

Summary
Emerging evidence indicates a link between the increased proportion of regulatory T cells (Tregs) and reduced survival in patients who have been diagnosed with cancer. Cancer stem cells (CSCs) have been indicated to play a vital role in tumour initiation, drug resistance and recurrence. However, the relationship between Tregs and CSCs remains largely unknown. Here, we sorted out ovarian cancer stem‐like side population (SP) cells and CD133+ cells to investigate the influence of ovarian CSCs on Tregs. Among the various immune‐related molecules that we assessed, C‐C motif chemokine ligand 5 (CCL5) was the most elevated in ovarian CSCs relative to that in the non‐CSCs. The expression of its receptor, C‐C motif chemokine receptor 5 (CCR5), was also increased on the surface of Tregs in ovarian cancer patients. This receptor‐ligand expression profile indicated that ovarian CSCs recruit Tregs via CCL5–CCR5 interactions. We further assessed the expression of interleukin (IL)‐10 in Tregs cultured with different cancer cells. Tregs cultured in conditioned medium (CM) from ovarian CD133+ cells expressed a higher level of IL‐10 than Tregs cultured in CM from CD133– cells, indicating that Tregs exert pronounced immune‐inhibitory functions in CSC‐rich environments. Furthermore, co‐culture with ovarian cancer cell lines induced the expression of matrix metalloproteinase‐9 (MMP9) in Tregs which, in turn, enhanced the degradation of the extracellular matrix and enabled the invasion of tumour cells, thereby facilitating tumour metastasis. For the first time, to our knowledge, our findings describe the relationship between ovarian CSCs and Tregs, and demonstrated that these two cell populations co‐operate to promote tumour immune tolerance and enhance tumour progression.
Keywords: immune tolerance, invasion, ovarian cancer stem‐like cells, regulatory T cells
Introduction
Ovarian cancer is the most lethal gynaecological malignancy, accounting for 50% of mortality 1. Due to the lack of typical clinical symptoms in early stage disease, approximately 75% of patients are diagnosed at an advanced stage 2. Because of the high recurrence rate, the median overall survival of ovarian cancer patients is still less than 65 months after treatment with surgery and chemotherapy 1. The concept of cancer stem cells (CSCs) was first proposed due to the similarities between tumour cells and stem cells, with properties such as self‐renewal, multi‐lineage differentiation potential and capacity to produce organs 3, 4. CSCs have been defined as a rare subpopulation of cancer cells with the ability to form tumours, self‐renew and undergo differentiation 5. The existence of CSC was first described in acute myeloid leukaemia 6, breast cancer 7 and brain tumours 8. Ovarian CSCs can be separated from tumour cells using surface protein markers, such as c‐Kit 9, CD133 10, CD24 11, CD44 and CD117 12, or via functional assays, such as by taking advantage of their ability to export the DNA‐binding dye Hoechst 33342 13, 14. CD133, a surface marker of neural stem cells 15, has been used recently to isolate CSCs from tumours with properties such as self‐renewal, tumour initiation and tumour reconstitution 16, 17. Side population (SP) cells are cells that can export Hoechst 33342. We and others have demonstrated previously that ovarian cancer SP cells exhibit enhanced tumour formation capacity, colony formation ability, chemoresistance and pluripotency 13, 14. With high levels of expression of stem cell markers, CSCs have been proposed to cause tumour chemoresistance and relapse 18. Therefore, understanding the role of CSCs in tumour immunity is of great significance for cancer immunotherapy.
Regulatory T cells (Tregs) modulate the immune system negatively under both physiological and pathological conditions 19. There are four mechanisms by which Tregs suppress responder T cells 20, 21, 22, 23, 24. First, Tregs secrete inhibitory cytokines such as interleukin (IL)‐35, IL‐10 and transforming growth factor (TGF)‐β to inhibit T cells directly. Secondly, Tregs in the tumour microenvironment produce granzyme B and perforin to suppress responder cells, in the manner of cytotoxic T cells. Thirdly, Tregs induce apoptosis in effector cells by metabolic deprivation of the cytokine IL‐2, using the specific affinity of IL‐2 towards CD25. Lastly, Tregs suppress dysfunctional dendritic cells (DCs) and T cells by directly binding with them through surface molecules such as cytotoxic T lymphocyte antigen‐4 (CTLA‐4) and lymphocyte activation gene‐3 (LAG‐3).
Tumour‐associated antigen (TAA)‐specific T cells can infiltrate the tumour microenvironment 25, and these T cells can also be induced via tumour vaccines 26. However, the immune system often fails to eliminate cancer cells. It has been demonstrated that tumour cells develop potent, overlapping mechanisms to foster immune privilege by including the infiltration of Tregs into the tumour microenvironment 27, 28, 29, 30. Tregs enriched in ovarian carcinoma lead to reduced survival 27 and this has also been observed in many other solid tumours, including melanomas, renal cancer, breast cancer, cervical cancer 31 and colorectal cancer 32. Tregs infiltrate into tumour sites via various mechanisms, such as trafficking, differentiation, expansion and conversion 33. C‐C motif chemokine ligand 22 (CCL22) 27, 34, CCL5 30, 35 and CCL28 28 are responsible for trafficking, while TGF‐β and DCs are associated with the activation of Tregs induction in the tumour microenvironment 33.
Currently, many therapeutic strategies targeting Tregs, including CD25‐specific antibody, CTLA‐4‐specific antibody and IL‐2 immunotoxin, are being developed 33, 36. However, antibodies targeting Tregs fail to induce tumour rejection by the immune system, and thus the mechanisms by which Tregs infiltrate into tumour sites need to be elucidated further. CSCs have been reported to inhibit T cell activation and induce Treg recruitment from peripheral blood mononuclear cells (PBMCs) in melanoma and glioma patients 37. However, only a few researchers have addressed the influence of ovarian CSCs on the recruitment and inhibitory function of Tregs. This study aimed to elucidate the effects of ovarian CSCs on Tregs in vitro. In this study, we compared the expression of a series of immunosuppression‐associated molecules in CSCs and non‐CSCs, and found that the expression of indoleamine 2,3‐dioxygenase 1 (IDO1), CCL5 and C‐X‐C motif chemokine ligand 2 (CXCL2) was up‐regulated in CSCs, with CCL5 being the most affected. The expression of C‐C motif chemokine receptor 5 (CCR5), the receptor of CCL5, was increased in the Tregs isolated from ovarian cancer patients, enabling ovarian CSCs to recruit more Tregs through the CCL5–CCR5 interactions. We also found that Tregs incubated in conditioned medium (CM) from ovarian CSCs expressed higher levels of the inhibitory cytokines IL‐10, indicating that Tregs in CSC‐rich microenvironments display enhanced inhibitory properties. Furthermore, co‐culture with ovarian cancer cell lines promoted the expression of matrix metalloproteinase‐9 (MMP9) in Tregs which, in turn, enhanced the invasion of tumour cells. Altogether, for the first time, we demonstrated the effect of ovarian CSCs on the recruitment and inhibitory function of Tregs and found that ovarian cancer cells and Tregs co‐operate to promote tumour progression.
