

Key Points
LMP2A enhances MYC expression, resulting in the rapid degradation of the p27kip1 tumor suppressor via Cks1, a member of the SCFSkp2 complex.
Loss of Cks1 restores levels of the p27kip1 tumor suppressor and prolongs LMP2A-mediated lymphomagenesis.
Abstract
Epstein-Barr virus (EBV) establishes lifelong infection in B lymphocytes of most human hosts and is associated with several B lymphomas. During latent infection, EBV encodes latent membrane protein 2A (LMP2A) to promote the survival of B cells by mimicking host B-cell receptor signaling. By studying the roles of LMP2A during lymphoma development in vivo, we found that LMP2A mediates rapid MYC-driven lymphoma onset by allowing B cells to bypass MYC-induced apoptosis mediated by the p53 pathway in our transgenic mouse model. However, the mechanisms used by LMP2A to facilitate transformation remain elusive. In this study, we demonstrate a key role of LMP2A in promoting hyperproliferation of B cells by enhancing MYC expression and MYC-dependent degradation of the p27kip1 tumor suppressor. Loss of the adaptor protein cyclin-dependent kinase regulatory subunit 1 (Cks1), a cofactor of the SCFSkp2 ubiquitin ligase complex and a downstream target of MYC, increases p27kip1 expression during a premalignant stage. In mice that express LMP2A, Cks1 deficiency reduces spleen weights, restores B-cell follicle formation, impedes cell cycle progression of pretumor B cells, and eventually prolongs MYC-driven tumor onset. This study demonstrates that LMP2A uses the role of MYC in the cell cycle, particularly in the p27kip1 degradation process, to accelerate lymphomagenesis in vivo. Thus, our results reveal a novel mechanism of EBV in diverting the functions of MYC in malignant transformation and provide a rationale for targeting EBV’s roles in cell cycle modulation.
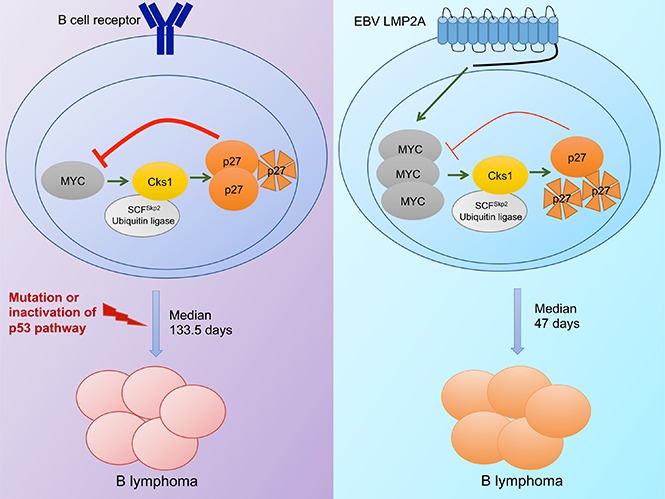
Visual Abstract
Introduction
Epstein-Barr virus (EBV) is a lymphocryptic herpesvirus that ubiquitously infects >95% of the human population by adulthood and persists in B lymphocytes throughout life.1 EBV was first discovered in Burkitt lymphoma (BL).2 To date, several lymphomas are associated with EBV latent infection, including Hodgkin lymphoma (HL), non-Hodgkin lymphoma, AIDS-associated lymphoma, and posttransplant lymphoproliferative diseases.1 However, the degrees of EBV association and patterns of viral gene expression vary among lymphoma types, highlighting the complexity of mechanisms that EBV contributes to malignant transformation. EBV can readily transform B cells in vitro by expressing all latency gene products (growth program or latency III), including all EBV nuclear antigens (EBNA1, –2, –3A, –3B, –3C, and -LP), latent membrane proteins (LMP1, LMP2), and viral noncoding RNA transcripts. Latency III is found in posttransplant lymphoproliferative diseases and in some AIDS-associated lymphomas. Latency II is the gene expression pattern found in HL, which includes EBNA1, both LMPs, and the viral noncoding RNAs. Latency I is found in BL, which includes EBNA1 and, in some instances, LMP2A.1
LMP2A is encoded by EBV in most latency programs and is a viral mimic of host B-cell receptor signaling.3-5 The cytoplasmic tail of LMP2A contains an immunoreceptor tyrosine-based activation motif similar to that of CD79a and CD79b found in the BCR complex.6 Without extracellular ligands, LMP2A can mimic BCR proximal signaling via association with Src family kinases and spleen tyrosine kinase.7-9 LMP2A provides survival signals to B cells, including when the BCR is not functional or not present. Signal transduction of LMP2A occurs predominantly via the PI3K/Akt pathway,10,11 similar to tonic BCR signaling.12
The transcription factor c-MYC (or MYC hereafter) is arguably the most important driving force of cancer development, with abundant functions controlling many cellular processes. Most, if not all, cancer types show the dysregulation of MYC expression via chromosomal translocation, amplification of transcription, and protein stabilization. However, recent studies have indicated that MYC alone is not sufficient to drive malignancies13,14 because MYC target genes include those promoting both proliferation and apoptosis. Due to the seemingly paradoxical roles of MYC, cells must acquire additional genetic mutations or disruptions of tumor suppressor functions in order for tumors to develop. In addition, genetic studies of human BL (a MYC-driven lymphoma) showed that endemic BL cases, which are virtually all EBV+, have different mutational landscapes compared with other subtypes in which EBV association is lower, suggesting that EBV infection diverts B cells to distinct pathological pathways.14,15 Murine models of MYC-induced lymphomagenesis have revealed several antiapoptotic genes, such as p53,16 Cdkn2a,17 Bax,18 Bim,19 and Puma,20 which are important for counteracting MYC’s proliferative functions. Studies in human lymphoma cell lines and in mouse models have demonstrated the cooperation of BCR and MYC in lymphoma development and survival.14,21-23 Consistent with the pathogenic roles of BCR, our previous studies showed that LMP2A accelerates lymphomagenesis without major perturbation of the p53 tumor suppressor pathway in a MYC model.24
