

Abstract
Evolutionary studies of DNA methylation offer insights into the mechanisms governing the variation of genomic DNA methylation across different species. Comparisons of gross levels of DNA methylation between distantly related species indicate that the size of the genome and the level of genomic DNA methylation are positively correlated. In plant genomes, this can be reliably explained by the genomic contents of repetitive sequences. In animal genomes, the role of repetitive sequences on genomic DNA methylation is less clear. On a shorter timescale, population-level comparisons demonstrate that genetic variation can explain the observed variability of DNA methylation to some degree. The amount of DNA methylation variation that has been attributed to genetic variation in the human population studies so far is substantially lower than that from Arabidopsis population studies, but this disparity might reflect the differences in the computational and experimental techniques used. The effect of genetic variation on DNA methylation has been directly examined in mammalian systems, revealing several causative factors that govern DNA methylation. On the other hand, studies from Arabidopsis have furthered our understanding of spontaneous mutations of DNA methylation, termed “epimutations.” Arabidopsis has an extremely high rate of spontaneous epimutations, which may play a major role in shaping the global DNA methylation landscape in this genome. Key missing information includes the frequencies of spontaneous epimutations in other lineages, in particular animal genomes, and how population-level variation of DNA methylation leads to species-level differences.
Keywords: DNA methylation, epimutation, whole-genome bisulfite sequencing, gene body methylation, CpG islands, differentially methylated regions, mQTLs
Introduction: Evolution of Epigenetics
Epigenetic modifications are essential chemical modifications that affect the packaging of genomic DNA in the nucleus of each cell. As such, epigenetic modifications influence how cell lineages are defined during developmental processes. One of the earliest uses of the term “epigenetics” was by Waddington (Waddington 1957) to depict how totipotent cells in early development differentiate into more specialized cell lineages, a concept termed “epigenetic landscape” (box 1). The molecular mechanisms of the epigenetic landscape remained largely unknown for many decades, but they are now being uncovered at an unprecedented rate. Epigenetics is an especially active area in current research.
Box 1.
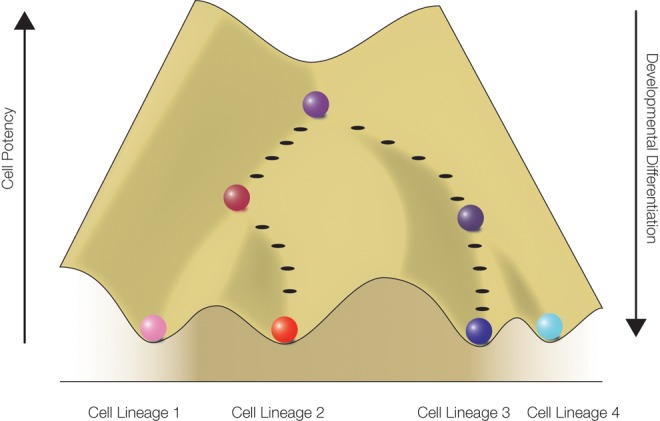
The classic figure that appeared in Waddington (1957) depicted a ball sitting on top of a landscape consisting of several “valleys,” similar to the landscape redrawn here. In Waddington’s figure, the ball depicts an immature cell, and when it has made its descent downward to any of the several valleys to reach the bottom, it would have become a terminally differentiated cell. In the current figure, the landscape visualizes different epigenetic modifications occurring in different cell lineages. The epigenetic landscape implicitly assumes that these modifications occur hierarchically so that the cell gradually loses its potency and becomes more developmentally specialized as it navigates different epigenetic paths to reach distinctive cellular lineages.
Specifically, epigenetics investigates molecular alterations in chromatin (fig. 1). The genomic DNA in each cell is elaborately packaged as chromatins in the nucleus, allowing for the activation or repression of specific regions of the DNA, which ultimately determines cellular identities. The basic unit of this packaging is the nucleosome, which consists of ∼150 bp of genomic DNA wrapped around eight histone “cores.” The major components of the nucleosome, the genomic DNA template and the histone cores, harbor extensive and distinctive chemical modifications.
Fig. 1.—

Nucleosomes are basic units of chromatin. Both the DNA templates and the histone cores of the nucleosomes exhibit extensive chemical modifications. Some of the modifications observed in histone tails are shown, including acetylation (Ac), methylation (Me), phosphorylation (Ph), and Ubiquitination (Ub). These modifications can occur in many different amino acids of the tail, and the total combinatorial space of the possible modifications is extremely large, of which only a very small portion is currently known. Genomic DNA template is often modified by methylation. Three types of DNA methylation are commonly observed in nature: methylation of the fifth position of the cytosine base (C5 methylation), the fourth position of cytosine (C4 methylation), and the sixth position of adenine (N6 methylation).