Materials and methods
Ovarian cancer cells culture
The human ovarian cancer cell line highly invasive ovarian cancer cells (HEY) was acquired from the MD Anderson Cancer Center. The cell line HEY A8 was provided by Professor Gong Yang from Fudan University Shang Hai Cancer Center (FUSCC). These two cancer cell lines were cultured in RPMI‐1640 with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco, Carlsbad, CA, USA). Another ovarian cancer cell line, HO‐8910PM, was obtained from Shanghai Institute of Biochemistry and Cell Biology Cell Bank and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. All the cells were incubated at 37°C with 5% CO2.
Sorting of cancer stem‐like cells
Ovarian cancer cell lines HEY and HEY A8 were stained with specific antibodies against human CD133 [phycoerythrin (PE), mouse immunoglobulin (IgG); Miltenyi Biotec, Bergisch Gladbach, Germany] at a density of 1 × 107/10 µl on ice in the dark for 30 min and purified by fluorescence‐activated cell sorting (FACS) (FACSAria III; BD Biosciences, San Jose, CA, USA). The SP cells which pumped out Hoechst 33342 dye were isolated by ultraviolet laser cytometry, as described previously 14. Briefly, the ovarian cancer cell line HO8910‐PM was stained with 5 µg Hoechst 33342 (Sigma, Hiroshima City, Japan) per 1 × 106 cells in DMEM with 2% FBS for 90 min at 37°C. Verapamil, which inhibits Hoechst 33342 efflux by SP cells, was added as a control. Cells were sorted and collected by ultraviolet laser cytometry (Beckman Coulter, Brea, CA, USA).
Isolation and flow cytometric analysis of Tregs
Tregs were isolated by FACS, and membrane‐bound or intracellular factors of Tregs were analysed by flow cytometer. PBMC were separated via gradient centrifugation using Ficoll from the peripheral blood of healthy volunteers. The PBMCs were labelled with mouse anti‐human CD4‐fluorescein isothiocyanate (FITC; Biolegend, San Diego, CA, USA), mouse anti‐human CD25‐PE (Biolegend), mouse anti‐human CD127‐allophycocyanin (APC) (Biolegend), mouse anti‐human forkhead box protein 3 (FoxP3)‐BV421 (Biolegend) and mouse anti‐human CCR5‐PE‐CY7 (Biolegend). For intracellular FoxP3 staining, after staining cell surface markers, the cells were fixed and permeabilized on ice for 40 min and then labelled with anti‐FoxP3 antibody. The CD4+CD25+CD127–/low Tregs were purified via FACS (FACSAria III; BD Biosciences), and the results were analysed with FlowJo software (Tree Star Inc., Ashland, OR, USA). The CD4+ T cells and Tregs were maintained in RPMI‐1640 with 10% FBS.
For intracellular IL‐10 and TGF‐β analysis, cells were fixed and permeabilized on ice for 40 min and then labelled with mouse anti‐human IL‐10‐PE or TGF‐β‐APC (Biolegend) antibodies. To detect the expression of MMP9, permeabilized tumour cells were stained with rabbit anti‐human MMP9 (ab52625; Abcam, Cambridge, UK) antibody, followed by Alexa Fluor 488‐conjugated mouse anti‐rabbit antibodies (Jackson ImmunoResearch, West Grove, PA, USA) at 4°C. As Tregs were collected from different samples, the expression of the positive group was analysed relative to that of the control group in the same sample. To assess IDO1 expression in CD133– and CD133+ ovarian cancer cells, Hey‐A8 cells were harvested and stained with surface marker CD133‐APC for 30 min, and fixed and permeabilized for 40 min on ice. Subsequently, these cells were stained with anti‐IDO1 antibody (ab211017; Abcam). After incubation for 30 min at 4°C, the cells were washed twice and then labelled with Alexa Fluor 488‐conjugated antibodies (Jackson ImmunoResearch). The cells were analysed with flow cytometry. All subjects gave informed consent, and the study was approved by the ethics committee of the Obstetrics and Gynecology Hospital of Fudan University.
Real‐time polymerase chain reaction (PCR)
Total RNA was isolated using TRIzol reagent, according to the manufacturer's instructions, and then reverse‐transcribed to cDNA using an RT kit (360A; Takara, Tokyo, Japan). The mRNA expression of interest gene was quantified using the Applied Biosystems real‐time PCR system (Life Technologies, Camarillo, CA, USA) with SYBR Green, and the housekeeping gene β‐actin served as the control. The relative expression was analysed using the comparative Ct method (ΔΔCt), and each experiment was repeated at least three times.
Enzyme‐linked immunosorbent assay (ELISA)
Isolated CD133+/– cancer cells or SP/non‐SP cells were cultured at 2 × 104/500 µl l for 48 h in 24‐well plates, and the conditioned media were collected and stored at −80°C. Freshly isolated Tregs were cultured in CD133+ and CD133– conditioned medium at a concentration of 5 × 105/ml for 24 h, and phorbol myristate acetate (PMA) (PeproTech, Rocky Hill, NJ, USA) with ionomycin (PeproTech) were added to activate Tregs for another 12 h. Supernatants collected from the Tregs cultured in CD133+ and CD133– conditioned media were defined as ‐ and ‐conditioned media. CCL5 (DRN00B; R&D Systems, Minneapolis, MN, USA) and CXCL2 (DL‐GROb‐Hu, DLDEVELOP, CN) levels were measured in the CSC and non‐CSC conditioned media; IL‐10 (D1000B; R&D Systems) levels were measured in ‐conditioned media using the corresponding ELISA kit according to the manufacturers' instructions.
Cell proliferation assay
Tregs were precultured in 96‐well plates at a density of 2 × 104/well for 24 h. Cell Counting Kit‐8 (CCK8; Dojindo, Kumamoto, Japan) solution was added to each well, and the reaction mixture was incubated for 5 h at 37°C. The CD133+ and CD133– cells were grown in 96‐well plates at a concentration of 4 × 103/100 µl for 24 h. Then, 10 µl CCK8 solution was added to each well for 2·5 h. The proliferation ability of the different cells was measured based on their optical density at 450 nm.
Chemotaxis assay of Tregs
Chemotaxis assay was performed in 24‐well plates with permeable membranes (5‐µm pore, Corning) (Fig. 1a,b). Mixtures containing recombinant human CCL5 (PeproTech) with 0·1% bovine serum albumin (BSA) (Sigma) in RPMI‐1640, CD133+‐CM and CD133–‐CM or CD133+‐CM with different concentrations of anti‐CCL5 antibodies (PeproTech) were placed into the lower chamber. The chemotaxis assays were performed with this set‐up in triplicate and were repeated several times. The chemotaxis assay was performed using two methods. First, purified Tregs (100 µl, 1 × 105) suspended in RPMI containing 0·1% BSA were added to the upper chambers and incubated for 4 or 6 h at 37°C with 5% CO2. The Tregs that had migrated to the lower chamber were counted with an inverted microscope 38, 39 (Fig. 1a). For the second method, CD4+ T cells (100 µl, 5 × 105) were plated in the top chamber, and the percentage of Tregs in the lower chamber among the CD4+ T cells that had migrated to the lower chamber was analysed with a flow cytometer 28. The chemotactic index was represented as the ratio of the number of migrated Tregs in the positive group relative to that in the control group 39 (Fig. 1b).
