasts (SEMFs) are α‐smooth muscle actin (SMA)‐positive cells, which are located subjacent to the basement membrane of the intestinal mucosa 15. We have reported previously that colonic SEMFs play important roles in the physiological processes associated with inflammatory response and mucosal repair in the intestine 16.
In the present study, to explore the alteration of anti‐microbial activity in IBD we investigated LL‐37 expression in the inflamed mucosa of patients with IBD. In addition, the regulatory mechanisms underlying LL‐37 induction were analysed in human colonic SEMFs.
Materials and methods
Reagents and antibodies
All reagents and antibodies used in this study are listed in Supporting information, Table S1.
Tissue samples
The diagnosis of IBD was based on conventional clinical and endoscopic criteria. Clinical activity of patients was determined based on the colitis activity index for UC and Crohn's activity index 17, 18. Tissue samples were obtained by colonoscopy or surgery. Tissue samples of active and inactive lesions were collected from endoscopically active and inactive lesions of patients, respectively. Normal colorectal tissues were obtained by the surgical resection of colon cancer at distal tumour sites. The ethics committee of the Shiga University of Medical Science approved this project, and written informed consent was obtained from all patients (Permit number: 27‐27).
Culture of colonic SEMFs
The primary culture of human colonic SEMFs was prepared according to the method reported by Mahida et al. 19. The cellular characteristics and culture conditions have been described in our previous report 20. The purity of SEMFs was confirmed by α‐SMA‐positivity, and was greater than 95%. All studies were performed on passages 3–6.
Immunohistochemistry
Immunohistochemical analyses were performed according to the method described in our previous report 21. Briefly, mouse anti‐LL‐37 antibody was used as the primary antibody, then sections were treated with horseradish peroxidase (HRP)‐labelled anti‐mouse antibody. Diaminobenzidine was used as a substrate for colour development.
For the double‐staining procedures, anti‐LL‐37 antibody was applied and incubated overnight at 4°C in a humidified chamber. Subsequently, anti‐α‐SMA antibody was applied and incubated overnight at 4°C. Dylight488‐labelled anti‐mouse IgG and Dylight549‐labelled anti‐rabbit immunoglobulin (Ig)G were used as secondary antibodies. Images were obtained with a digital confocal laser scanning microscope, LSM510 version 3.0 (Carl Zeiss Meditec, Tokyo, Japan).
Real‐time polymerase chain reaction (PCR) analysis
Total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Total RNA was converted to cDNA using Superscript II (Invitrogen). Real‐time PCR was performed using the LightCycler480 System II (Roche Diagnostics, Basel, Switzerland) with specific primers for target genes. The mRNA expression was converted to a value relative to β‐actin mRNA expression and presented as fold increase relative to the results for medium alone. The oligonucleotide primers used in this study are listed in Supporting information, Table S1.
Enzyme‐linked immunosorbent assay
LL‐37 protein levels in culture supernatant were measured by an enzyme‐linked immunosorbent assay (ELISA) kit purchased from HycultBiotech (Plymouth Meeting, PA, USA). Interleukin (IL)‐6 and IL‐8 levels in culture supernatant were measured by an ELISA kit purchased from R&D Systems (Minneapolis, MN, USA).
Silencing of gene expression in cultured cells
Colonic SEMFs were transfected with siRNAs specific for intracellular adaptor proteins [Toll/IL‐1R domain‐containing adaptor‐inducing interferon (IFN) (TRIF), tumour necrosis factor receptor‐associated factor (TRAF)3, TRAF6, transforming growth factor β‐activated kinase 1 (TAK1), NF‐κBp65 and c‐Jun] using lipofectamine RNAiMAX (Invitrogen). Briefly, cells were cultured in complete medium in the presence of a mixture of an RNAi duplex and lipofectamine RNAiMAX for 24 h.
Nuclear and cytoplasmic protein extraction and immunoblot analysis
Cytoplasmic protein was extracted using a lysis buffer [50 mM Tris pH 8·0, 0·5% Nonidet P‐40, 1 mM ethylenediamine tetraacetic acid (EDTA), 150 mM NaCl, 2 mM Na3VO4, 1 mM NaF, 20 mM Na4P2O7, 1 mM phenylmethane sulphonyl fluoride (PMSF), 10% glycerol and complete Mini Protease Inhibitor Cocktail (Roche Diagnostics)]. Extracted protein was subjected to immunoblotting with antibodies against phospho‐p44/42 mitogen‐activated protein kinase (MAPK) [extracellular‐regulated kinase (ERK)1/2, p38 MAPK or stress‐activated protein kinase/c‐Jun NH(2)‐terminal kinase (SAPK/JNK)], followed by incubation with HRP‐labelled anti‐rabbit antibodies or HRP‐labelled anti‐mouse antibodies. After detection, the membrane was stripped using Restore Western Blot Stripping Buffer (Thermo Scientific Inc., Waltham, MA, USA) and then incubated with antibodies against total‐p44/42 MAPK (ERK1/2), p38 MAPK, SAPK/JNK or glyceraldehyde 3‐phosphate dehydrogenase (GAPDH).
The extraction of nuclear protein was performed according to a previous report 22. Nuclear proteins were extracted using the CelLytic NuCLEAR Extraction Kit (Sigma‐Aldrich, St Louis, MO, USA). Extracted nuclear proteins were subjected to immunoblotting with rabbit anti‐p65NF‐κB (C‐20) antibody or mouse anti‐phospho (P)‐c‐Jun (KM‐1) antibody. These were followed by incubation with HRP‐labelled anti‐rabbit antibody or HRP‐labelled anti‐mouse antibody. Signal detection was performed using the enhanced chemiluminescence Western blot system (GE Healthcare, Little Chalfont, UK).