The genomic DNA template is often modified by the addition of a methyl (-CH3) group, which is referred to as “DNA methylation.” In eukaryotes, methylation of the fifth position of the cytosine base (C5 methylation) is the major mode of DNA modification, but methylation of the sixth position of adenine (N6 methylation) has also been reported in some species (Dabe et al. 2015; Greer et al. 2015). In some prokaryotes, methylation of the fourth position of cytosine (C4 methylation), in addition to C5 and N6 methylation, has been observed (Ouellette et al. 2015; Blow et al. 2016). Among these different types of DNA methylation, eukaryotic C5 methylation is the best understood. Many aspects of the molecular mechanics and regulators of eukaryotic C5 methylation have been identified (Allis and Jenuwein 2016). Unless otherwise defined, DNA methylation typically refers to C5 methylation. This article will focus on C5 methylation and will refer to it simply as DNA methylation henceforth.
DNA methylation occurs in different nucleotide contexts in animal and plant genomes, in that cytosines in CpG contexts are major targets of DNA methylation in animal genomes, whereas, in plants, Cs in all nucleotide contexts are subject to methylation. Recently, several oxidated forms of 5-methyl-cytosines have been also discovered, including 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). The abundance and functional roles of these derivatives, which are currently less understood, are gaining much attention (Jeschke et al. 2016; Lunnon et al. 2016; Cimmino and Aifantis 2017; Wu and Zhang 2017).
Histone cores of nucleosomes also bear diverse epigenetic modifications. Each of the eight histone cores has “tails” consisting of dozens of amino acids, which are often chemically modified via dedicated enzymes (fig. 1). For example, the tails are particularly enriched with positively charged lysine (K) residues, which are frequently found methylated. Functions of some histone modifications, particularly those involving the methylation of lysine residues, have been extensively studied. For instance, the trimethylation of H3 lysine 4, often written simply as H3K4me3, is associated with an active transcription start site and is considered to be a marker of promoters. On the other hand, the trimethylation of the H3 lysine 27 (H3K27me3) is considered to be an indicator of repression of gene expression. However, the total diversity and complexity of histone modifications are staggering: both the target residues and the possible modifications range widely. To date, more than 100 distinctive histone tail modifications have been discovered, and this number is likely to continue to increase for some time. Our understanding of the function of the majority of histone tail modifications is still in its infancy.
As the knowledge of epigenetic modifications and their regulatory roles accumulates, questions on how epigenetic components evolve, and how they affect evolution, are also gaining much interest. Among the epigenetic components, DNA methylation is the most extensively characterized and understood. During the last decade, DNA methylation research has experienced a particularly pronounced growth thanks to the developments of powerful techniques that measure DNA methylation of nearly every nucleotide in the genome with high precision (table 1). These techniques also facilitated analyses of DNA methylation in nonmodel species. As a result, the phylogenetic scopes of DNA methylation have dramatically expanded during the past decade. In this article, we will discuss newly gained evolutionary insights from studies of DNA methylation from diverse taxa. Even though we will only discuss findings from eukaryotic DNA methylation, it is worth noting that epigenomic components have deep evolutionary origins (box 2).
Table 1.
Commonly Used Methods to Characterize DNA Methylation
| Technique | Mechanism | Genomic Targets | Advantages | Disadvantages |
|---|---|---|---|---|
| Methylation array (e.g., Illumina Infinium Methylation Assay) |
|
Preselected CpG sites via the source company |
|
|
| Methylated DNA immunoprecipitation chip (MeDIP-chip) |
|
Genomic fragments with substantial DNA methylation |
|
|
| Methylated DNA immunoprecipitation sequencing (MeDIP-seq) |
|
Genomic fragments with substantial DNA methylation |
|
|
| CXXC affinity purification (CAP)-seq |
|
Genomic fragments devoid of DNA methylation |
|
|
| Reduced representation Bisulfite Sequencing (RRBS) |
|
Genomic fragments within pairs of certain recognition sites |
|
|
| Whole genome bisulfite sequencing (WGBS) |
|
All nucleotides |
|
|
Box 2.
Major components of the epigenome have deep evolutionary origins. DNA methylation, and the proteins that perform DNA methylation, are present in all three domains of life. In prokaryotes, DNA methylation is considered a part of the restriction modification system (Roberts et al. 2015), which is involved in defense against viral and other infectious agents. However, DNA methylation is widespread in prokaryotic and archaeal lineages, often independent of the restriction modification system, suggesting that there exists a yet uncharacterized potential regulatory role of DNA methylation in these domains (Blow et al. 2016). Nucleosomes and chemical modifications of nucleosome proteins are also not limited to eukaryotes. Archaeal species have primitive nucleosomes that consist of two of the four core eukaryotic histones (Reeve et al. 1997; Slesarev et al. 1998; Sandman and Reeve 2006). Genes performing modifications to histone tails that are critical to the regulation of gene expression, such as histone acetylase and deacetylases, are also found in Archaea (Gregoretti et al. 2004; Marsh et al. 2005). Furthermore, proteins that bind to DNA and share some characteristics of eukaryotic histones are also present in the genomes of some prokaryotes and are referred to as “histone-like proteins” (Dorman and Deighan 2003). These observations indicate that packaging and interacting with genomic DNA is an essential cellular task in all three domains of life.