Figure 1.
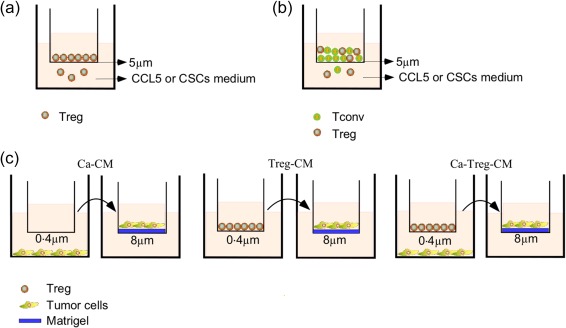
Transwell chemotaxis assay, co‐culture model, and invasion assay. (a,b) For chemotaxis assay, recombinant human C‐C motif chemokine ligand 5 (CCL5), CD133+‐conditioned medium (CM), CD133–‐CM or CD133+‐CM with anti‐CCL5 antibodies were added to the lower chambers of the chemotaxis apparatus with 5 µm pore‐containing permeable membranes. Regulatory T cells (Tregs) (a) or CD4+ T cells (b) isolated from peripheral blood mononuclear cells (PBMCs) were added to the upper chamber to migrate towards CCL5 or various types of CM. (c) For the co‐culture experiments, Tregs were added to the upper chamber Transwell plates with 0·4 µm membrane, and ovarian carcinoma cells were cultured in the lower chamber. For invasion assay, Ca‐CM, Treg‐CM and Ca‐Treg‐CM were diluted 1:1 with complete medium and added to the lower chamber, while ovarian cancer cell lines HEY A8 or HO‐8910PM were added the upper chamber to migrate towards the CM.
Invasion assay and co‐culture model
For the invasion assay (Fig. 1c), cells in 24‐well culture inserts (8 µm; Corning, New York, NY, USA) coated with diluted Matrigel (1 : 4, in serum‐free medium; 40 µl/well) (BD Biosciences) were incubated at 37°C for 3h. For the co‐culture model, ovarian carcinoma cells were co‐cultured with 1 × 105 Tregs in 12‐well plates, which were segregated with a 0·4‐µm polycarbonate membrane to facilitate the exchange of the soluble factors but not cells, and the wells that contained cancer cells or Tregs alone were set as the controls. The CM of the co‐culture cells (Ca‐Treg‐CM), the control cancer cells (Ca‐CM) or the control Tregs (Treg‐CM) was collected and diluted 1 : 1 with complete medium and added to the lower chamber of the invasion apparatus 40, while 3 × 104 of HEY A8 or 5 × 104 of HO‐8910PM cells were placed into the upper chamber. After migration for 24 or 36 h at 37°C, the invaded cells were stained with crystal violet and observed under a microscope.
Statistical analysis
The data are presented as the mean ± standard deviation (s.d.). Real‐time PCR and chemotaxis assays were performed in triplicate, and experiments were repeated at least three times. Comparisons were performed using the two‐tailed Student's t‐test or paired t‐test. P < 0·05 was considered significant.
Results
Characterization of immune‐associated molecules in CSCs
CSCs have been thought to be responsible for drug resistance and recurrence in cancer. However, the relationship between ovarian CSCs and immune suppression in tumours remains largely unknown. Here, we isolated cancer stem‐like SP cells and CD133+ cells from ovarian carcinoma cell lines HO‐8910PM, HEY and HEY A8 by FACS, as described previously 10, 14 (Fig. 2a). To demonstrate the stemness‐associated features of the sorted cells, the expression of pluripotency‐related genes, such as Nanog homeobox (Nanog), octamer‐binding transcription factor 4 (Oct4) and sex determining region Y box 2 (Sox2), was analysed by real‐time PCR. Both CD133+ cells and SP cells expressed elevated levels of pluripotent genes compared with CD133– cells and non‐SP cells, respectively (Fig. 2b).
Figure 2.
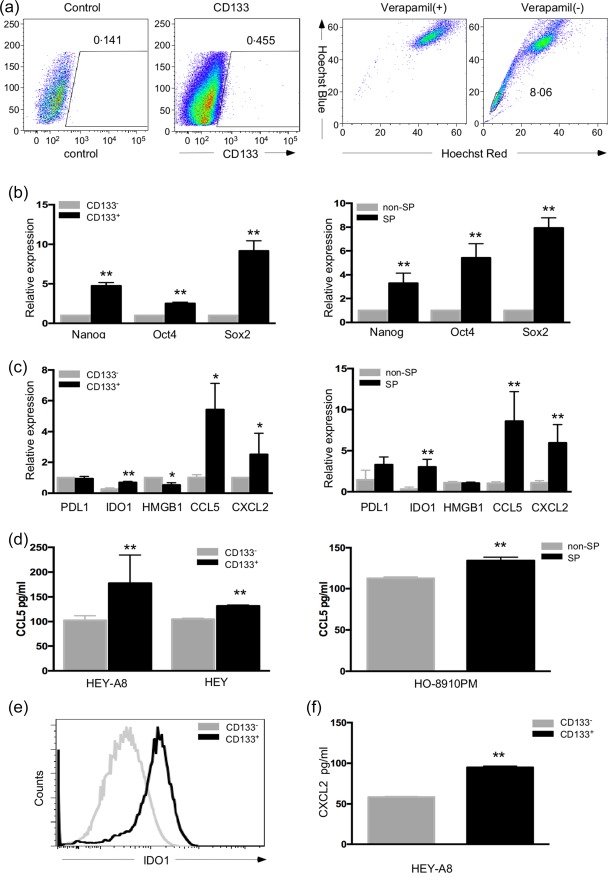
C‐C motif chemokine ligand 5 (CCL5) was elevated in ovarian cancer stem‐like (SP) and CD133+ cells. (a) Two methods were used to isolate cancer stem cells (CSCs) from ovarian cancer cell lines‐CD133+ CSCs were isolated from highly invasive ovarian cancer cells (HEY) A8 and HEY cells, and the Hoechst 33342‐low SP cells were isolated from HO‐8910PM cells. (b) The stem‐like properties of the two groups of isolated CSCs were demonstrated by elevated expression of the pluripotency‐related genes Nanog homeobox (Nanog), octamer‐binding transcription factor 4 (Oct4) and sex determining region Y box 2 (Sox2). (c) Comparison of the immune‐associated molecules programmed death‐ligand 1 (PDL1), indoleamine 2,3‐dioxygenase 1 (IDO1), high mobility group box 1 (HMGB1), CCL5 and chemokine (C‐X‐C motif) ligand 2 (CXCL2) in two kinds of ovarian CSCs and non‐CSCs by polymerase chain reaction (PCR). (d) Supernatants from CD133+/– and SP/non‐SP cells were collected and analysed for CCL5 expression by enzyme‐linked immunosorbent assay (ELISA). (e) IDO1 expression was compared between CD133+ cells and CD133– cells by fluorescence activated cell sorter (FACS). (f) CXCL2 protein expression was measured with ELISA in supernatants from CD133+ and CD133– cells. *P < 0·05 and **P < 0·01
Tumour cells can interact with immune cells in the tumour microenvironment through the expression of immune‐associated molecules, such as programmed cell death ligand 1 (PD‐L1), IDO1, high‐mobility group box‐1 protein (HMGB1), CCL5 and CXCL2. Among these molecules, PD‐L1 and IDO1 can directly suppress T cells 41, 42, HMGB1 and CCL5 can enhance Treg‐mediated immunosuppression 43, 44 and CXCL2 can lead to immune tolerance by attracting myeloid‐derived suppressor cells (MDSCs) to the tumour microenvironment 45. To explore the role of ovarian CSCs in tumour immune evasion, these immunosuppression‐associated molecules were assessed in CSCs and non‐CSCs by real‐time PCR (Fig. 2c). There was no difference in the expression of PDL1 between CSCs and non‐CSCs. HMGB1 expression was lower in the CD133+ cancer cells than that in the CD133– cells, while no difference was found between the SP and non‐SP cells. IDO1, CCL5 and CXCL2 were elevated in the CD133+ cancer cells and SP cells, and CCL5 exhibited the highest increase among these factors. The expression of CCL5 and CXCL2 in the supernatants of CSCs and non‐CSCs was analysed further by ELISA. It was found that the CCL5 protein level was higher in the supernatant from CD133+ cells than that in the supernatant from CD133– cells. Similar results were obtained for the supernatant from SP cells, indicating that the SP cells secrete a higher level of CCL5 than do the non‐SP cells (Fig. 2d). Using flow cytometry, we found that IDO1 expression was higher in the CD133+ cells than that in the CD133– cells (Fig. 2e). The level of CXCL2 was also higher in the supernatant from CD133+ cells than that in the supernatant from CD133– cells (Fig. 2f).