Immunoprecipitation
The cells were stimulated for 5 min with polyinosinic‐polycytidylic acid [poly(I:C)], an analogue of viral dsRNA. After stimulation, the cells were lysed using a lysis buffer. The lysates were incubated with human anti‐TRAF6 antibody and µMACS MicroBeads (Miltenyi Biotec, Bergsch Gladbach, Germany) on ice for 30 min, and then applied onto a µ column (Miltenyi Biotec). The control was incubated with normal IgG and beads. After vigorous washing steps, proteins were eluted from the beads by heating in a loading buffer and were then subjected to immunoblot for detection of TRIF and TAK1.
Statistical analysis
Differences between samples were evaluated using the non‐parametric Mann–Whitney U‐test. Differences resulting in P‐values of lower than 0·05 were considered to be statistically significant.
Results
Expression of LL‐37 in patients with IBD
To evaluate the expression of LL‐37 in the inflamed mucosa of patients with IBD, we investigated the mRNA expression of LL‐37 in biopsy specimens using real‐time PCR. As shown in Fig. 1a, the mRNA expression of LL‐37 was increased significantly in the active mucosa of UC and CD patients compared to normal mucosa. There were no significant differences between inactive and normal mucosa.
Figure 1.
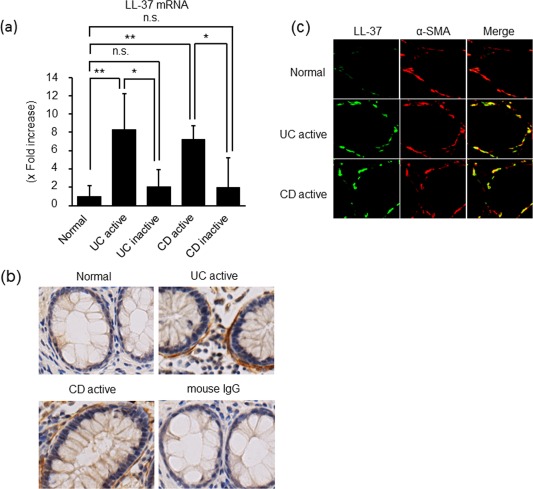
LL‐37 expression in the inflamed mucosa of patients with inflammatory bowel disease (IBD). (a) The mRNA expression of LL‐37 in the inflamed mucosa of patients with IBD. Total RNA was extracted from biopsied samples, and the mRNA expression of LL‐37 was evaluated using real‐time polymerase chain reaction (PCR). LL‐37 mRNA expression was converted to a value relative to β‐actin mRNA expression, and presented as an increase relative to normal mucosa. Data are expressed as mean values ± standard error of the mean (s.e.m.). Normal mucosa, n = 20; active ulcerative colitis (UC), n = 23; inactive UC, n = 27; active Crohn's disease (CD), n = 20; inactive CD, n = 20. **P < 0·01, *P < 0·05; not significant (n.s.). (b) Immunostaining for LL‐37 in normal mucosa and inflamed mucosa of UC and CD. Normal mouse immunoglobulin (Ig)G was used as the primary antibody for active UC samples. Pictures are shown from one of six independent samples with similar results. Magnification ×200. (c) Dual‐coloured immunofluorescence was used to determine expression of LL‐37 (green fluorescence) and α‐smooth muscle actin (SMA) (red fluorescence) in the inflamed mucosa of patients with UC and CD. Representative pictures are shown from one of six independent samples. Double‐positive cells were detected by yellow fluorescence. [Colour figure can be viewed at wileyonlinelibrary.com]
As shown in Fig. 1b and Supporting information, Fig. S1, the expression of LL‐37 was detected in the cells located below the basement membrane of the epithelial cells. To characterize the cellular origin of LL‐37 in the inflamed mucosa, the samples were double‐stained with anti‐α‐SMA, a marker for myofibroblasts, and anti‐human LL‐37 antibody (Fig. 1c). In the normal mucosa, α‐SMA (red fluorescence) was clearly stained at the subepithelial regions, but LL‐37 (green fluorescence) was not detected. Conversely, LL‐37 was detected clearly in the subepithelial regions in the active mucosa of patients with UC and CD. The α‐SMA/LL‐37 double‐positive cells were detected as yellow, and the LL‐37‐positive cells coincided with parts of the α‐SMA‐positive cells. These findings indicate that colonic SEMFs are a major source of LL‐37 in the inflamed mucosa of IBD patients.
The regulation of LL‐37 expression in human colonic SEMFs
Regulation of LL‐37 expression was investigated in isolated colonic SEMFs. The cells were cultured for 12 h with various cytokines, and LL‐37mRNA expression was measured using real‐time PCR. None of the cytokines affected the mRNA expression of LL‐37 significantly (Fig. 2a). Of the Toll‐like receptor (TLR) ligands used [lipopolysaccharide (LPS; TLR‐4 ligand), poly(I:C); TLR‐3 ligand, cytosine–phosphate–guanine (CpG); TLR‐9 ligand and Fragellin; TLR‐5 ligand and N‐formyl‐L‐methionyl‐L‐leucyl‐phenylalanine (fMLP)], only poly(I:C) enhanced the mRNA expression of LL‐37 significantly (Fig. 2b). Immunoblot analysis showed that the expression of LL‐37 protein in the cells was enhanced by poly(I:C) compared to medium alone (Fig. 2c). Stimulation by poly(I:C) enhanced the secretion of LL‐37 in the supernatant (Fig. 2d). The induction of LL‐37 secretion by poly(I:C) was observed in both dose‐ and time‐dependent manners (Fig. 2e,f).
Figure 2.