Evolutionary studies of DNA methylation in animals and plants have offered many interesting points of comparison, providing complementary insights into the evolution of DNA methylation. For example, many resources for studying epigenetic variation are available for the model plant Arabidopsis thaliana, including numerous mutation accumulation lines with fully characterized methylomes. Consequently, A. thaliana has been extremely useful in understanding how spontaneous epigenetic mutations arise and are inherited. On the other hand, human and mouse genomes have been extensively used in functional studies, offering rich experimental tools and conceptual frameworks to investigate detailed molecular mechanisms of DNA methylation. For example, a series of studies have pinpointed nucleotide-level drivers of DNA methylation using human and mouse systems. In the present article, we will discuss complementary findings from plants and animals to provide an integrative perspective on the evolution of DNA methylation at different timescales. We first review variation of DNA methylation across large-scale phylogenies in animals and plants. We then discuss the findings from population-level studies in two model species for epigenetic variation—namely, A. thaliana and humans. These studies offer insights into the impact of genetic contribution on epigenetic variation. We then discuss a series of studies furthering the mechanistic understanding of genetic determinants of DNA methylation in mammals. Finally, we discuss recent studies on the rate of spontaneous mutations of DNA methylation—termed epimutations—in A. thaliana, and their evolutionary nature.
Variation of DNA Methylation in Animal and Plant Genomes
Even though DNA methylation has long been recognized as a significant regulator of gene expression in animals, its roles in animal species other than humans and mice received relatively little attention until the past decade. This view was to a large degree reinforced by the lack of canonical DNA methylation in widely used lab animal species, namely, Drosophila melanogaster and Caenorhabditis elegans. However, interests in DNA methylation in nonmammalian animals were reignited when the honey bee genome consortium discovered a fully functional DNA methylation machinery in the honey bee genome (The Honeybee Genome Sequencing Consortium 2006; Wang et al. 2006). Following this inspiring discovery, the analysis of DNA methylation machineries has become a common task of new genome consortia, and discoveries of DNA methylation from many animal taxa followed, including those of basal invertebrate lineages such as sponges and ctenophores (Srivastava et al. 2010; Dabe et al. 2015). These studies have shown that DNA methylation is widespread among animal taxa and that it was lost in some lineages, including those of common model organisms.
In animal genomes, DNA methylation typically occurs at cytosines that are followed by guanines, or “CpGs.” Two commonly observed targets of DNA methylation in animal genomes are genes and repetitive sequences (such as transposable elements, or TEs). The DNA methylation of genes typically encompasses both exons and introns and is referred to as “gene body DNA methylation.” DNA methylation of repetitive sequences is not as widespread as gene body DNA methylation in animal genomes (see below). Genomic patterns of DNA methylation are highly divergent among animal taxa (fig. 2) (Simmen et al. 1999; Suzuki et al. 2007; Suzuki and Bird 2008; Lyko et al. 2010; Zemach et al. 2010; Sarda et al. 2012; Bewick et al. 2017). In the majority of invertebrate genomes, DNA methylation is observed in a “mosaic” pattern, where gene body methylation is the most prominent (Tweedie et al. 1997; Simmen et al. 1999; Suzuki et al. 2007; Suzuki and Bird 2008; Lyko et al. 2010; Hunt et al. 2013a). In a mosaically methylated genome, a subset of genes, typically those that are evolutionarily conserved and constitutively expressed, are methylated (Suzuki et al. 2007; Elango et al. 2009; Zeng and Yi 2010; Sarda et al. 2012; Gavery and Roberts 2013; Hunt et al. 2013b), whereas the rest of the genes and other genomic regions are devoid of DNA methylation. Interestingly, the methylation of repetitive sequences, such as TEs, is absent or limited to specific classes of TEs in invertebrates (Simmen et al. 1999; Lyko et al. 2010; Keller et al. 2016a). Promoter methylation is found in some invertebrates but is not extensive (Saint-Carlier and Riviere 2015; Keller et al. 2016a). The genomic DNA methylation patterns of vertebrates are unique in that they exhibit “global” DNA methylation, where nearly all CpGs in the genome are methylated (Suzuki and Bird 2008). The transition from mosaic to global DNA methylation occurred during early vertebrate evolution (Tweedie et al. 1997; Elango and Yi 2008; Zhang et al. 2015), although the nature of the factors driving such a dramatic transition is debated (Bird 1995; Yoder et al. 1997). Recent methylome analyses of birds indicate that they tend to exhibit reduced TE methylation (Li et al. 2015; Derks et al. 2016), suggesting that more complexities in animal genome methylation await discovery.
Fig. 2.—

Variation of genomic DNA methylation patterns in different species. The methylomes of invertebrates and vertebrates are drastically different as the former typically harbor much lower levels of DNA methylation than the latter. Limited amount of DNA methylation in invertebrate genomes is found in a subset of genes, and very rarely in promoters. In contrast, vertebrate genomes are heavily methylated across the board, although some variation exists in different taxa. Plant genomes generally have methylated transposable elements and some gene bodies. Recent studies showed that gene body methylation is lacking in some of the plant lineages.