Overexpression of cyclins or downregulation of cyclin-dependent kinase (CDK) inhibitors is often observed in a variety of human cancers. The cyclin-dependent kinase inhibitor p27kip1 is a mammalian cell cycle regulator that negatively regulates gap 1 (G1) progression by binding to G1-specific cyclin/CDK complexes and preventing their activity. Thus, p27kip1 acts as a cell cycle checkpoint regulator and a potent tumor suppressor that prevents untimed cell cycle progression. P27kip1 is downregulated in several human cancers, but genetic mutations or deletions in the gene encoding p27kip1 are rare.25 The concentration of p27kip1 is largely controlled at the protein level by protein phosphorylation and ubiquitin-proteasome pathways throughout the cell cycle. Two main pathways have been described for p27kip1 proteolysis. In gap 0 (G0) and early G1 phases, Ser10-phosphorylated p27kip1 is translocated from the nucleus to the cytoplasm, where p27kip1 is degraded by the Kip1 ubiquitination-promoting complex (KPC).26,27 In late G1 and synthesis (S) phases, phosphorylation of Thr187 on p27kip1 recruits CDK regulatory subunit 1 (Cks1), a cofactor of the SCFSkp2 ubiquitin-ligase complex, and thereby promotes p27kip1 ubiquitination by SCFSkp2 in the nucleus.28,29
Our previous study identified p27kip1 as an important checkpoint regulator in the early stage of LMP2A-mediated B-cell hyperproliferation, because LMP2A expression correlates with the enhancement of MYC-induced degradation of p27kip1 30. However, we found that p27kip1 with a mutation in Ser10 (p27S10A) had a minimal effect on altering LMP2A roles in driving the cell cycle, despite previous studies that indicated this mutant can reduce K-Ras–driven tumorigenesis.30 A previous study described Cks1 as a MYC target important for p27kip1 regulation.31 To investigate whether another pathway is important for LMP2A-mediated p27kip1 degradation and lymphomagenesis, Cks1 knockout mice were used in our EBV-driven murine lymphoma model. Here, we found that Cks1 is important for the degradation of p27kip1 in LMP2A-expressing B cells. Knockout of Cks1 rescues p27kip1 levels and delays LMP2A-mediated lymphoma development without increasing the frequency of p53 pathway abnormalities. This study highlights the synergistic function of LMP2A and a host oncogene, such as MYC, and provides a basis for understanding how viruses mechanistically contribute to human cancers.
Materials and methods
Mice
The Tg6 line of Eµ-LMP2A transgenic mice, in which LMP2A is expressed under immunoglobulin (Ig) heavy chain promoter and intronic enhancer (Eμ),32 and λ-MYC mice, in which human MYC is overexpressed,33 have been described and are in the C57BL/6 background. Cks1−/− mice29 were constructed by the deletion of exon 2 of the murine Cks1 locus and were obtained from Steven Reed Laboratory at The Scripps Research Institute (La Jolla, CA). Tumor mice were sacrificed when lymph node tumors could be observed externally or when mice were moribund. Animals were maintained at Northwestern University’s Center for Comparative Medicine in accordance with the University’s animal welfare guidelines.
Tumor, spleen, and bone marrow cell isolation
Pretumor spleens and bone marrow were isolated from 4-week-old mice. The Mouse Pan-B Cell Isolation Kit (StemCell Technologies) was used to purify splenic B cells. Bone marrow cells were flushed from femurs and tibia. Tumor-bearing lymph nodes were prepared as previously described.24,34,35 More than 90% of tumor cells in tumor-bearing lymph nodes were of B-cell lineage and thus did not require further cell sorting.
Flow cytometry and cell cycle analysis
For flow cytometry, isolated cells were stained with the indicated antibodies and analyzed with the FACS-CantoII flow cytometer (BD Biosciences). IgM- fluorescein isothiocyanate (FITC), CD3ε-phycoerythrin, and B220-allophycocyanin antibodies were from BD Biosciences. For cell cycle analysis, purified pretumor B cells were fixed in 70% ethanol and stained with a Ki-67–FITC antibody. DNA was stained with propidium iodide/ribonuclease staining buffer according to the manufacturer’s instructions (BD Biosciences). All results were analyzed with FlowJo software (FlowJo, LLC).
Immunohistochemistry
Spleens and tumor-bearing lymph nodes were fixed in 10% buffered formalin phosphate, stored in 70% ethanol, and then embedded in paraffin. Samples were serially sectioned and stained with hematoxylin and eosin, anti-p27kip1 (Abgent), anti-p27kip1 (phospho T187) (Abcam), or anti-B220 (BD Biosciences) antibody. Stained tissue slides were imaged using EVOS XL Core and Nikon Eclipse E600 microscopes.
Immunoblots
Isolated pretumor B cells or tumor cells were lysed, and immunoblotting was performed as described.34 The primary antibodies used in the immunoblots included p27kip1 (Santa Cruz), p53 (Cell Signaling), p19ARF and Gapdh (Abcam), and Calnexin (Stressgen).
Statistics
One-way analysis of variance (ANOVA), 2-way ANOVA, Fisher’s exact test, survival analysis, and log-rank (Mantel-Cox) test were used to calculate with Prism 7 (GraphPad Software). P < .05 was considered statistically significant.