Tregs from ovarian cancer patients expressed higher levels of CCR5 than Tregs from healthy controls
Cells expressing CCL5 and its receptor CCR5 were attracted via chemokine–receptor interactions 30, 35. It has been demonstrated that CCR5 is involved in the recruitment of Tregs in mice 35. To determine the expression of CCR5 on Tregs, we isolated Tregs from ovarian cancer patients. Figure 3a shows the gating strategy and the identification of Tregs. First, the CD4+ T cells were gated from the PBMCs. Subsequently, the CD25+CD127–/low Tregs, representing a distinct fraction, were gated within the CD4+ T cells. Then, a four‐colour staining strategy was used to measure the intracellular FoxP3 expression in the CD4+CD25+CD127–/low Tregs, and we found that the majority of these Tregs expressed FoxP3. We then analysed CCR5 expression on Tregs from the PBMCs isolated from healthy volunteers (n = 10) and ovarian cancer patients (n = 10) with a flow cytometer (Fig. 3b,c). The level of CCR5 was increased in the Tregs from the ovarian cancer patients (P‐Tregs) relative to that in the Tregs from healthy controls (H‐Tregs). However, the conventional T cells (Tconv) from the healthy volunteers (H‐Tconv) and ovarian cancer patients (P‐Tconv) displayed a lower degree of CCR5 expression compared with the H‐Tregs and P‐Tregs, respectively. Thus, these results suggested that Tregs of ovarian cancer patients displayed increased CCR5 expression, which might enable the ovarian CSCs to recruit Tregs preferentially to the tumour microenvironment through ligand–receptor interaction.
Figure 3.
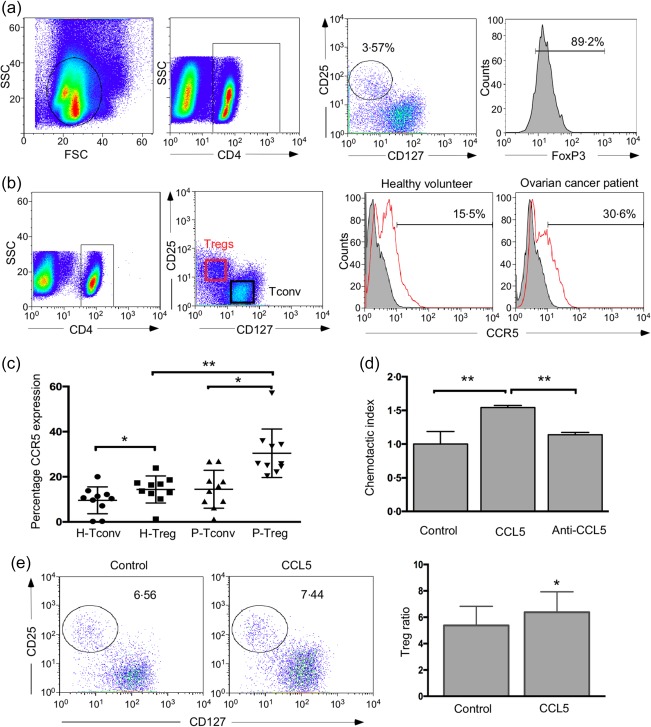
C‐C motif chemokine receptor 5 (CCR5) expression was increased on regulatory T cells (Tregs) from ovarian cancer patients. (a) Gating strategy of CD4+CD25+CD127–/low Tregs and fluorescence activated cell sorter (FACS) analysis of forkhead box protein 3 (FoxP3) expression in CD4+CD25+CD127–/low Tregs. Tregs were gated with CD25 and CD127 in CD4+ T cell population. FoxP3 expression in the CD4+CD25+CD127–/low Tregs was measured by FACS. (b) CCR5 expression on the Tregs and conventional T cells (Tconv) sorted from peripheral blood mononuclear cells (PBMCs) of both ovarian cancer patients and healthy volunteers was analysed by flow cytometry. Tregs were isolated by gating on CD4+CD25+CD127–/low cells, and Tconv cells were gated as CD4+CD25– cells. Histograms showing CCR5 levels on Tregs and Tconv from healthy volunteers and ovarian cancer patients. (c) Scatter‐plots showing CCR5 levels on Tregs and Tconv from 10 pairs of healthy volunteers and ovarian cancer patients. P represents ovarian cancer patients, and H represents healthy volunteers. (d) Recombinant human C‐C motif chemokine ligand 5 (CCL5) recruited Tregs, and CCL5‐neutralization antibody could block this recruitment. The y‐axis represents the chemotactic index. (e) Percentage of Tregs migrated towards CCL5 was analysed by flow cytometry (n = 4, paired t‐test). *P < 0·05 and **P < 0·01.
Next, the effect of CCL5 on the recruitment of Tregs was examined by two chemotaxis assays using culture plates with 5 µm pore‐containing permeable membranes. For the first method, the CD4+CD25+CD127–/low Tregs sorted freshly from PBMCs were added to the upper chamber of the chemotaxis assay set‐up to test their migration towards recombinant human CCL5, and the migrated Tregs in the lower chamber were counted with an inverted microscope. The chemotactic index of Tregs in the group with CCL5 was approximately two‐fold higher than that in the group with blank control. Additionally, this migration of the Tregs towards CCL5 could be blocked by anti‐CCL5 antibodies (Fig. 3d). For the second method, we added CD4+ T cells sorted from PBMCs to the upper chamber to migrate towards CCL5 in the lower chamber, and the percentage of Tregs in the lower chamber was measured by flow cytometry. The percentage of Tregs in the lower chamber was higher in the CCL5 group than that in the control group, indicating that Tregs were the predominant population among the CD4+ T cells that interact with CCL5 in vitro (Fig. 3e).