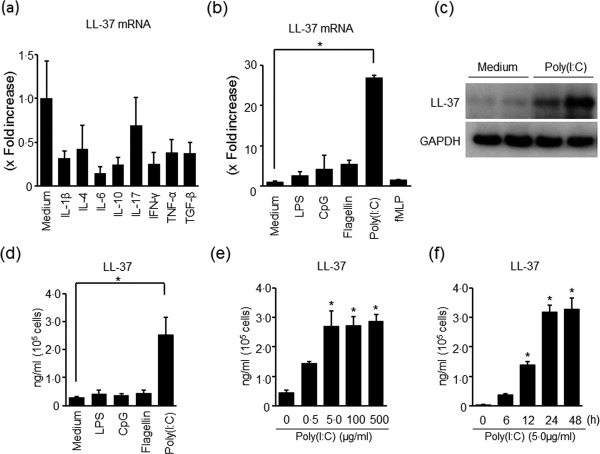
LL‐37 expression in colonic subepithelial myofibroblasts (SEMFs). (a) The expression of LL‐37 mRNA in response to cytokines. The cells were stimulated for 12 h with cytokines [interleukin (IL)‐1β: 10 ng/ml, IL‐4: 100 ng/ml, IL‐6: 100 ng/ml, IL‐10: 100 ng/ml, IL‐17: 100 ng/ml, interferon (IFN)‐γ: 100 ng/ml, TNF‐α: 100 ng/ml and transforming growth factor (TGF)‐β: 100 ng/ml], and the mRNA expression of LL‐37 was determined using real‐time polymerase chain reaction (PCR). LL‐37 mRNA expression was converted to a value relative to β‐actin mRNA expression, and presented as fold increase relative to the results for medium alone (no stimulation). Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. (b) The expression of LL‐37 mRNA in response to Toll‐like receptor (TLR) ligands and N‐formyl‐L‐methionyl‐L‐leucyl‐phenylalanine (fMLP). The cells were stimulated for 12 h with TLR ligands [poly(I:C)]: 5 µg/ml, lipopolysaccharide (LPS): 100 ng/ml, fragellin: 5 µg/ml and cytosine–phosphate–guanosine (CpG): 1 µg/ml) and N‐formyl‐L‐methionyl‐L‐leucyl‐phenylalanine (fMLP) (100 nM), and the mRNA expression of LL‐37 was determined using real‐time PCR. LL‐37 mRNA expression was converted to a value relative to β‐actin mRNA expression, and presented as fold increase relative to the results for medium alone (no stimulation). Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. *P < 0·05. (c) The expression of LL‐37 protein in response to poly(I:C) stimulation. The cells were stimulated for 24 h with 5 µg/ml of poly(I:C), and the expression of LL‐37 protein in the cells was evaluated using immunoblot analysis. The expression of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) used as a loading control is shown. The data are representative of two independent experiments. (d) The secretion of LL‐37 protein in response to poly(I:C) stimulation. The cells were stimulated for 24 h with 5 µg/ml of poly(I:C), and the level of LL‐37 in the supernatants was determined using enzyme‐linked immunosorbent assay (ELISA). Data are expressed as means ± s.e.m. of four independent experiments. *P < 0·05. (e) Dose‐dependent effects of poly(I:C) on LL‐37 secretion. The cells were stimulated for 24 h with increasing concentrations of poly(I:C), and the level of LL‐37 in the supernatants was determined using ELISA. Data are expressed as means ± s.e.m. of four independent experiments. *P < 0·05. (f) The kinetics of LL‐37 secretion in response to poly(I:C) stimulation. The cells were stimulated with 5 µg/ml of poly(I:C) for the indicated predetermined times, and LL‐37 levels in the supernatant were determined using ELISA. Data are expressed as means ± s.e.m. of four independent experiments. *P < 0·05.
Assembly of intracellular adaptor proteins (TRIF, TRAF6 and TAK1) by poly(I:C) stimulation
We next examined the signal transduction involved in the induction of LL‐37 by poly(I:C) stimulation. Most TLR‐mediated responses are controlled mainly by the myeloid differentiation factor 88 (MyD88)‐dependent signalling pathway, but TLR‐3 uses the TRIF‐dependent pathway 23. As shown in Fig. 3a, the transfection of siRNA specific for TRIF suppressed the expression of LL‐37mRNA by poly(I:C) stimulation significantly, indicating that the TRIF‐dependent pathway is involved in poly(I:C)‐induced LL‐37 induction in colonic SEMFs. Efficacy of siRNAs for targeted genes was shown in Supporting information, Fig. S2a.
Figure 3.
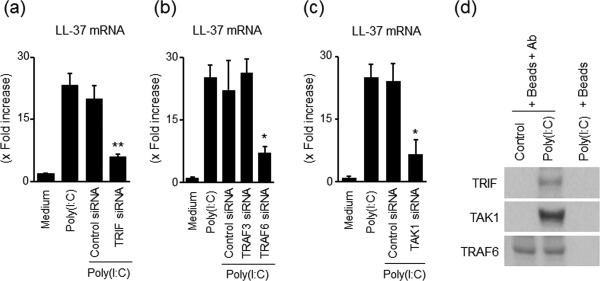
Role of Toll‐like receptor (TLR)‐3‐associated adaptor proteins in the induction of LL‐37 in response to poly(I:C) stimulation. (a) The effects of siRNA for Toll/interleukin (IL)‐1R domain‐containing adaptor inducing interferon (TRIF) small interference RNA (siRNA) on poly(I:C)‐induced LL‐37 mRNA expression. The cells were transfected with siRNA for TRIF or with control siRNA, and were then incubated for 12 h with or without 5 µg/ml of poly(I:C). LL‐37mRNA expression was expressed as values relative to β‐actin mRNA expression, and presented as fold increase relative to the results for medium alone (no stimulation). Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. **P < 0·01. (b) The effects of siRNA for tumour necrosis factor receptor‐associated factor (TRAF)6 and TRAF3 on poly(I:C)‐induced LL‐37 mRNA expression. The cells were transfected with siRNA for TRAF6, TRAF3 or with control siRNA, and then incubated for 12 h with or without 5 µg/ml of poly(I:C). Data are expressed as means ± s.e.m. of four independent experiments. *P < 0·05. (c) The effects of siRNA for transforming growth factor β‐activated kinase (TAK)1 on poly(I:C)‐induced LL‐37 mRNA expression. The cells were transfected with siRNA for TAK1 or with control siRNA, and incubated for 12 h with or without 5 µg/ml of poly(I:C). Data are expressed as means ± s.e.m. of four independent experiments. *P < 0·05. (d) Immunoprecipitation analysis of TLR‐3‐associated adaptor proteins. The cells were stimulated with 5 µg/ml of poly(I:C) for 15 min, and were then lysed with a lysis buffer. Lysates were incubated with magnetic beads (Beads) coupled with anti‐TRAF6 antibodies (Ab) or with control beads, and the precipitated proteins were then subjected to sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and immunoblotting to detect co‐precipitated TRIF1 and TAK1. The bottom row reveals that the precipitated TRAF6 was detected by anti‐TRAF6 antibodies, indicating equal loading dose and specificity of this system (no reaction without anti‐TRAF6 antibodies). The data are representative of two independent experiments.