Results
Loss of Cks1 dampens LMP2A-mediated expansion of pretumor B cells
Using a mouse model of EBV latent infection, mice expressing LMP2A and human c-MYC transgene (LMP2A/λ-MYC) showed increases in normalized spleen weights (Figure 1A) and B-cell populations in the spleen and bone marrow (Figure 1B) compared with λ-MYC and wild-type (WT) mice, which is consistent with our previous studies.24,36 To examine the importance of p27kip1 in LMP2A-mediated B-cell hyperproliferation, we crossed our mice with Cks1−/− mice. Knockout of Cks1 exhibited minimal effect on spleen weights and the number of B cells in λ-MYC mice. In contrast, Cks1 loss in LMP2A/λ-MYC mice significantly reduced the normalized percentage of spleen weights similar to those of λ-MYC and WT mice (Figure 1A). This phenotype was specific to B cells as both the percentage and absolute number of B cells in LMP2A/λ-MYC/Cks1−/− mice were reduced to the WT level, but the absolute number of splenic T cells was unchanged (Figure 1B).
Figure 1.

Cks1 knockout decreases the pretumor B-cell population in LMP2A/λ-MYC mice. (A) Spleens were isolated from 4-week-old mice at the pretumor stage, and spleen weights as a percentage of whole body weight are indicated. (B) Lymphocytes were isolated from the bone marrow (BM) and spleens (SP) of pretumor mice, and their B- and T-cell numbers and percentages were calculated by flow cytometry stained with B220-allophycocyanin and CD3ε- phycoerythrin antibodies. Data represent the mean ± standard deviation. (C) The percentage of IgM-positive cells to total splenic B cells were calculated by staining with IgM-FITC antibody. (D) Representative immunohistochemical analysis of 4-week-old mouse spleens from 3 mice of each genotype stained with B220 (original magnification ×4). The statistic differences of Cks1 knockouts against their controls are indicated with thick lines. *P < .05, **P < .01, ***P < .001, ****P < .0001, calculated by 1-way ANOVA with post hoc multiple comparison tests. MYC, λ-MYC mice; 2A MYC, LMP2A/λ-MYC mice.
Previous studies have shown that LMP2A expression during early B-cell development can replace the expression of surface BCRs in the periphery,3,4 presumably due to the bypass of the requirement of Ig gene rearrangement for generating a functional BCR. λ-MYC B cells showed significant reduction of surface IgM+ B cells compared with WT mice, similar to previous Eµ-Myc studies.37,38 In contrast, LMP2A/λ-MYC B cells are exclusively IgM−, as previously shown.35 Loss of Cks1 did not significantly change the status of surface IgM in both LMP2A/λ-MYC and λ-MYC B cells (Figure 1C). There was a slight increase in the IgM+ population in LMP2A/λ-MYC/Cks1−/− B cells, although the increase was not statistically significant (Figure 1C). In the secondary lymphoid organs, such as the spleen, B cells form follicles after exiting the bone marrow. Immunohistochemistry of B220 (a pan–B-cell marker) showed that λ-MYC B cells still formed apparent B-cell follicles, whereas such B-cell localization was completely absent in LMP2A/λ-MYC spleens. Interestingly, knockout of Cks1 restored the organization of B-cell follicles in both LMP2A/λ-MYC and λ-MYC spleens to a near-WT phenotype (Figure 1D). The effect of Cks1 knockout on B-cell localization was more prominent in LMP2A/λ-MYC than λ-MYC mice. Despite the fact that B cells from LMP2A/λ-MYC/Cks1−/− mice largely lacked surface IgM (Figure 1C), these B cells were able to form follicles in the periphery (Figure 1D), suggesting that they still maintained hallmarks of functional peripheral B cells.
Cks1 deficiency rescues p27kip1 expression in pretumor B cells
Cks1 is an adaptor protein that binds and brings p27kip1 to an E3 ubiquitin ligase complex for degradation via proteasome.29 To elucidate Cks1 status on p27kip1 levels, immunohistochemistry of p27kip1 in spleens was performed. LMP2A/λ-MYC/Cks1−/− and λ-MYC/Cks1−/− spleens showed increased staining intensity in white pulp areas compared with their Cks1+/+ counterparts (Figure 2A). To specifically examine p27kip1 levels in B cells, purified B cells were subjected to western blotting. P27kip1 expression in both LMP2A/λ-MYC and λ-MYC pretumor B cells was significantly lower than that in WT (Figure 2B-C), similar to our previous report.34 Next, we examined MYC expression to delineate the role of LMP2A on p27kip1 in MYC-mediated proliferation. MYC levels are highest in LMP2A/λ-MYC B cells (Figure 2B,D). Knockout of Cks1 rescued p27kip1 levels in both LMP2A/λ-MYC and λ-MYC close to WT levels and suppressed MYC expression. These data indicate that LMP2A enhances MYC expression and downregulates p27kip1 via a Cks1-dependent mechanism. In turn, the increased p27kip1 levels in Cks1−/− backgrounds can negatively regulate MYC levels.39
Figure 2.

Loss of Cks1 restores p27kipexpression in pretumor spleens of LMP2A/λ-MYC and λ-MYC mice. (A) Representative immunohistochemical analysis of spleens from 4-week-old mice stained with p27kip1 (original magnification ×40). Representative B- and T-cell areas in white pulps are shown from a total of 3 mice of each genotypes. (B) Representative immunoblots of p27kip1 and MYC expression in purified splenic B cells from 4-week-old mice. (C) P27kip1 and (D) MYC expression were normalized to Gapdh (a loading control). The statistical differences of Cks1 knockouts against their controls are indicated with thick lines. Data represent the mean ± standard deviation. **P < .01, ****P < .0001, calculated by 1-way ANOVA with post hoc multiple comparison tests.