Ovarian CSCs recruited Tregs through chemokine CCL5
To determine the relationship between ovarian CSCs and Treg recruitment via the CCL5–CCR5 axis, we performed a chemotaxis assay using two methods with CMs collected from different types of cancer cell populations. First, isolated Tregs were added to the upper chamber of the chemotactic apparatus, and the chemotactic index of Tregs in the CD133+‐CM group was found to be (1·7 ± 0·033)‐ and (1·3 ± 0·077)‐fold higher than the chemotactic indices of Tregs in the CD133–‐CM groups using CM from HEY and HEY A8 cell lines, respectively, indicating that ovarian CSCs exert a greater chemotactic effect on Tregs than do non‐CSCs (Fig. 4a). Secondly, to confirm the preferential recruitment of Tregs by CD133+‐CM, we added CD4+ T cells into the upper compartments of the chemotaxis apparatus and assessed their migration towards CD133+‐CM and CD133–‐CM in the lower chamber. After 6 h, the CD4+CD25+CD127–/low Tregs in the lower and upper chambers were analysed via flow cytometry. The results showed that the CD133+‐CM recruited more Tregs to the lower chamber than did CD133–‐CM, while the percentage of Tregs in the upper chamber of the CD133+ group was lower than that of the CD133– group (Fig. 4b,c). The percentage of Tregs in the lower chamber was higher than that in the upper chamber for both the CD133–‐CM and CD133+‐CM groups, indicating that the Tregs were attracted preferentially to the CM of CD133+ and CD133– cells than the other CD4+ T cells.
Figure 4.
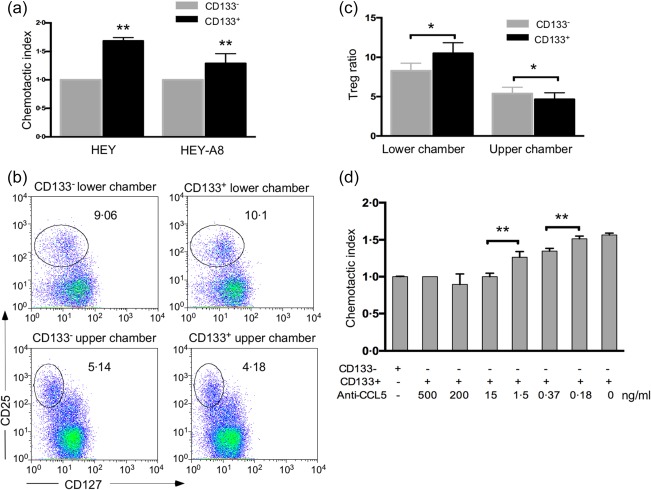
Ovarian cancer stem cells (CSCs) recruited regulatory T cells (Tregs) through the chemokine C‐C motif chemokine ligand 5 (CCL5). (a) CD133+‐ conditioned medium (CM) recruited more Tregs than did CD133–‐CM, which were obtained using the cancer cell lines highly invasive ovarian cancer cells (HEY) and HEY A8, respectively. The number of Tregs that had migrated to the CD133–‐CM was set as the control for the chemotactic index in each group. (b) Fluorescence activated cell sorter (FACS) analysis was performed on Tregs in both lower and upper chamber in the CD133+‐CM and CD133–‐CM groups, after the CD4+ T cells in the upper chambers had migrated toward the CM. (c) The ratio of Tregs in the lower and upper chambers of the CD133+‐CM and CD133–‐CM groups. (d) Treg migration towards CD133+‐CM was abrogated by different concentrations of anti‐CCL5 antibody. The group with 500 ng/ml anti‐CCL5 antibody was used to define the reference chemotactic index in this panel. *P < 0·05 and **P < 0·01.
To identify whether CCL5 plays a role in the CSC‐dependent preferential recruitment of Tregs, different concentrations of human CCL5‐neutralizing antibodies were added to the CD133+‐CM to block the CCL5‐related chemotaxis. The results showed that the recruitment of Tregs by CD133+‐CM could be abrogated completely when the concentration of anti‐CCL5 antibody was higher than 15 ng/ml, and the blocking effect was dose‐dependent below 15 ng/ml (Fig. 4d), suggesting that the recruitment of Tregs by CD133+ cells was dependent upon CCL5. Taken together, a direct link between ovarian CSCs and Treg recruitment through CCL5 was established in this study.
Ovarian CSCs promoted the inhibitory function and proliferation of Tregs
As Tregs can exert their function on effector T cells through inhibitory cytokines, such as IL‐10 and TGF‐β 24, the effect of CSCs on the function of Tregs was assessed by measuring IL‐10 and TGF‐β levels using flow cytometry and ELISA in Tregs that were incubated in CD133–‐CM and CD133+‐CM. CD133–‐CM and CD133+‐CM were collected, respectively, from isolated CD133– and CD133+ cells that were cultured for 48 h. Treatment with CD133+‐CM increased the expression of IL‐10 significantly, but not TGF‐β (data not shown) in Tregs (Fig. 5a). Additionally, the level of IL‐10 was higher in CM than that in CM. IL‐10 expression in Tregs incubated in the CD133+‐CM was 1·8 (± 0·2)‐fold than that in Tregs incubated in CD133–‐CM. These results suggested that IL‐10 was involved in the immunosuppressive function of Tregs induced by ovarian CSCs.
Figure 5.
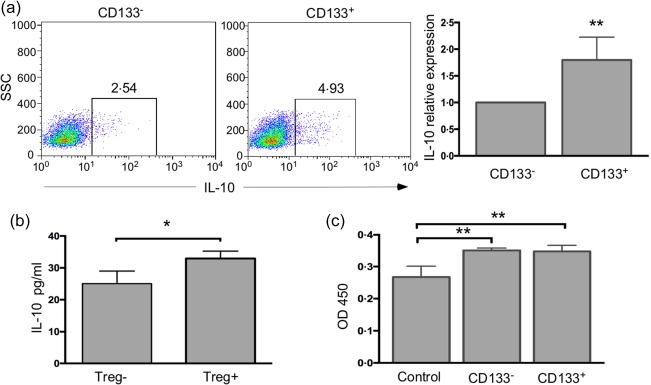
Cancer stem cells (CSCs) promoted interleukin (IL)‐10 expression and proliferation of regulatory T cells (Tregs). (a) Flow cytometric analysis of IL‐10 expression in Tregs incubated with different conditioned medium (CM) obtained from CD133– cells and CD133+ cells for 48 h. The data in the right histogram represent the relative expression of IL‐10 in Tregs cultured in CD133+‐CM to that of Tregs cultured in CD133–‐CM, because the Tregs isolated from different samples had different baseline IL‐10 expression. (b) IL‐10 secretion by Tregs cultured in CD133+‐CM and CD133–‐CM was assessed by enzyme‐linked immunosorbent assay (ELISA). (c) The optical density (OD)450 values from the cell counting kit 8 (CCK8) assay represent the proliferation of Tregs after incubation in control CM, CD133–‐CM and CD133+‐CM. *P < 0·05 and **P < 0·01.