In plants, the model organism A. thaliana has also served as a great system to study DNA methylation. Both gene body and TE methylation are present in the A. thaliana genome (Zhang et al. 2006; Lister et al. 2008). Cytosine methylations are found in substantial frequencies in all sequence contexts in plants; in addition to cytosines in CpGs, cytosines in CpHpG and CpHpHs (H stands for A, T, or C) contexts are also frequently methylated in plant genomes. DNA methylation in all three contexts is found in TEs, whereas CpG methylation dominates gene body methylation (Lister et al. 2008; Takuno et al. 2016; Vidalis et al. 2016). Genome-wide patterns of DNA methylation across different plant lineages also vary substantially (fig. 2) (Amborella Genome Project 2013; Seymour et al. 2014; Bewick et al. 2016; Niederhuth et al. 2016; Takuno et al. 2016). Gene body DNA methylation, which, similar to the case in animals, is associated with evolutionarily conserved genes (Takuno and Gaut 2012, 2013), has been independently lost in some angiosperm lineages (Bewick et al. 2016; Niederhuth et al. 2016).
Genome Size Covaries with Large-Scale Phylogenetic Variations in DNA Methylation
Why does DNA methylation vary so widely between lineages? Deletion of DNA methylation enzymes from the genome is certainly linked to the absence of DNA methylation in specific lineages. For example, the lack of DNA methylation in D. melanogaster and C. elegans can be linked to the deletion of key DNA methyltransferases in the lineages leading to these species (Yi and Goodisman 2009). Similarly, in plants, lineage-specific loss of gene body DNA methylation is linked to the loss of a key DNA methylation enzyme (Bewick et al. 2016; Niederhuth et al. 2016), although some nonvascular plants lack gene body DNA methylation despite harboring methylation enzymes in their genomes (Takuno et al. 2016).
When measured as a per nucleotide level DNA methylation, variation of DNA methylation is significantly positively correlated with genome size in plants (Alonso et al. 2015; Niederhuth et al. 2016). Genome size was able to explain ∼14% of the variation in genome-wide methylation levels of 34 angiosperm plants (Niederhuth et al. 2016; Vidalis et al. 2016). Deeper analyses of this relationship revealed that it is robust between CHG methylation and genome size, but not necessarily in the context of CpG methylation, which is largely restricted to gene bodies (Niederhuth et al. 2016; Takuno et al. 2016). Consequently, the relationship between genome size and DNA methylation in plants can be largely explained by differences in TE content (Niederhuth et al. 2016; Takuno et al. 2016; Vidalis et al. 2016), suggesting that the suppression of TEs may be a major genomic benefit at the expense of maintaining DNA methylation in plants. At the time of this writing, although some examples of gene body DNA methylation loss have been reported, a loss of TE methylation has yet to be observed in plants.
On the animal side, Lechner et al. (2013) analyzed the relationship between genome size and DNA methylation using 78 metazoan species. Due to the lack of experimentally determined DNA methylation data at that time, the authors used dinucleotide frequencies as an indirect measure of DNA methylation. Specifically, the methylation of C in the CpG context makes CpGs vulnerable to deamination-mediated point mutations. Methylated CpGs tend to mutate to either TpG or CpA (depending on the strand in which the deamination occurred) much more frequently than point mutation rates in a non-CpG context (Elango et al. 2008). Lechner et al. (2013) used a composite measure of the depletion of CpG and the enrichment of TpG/CpA as a proxy of germline DNA methylation. They observed a significant positive correlation between genome size and their computational measure of DNA methylation. In other words, the larger the genome sizes were, the more there was evidence of germline DNA methylation. However, unlike in the case of plants, it is not clear whether the correlation between genome size and DNA methylation in animals can be attributed to TE methylation. As discussed earlier, TEs are not uniformly methylated in different animal lineages (Simmen et al. 1999; Lyko et al. 2010; Keller et al. 2016a). Lechner et al. (2013) also inferred that the degree of TE methylation varied between different animal genomes, and that TEs tended to be more heavily methylated in tetrapods than in other taxa. As there are many factors that correlate with genome size (Lynch and Conery 2003; Charlesworth and Barton 2004), the underlying driver of the relationship between genome size and DNA methylation in animal genomes remains to be resolved.