In addition, it has been reported that TRIF recruits TRAF6, leading to the activation of MAPKs and nuclear factor kappa B (NF‐κB) 24. Moreover, it has been reported that TRIF interacts not only with TRAF6 but also with TRAF3, followed by the activation of TANK binding kinase 1, leading to the phosphorylation of interferon regulatory factor 3 24. In colonic SEMFs, the transfection of siRNA specific for TRAF6, but not TRAF3, suppressed the expression of LL‐37mRNA significantly (Fig. 3b). TAK1 is located downstream of TRIF and TRAF6. The transfection of siRNA specific for TAK1 suppressed the expression of LL‐37mRNA by poly(I:C) significantly (Fig. 3c).
Next, we examined whether poly(I:C) stimulation assembles these adaptor proteins (TRIF, TRAF6 and TAK1) into a complex using immunoprecipitation. The cells were stimulated with 5·0 µg/ml of poly(I:C) for 5 min and then lysed with a lysis buffer. The lysate was immunoprecipitated with an anti‐TRAF6 antibody and the proteins that co‐precipitated with TRAF6 were analysed by immunoblotting. As shown in Fig. 3d, TRIF and TAK1 were detected clearly in the protein complex immunoprecipitated by the anti‐TRAF6 antibody. These findings indicate that TLR‐3‐associated intracellular adaptor proteins, including TRIF, TRAF6 and TAK1, are involved in the induction of LL‐37 mRNA by poly(I:C) in colonic SEMFs.
Poly(I:C) activates MAPKs, NF‐κB and activator factor protein (AP)‐1
It has been reported that TAK1 activation leads to the activation of MAPK and NF‐κB in TLR‐3 signalling 25, 26, 27. In colonic SEMFs, as shown in Fig. 4a, poly(I:C) induced phosphorylation of MAPKs, including p38MAPK, p42/44MAPK and JNK, as early as 5 min after the stimulation of poly(I:C). MEK1/2 inhibitors (U0126 and PD98059), a p38 MAPK inhibitor (SB239065) and a JNK inhibitor (JNK1) suppressed the LL‐37mRNA expression induced by poly(I:C) stimulation significantly (Fig. 4b). These findings indicate that the activation of MAPKs is required for LL‐37 induction by poly(I:C) in colonic SEMFs.
Figure 4.
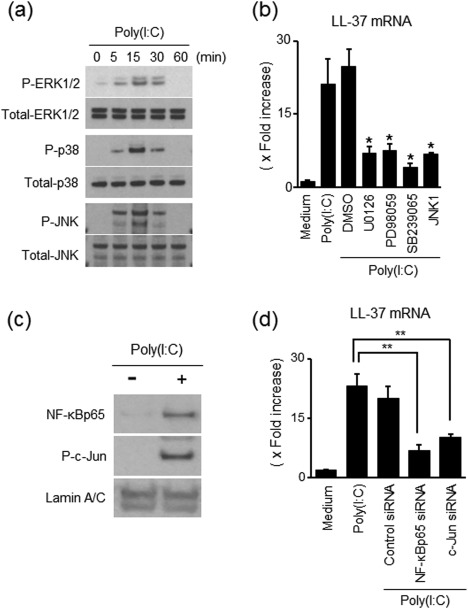
Involvement of mitogen‐activated protein kinase (MAPK), nuclear factor (NF)‐κB and activating factor protein (AP)‐1 activation in poly(I:C)‐stimulated LL‐37 induction. (a) MAPK activation in response to poly(I:C). The cells were stimulated with 5 µg/ml of poly(I:C) for the indicated predetermined times, and the phosphorylation (P) of MAPKs was evaluated by immunoblot analyses. The data are representative of two independent experiments. (b) Effects of inhibitors of MAPKs on LL‐37 induction in response to poly(I:C). The cells were pretreated with 10 µM of a p38 MAPK inhibitor (SB203580), MAP/ERK kinase (MEK)1/2 inhibitor (U02016 or PD098059) or 3 µM of c‐Jun N‐terminal kinase (JNK) inhibitor (JNK inhibitor 1) for 30 min, and then incubated with or without 5 µg/ml of poly(I:C) for 12 h. LL‐37 mRNA expression was expressed as a value relative to β‐actin mRNA expression, and presented as fold increase relative to the results for medium alone (no stimulation). Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. *P < 0·05. (c) NF‐κB and AP‐1 activation in response to poly(I:C). The cells were stimulated with 5·0 µg/ml of poly(I:C) for 15 min, and nuclear proteins were then extracted and analysed using immunoblotting for NF‐κBp65 and phosphorylated (P‐)c‐Jun. Lamin A/C was used as a loading control. The data are representative of one of two independent experiments. (d) The effects of siRNAs for NF‐κBp65 and AP‐1 (c‐Jun) on poly(I:C)‐induced LL‐37mRNA expression. The cells were transfected with siRNA for NF‐κBp65, AP‐1 (c‐Jun) or control siRNA, and were then incubated for 12 h with or without 5·0 µg/ml of poly(I:C). LL‐37 mRNA expression was expressed as a value relative to β‐actin mRNA expression. Data are expressed as means ± s.e.m. of four independent experiments. **P < 0·01.
TAK1 activation leads to NF‐κB activation 24. Moreover, TAK1 also activates MAPKs leading to the activation of activator protein 1 (AP‐1) 24, 26, 28. In the nuclear proteins, the expression of NF‐κBp65 and phosphorylated c‐Jun (AP‐1) was detected clearly after poly(I:C) stimulation (Fig. 4c). As shown in Fig. 4d, the silencing of NF‐κBp65 and c‐Jun suppressed the expression of LL‐37mRNA significantly by poly(I:C) stimulation. Efficacy of siRNAs for targeted genes is shown in the Supporting information, Fig. S2b.