Tregs have been proposed to be enriched in the tumour microenvironment via four mechanisms, including trafficking, differentiation, expansion and conversion 33. To investigate whether CSCs can stimulate the expansion of Tregs, we pre‐incubated the sorted Tregs in CD133–‐CM or CD133+‐CM in 96‐well plates for 24 h, and the Tregs incubated in the RPMI (with 10% FBS) were used as controls. Then, CCK8 reagent was added to the cells, and the cells were incubated for another 5 h. Compared with the control medium, CD133–‐CM and CD133+‐CM both promoted proliferation in Tregs (Fig. 5c).
Tregs enhanced cancer cell invasion by increasing MMP9 expression
In our present study, Tregs have been demonstrated to be recruited to the tumour microenvironment partly by ovarian CSCs. To determine whether these Tregs affected tumour progression, we used co‐culture experiments in which isolated Tregs were placed into the upper chamber, while ovarian cancer cells were in the lower chamber, segregated by a 0·4 µm polycarbonate membrane to facilitate the exchange of soluble factors but not of cells. We first focused on the expression of MMPs, and found no obvious change in the MMP9 expression in HEY cells in the presence of Tregs; however, MMP9 expression was elevated in the Tregs after co‐culturing (Fig. 6a). Similar results were found when other cell lines, including HEY A8(A8) and HO‐8910PM (PM), Treg‐A, Treg‐H and Treg‐P represented Tregs that interacted with the cancer cell lines HEY A8, HEY and HO‐8910‐PM, respectively. To identify further the changes in MMP9 expression in Tregs after culturing them with cancer cells, indirect fluorescence immunoassay‐based flow cytometry was used to evaluate MMP9 expression at the protein level, and the relative expression of MMP9 in the co‐cultured Tregs was 1·74 (± 0·1)‐fold higher than that of mono‐cultured Tregs (Fig. 6b,d).
Figure 6.
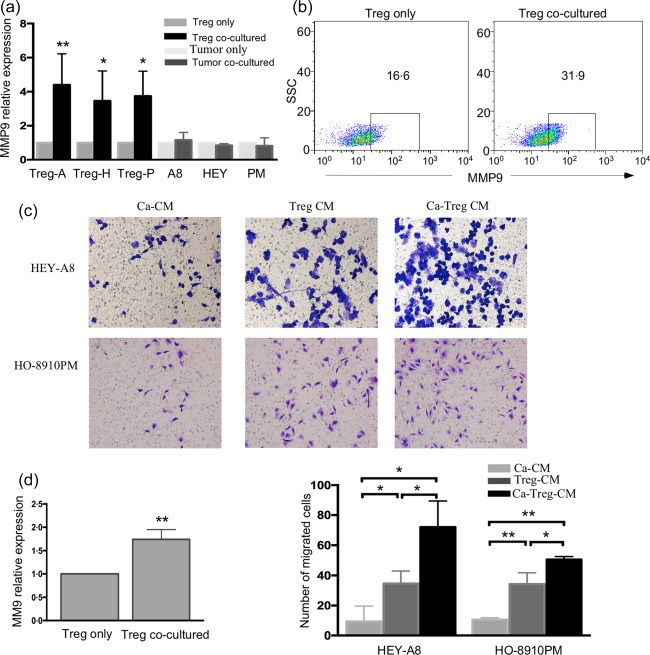
Regulatory T cells (Tregs) enhanced the invasion of tumour cells through increased matrix metalloproteinase 9 (MMP9) expression by cancer cells. (a) MMP9 expression in the Tregs co‐cultured with ovarian cancer cell lines was determined by real‐time polymerase chain reaction (PCR). Treg‐A, Treg‐H and Treg‐P represent the Tregs that interacted with cancer cell lines highly invasive ovarian cancer cells (HEY) A8, HEY and HO‐8910‐PM, respectively. (b,d) Flow cytometric analysis of MMP9 expression in Tregs after they were co‐cultured with cancer cells. The left panel in (d) represents the relative expression of MMP9 in the co‐cultured Tregs to monocultured Tregs. (c,d) Crystal violet staining showed the Ca‐Treg‐CM and Treg‐conditioned medium (CM) promoted tumour cells invasion in vitro. The right panel in (d) represents the number of cells that had invaded the membrane. *P < 0·05 and **P < 0·01.
To investigate whether the increased MMP9 levels in the co‐cultured Tregs could affect the invasion of cancer cells, we collected the conditioned medium from co‐cultured cancer cells and Tregs (Ca‐Treg‐CM) and conditioned medium from mono‐cultured cancer cells (Ca‐CM) to add to the lower chamber of the invasion apparatus, which was equipped with a 8‐µm pore polycarbonate membrane and coated with Matrigel, and cancer cells were added to the upper chamber to migrate towards the CMs to test the influence of Tregs on the invasion of cancer cells. The data showed that compared with Ca‐CM, Ca‐Treg‐CM promoted the invasion of the cancer cell lines HEY A8 and HO‐8910PM (Fig. 6c,d). To identify whether Tregs themselves induced the invasion of ovarian tumour cells, we collected medium from mono‐cultured Tregs (Treg‐CM) to add to the lower chamber of the invasion apparatus and tested the invasion of cancer cells. The results showed that Tregs‐CM enhanced the invasion of cancer cells to a much greater extent than did Ca‐CM. Taken together, cancer cells up‐regulated MMP9 expression in Tregs which, in turn, promoted the invasion of tumour cells via MMP9.
Discussion
Being at the top of the differentiation hierarchy, CSCs, which play an important role in tumorigenesis, may evade immune rejection preferentially, but the role of CSCs in tumour immune tolerance remains largely unclear. In the present study, we showed that both the CD133+ cancer stem‐like cells and the SP cells isolated from ovarian cancer cell lines displayed elevated expression of immunosuppression‐associated molecules such as CCL5, CXCL2 and IDO1, which enabled the CSCs to evade immune response. Interestingly, some recent studies also indicated that the expression of tumour‐associated antigens (TAA) is decreased in CSCs, and major histocompatibility complex (MHC) molecules and natural killer group 2, member D (NKG2D) ligand molecules are also reduced or absent in CSCs 37, 46, 47. Other than the aberrant expression of immune‐associated molecules, CSCs can also dampen the proliferation of T cells and induce the infiltration of Tregs in melanoma 37. All these studies revealed the role of CSCs in promoting immune tolerance.
CD4+CD25+CD127–/low Tregs, a subpopulation of CD4+ T cells, help to maintain immune homeostasis by inhibiting immune responses under physiological conditions 19. Recently, emerging evidence indicated that the proportion of Tregs is increased in various types of solid tumour, which is associated with poor prognosis 31. However, the effect of CSCs on the recruitment of Tregs remains unknown. We found that the CD133+ cancer cells expressed high levels of CCL5, a molecule that has been reported previously to be expressed highly in ovarian CSCs to promote cancer invasion and migration by the up‐regulation of MMP9 and induction of epithelial–mesenchymal transition (EMT) 10, 48. However, the relationship between CCL5 and Tregs recruitment has not been explored. The chemokines CCL5 and CCL22 have been reported to be responsible for Treg recruitment 27, 30, 35, 44. In this study, we showed that CCL5 could attract Tregs, and CD133+‐CM recruited more Tregs than did CD133–‐CM, which could be blocked by antibodies against human CCL5, suggesting that the recruitment of Tregs by CSCs rely upon CCL5. Our results, combined with those from former reports, indicate that CCL5 expression in ovarian carcinoma CSCs has a profound effect on both Tregs recruitment and tumour metastasis, and anti‐CCL5 antibodies might thus provide a novel strategy for immunotherapy in ovarian cancer patients.