LMP2A enhances MYC-induced cell cycle progression via Cks1
We previously demonstrated that LMP2A cooperates with MYC to promote G1-S transition of pretumor B cells.34 Homozygous, but not heterozygous, knockout of Cks1 significantly increased G1-phase and decreased S-phase B cells in the LMP2A/λ-MYC background (Figure 3A-B). Although Cks1 deficiency also led to a decrease in S-phase B cells in λ-MYC mice, the effect was not statistically significant and was milder compared with that in LMP2A/λ-MYC B cells. Consistent with the cell cycle analyses, Cks1 knockout significantly reduced the percentage of Ki67+ B cells in both LMP2A/λ-MYC and λ-MYC mice, although the reduction of Ki67+ B cells was more pronounced in the LMP2A/λ-MYC background (Figure 3C). This finding suggests that the loss of Cks1 reduces the number of B cells in the proliferative phases of the cell cycle and increases cell cycle exit. Taken together, our data indicate that LMP2A cooperates with MYC to downregulate p27kip1 and enhance B-cell hyperproliferation via Cks1.
Figure 3.

Cks1 deficiency inhibits pretumor B-cell proliferation in LMP2A/λ-MYC mice. The effects of Cks1 status on the cell cycle phases of purified splenic pretumor B cells from 4-week-old mice are indicated in (A) pie charts and (B) column graphs. (C) The percentages of Ki-67–positive cells in purified splenic pretumor B cells. Data were calculated by 1-way ANOVA with post hoc multiple comparison tests. The statistical differences of Cks1 knockouts against their controls are indicated with thick lines. Data represent the mean ± standard deviation. *P < .05, **P < .01, ***P < .001, and ****P < .0001.
LMP2A increases Cks1-mediated p27kip1 proteolysis
We previously showed that LMP2A expression enhances MYC-dependent proteasomal degradation of p27kip1.34 To examine the role of Cks1 on the p27kip1 turnover rate in our model, purified pretumor B cells were treated with cycloheximide (CHX), a protein translational inhibitor, for 3 hours (Figure 4A). The fold reduction of p27kip1 was delayed in Cks1−/− B cells compared with those with Cks1+/+ in the LMP2A/λ-MYC background, but not in the λ-MYC background (Figure 4B, filled circles and filled squares). This result suggests that Cks1 strongly contributes to the p27kip1 degradation process in LMP2A/λ-MYC B cells compared with λ-MYC alone. In addition, it is likely that there is another Cks1-independent pathway regulating the turnover of p27kip1 in both LMP2A/λ-MYC/Cks1−/− and λ-MYC/Cks1−/− B cells, because these B cells showed a considerable decrease in p27kip1 levels 3 hours after CHX treatment (Figure 4B, filled squares).
Figure 4.

Loss of Cks1 stabilizes p27kip1in a proteasome-dependent manner. (A) Representative immunoblot analyses of p27kip1 expression in purified splenic pretumor B cells from 4-week-old mice after treatment with 25 μg/mL CHX with or without 40 μM MG-132 (MG) at indicated time points at 37°C. (B) The fold reduction of p27kip1 after CHX treatment was normalized to calnexin (a loading control), and the fold reduction of p27kip1 was calculated using cells at 0 hour of each genotype as the control. Combined data from 3 to 4 different mice represent the mean ± standard deviation. **P < .01, ****P < .0001 were calculated by 2-way ANOVA with multiple comparison tests.
To examine the proteasome-mediated proteolysis of p27kip1 over time, we treated pretumor B cells with both cycloheximide and MG-132, a proteasome inhibitor. The addition of MG-132 significantly prolonged the p27kip1 turnover rate in both LMP2A/λ-MYC and λ-MYC B cells compared with CHX treatment alone (Figure 4B, filled circles and open circles), indicating that p27kip1 degradation is proteasome-dependent, similar to our previous results.34 Interestingly, despite the loss of Cks1, the addition of MG-132 significantly increased the stability of p27kip1 in both LMP2A/λ-MYC/Cks1−/− and λ-MYC/Cks1−/− B cells compared with CHX treatment alone. This finding suggests that the Cks1-independent mechanism also uses the proteasome to degrade p27kip1 (Figure 4B, filled squares and open squares). Because the amount of p27kip1 at time 0 was higher in Cks1-deficient B cells, it is possible that the delay in p27kip1 turnover in these cells may rescue p27kip1 to WT levels (Figure 2A) and may result in the hindrance of cell cycle progression over time (Figure 3A). Overall, these results indicate that Cks1 is important for LMP2A-promoted p27kip1 degradation.
LMP2A accelerates MYC-driven lymphomagenesis through Cks1
A previous study showed that human BL cells have elevated Cks1 levels compared with normal B cells. In the Eµ-Myc model of lymphoma, overexpression of Myc increases Cks1 expression, whereas the loss of Cks1 significantly prolongs Myc-induced lymphoma development.31 Therefore, we examined the effect of Cks1 loss on tumor-free survival (Figure 5). λ-MYC/Cks1−/− mice (median survival time, 419 days) had a significant increase in tumor-free survival compared with λ-MYC mice (median survival time, 133.5 days), confirming the role of Cks1 as a MYC target. By depleting Cks1 to block p27kip1 degradation, we found a significant delay in tumor onset in LMP2A/λ-MYC/Cks1−/− mice (median survival time, 108.5 days) compared with LMP2A/λ-MYC mice (median survival time, 47 days). The delayed lymphoma development is in line with the results showing increased p27kip1 levels (Figure 2A) and prolonged p27kip1 turnover rate (Figure 4) at the pretumor stage of LMP2A/λ-MYC/Cks1−/− compared with LMP2A/λ-MYC B cells. However, delayed tumor onset in these mice is partial, and loss of Cks1 did not fully rescue the tumor onset of these mice to that of λ-MYC mice (Figure 5). Heterozygous knockout of Cks1 did not change tumor-free survival in LMP2A/λ-MYC mice, but significantly delayed lymphoma development in λ-MYC mice. This finding indicates that there are other pathways independent of Cks1 that LMP2A uses to promote lymphomagenesis.