Association between Genetic Variation and DNA Methylation from Population Epigenomic Studies
There are now several studies that have investigated population-level variation of DNA methylation from humans and plants. These population epigenomic studies consistently reveal that genetic variation contributes to DNA methylation in both taxa. For example, many single nucleotide polymorphisms (SNPs) that are significantly associated with variation in the DNA methylation, referred to as “methylation quantitative trait loci” or “mQTLs,” have been reported in human and plant populations (Gibbs et al. 2010; Zhang et al. 2010; Bell et al. 2011, 2012; Eichten et al. 2013; Schmitz et al. 2013; McRae et al. 2014; Dubin et al. 2015; Fagny et al. 2015; Hannon et al. 2015; Gaunt et al. 2016). However, the estimated degrees to which genetic variation affect DNA methylation are highly divergent between humans and plants. For instance, in the majority of human population epigenomic studies, the number of CpGs for which methylation levels could be associated with mQTLs is at most a few percent of the total CpGs surveyed (Taudt et al. 2016). In contrast, plant population epigenomic studies often report a much higher proportion of sites for which methylation could be explained by genetic variation, from 18% (Dubin et al. 2015) to over 50% (Eichten et al. 2013). If taken at face value, this discrepancy could indicate fundamental differences in the genetic contributions to epigenetic variation between humans and plants. However, given the following differences in technical and statistical methods used in these studies, the estimates of genetic contributions for human and plant population epigenomic data are likely have been under- and overestimated, respectively.
In terms of experimental techniques, most human population epigenomic studies to date have used arrays such as the Illumina Infinium Human Methylation450 BeadChip array and the earlier 29 K chip (table 1). These chips allow for the detection of DNA methylation at preselected CpG sites at low cost. An important limitation of these arrays, however, is that most of the preselected CpG sites are from “traditional” CpG islands and promoters, because earlier studies often assumed that DNA methylation variations at those sites were functionally relevant. However, as more data on whole-genome methylomes accumulate, it is now clear that CpGs residing in traditional CpG islands, most of which are located in promoters of broadly expressed genes, harbor the least amount of DNA methylation variation. CpGs that are differentially methylated across tissues or cell types tend to localize outside of traditional CpG islands or promoters (Doi et al. 2009; Ziller et al. 2013). Consequently, the arrays based on CpG islands and promoters target cytosines that are least epigenetically variable (Zeng et al. 2014; Taudt et al. 2016), making themselves inherently underpowered to capture the true variation of CpG methylation. In contrast, plant population epigenomic studies have typically used next-generation sequencing based methods. For example, Schmitz et al. (2013) and Dubin et al. (2015) both used whole-genome bisulfite sequencing (WGBS). Other studies combined reduced sampling with next-generation sequencing approaches (e.g., Eichten et al. 2013 used MeDIP-seq). Unlike arrays, these studies are not biased toward preselected cytosines, thus capturing more realistic patterns of population epigenomic variation. This difference in experimental techniques likely led to underestimation of genetic contribution to epigenetic variation in human studies.
Another important consideration is the differences in statistical methods employed to analyze variation of DNA methylation. Array-based studies usually test association between methylation and genetic variation for individual cytosines. However, next-generation sequencing-based studies (such as WGBS) rarely analyze individual cytosines. One reason for this is because of the extremely large numbers of total cytosines surveyed in such studies. For example, the total number of CpGs in the human genome is approximately 30 million sites. In plants, despite the much smaller genome size compared with humans, the numbers of target cytosines are similar or even greater than that in the human genome, since in plants DNA methylation targets nearly all cytosines. It is challenging to investigate the statistical significance of individual cytosines for such a large number of sites (Huh et al. 2017). One way to avoid this issue is to analyze DNA methylation of preannotated genomic regions such as promoters and gene bodies (Zeng et al. 2012; Roessler et al. 2016). However, meaningful variation occurring in yet unannotated functional regions of the genome could go undetected this way. Another popular method has been to investigate DNA methylation variations in clusters of cytosines, by identifying and characterizing “differentially methylated regions (DMRs).” Compared with analyses of individual sites, using clusters of CpGs can increase the statistical power to detect genetic determinants (Dubin et al. 2015; Keller et al. 2016b). However, methods to identify DMRs are not well defined and vary between researchers and specific tools used (Roessler et al. 2016; Huh et al. 2017). Consequently, it is difficult to directly compare results from different studies. Importantly, when used in a setting without proper biological replicates, DMRs can have high false positive rates (Roessler et al. 2016). Thus, studies relying on DMRs without robust biological replicates could lead to overestimation of genetic contributions to epigenetic variation.
Given these technical considerations, comparing the degree of genetic contributions between taxa needs to wait until studies using comparable experimental and computational methods become available. Indeed, researchers began to employ next-generation sequencing based methods to examine variation of DNA methylation from a relatively large number of human samples (Busche et al. 2015; McClay et al. 2015), and we expect to see more of such studies soon. Nevertheless, despite the technical differences, population epigenomic studies from humans and plants both indicate that mQTLs mainly function in cis- (typically defined as within 100 kb of the target CpG or DMR). The number of trans-mQTLs is much smaller than cis-QTLs. Even though this observation should be taken with caution given that the statistical power to detect trans-QTLs is generally low, it is consistent with the results of direct functional studies of genetic determinants as discussed below.