Tregs have been reported to display higher levels of CCR5, a receptor of CCL5, than effector T cells. CCR5 is responsible for the migration of Tregs in murine pancreatic cancer and in fungal infections 35, 49. However, the expression of CCR5 on Tregs isolated from ovarian cancer patients was not understood clearly. Here, we found that Tregs expressed more CCR5 than did the effector T cells, which was consistent with previous reports. CCR5 expression on Tregs isolated from ovarian cancer patients was significantly higher than that of Tregs isolated from healthy volunteers. Furthermore, CCL5 has been reported to be elevated in ovarian cancer patients 50, which might account for the accumulation of Tregs in the tumour microenvironment of ovarian cancer patients.
Inhibitory cytokines, such as IL‐10 and TGF‐β, are attributable for the inhibitory function of Tregs, but it is unclear whether ovarian CSCs are involved in affecting the secretion of these inhibitory cytokines by Tregs. CSCs in melanoma have been demonstrated to promote PBMCs to secrete IL‐10 in a B7.2‐dependent manner 37. In this study, we showed that CD133+‐CM increased IL‐10 expression significantly by Tregs in vitro, but had no effect on TGF‐β secretion, suggesting that cancer stem cells probably promoted immune privilege partly by the induction of IL‐10 secretion in Tregs.
Tregs inhibit immune response by inducing apoptosis in T cells and deregulating antigen‐presenting cell dysfunction in the tumour microenvironment 33. However, whether Tregs recruited by CSCs can affect tumour cells directly is still unclear. Previous studies have suggested that tumour‐infiltrating immune cells, such as myeloid immune suppressor cells, neutrophils, mast cells and macrophages, secrete MMP9 to promote tumour survival, metastasis, angiogenesis and intravasation 51, 52, 53, 54, 55. Tumour‐specific T cells were reported to promote tumour cells to express MMP9 40, and MMP9 was correlated with the proportion of Tregs in laryngeal cancer 56. Here, we provided evidence that co‐culture with tumour cells induced MMP9 expression in Tregs. Our results also showed that Ca‐Treg‐CM promoted a higher degree of invasion in tumour cells compared with Ca‐CM and Treg‐CM, which might be attributed to the increased MMP9 expression in Tregs. However, the effect of tumour cells on MMP9 expression in Tregs should be elucidated further with clinical cancer tissues to investigate whether the same phenomemon is involved in the tumour microenvironment in vivo.
In summary (Fig. 7), our data provide evidence for the first time that ovarian cancer stem‐like cells express higher levels of immunosuppression‐associated molecules, such as CCL5, CXCL2 and IDO1, and recruit Tregs through CCL5. We also show that Tregs from ovarian cancer patients express high levels of CCR5, the receptor for CCL5, which facilitates the recruitment of Tregs by ovarian cancer stem‐like cells. Cancer stem‐like cells can also increase IL‐10 expression in Tregs. In exchange, Tregs promote the invasion of ovarian cancer cells through MMP9, the expression of which can be enhanced by co‐culture of Tregs with cancer cells.
Figure 7.
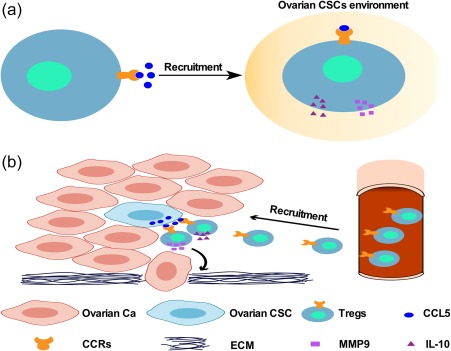
Schematic representation of the relationship between cancer stem‐like cells and regulatory T cells (Tregs). (a,b) Ovarian cancer stem cells (CSCs) affected immune tolerance by recruiting more Tregs to the tumour microenvironment than did non‐CSCs, due to their high expression of C‐C motif chemokine ligand 5 (CCL5) and the high expression of CCR5 on the Tregs of ovarian cancer patients. The Tregs infiltrated the microenvironment enriched in ovarian CSCs and secreted high levels of interleukin (IL)‐10 to inhibit anti‐tumour immunity. Ovarian cancer cells (Ca) promoted Tregs to express MMP9, which can degrade the extracellular matrix (ECM) and lead to the invasion of tumour cells.
Disclosure
The authors indicate no potential conflicts of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos 91542109, 81561138002 and 31371452) and Foundation from Science and Technology Commission of Shanghai Municipality (15JC1403200 and 16XD1400600).
Contributor Information
T. Chen, Email: chentong@fudan.edu.cn
H. Jiang, Email: jianghua@fudan.edu.cn
References
- 1. Salani R, Backes FJ, Fung MF et al Posttreatment surveillance and diagnosis of recurrence in women with gynecologic malignancies: Society of Gynecologic Oncologists recommendations. Am J Obstet Gynecol 2011; 204:466–78. [DOI] [PubMed] [Google Scholar]
- 2. Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet 2014; 384:1376–88. [DOI] [PubMed] [Google Scholar]
- 3. Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem‐cell biology to cancer. Nat Rev Cancer 2003; 3:895–902. [DOI] [PubMed] [Google Scholar]
- 4. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105–11. [DOI] [PubMed] [Google Scholar]
- 5. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med 2009; 15:1010–2. [DOI] [PubMed] [Google Scholar]
- 6. Lapidot T, Sirard C, Vormoor J et al A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367:645–8. [DOI] [PubMed] [Google Scholar]
- 7. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100:3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh SK, Hawkins C, Clarke ID et al Identification of human brain tumour initiating cells. Nature 2004; 432:396–401. [DOI] [PubMed] [Google Scholar]
- 9. Chau WK, Ip CK, Mak AS, Lai HC, Wong AS. c‐Kit mediates chemoresistance and tumor‐initiating capacity of ovarian cancer cells through activation of Wnt/beta‐catenin‐ATP‐binding cassette G2 signaling. Oncogene 2013; 32:2767–81. [DOI] [PubMed] [Google Scholar]
- 10. Long H, Xiang T, Qi W et al CD133+ ovarian cancer stem‐like cells promote non‐stem cancer cell metastasis via CCL5 induced epithelial–mesenchymal transition. Oncotarget 2015; 6:5846–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010; 29:2672–80. [DOI] [PubMed] [Google Scholar]
- 12. Zhang S, Balch C, Chan MW et al Identification and characterization of ovarian cancer‐initiating cells from primary human tumors. Cancer Res 2008; 68:4311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szotek PP, Pieretti‐Vanmarcke R, Masiakos PT et al Ovarian cancer side population defines cells with stem cell‐like characteristics and Mullerian inhibiting substance responsiveness. Proc Natl Acad Sci USA 2006; 103:11154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang H, Lin X, Liu Y et al Transformation of epithelial ovarian cancer stemlike cells into mesenchymal lineage via EMT results in cellular heterogeneity and supports tumor engraftment. Mol Med 2012; 18:1197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Uchida N, Buck DW, He D et al Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA 2000; 97:14720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singh SK, Clarke ID, Terasaki M et al Identification of a cancer stem cell in human brain tumors. Cancer Res 2003; 63:5821–8. [PubMed] [Google Scholar]