The effect of LL‐37 on LPS‐induced expression of inflammatory mediators
LL‐37 has been reported to suppress the expression of LPS‐induced inflammatory mediators in human macrophages 29, 30, so we investigated whether or not similar responses occurred in colonic SEMFs. The cells were stimulated with LL‐37 (5·0 µg/ml), LPS (100 ng/ml) or LL‐37 plus LPS for 24 h, and then secretion of IL‐6 and IL‐8 in the supernatant was evaluated using ELISA. As shown in Fig. 5a, the IL‐6 and IL‐8 secretion induced by LPS was suppressed significantly by LL‐37. These responses were confirmed at the mRNA levels (Supporting information, Fig. S3a), and LL‐37 suppressed LPS‐induced IL‐6 and IL‐8 expression dose‐dependently (Fig. 5b and Supporting information, Fig. S3b). These responses were also evaluated by NF‐κB activation. For the washing procedure, colonic SEMFs were pretreated with LL‐37 (5·0 µg/ml) for 1 h, washed with sterile PBS several times to remove exogenous LL‐37, and then stimulated with LPS (100 ng/ml) for 15 min. As shown in Fig. 5c, the presence of LL‐37 suppressed the LPS‐induced translocation of NF‐κBp65 into the nucleus, but removal of LL‐37 by washing attenuated the inhibitory effects of LL‐37 on NF‐κBp65 activation. These findings suggest that LL‐37 interacts with LPS at the fluid level and exerts anti‐LPS activity, but does not modulate the intracellular signal pathway of LPS.
Figure 5.
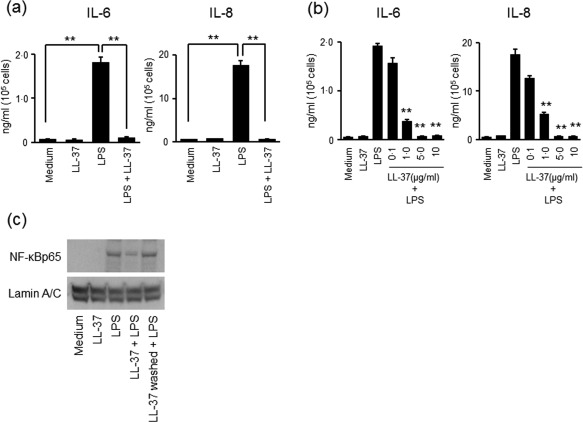
The effect of LL‐37 on lipopolysaccharide (LPS)‐induced expression of interleukin (IL)‐6 and IL‐8. (a) The effect of LL‐37 on LPS‐induced expression of IL‐6 and IL‐8. The cells were incubated for 24 h with or without LL‐37 (5·0 µg/ml), LPS (100 ng/ml) or LL‐37 (5·0 µg/ml) plus LPS (100 ng/ml). The levels of IL‐6 and IL‐8 in the supernatants were measured using enzyme‐linked immunosorbent assay (ELISA). Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. **P < 0·01. (b) Dose‐dependent effects of LL‐37 on LPS‐induced secretion of IL‐6 and IL‐8. The cells were incubated for 24 h with a combination of LPS (100 ng/ml) and increasing concentrations of LL‐37. The levels of IL‐6 and IL‐8 in the supernatants were measured using ELISA. Data are expressed as means ± s.e.m. of four independent experiments. **P < 0·01. (c) The effect of LL‐37 on LPS‐induced translocation of nuclear factor kappa B (NF‐κB)p65 into the nucleus. The cells were incubated for 15 min with medium alone, LL‐37 (5·0 µg/ml), LPS (100 ng/ml) or LL‐37 (5·0 µg/ml) plus LPS (100 ng/ml). Another group of cells was pretreated with LL‐37 (5·0 µg/ml) for 1h and then washed with phosphate‐buffered saline (PBS), and then LPS (100 ng/ml) was added and incubated for 15 min. NF‐κBp65 in the nuclear protein was analysed using immunoblotting. The data are representative of two independent experiments.
Discussion
In the present study we demonstrated that: (i) LL‐37 was up‐regulated in the inflamed mucosa of patients with UC and CD; (ii) colonic SEMFs were a major cellular source of LL‐37 in the inflamed mucosa of IBD patients; (iii) LL‐37 expression was induced by TLR‐3 stimulation via the assembly of intracellular adaptor proteins including TRIF, TRAF6 and TAK1; and (iv) LL‐37 interacted with LPS and attenuated proinflammatory activity of LPS. To our knowledge, this is the first extensive study of LL‐37 expression in human colonic tissue and the mechanisms controlling its induction in primary cells isolated from human colonic mucosa.
In the small intestine, Paneth cells produce anti‐microbial peptides, principally human α‐defensin 5 (HD5) and HD6. Patients with CD of the ileum are characterized by reduced constitutive expression of HD5 and HD 6 and, accordingly, compromised anti‐microbial barrier function 31, 32. Epithelial cells have been reported to be a major expression site of LL‐37 in various tissues 33. However, there are few reports of extensive study regarding LL‐37 expression in human colonic tissue. Initial study of IBD showed that mucosal mRNA expression of LL‐37 was increased in UC patients, but not in CD patients 12. In the current study, we observed that LL‐37 expression was increased significantly in the inflamed mucosa of both UC and CD patients. Moreover, we observed that colonic SEMFs, but not epithelial cells, are the main cellular source of LL‐37 in the inflamed mucosa of IBD, although a previous study showed a limited expression of LL‐37 in the inflamed mucosa of IBD patients 12. The reason for different observations between the previous and current studies remains unclear, but our findings indicate that LL‐37 is induced in the inflamed mucosa of IBD patients and suggest that LL‐37 may act as one of the protective factors against microbial invasion which aggravate gut inflammation. In contrast to reduced expression of HD5 and HD6 in the small intestine in patients with ileal CD 31, 32, anti‐microbial activity in the colon might be enhanced through increased expression of LL‐37 in colonic SEMFs.