Figure 5.

Cks1 knockout results in the delay of tumor onset in both LMP2A/λ-MYC and λ-MYC mice. Kaplan-Meier curves indicating the percentage of tumor-free survival of indicated genotypes. P values were calculated by comparing 2 genotypes by log-rank (Mantel-Cox) test.
Independent of when tumors arose in the genotypes analyzed, we found that tumors appeared histologically similar. Tumor-bearing lymph nodes were virtually all of B-cell lineage because they were almost all positive for B220 staining (Figure 6A). All LMP2A/λ-MYC tumors expressed very low levels of p27kip1, whereas λ-MYC tumors showed weak p27kip1 expression. Cks1−/− tumors exhibited high p27kip1 expression in both LMP2A/λ-MYC and λ-MYC groups with intense p27kip1 staining in the nucleus (Figure 6A,C-D). Regardless of LMP2A status, the percentage of cells that stained positive for Thr187-phosphorylated p27kip1 was increased in Cks1-deficient tumors (Figure 6A-B), possibly because p27kip1 is not directed to the proteasome. Despite increased p27kip1 levels in Cks1−/− tumors and a significant reduction of MYC at the pretumor stage (Figure 2B), MYC levels were as high as those in Cks1+/+ tumors (Figure 6C,E). These data suggest that other mutations or cellular changes likely occur at the later stages of malignant transformation to overcome increased p27kip1 levels and upregulate MYC in Cks1-knockout animals. In addition, downregulation of p27kip1, especially at the pretumor stage, is an important step in MYC-driven tumorigenesis.
Figure 6.

P27kip1levels remain high as observed in pretumor cells of tumors from Cks1 knockout LMP2A/λ-MYC and λ-MYC mice. (A) Representative immunohistochemical analysis of lymph node tumors from 3 to 6 mice of each genotype stained with hematoxylin and eosin, B220, p27kip1, and Thr187-phosphorylated p27kip1, using an EVOS XL core microscope with ×40 magnification. Insets are tumors imaged by a Nikon Eclipse E600 microscope with ×100 magnification and oil immersion. (B) Percentage of lymphoma cells with intense nuclear staining of Thr187-phosphorylated p27kip1 in total lymphoma cells per microscopic field (×40 magnification) analyzed by ImageJ software (National Institutes of Health). (C) Immunoblots of p27kip1 and MYC expression in tumor cells. (D) P27kip1 and (E) MYC expression were normalized to Gapdh (a loading control). Data represent the mean ± standard deviation. ***P < .001, ****P < .0001, calculated by 1-way ANOVA with post hoc multiple comparison tests.
LMP2A allows lymphoma cells to bypass p53-mediated apoptosis regardless of Cks1 status
Increased expression of MYC induces cell cycle progression but also activates apoptosis via the p53 pathway. Thus, mutating or inhibiting p53 pathway function is a major mechanism for dysregulated MYC B cells to establish malignancy.16,24 Aberrant accumulation or stabilization of p19ARF and/or p53 on western blots has been used as a readout for inactivation of the p53 pathway due to the disruption of negative feedback mechanisms.16,18-20,24 We examined whether increased p27kip1 expression and the loss of Cks1 would change the status of the p53 pathway and found that very few tumors from LMP2A/λ-MYC and LMP2A/λ-MYC/Cks1−/− mice showed abnormal expression of p53 and p19ARF. However, the aberrant statuses of p53 and p19ARF (abnormal accumulation and/or undetected levels) were more frequent in λ-MYC mice than in LMP2A/λ-MYC mice (Figure 7A-B), which is consistent with a previous study.24 Knockout of Cks1 did not change the frequency of p53 and p19ARF abnormalities on western blot analyses in both λ-MYC and LMP2A/λ-MYC groups (Figure 7B; supplemental Table 1, available on the Blood Web site).
Figure 7.

Cks1 knockout does not change the occurrence of p53 and/or p19ARFabnormalities in LMP2A/λ-MYC and λ-MYC tumors. The status of p53 and p19ARF expression in lymph node tumors from the indicated mouse genotype. (A) Representative immunoblots of p53 and p19ARF. (B) Combined data from several independent experiments. Statistical analyses were performed with Fisher’s exact test by comparing the frequency of p53 and p19ARF abnormalities in Cks1+/+ vs Cks1−/− within the LMP2A/λ-MYC or λ-MYC background. p53+ and p19−, baseline p53 and p19ARF levels as indicated by immunoblots; p53−, undetected baseline level of p53; p53++, aberrant p53 accumulation at ∼53 kDa; p19+, abnormal accumulation of p19ARF. (C) The number of tumor-free days in each indicated genotype. Medians with 95% confident intervals of ratios are indicated. Survival analyses were performed with both the log-rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test by comparing the tumor-free days of the subgroups with (+) vs without (–) p53 and p19ARF abnormalities for each genotype. NS, not significant.
When stratifying the numbers of tumor-free days by the presence or absence of p53 and/or p19ARF abnormalities, there was no significant increase in tumor-free survival of abnormal p53 and/or p19ARF groups compared with their normal counterparts within each genotype (Figure 7B), suggesting that p53 pathway inactivation is a random process. Despite LMP2A/λ-MYC/Cks1−/− mice showing prolonged tumor onset compared with LMP2A/λ-MYC mice, Cks1 deficiency did not change p53 pathway status in our analysis, similar to a previous report.31 These results suggest that LMP2A is still able to allow B cells to bypass p53 pathway inactivation during lymphomagenesis. Taken together, our data indicate that LMP2A enhances MYC-mediated p27kip1 degradation partially through the Cks1 adaptor protein and that increased p27kip1 levels in LMP2A/λ-MYC/Cks1−/− mice in the pretumor stage prolongs lymphoma onset without significantly changing p53 pathway status.