Identifying Nucleotide-Level Determinants of DNA Methylation in Mammalian Genomes
Computational Prediction of Sequence Determinants
Complementary to population-level analyses of DNA methylation, a number of studies in mammals have investigated the nucleotide-level, sequence-encoded determinants of DNA methylation. The majority of these studies have focused on the DNA methylation of CpG islands. CpG islands were originally identified as short stretches of CpGs that are devoid of DNA methylation in the human genome (Cooper et al. 1983). Noting that many of these genomic regions had higher than expected numbers of CpG dinucleotides and high GC content, the term “CpG islands” was coined (Bird 1986; Antequera and Bird 1988). The concept of CpG islands underwent phenomenal evolution as genomic and epigenomic methods advanced (Bock et al. 2007; Illingworth and Bird 2009; Mendizabal and Yi 2017). Earlier studies relied on computational algorithms to define CpG islands based on their traits, such as high CpG density and high GC content (Gardiner-Garden and Frommer 1987; Takai and Jones 2002). However, CpG islands defined in this way had many “false positives”—in addition to the originally intended unmethylated regions, some CpG islands were methylated when examined in detail (Larsen et al. 1993; Yamada et al. 2004; Weber et al. 2005). Recent genome-scale epigenetic studies demonstrated that 30∼40% of human and mouse CpG islands, as determined by computational methods alone, are methylated in normal tissues (Illingworth et al. 2008, 2010; Mendizabal and Yi 2016).
The methylation of some CpG islands has motivated researchers to search for local sequence traits (i.e., sequence traits of CpG islands themselves) that may be used to predict the methylation status of specific CpG islands. For example, Feltus et al. (2003) showed that a handful of sequence features could be used to discriminate between CpG islands that were prone to methylation (vs. those that were not) in a human fibroblast cell line with DNA methyltransferase overexpression. Other studies subsequently demonstrated that a number of sequence attributes were able to predict substantial amounts of variation in CpG island DNA methylation (Bhasin et al. 2005; Bock et al. 2006). More recently, Gaidatzis et al. (2014) focused on “partially methylated domains,” which are large genomic regions with intermediate levels of DNA methylation. They showed that nearly one-third of the variation in the partially methylated domains could be explained by a computational model using a dinucleotide context of 80 bps sequences flanking specific CpGs (Gaidatzis et al. 2014). Furthermore, predictive sequence models of DNA methylation could be constructed in distant vertebrate species, such as Gallus gallus, Anolis carolinensis, Xenopus tropicalis, and Danio rerio (Huska and Vingron 2016). Together, these studies point to the existence of specific sequence features that encode information on DNA methylation.
Experimental Analyses Reveal DNA Methylation Grammar
Indeed, our understanding of the relationship between DNA sequence features and methylation has improved greatly thanks to ingenious experiments directly interrogating the effects of DNA sequence features on their methylation. This line of research has the potential to distinguish between direct causes and effects of genetic changes and epigenetic changes, something that association studies are not capable of doing.
In a pioneering experiment, Lienert et al. (2011) developed a system to stably insert specific sequences into a defined genomic position in mouse embryonic stem cells. As the methylation state of the insert sequence could be affected by the chromosomal environment where the insert lands, Lienert et al. (2011) controlled the insertion site to be exactly the same location for different inserts. Then, by changing the sequence composition of the inserts, they could directly measure the methylation levels of inserted sequences. This experiment tested dozens of different inserts and demonstrated that most of the inserts could faithfully recapitulate their own methylation profiles in their native cellular environment, indicating that sequence fragments themselves encode information to guide DNA methylation. Krebs et al. (2014) and Wachter et al. (2014) furthered this method to test hundreds of DNA inserts. These experiments enabled researchers to extract some general rules as to how sequence features dictate their methylation profiles. On the one hand, sequence fragments with high GC content and high CpG density nearly always remained hypomethylated. On the other hand, some sequences that have low CpG density and GC content could still be hypomethylated, especially when they encoded binding sites for specific transcription factors.
In a complementary approach, Long et al. (2016) investigated the genomic methylation of a mouse strain carrying almost the entire human chromosome 21 as a stably transmitting separate chromosome (O’Doherty et al. 2005). They showed that the DNA methylation patterns of hypomethylated regions in human chromosome 21 were nearly entirely (>80%) recapitulated in this mouse model. Importantly, hypomethylated regions that maintained the native methylation status tended to be CpG- and GC-rich. Moreover, Long et al. (2016) also showed that bacterial artificial chromosomes (BACs) of mouse sequences, when injected into zebrafish embryos, could nearly recapitulate the native mouse DNA methylation patterns. These findings show that sequence contexts themselves were strong enough to drive DNA methylation in a completely different cellular environment, separated by tens and hundreds of millions of years of evolution.
Consequently, these complementary experiments solidify the role of sequence determinants in local DNA hypomethylation and show that some of these “methylation grammars” are conserved in humans, mice, and in zebrafish. High GC content and high CpG density, features often found in traditional CpG islands located in the promoters, can drive hypomethylation. On the other hand, the methylation grammars of low-GC and low-CpG regions are intertwined with transcription factor binding. In Long et al.’s (2016) experiment, regions that gained discordant methylation profiles compared with those in their native cellular environment were often found in distal regulatory regions. Transcription factor binding and DNA methylation can compete for the same sequence motifs (Domcke et al. 2015), and mutations at transcription factor binding sites can disrupt the binding of specific TFs and lead to differential DNA methylation (Lienert et al. 2011; Stadler et al. 2011; Krebs et al. 2014; Wachter et al. 2014). Additionally, the methylation profiles of a specific region may be determined by the availabilities of different transcription factors specific to each cell type.