The molecular mechanisms responsible for LL‐37 induction are not yet understood fully. We found that a TLR‐3 ligand stimulates the expression of LL‐37 in colonic SEMFs. TLR‐3 is a major mediator of the cellular response to viral infection because it responds to dsRNA, which is produced by viral replication 34. Recent studies have suggested that bacterial and viral infection co‐exist mainly at inflammatory sites 35 and that the cross‐talk between TLR‐4 and TLR‐3 enhances synergistically the inflammatory or protective response in cases of mixed infection 35. In such situations, TLR‐3 stimulation by viral infection may contribute to an increase in subsequent anti‐bacterial activity mediated by induced LL‐37.
In the cases of cell injury or tissue damage, cells die by necrosis and the heterologous RNA released from necrotic cells stimulates TLR‐3 signalling and induces immune activation without the involvement of pathogens. The response to self‐RNA is known as one of the ‘danger’ signals 35. In addition, TLR‐3 serves as a regulator of the amplification of the immune response as well as an endogenous sensor of necrosis, independent of viral activation 34. LL‐37 promotes anti‐bacterial activity by binding to nucleic acids to facilitate recognition of self‐DNA by TLR‐9, self‐RNA by TLR‐7 and TLR‐8 or double‐stranded RNA by TLR‐3 34. These findings suggest that endogenous RNA released from necrotic cells may be involved in LL‐37 induction and may contribute to amplification of TLR‐3 signalling, leading to enhancement of anti‐bacterial activity against microbial infection after tissue injury.
The intracellular signalling pathway for LL‐37 induction remains unclear, as the stimulatory molecules that induce LL‐37 expression have not been identified. A previous study reported that butyrate induces LL‐37 expression in colon cancer cell lines 14, but precise study has not been conducted in primary cells isolated from human colonic mucosa. In colonic SEMFs, the TLR‐3 ligand [poly(I:C)] stimulated LL‐37 expression. Previous studies reported that the TLR adaptor molecule (TRIF) facilitates TLR‐3 signalling and concomitant activation of the transcription factors, NF‐κB and AP‐1, leading to proinflammatory cytokine production 28, 36. In this study, we demonstrated for the first time that the TLR‐3‐mediated intracellular signalling pathway leading to LL‐37 expression required the formation of a complex of adaptor proteins, TRIF, TRAF6 and TAK1 in human colonic SEMFs. Furthermore, we found that LL‐37 induction is mediated by activation of transcription factors NF‐κB and AP‐1, although the roles of NF‐κB and AP‐1 have not been investigated previously.
A variety of biological functions, including anti‐microbial activity and a variety of immunomodulatory properties, have been attributed to LL‐37 37. For example, LL‐37 has been reported to down‐regulate TLR‐4 signalling in the macrophage cell line via binding for LPS and CD 14 38. To investigate the role of LL‐37 under the condition of bacterial and viral mixed infection, we examined the interaction between LL‐37 and an endotoxin in colonic SEMFs. LL‐37 suppressed LPS‐induced expression of IL‐6 and IL‐8 significantly, and this inhibitory effect was also observed in LPS‐induced NF‐κB activation. Interestingly, pretreatment with LL‐37 and the washing process cancelled the inhibitory effect of LPS on NF‐κB activation. This indicates that the co‐existence of LL‐37 and LPS is required to exert the inhibitory effect of LL‐37 and that LL‐37 does not modulate intracellular LPS‐TLR‐4 signalling. These findings suggest that LL‐37 might interact directly with LPS in the fluid phase and might attenuate the biological activity of LPS. In general, a major function of AMPs is to respond rapidly to mucosal damage and establish a temporary protective shield against infection 7. Therefore, LL‐37 might play a protective role in bacterial and viral mixed infection through the formation of an anti‐bacterial shield and attenuation of LPS activity to quell excess gut inflammation.
In summary, LL‐37 expression was increased in the inflamed mucosa of UC and CD patients. In such conditions, colonic SEMFs were identified as the primary cellular source. In‐vitro experiments using isolated SEMFs suggest that the major signalling pathway leading to LL‐37 expression was mediated by TLR‐3. As TLR‐3 responds to viral and heterologous RNA from necrotic cells, LL‐37 might play a role in viral infection as well as tissue injury with necrosis. In addition, LL‐37 attenuated the LPS‐induced inflammatory response in colonic SEMFs strongly. In the inflamed mucosa of IBD, LL‐37 might enhance anti‐bacterial and anti‐LPS activities to protect tissue from bacterial infection and excess inflammatory response.
Disclosure
The authors disclose no conflicts of interest.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Antibodies, reagents and polymerase chain reaction (PCR) primers used in this study
Fig. S1. Additional pictures of immunostaining for LL‐37 in normal mucosa and inflamed mucosa of inflammatory bowel disease (IBD) patients. Pictures are shown from two of six independent samples. Magnification × 200.
Fig. S2. Efficacy of small‐interference RNAs (siRNAs). (a) Immunoblot analyses of the efficacy of siRNAs for knock‐down of intracellular adaptor proteins [Toll/IL‐1R domain‐containing adaptor inducing interferon (TRIF), tumour necrosis factor receptor‐associated factor (TRAF)3, TRAF6, interleukin‐1 (IL‐1) receptor‐associated kinase (IRAK)1 and transforming growth factor β‐activated kinase (TAK)1] in human colonic SEMFs. The cells were transfected with control siRNA or siRNA specific for intracellular adaptor proteins, and incubated for 12 h. The expression of intracellular adaptor proteins was evaluated by immunoblotting. (b) Effect of control siRNA and siRNAs specific for nuclear factor (NF)‐κBp65 and c‐Jun in human colonic subepithelial myofibroblasts (SEMFs). The cells were transfected with control siRNA and siRNAs specific for NF‐κBP65 and c‐jun. The effect of siRNAs was evaluated using real‐time polymerase chain reaction (PCR). NF‐κBp65 and c‐Jun mRNA expression was converted to a value relative to β‐actin mRNA expression and presented as fold increase relative to the results for medium alone. Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. *P < 0·05.