Discussion
The causative roles of EBV latent antigens like LMP2A in EBV-associated cancers have been highly studied. Studies from our group and others using transgenic mouse models demonstrate the roles of LMP2A in cancer development in combination with dysregulated MYC,24,36 mutated Ras,40 and EBV LMP1.11,41 These studies indicate that LMP2A takes advantage of different pathological changes inside cells to promote malignancy. Transgenic models have proven valuable in establishing causal roles of LMP2A in tumorigenesis. In this study, we demonstrated a novel role of LMP2A in the modulation of the cell cycle to promote lymphoma development.
Our previous studies demonstrated the cooperative roles of LMP2A and MYC in lymphomagenesis.24,34,35,36 The data presented in this study highlight the role of LMP2A in tipping the equilibrium between proliferation and apoptosis in the context of MYC overexpression. Our results, at least in part, recapitulated a correlation of LMP2A expression with the enrichment of genes involved in the G1-S transition in endemic BL.15 The roles of Cks1 in the cell cycle and protein regulation of p27kip1 were demonstrated previously. Cks1−/− mice show increased p27kip1 levels in tissues, whereas p27−/− mice exhibited the opposite phenotypes.29,42 Furthermore, p27kip1 is a critical checkpoint regulator that harnesses cell cycle progression induced by MYC, because the knockout of p27kip1 accelerates lymphoma in the Eμ-Myc model.43 On the other hand, Cks1 knockout in Eμ-Myc31 as well as in λ-MYC mice in this study delayed tumor onset. Our results demonstrate for the first time that blocking the role of LMP2A in cell cycle progression can significantly impede tumor development (Figure 5), partially through stabilization of p27kip1 via Cks1. These data also underscore that the disruption of cell cycle regulation is an important feature for MYC- and LMP2A-mediated lymphoma development.
During EBV infection and transformation of primary human B cells, EBV products, such as EBNA2,44,45 EBNALP,46 EBNA347,48 and LMP1,49 contribute to MYC upregulation and cell cycle progression. It is possible that LMP2A acts in concert with these EBV proteins to use the MYC-Cks1-p27kip1 axis to promote B-cell proliferation, because LMP2A loss delayed EBV-mediated B-cell proliferation during early infection and long-term outgrowth.50 BCR signaling is important for the survival of EBV-transformed B cells,51 and BCRs may modulate Cks1 and p27kip1 similarly to LMP2A. Furthermore, LMP2A is necessary for EBV transformation of human B cells in the absence of BCRs,5 indicating the critical ability of LMP2A in mimicking the BCR signal. Our model suggests that LMP2A accelerates tumor onset through BCR-like signals because Lyn and mTOR inhibitors block LMP2A/λ-MYC tumor growth.52,53 This study revealed another point of intervention: blocking a cell cycle regulator, such as Cks1, can prevent LMP2A function in lymphomagenesis. However, the detailed mechanisms of cell cycle regulation by LMP2A are still unclear and warrant further study.
P27kip1 levels from λ-MYC/Cks1−/− and LMP2A/λ-MYC/Cks1−/− mice remained high from premalignant (Figure 2) through malignant stages (Figure 6), whereas MYC levels were low in the pretumor stage, but were as high as those of Cks1+/+ counterparts in the tumor stage. These data suggest that B cells in these mice developed other cellular changes to overcome WT-level p27kip1 and regain MYC levels to establish malignancy. It is unknown whether B cells from LMP2A/λ-MYC/Cks1−/− mice acquire the same or unique changes compared with λ-MYC/Cks1−/− mice. We examined the p53 pathway to explore whether high p27kip1 levels would alter p53 pathway status. Consistent with the results from Keller and colleagues, loss of Cks1 or an increase in p27kip1 did not change p53 pathway abnormalities.31 Although p53 and p27kip1 are both evidently rate-limiting factors in MYC-induced lymphomagenesis, the downmodulations of these 2 tumor suppressors may occur independently. Nevertheless, in LMP2A-expressing tumors, inactivation of the p53 pathway is rare. Taken together, our results are the first to show that LMP2A uses a MYC target, Cks1, to downregulate p27kip1 and accelerate lymphomagenesis without changing apoptosis processes.
Although we showed that loss of Cks1 impedes LMP2A-mediated lymphoma development (Figure 6), Cks1 is likely not a direct LMP2A signaling constituent, and the process LMP2A exploits to downregulate p27kip1 via Cks1 is MYC dependent. LMP2A alone does not drastically affect p27kip1 levels or the cell cycle profile in B cells.34 Combined with dysregulated MYC, LMP2A promotes MYC-mediated p27kip1 degradation (Figure 3). For B cells in which LMP2A almost entirely replaces BCR expression, knockout of proximal BCR effectors, including Lyn,9 Btk,54 SLP-65 (BLNK),55 or tyrosine mutation of LMP2A ITAM,8 restores BCR expression because LMP2A function is lost and these cells need the BCR signal to survive. Knockout of Cks1 in LMP2A/λ-MYC mice did not significantly restore surface IgM expression (Figure 1C), suggesting that Cks1 is a proximal target of MYC rather than LMP2A. Although pretumor B cells of LMP2A/λ-MYC/Cks1−/− and λ-MYC/Cks1−/− mice showed similar p27kip1 levels (Figure 2) and cell cycle distributions (Figure 3), the rate of tumor onset in LMP2A/λ-MYC/Cks1−/− mice was much more rapid than that in λ-MYC and λ-MYC/Cks1−/− mice (Figure 5). These results indicate that although LMP2A replaces the BCR requirement in LMP2A/λ-MYC/Cks1−/− B cells, LMP2A may have distinct functions compared with host BCRs.