Sequence Evolution and Methylome Evolution
The aforementioned studies show that DNA sequences themselves have the ability to dictate DNA methylation, and provide mechanistic understanding on the presence of genetic determinants of DNA methylation. Consequently, we can infer that sequence evolution can affect mammalian methylome evolution in the following ways. First, mutations that affect the GC content and CpG density can cause hypomethylation or hypermethylation. As discussed earlier, genomic regions with high GC content and high CpG density are themselves refractory to DNA methylation in mammals. Mutations in those regions will not affect their hypomethylation as long as high GC content and high CpG density are maintained. However, a CpG to non-CpG point mutation would reduce CpG density, which can make the region more susceptible to DNA methylation. Other GC to AT mutations will also increase the likelihood of hypermethylation of such regions. In fact, insertions or deletions that reduce GC content and CpG density were shown to increase DNA methylation (Takahashi et al. 2017). Conversely, mutations that increase GC content and CpG density could drive hypomethylation.
Biased gene conversion can also affect methylation. Gene conversion occurs when a homologous recombination results in a mismatch base pair, which in turn is corrected to one of the two nucleotides that were mismatched. In many genomes, gene conversion is biased so that the mismatch is more likely to be converted to a G/C allele than to an A/T allele (Birdsell 2002; Pessia et al. 2012; Lassalle et al. 2015). Such biased gene conversion (BGC) will increase GC content and could also directly increase CpG density. Consequently, the BGC process could increase the likelihood of hypomethylation. Indeed, Cohen et al. (2011) showed that a substantial number of CpG islands in the human genome has been under the influence of BGC.
Another important factor that could affect DNA methylation is transcription factor binding sites turnover. The nucleotide sequence of transcription factor binding sites (TFBS) evolves rapidly, a phenomenon referred to as “transcription factor binding site turnover” (Dermitzakis and Clark 2002; Moses et al. 2006; Borneman et al. 2007). Since transcription factors and DNA methylation can utilize and compete for the same genomic sequences (Stadler et al. 2011), transcription factor turnover could influence regional DNA methylation, and potentially other epigenetic components (Lowdon et al. 2016). In the simplest case, a point mutation that generates a new TFBS might increase the likelihood of hypomethylation. On the other hand, a mutation that disrupts an existing TFBS could increase the methylation of target sequences. Comparative methylome studies provide some evidence for this model: a comparison of human and chimpanzee brain methylomes has identified several SNPs that fit the expectations of the TFBS turnover model of evolution of DNA methylation (Mendizabal et al. 2016). In addition, changes in the blood methylome across the primate lineage often involved sites that encode TFBS (Hernando-Herraez et al. 2015). A recent analysis of sperm methylomes in humans, chimpanzees, and macaques also found consistent results (Fukuda et al. 2017).
However, there are several complicating factors in applying the TFBS turnover model. For example, the abundance of other transcription factors and the sequence context both play roles in the competition between DNA methylation and transcription factor binding. In GC-rich and CpG-dense regions, sequence context could overwrite the absence of TFBS and drive hypomethylation (Krebs et al. 2014; Wachter et al. 2014). The TFBS turnover model may be better realized in distal regulatory regions, which are often relatively GC- and CpG-poor. Another complicating factor is that a substantial number of transcription factors bind differentially to methylated versus unmethylated CpGs (Hu et al. 2013). For example, some TFs, particularly those harboring homeodomains, preferentially bind to methylated CpGs (Yin et al. 2017). The nature of the interaction between TFs and methylated CpGs is still debated (Zhu et al. 2016). Mutations that generate a TFBS with the affinity to methylated CpGs may not necessarily have an effect on DNA methylation unless it occurs in methylated regions.
Spontaneous Epimutations
An intriguing aspect of the epigenome is that it can diverge without genetic changes. For example, DNA methylation can be stochastically gained or lost during the lifetime of somatic cell lineages (Fraga et al. 2005; Teschendorff et al. 2013). DNA methylation may also change as a consequence of environmental perturbation (Dowen et al. 2012; Zheng et al. 2013). If the DNA methylation of a specific nucleotide changes in the germline, it could be transmitted to the next generation. Direct inheritance of epigenetic changes, without genetic sequence changes, is referred to as “transgenerational inheritance.”
There is much current interest in transgenerational inheritance and how it could affect evolution. The relevant and important questions for evolution are whether epigenetic mutations (herein referred to as “epimutations”) can be adaptive, and if the epimutations can be stably transmitted entirely via transgenerational inheritance. Answering these questions has been extremely challenging, in large part because decisively ruling out genetic effects or maternal effects is very difficult (Daxinger and Whitelaw 2012; Heard and Martienssen 2014). In this regard, studies from plants provide unique insights into the prevalence of epimutations and their evolutionary potential. Unlike animal germlines that originate and are maintained separately from somatic lineages, the plant germline can originate from somatic cells, and clonal propagation is common in plants. Accordingly, the transgenerational inheritance of DNA methylation has been relatively frequently observed in plants.