Fig. S3. The effect of LL‐37 on lipopolysaccharide (LPS)‐induced mRNA expression of interleukin (IL)‐6 and IL‐8. (a) The effect of LL‐37 on LPS‐induced mRNA expression of IL‐6 and IL‐8. The cells were incubated for 12 h with LL‐37 (5·0 µg/ml), LPS (100 ng/ml) or LL‐37 (5·0 µg/ml) plus LPS (100 ng/ml). The mRNA expression of IL‐6 and IL‐8 was evaluted using real‐time polymerase chain reaction (PCR). The mRNA expression of IL‐6 and IL‐8 was converted to a value relative to β‐actin mRNA expression, and presented as fold increase relative to the results for medium alone (no stimulation). Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. **P < 0·01. (b) Dose‐dependent effects of LL‐37 on LPS‐induced mRNA expression of IL‐6 and IL‐8. The cells were incubated for 12 h with LPS (100 ng/ml) plus increasing concentrations of LL‐37. The mRNA expression of IL‐6 and IL‐8 was evaluated using real‐time PCR. The mRNA expression of IL‐6 and IL‐8 was converted to a value relative to β‐actin mRNA expression, and presented as fold increase relative to the results for medium alone (no stimulation). Data are expressed as means ± s.e.m. of four independent experiments. **P < 0·01.
Acknowledgements
This study was supported in part by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (15K08967, 15K19322), a grant for the Intractable Diseases from the Ministry of Health, Labor and Welfare of Japan (067), a grant from the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED (15AeK0109047h0002) and a grant from the Smoking Research Foundation (1848).
References
- 1. Goldsmith JR, Sartor RB. Response to the letter by Brotherton regarding ‘insoluble fiber and intestinal microbiota metabolism’. J Gastroenterol 2015; 50:492–3. [DOI] [PubMed] [Google Scholar]
- 2. Sheehan D, Moran C, Shanahan F. The microbiota in inflammatory bowel disease. J Gastroenterol 2015; 50:495–507. [DOI] [PubMed] [Google Scholar]
- 3. Caballero S, Pamer EG. Microbiota‐mediated inflammation and antimicrobial defense in the intestine. Annu Rev Immunol 2015; 33:227–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol 2012; 30:759–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun M, He C, Cong Y, Liu Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol 2015; 8:969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamasaki K, Schauber J, Coda A et al Kallikrein‐mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J 2006; 20:2068–80. [DOI] [PubMed] [Google Scholar]
- 7. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol 2012; 12:503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Travis SM, Anderson NN, Forsyth WR et al Bactericidal activity of mammalian cathelicidin‐derived peptides. Infect Immun 2000; 68:2748–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hou M, Zhang N, Yang J et al Antimicrobial peptide LL‐37 and IDR‐1 ameliorate MRSA pneumonia in vivo . Cell Physiol Biochem 2013; 32:614–23. [DOI] [PubMed] [Google Scholar]
- 10. Huang LC, Reins RY, Gallo RL, McDermott AM. Cathelicidin‐deficient (Cnlp –/–) mice show increased susceptibility to Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci 2007; 48:4498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ong PY, Ohtake T, Brandt C et al Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 2002; 347:1151–60. [DOI] [PubMed] [Google Scholar]
- 12. Schauber J, Rieger D, Weiler F et al Heterogeneous expression of human cathelicidin hCAP18/LL‐37 in inflammatory bowel diseases. Eur J Gastroenterol Hepatol 2006; 18:615–21. [DOI] [PubMed] [Google Scholar]
- 13. Gutiérrez A, Holler E, Zapater P et al Antimicrobial peptide response to blood translocation of bacterial DNA in Crohn's disease is affected by NOD2/CARD15 genotype. Inflamm Bowel Dis 2011; 17:1641–50. [DOI] [PubMed] [Google Scholar]
- 14. Schauber J, Svanholm C, Termen S et al Expression of the cathelicidin LL‐37 is modulated by short chain fatty acids in colonocytes: relevance of signalling pathways. Gut 2003; 52:735–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol 2011; 73:213–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andoh A, Bamba S, Brittan M, Fujiyama Y, Wright NA. Role of intestinal subepithelial myofibroblasts in inflammation and regenerative response in the gut. Pharmacol Ther 2007; 114:94–106. [DOI] [PubMed] [Google Scholar]
- 17. Best WR, Becktel JM, Singleton JW. Rederived values of the eight coefficients of the Crohn's Disease Activity Index (CDAI). Gastroenterology 1979; 77:843–6. [PubMed] [Google Scholar]
- 18. Rachmilewitz D. Coated mesalazine (5‐aminosalicylic acid) versus sulphasalazine in the treatment of active ulcerative colitis: a randomised trial. BMJ 1989; 298:82–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahida YR, Beltinger J, Makh S, Goke M, Gray T, Podolsky DK, Hawkey CJ. Adult human colonic subepithelial myofibroblasts express extracellular matrix proteins and cyclooxygenase‐1 and −2. Am J Physiol 1997; 273:G1341–8. [DOI] [PubMed] [Google Scholar]
- 20. Okuno T, Andoh A, Bamba S et al Interleukin‐1beta and tumor necrosis factor‐alpha induce chemokine and matrix metalloproteinase gene expression in human colonic subepithelial myofibroblasts. Scand J Gastroenterol 2002; 37:317–24. [DOI] [PubMed] [Google Scholar]
- 21. Nishida A, Hidaka K, Kanda T et al Increased expression of interleukin‐36, a member of the interleukin‐1 cytokine family, in inflammatory bowel disease. Inflamm Bowel Dis 2016; 22:303–14. [DOI] [PubMed] [Google Scholar]