Our results shed light on the unique pathological pathways mediated by EBV infection or LMP2A expression. The roles of BCRs in B-cell lymphomas have gained attention in recent years, because these malignant cells still maintain BCR expression.12,56 This study revealed the possibility that LMP2A may provide unique reprogramming and signal transduction in addition to mimicking the BCR signal. Likewise, inhibiting MYC or its targets is also an appealing approach to cancer treatment because elevated MYC levels are found in many cancers.57 However, our results showed that blocking one of the MYC targets, Cks1, only partially rescued LMP2A-mediated tumor development and still did not fully phenocopy the effect seen in λ-MYC mice. A more comprehensive study of LMP2A signals would reveal novel ways to specifically treat EBV-associated lymphoma. In summary, we provided mechanistic evidence that EBV LMP2A enhances MYC-dependent p27kip1 degradation, partially through Cks1, to accelerate hyperproliferation of B cells and lymphoma development. Our study highlights a role of EBV infection or LMP2A expression in conferring distinct contributions to the pathogenesis of lymphoma.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank members of the Longnecker laboratory for help in the completion of this study and the Steven Reed laboratory for the Cks1−/− mice. Histology services were provided by the Northwestern University Mouse Histology and Phenotyping Laboratory, which is supported by NCI P30-CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center.
This work was supported by the Northwestern University Interdepartmental Immunobiology Flow Cytometry Core Facility, National Institutes of Health, National Cancer Institute grant R01 CA073507 (R.L.), Training Program in Viral Replication grant T32AI060523 (K.F.), and Carcinogenesis Training Program grant T32CA009560 (R.P.S.). R.L. is a Dan and Bertha Spear Research Professor.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.F., R.L., and M.I. conceived the study and wrote the manuscript; M.I. supervised and performed the experiments with K.F., R.P.S., and S.J.S.; and M.I. and K.F. analyzed the experimental data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Richard Longnecker, Department of Microbiology and Immunology, Northwestern University Feinberg School of Medicine, 303 E. Chicago Ave, Ward Building 6-255, Chicago, IL 60611; e-mail: r-longnecker@northwestern.edu.
References
- 1.Longnecker R, Kieff E, Cohen JI. Epstein-Barr Virus. In: Knipe DM, Howley PM, eds. Fields Virology. 6th ed Philadelphia, PA: Lippincott Williams & Wilkins; 2013:1898-1959 [Google Scholar]
- 2.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet. 1964;283(7335):702-703. [DOI] [PubMed] [Google Scholar]
- 3.Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9(3):405-411. [DOI] [PubMed] [Google Scholar]
- 4.Casola S, Otipoby KL, Alimzhanov M, et al. . B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5(3):317-327. [DOI] [PubMed] [Google Scholar]
- 5.Mancao C, Hammerschmidt W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood. 2007;110(10):3715-3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fruehling S, Longnecker R. The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction. Virology. 1997;235(2):241-251. [DOI] [PubMed] [Google Scholar]
- 7.Fruehling S, Swart R, Dolwick KM, Kremmer E, Longnecker R. Tyrosine 112 of latent membrane protein 2A is essential for protein tyrosine kinase loading and regulation of Epstein-Barr virus latency. J Virol. 1998;72(10):7796-7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merchant M, Caldwell RG, Longnecker R. The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo. J Virol. 2000;74(19):9115-9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rovedo M, Longnecker R. Epstein-Barr virus latent membrane protein 2A preferentially signals through the Src family kinase Lyn. J Virol. 2008;82(17):8520-8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swart R, Ruf IK, Sample J, Longnecker R. Latent membrane protein 2A-mediated effects on the phosphatidylinositol 3-Kinase/Akt pathway. J Virol. 2000;74(22):10838-10845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wirtz T, Weber T, Kracker S, Sommermann T, Rajewsky K, Yasuda T. Mouse model for acute Epstein-Barr virus infection. Proc Natl Acad Sci USA. 2016;113(48):13821-13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sander S, Calado DP, Srinivasan L, et al. . Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22(2):167-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitz R, Young RM, Ceribelli M, et al. . Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abate F, Ambrosio MR, Mundo L, et al. . Distinct viral and mutational spectrum of endemic Burkitt lymphoma. PLoS Pathog. 2015;11(10):e1005158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13(20):2658-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999;13(20):2670-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eischen CM, Roussel MF, Korsmeyer SJ, Cleveland JL. Bax loss impairs Myc-induced apoptosis and circumvents the selection of p53 mutations during Myc-mediated lymphomagenesis. Mol Cell Biol. 2001;21(22):7653-7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA. 2004;101(16):6164-6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garrison SP, Jeffers JR, Yang C, et al. . Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. 2008;28(17):5391-5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Refaeli Y, Young RM, Turner BC, Duda J, Field KA, Bishop JM. The B cell antigen receptor and overexpression of MYC can cooperate in the genesis of B cell lymphomas. PLoS Biol. 2008;6(6):e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corso J, Pan KT, Walter R, et al. . Elucidation of tonic and activated B-cell receptor signaling in Burkitt’s lymphoma provides insights into regulation of cell survival. Proc Natl Acad Sci USA. 2016;113(20):5688-5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varano G, Raffel S, Sormani M, et al. . The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3β inhibition. Nature. 2017;546(7657):302-306. [DOI] [PubMed] [Google Scholar]
- 24.Bieging KT, Amick AC, Longnecker R. Epstein-Barr virus LMP2A bypasses p53 inactivation in a MYC model of lymphomagenesis. Proc Natl Acad Sci USA. 2009;106(42):17945-17950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8(4):253-267. [DOI] [PubMed] [Google Scholar]