- 17. Curley MD, Therrien VA, Cummings CL et al CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009; 27:2875–83. [DOI] [PubMed] [Google Scholar]
- 18. Shah MM, Landen CN. Ovarian cancer stem cells: are they real and why are they important? Gynecol Oncol 2014; 132:483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Josefowicz SZ, Lu L‐F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30:531–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Turnis ME, Sawant DV, Szymczak‐Workman AL et al Interleukin‐35 limits anti‐tumor immunity. Immunity 2016; 44:316–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cao X, Cai SF, Fehniger TA et al Granzyme B and perforin are important for regulatory T cell‐mediated suppression of tumor clearance. Immunity 2007; 27:635–46. [DOI] [PubMed] [Google Scholar]
- 22. Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008; 8:523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol 2005; 6:338–44. [DOI] [PubMed] [Google Scholar]
- 24. Shevach EM. Mechanisms of foxp3+ T regulatory cell‐mediated suppression. Immunity 2009; 30:636–45. [DOI] [PubMed] [Google Scholar]
- 25. Zhang L, Conejo‐Garcia JR, Katsaros D et al Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003; 348:203–13. [DOI] [PubMed] [Google Scholar]
- 26. Wefers C, Lambert LJ, Torensma R, Hato SV. Cellular immunotherapy in ovarian cancer: targeting the stem of recurrence. Gynecol Oncol 2015; 137:335–42. [DOI] [PubMed] [Google Scholar]
- 27. Curiel TJ, Coukos G, Zou L et al Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942–9. [DOI] [PubMed] [Google Scholar]
- 28. Facciabene A, Peng X, Hagemann IS et al Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011; 475:226–30. [DOI] [PubMed] [Google Scholar]
- 29. Yin Y, Cai X, Chen X et al Tumor‐secreted miR‐214 induces regulatory T cells: a major link between immune evasion and tumor growth. Cell Res 2014; 24:1164–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ward ST, Li KK, Hepburn E et al The effects of CCR5 inhibition on regulatory T‐cell recruitment to colorectal cancer. Br J Cancer 2015; 112:319–28. 25405854 [Google Scholar]
- 31. Shang B, Liu Y, Jiang S‐J, Liu Y. Prognostic value of tumor‐infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta‐analysis. Sci Rep 2015; 5:15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saito T, Nishikawa H, Wada H et al Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 2016; 22:679–84. [DOI] [PubMed] [Google Scholar]
- 33. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006; 6:295–307. [DOI] [PubMed] [Google Scholar]
- 34. Yang P, Li Q‐J, Feng Y et al TGF‐β‐miR‐34a‐CCL22 signaling‐induced Treg cell recruitment promotes venous metastases of HBV‐positive hepatocellular carcinoma. Cancer Cell 2012; 22:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tan MC, Goedegebuure PS, Belt BA et al Disruption of CCR5‐dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol 2009; 182:1746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu C, Workman CJ, Vignali DAA. Targeting regulatory T cells in tumors. FEBS J 2016; 283:2731–48. [DOI] [PubMed] [Google Scholar]
- 37. Schatton T, Schutte U, Frank NY et al Modulation of T‐cell activation by malignant melanoma initiating cells. Cancer Res 2010; 70:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tager AM, Bromley SK, Medoff BD et al Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol 2003; 4:982–90. [DOI] [PubMed] [Google Scholar]
- 39. Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol 2005; 6:895–901. [DOI] [PubMed] [Google Scholar]
- 40. Hu S, Li L, Yeh S et al Infiltrating T cells promote prostate cancer metastasis via modulation of FGF11→miRNA‐541→androgen receptor (AR)→MMP9 signaling. Mol Oncol 2015; 9:44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abiko K, Matsumura N, Hamanishi J et al IFN‐gamma from lymphocytes induces PD‐L1 expression and promotes progression of ovarian cancer. Br J Cancer 2015; 112:1501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Munn DH, Mellor AL. Indoleamine 2,3‐dioxygenase and tumor‐induced tolerance. J Clin Invest 2007; 117:1147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wild CA, Bergmann C, Fritz G et al HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol 2012; 24:485–94. [DOI] [PubMed] [Google Scholar]
- 44. Tan W, Zhang W, Strasner A et al Tumour‐infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL–RANK signalling. Nature 2011; 470:548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang H, Ye YL, Li MX et al CXCL2/MIF‐CXCR2 signaling promotes the recruitment of myeloid‐derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 2016; 36:2095–104. [DOI] [PubMed] [Google Scholar]
- 46. Schatton T, Frank MH. Antitumor immunity and cancer stem cells. Ann NY Acad Sci 2009; 1176:154–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Di Tomaso T, Mazzoleni S, Wang E et al Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res 2010; 16:800–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Long H, Xie R, Xiang T et al Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem‐like cells via NF‐kappaB‐mediated MMP‐9 upregulation. Stem Cells 2012; 30:2309–19. [DOI] [PubMed] [Google Scholar]
- 49. Cavassani KA, Campanelli AP, Moreira AP et al Systemic and local characterization of regulatory T cells in a chronic fungal infection in humans. J Immunol 2006; 177:5811–8. [DOI] [PubMed] [Google Scholar]
- 50. Tsukishiro S, Suzumori N, Nishikawa H, Arakawa A, Suzumori K. Elevated serum RANTES levels in patients with ovarian cancer correlate with the extent of the disorder. Gynecol Oncol 2006; 102:542–5. [DOI] [PubMed] [Google Scholar]
- 51. Bekes EM, Schweighofer B, Kupriyanova TA et al Tumor‐recruited neutrophils and neutrophil TIMP‐free MMP‐9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol 2011; 179:1455–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP‐9 supplied by bone marrow‐derived cells contributes to skin carcinogenesis. Cell 2000; 103:481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Acuff HB, Carter KJ, Fingleton B, Gorden DL, Matrisian LM. Matrix metalloproteinase‐9 from bone marrow‐derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res 2006; 66:259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hiratsuka S, Nakamura K, Iwai S et al MMP9 induction by vascular endothelial growth factor receptor‐1 is involved in lung‐specific metastasis. Cancer Cell 2002; 2:289–300. [DOI] [PubMed] [Google Scholar]
- 55. Yang L, DeBusk LM, Fukuda K et al Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor‐bearing host directly promotes tumor angiogenesis. Cancer Cell 2004; 6:409–21. [DOI] [PubMed] [Google Scholar]
- 56. Wang B‐Q, Zhang C‐M, Gao W, Wang X‐F, Zhang H‐L, Yang P‐C. Cancer‐derived matrix metalloproteinase‐9 contributes to tumor tolerance. J Cancer Res Clin Oncol 2011; 137:1525–33. [DOI] [PubMed] [Google Scholar]