- 22. Nishida A, Nagahama K, Imaeda H et al Inducible colitis‐associated glycome capable of stimulating the proliferation of memory CD4+ T cells. J Exp Med 2012; 209:2383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124:783–801. [DOI] [PubMed] [Google Scholar]
- 24. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 2010; 11:373–84. [DOI] [PubMed] [Google Scholar]
- 25. Ahmed AU, Williams BR, Hannigan GE. Transcriptional activation of inflammatory genes: mechanistic insight into selectivity and diversity. Biomolecules 2015; 5:3087–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 2012; 4:a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takatsuna H, Kato H, Gohda J et al Identification of TIFA as an adapter protein that links tumor necrosis factor receptor‐associated factor 6 (TRAF6) to interleukin‐1 (IL‐1) receptor‐associated kinase‐1 (IRAK‐1) in IL‐1 receptor signaling. J Biol Chem 2003; 278:12144–50. [DOI] [PubMed] [Google Scholar]
- 28. Kawai T, Akira S. Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011; 34:637–50. [DOI] [PubMed] [Google Scholar]
- 29. Mookherjee N, Brown KL, Bowdish DM et al Modulation of the TLR‐mediated inflammatory response by the endogenous human host defense peptide LL‐37. J Immunol 2006; 176:2455–64. [DOI] [PubMed] [Google Scholar]
- 30. Scott A, Weldon S, Buchanan PJ et al Evaluation of the ability of LL‐37 to neutralise LPS in vitro and ex vivo . PLOS ONE 2011; 6:e26525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Courth LF, Ostaff MJ, Mailander‐Sanchez D, Malek NP, Stange EF, Wehkamp J. Crohn's disease‐derived monocytes fail to induce Paneth cell defensins. Proc Natl Acad Sci USA 2015; 112:14000–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hayashi R, Tsuchiya K, Fukushima K et al Reduced human alpha‐defensin 6 in noninflamed Jejunal tissue of patients with Crohn's disease. Inflamm Bowel Dis 2016; 22:1119–28. [DOI] [PubMed] [Google Scholar]
- 33. Bals R, Wilson JM. Cathelicidins – a family of multifunctional antimicrobial peptides. Cell Mol Life Sci 2003; 60:711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang LJ, Gallo RL. Antimicrobial peptides. Curr Biol 2016; 26:R14–9. [DOI] [PubMed] [Google Scholar]
- 35. Rider P, Voronov E, Dinarello CA, Apte RN, Cohen I. Alarmins: feel the stress. J Immunol 2017; 198:1395–402. [DOI] [PubMed] [Google Scholar]
- 36. Ahmed S, Maratha A, Butt AQ, Shevlin E, Miggin SM. TRIF‐mediated TLR3 and TLR4 signaling is negatively regulated by ADAM15. J Immunol 2013; 190:2217–28. [DOI] [PubMed] [Google Scholar]
- 37. Mukherjee S, Hooper LV. Antimicrobial defense of the intestine. Immunity 2015; 42:28–39. [DOI] [PubMed] [Google Scholar]
- 38. Nagaoka I, Hirota S, Niyonsaba F et al Cathelicidin family of antibacterial peptides CAP18 and CAP11 inhibit the expression of TNF‐alpha by blocking the binding of LPS to CD14(+) cells. J Immunol 2001; 167:3329–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Antibodies, reagents and polymerase chain reaction (PCR) primers used in this study
Fig. S1. Additional pictures of immunostaining for LL‐37 in normal mucosa and inflamed mucosa of inflammatory bowel disease (IBD) patients. Pictures are shown from two of six independent samples. Magnification × 200.
Fig. S2. Efficacy of small‐interference RNAs (siRNAs). (a) Immunoblot analyses of the efficacy of siRNAs for knock‐down of intracellular adaptor proteins [Toll/IL‐1R domain‐containing adaptor inducing interferon (TRIF), tumour necrosis factor receptor‐associated factor (TRAF)3, TRAF6, interleukin‐1 (IL‐1) receptor‐associated kinase (IRAK)1 and transforming growth factor β‐activated kinase (TAK)1] in human colonic SEMFs. The cells were transfected with control siRNA or siRNA specific for intracellular adaptor proteins, and incubated for 12 h. The expression of intracellular adaptor proteins was evaluated by immunoblotting. (b) Effect of control siRNA and siRNAs specific for nuclear factor (NF)‐κBp65 and c‐Jun in human colonic subepithelial myofibroblasts (SEMFs). The cells were transfected with control siRNA and siRNAs specific for NF‐κBP65 and c‐jun. The effect of siRNAs was evaluated using real‐time polymerase chain reaction (PCR). NF‐κBp65 and c‐Jun mRNA expression was converted to a value relative to β‐actin mRNA expression and presented as fold increase relative to the results for medium alone. Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. *P < 0·05.
Fig. S3. The effect of LL‐37 on lipopolysaccharide (LPS)‐induced mRNA expression of interleukin (IL)‐6 and IL‐8. (a) The effect of LL‐37 on LPS‐induced mRNA expression of IL‐6 and IL‐8. The cells were incubated for 12 h with LL‐37 (5·0 µg/ml), LPS (100 ng/ml) or LL‐37 (5·0 µg/ml) plus LPS (100 ng/ml). The mRNA expression of IL‐6 and IL‐8 was evaluted using real‐time polymerase chain reaction (PCR). The mRNA expression of IL‐6 and IL‐8 was converted to a value relative to β‐actin mRNA expression, and presented as fold increase relative to the results for medium alone (no stimulation). Data are expressed as means ± standard error of the mean (s.e.m.) of four independent experiments. **P < 0·01. (b) Dose‐dependent effects of LL‐37 on LPS‐induced mRNA expression of IL‐6 and IL‐8. The cells were incubated for 12 h with LPS (100 ng/ml) plus increasing concentrations of LL‐37. The mRNA expression of IL‐6 and IL‐8 was evaluated using real‐time PCR. The mRNA expression of IL‐6 and IL‐8 was converted to a value relative to β‐actin mRNA expression, and presented as fold increase relative to the results for medium alone (no stimulation). Data are expressed as means ± s.e.m. of four independent experiments. **P < 0·01.