- 26.Kamura T, Hara T, Matsumoto M, et al. . Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6(12):1229-1235. [DOI] [PubMed] [Google Scholar]
- 27.Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277(17):14355-14358. [DOI] [PubMed] [Google Scholar]
- 28.Ganoth D, Bornstein G, Ko TK, et al. . The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;3(3):321-324. [DOI] [PubMed] [Google Scholar]
- 29.Spruck C, Strohmaier H, Watson M, et al. . A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell. 2001;7(3):639-650. [DOI] [PubMed] [Google Scholar]
- 30.Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20(1):47-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller UB, Old JB, Dorsey FC, et al. . Myc targets Cks1 to provoke the suppression of p27Kip1, proliferation and lymphomagenesis. EMBO J. 2007;26(10):2562-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caldwell RG, Brown RC, Longnecker R. Epstein-Barr virus LMP2A-induced B-cell survival in two unique classes of EmuLMP2A transgenic mice. J Virol. 2000;74(3):1101-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovalchuk AL, Qi CF, Torrey TA, et al. . Burkitt lymphoma in the mouse. J Exp Med. 2000;192(8):1183-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fish K, Chen J, Longnecker R. Epstein-Barr virus latent membrane protein 2A enhances MYC-driven cell cycle progression in a mouse model of B lymphoma. Blood. 2014;123(4):530-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bieging KT, Fish K, Bondada S, Longnecker R. A shared gene expression signature in mouse models of EBV-associated and non-EBV-associated Burkitt lymphoma. Blood. 2011;118(26):6849-6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bultema R, Longnecker R, Swanson-Mungerson M. Epstein-Barr virus LMP2A accelerates MYC-induced lymphomagenesis. Oncogene. 2009;28(11):1471-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams JM, Harris AW, Pinkert CA, et al. . The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318(6046):533-538. [DOI] [PubMed] [Google Scholar]
- 38.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in E-mu-myc transgenic mice. Cell. 1986;47(1):11-18. [DOI] [PubMed] [Google Scholar]
- 39.Bahram F, Hydbring P, Tronnersjö S, et al. . Interferon-γ-induced p27KIP1 binds to and targets MYC for proteasome-mediated degradation. Oncotarget. 2016;7(3):2837-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shair KH, Bendt KM, Edwards RH, Nielsen JN, Moore DT, Raab-Traub N. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) and LMP2A function cooperatively to promote carcinoma development in a mouse carcinogenesis model. J Virol. 2012;86(9):5352-5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minamitani T, Ma Y, Zhou H, et al. . Mouse model of Epstein-Barr virus LMP1- and LMP2A-driven germinal center B-cell lymphoproliferative disease. Proc Natl Acad Sci USA. 2017;114(18):4751-4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiyokawa H, Kineman RD, Manova-Todorova KO, et al. . Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell. 1996;85(5):721-732. [DOI] [PubMed] [Google Scholar]
- 43.Martins CP, Berns A. Loss of p27(Kip1) but not p21(Cip1) decreases survival and synergizes with MYC in murine lymphomagenesis. EMBO J. 2002;21(14):3739-3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang J, Zhou H, Gerdt C, et al. . Epstein-Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc Natl Acad Sci USA. 2016;113(49):14121-14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wood CD, Veenstra H, Khasnis S, et al. . MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife. 2016;5: e18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Portal D, Zhou H, Zhao B, et al. . Epstein-Barr virus nuclear antigen leader protein localizes to promoters and enhancers with cell transcription factors and EBNA2. Proc Natl Acad Sci USA. 2013;110(46):18537-18542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooper A, Johannsen E, Maruo S, et al. . EBNA3A association with RBP-Jkappa down-regulates c-myc and Epstein-Barr virus-transformed lymphoblast growth. J Virol. 2003;77(2):999-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt SC, Jiang S, Zhou H, et al. . Epstein-Barr virus nuclear antigen 3A partially coincides with EBNA3C genome-wide and is tethered to DNA through BATF complexes. Proc Natl Acad Sci USA. 2015;112(2):554-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dirmeier U, Hoffmann R, Kilger E, et al. . Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene. 2005;24(10):1711-1717. [DOI] [PubMed] [Google Scholar]
- 50.Wasil LR, Tomaszewski MJ, Hoji A, Rowe DT. The effect of Epstein-Barr virus Latent Membrane Protein 2 expression on the kinetics of early B cell infection. PLoS One. 2013;8(1):e54010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma Y, Walsh MJ, Bernhardt K, et al. . CRISPR/Cas9 screens reveal Epstein-Barr virus-transformed B cell host dependency factors. Cell Host Microbe. 2017;21(5):580-591 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dargart JL, Fish K, Gordon LI, Longnecker R, Cen O. Dasatinib therapy results in decreased B cell proliferation, splenomegaly, and tumor growth in a murine model of lymphoma expressing Myc and Epstein-Barr virus LMP2A. Antiviral Res. 2012;95(1):49-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cen O, Longnecker R. Rapamycin reverses splenomegaly and inhibits tumor development in a transgenic model of Epstein-Barr virus-related Burkitt’s lymphoma. Mol Cancer Ther. 2011;10(4):679-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merchant M, Longnecker R. LMP2A survival and developmental signals are transmitted through Btk-dependent and Btk-independent pathways. Virology. 2001;291(1):46-54. [DOI] [PubMed] [Google Scholar]
- 55.Engels N, Merchant M, Pappu R, Chan AC, Longnecker R, Wienands J. Epstein-Barr virus latent membrane protein 2A (LMP2A) employs the SLP-65 signaling module. J Exp Med. 2001;194(3):255-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Küppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med. 1999;341(20):1520-1529. [DOI] [PubMed] [Google Scholar]
- 57.McKeown MR, Bradner JE. Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med. 2014;4(10):a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.