In particular, analyses of DNA methylation in A. thaliana mutation–accumulation lines have been useful in estimating the background rates of epimutations and their evolutionary nature. Several studies have characterized whole-genome methylation maps of these A. thaliana mutation accumulation lines derived from the same reference Columbia strain (Becker et al. 2011; Schmitz et al. 2011; van der Graaf et al. 2015). Van der Graaf et al. (2015) analyzed these whole genome methylation maps to infer the epimutation rates of DNA methylation. They estimated that the spontaneous gain and loss of DNA methylation occurs at different rates, on an average 2.56×10−4 and 6.30×10−4 per individual CpG per generation, respectively (Van der Graaf et al. 2015). These rates are orders of magnitude higher than the estimated genetic mutation rate of 7×10−9 per site per generation (Ossowski et al. 2010), which was also derived from A. thaliana mutation accumulation lines. Such a high rate of epimutations implies that if they were functional, the genetic load due to deleterious mutations is too high to maintain the epimutations (Charlesworth et al. 2017). It follows that most of the spontaneous DNA methylation epimutations in A. thaliana are likely to be neutral. Vidalis et al. (2016) demonstrated that the site frequency spectra of CpGs in gene bodies are consistent with the idea that most CpG epimutations in gene bodies are functionally neutral. Some epimutations might be functional, but they appear to constitute a very small portion of the total number of epimutations in A. thaliana.
It was also shown that the current patterns of genomic DNA methylation in the A. thaliana genome are highly similar to what would be expected if the observed rates of gain and loss of DNA methylation were in equilibrium (Van der Graaf et al. 2015). In other words, in A. thaliana, neutral epimutations appear as the main factors shaping the current global DNA methylome. Whether the same is true in other lineages is currently unknown. It is an interesting possibility that different rates of epimutations in different taxa may contribute to the high variation of DNA methylation between species (e.g., fig. 2). For example, the rate of DNA methylation loss in the heavily methylated mammalian genomes is likely to be much lower than that observed in A. thaliana.
Conclusions
Comparative epigenomic analyses that have become available relatively recently provide snapshots of epigenome variation from the vantage points of differing evolutionary timescales. Literature from plants indicates that spontaneous epimutations play a large role in shaping the current pattern of DNA methylation in the A. thaliana genome. Rates of spontaneous epimutations are much higher than that of genetic mutations in A. thaliana, suggesting that on a short timescale, spontaneous epimutations dominate the DNA methylation landscape of the A. thaliana genome. One of the most significant questions on the evolution of DNA methylation is whether changes in DNA methylation itself could be adaptive. Given the extremely high rates of spontaneous epimutations, the genetic load argument indicates that the majority of A. thaliana spontaneous epimutations are neutral (Charlesworth et al. 2017). Population genetic analyses based on newly developed models that explicitly take into account high mutation rates (Charlesworth and Jain 2014) also indicate that the majority of epimutations in A. thaliana gene bodies are neutral (Vidalis et al. 2016). Consistent with this idea, methylomes of A. thaliana strains that have evolved in highly different natural environments show similar levels of divergence as those that were kept in relatively stable greenhouse environments (Hagmann et al. 2015). If epimutations played a role in adaptation to different environments, methylomes may have more drastically diverged in variable natural environments. On the other hand, on a large phylogenetic scale, the genomic content of repetitive sequences leads to a positive relationship between plant genome size and DNA methylation per nucleotide. Bookended by these observations at two different timescales, population epigenomic studies indicate that genetic variants can explain a substantial portion of DNA methylation variation across individuals in different plant species, predominantly in cis. This can explain the concordance between DNA methylome and genetic distance observed in many plant species (Eichten et al. 2013; Schmitz et al. 2013; Hagmann et al. 2015). A current challenge is how to link the evolutionary dynamics at short timescale (dominated by spontaneous epimutations) to the genomic diversity of DNA methylation at large timescale.
The prevalence and the inheritance of epimutations in animal genomes remain to be resolved. Lacking a relatively manageable and low-cost model system to study DNA methylation and its inheritance (D. melanogaster and C. elegans both lack the canonical DNA methylation system) is certainly a challenge in this regard. On the other hand, since DNA methylation is widespread in animal taxa, we may soon have access to reasonable model systems from which mutation accumulation lines and other resources could be developed to analyze DNA methylation. Regardless, studies using human and mouse illuminated how specific genetic change could cause methylation changes in cis. In principle, utilizing this information, we could test the selective effects of DNA methylation divergence by investigating the population genetic and evolutionary trajectories of causative mutations of DNA methylation changes. Such a study, combined with genome editing, could provide answers to the elusive question of the evolutionary significance of epigenetic variation.
Acknowledgments
The author thanks Xin Wu, Isabel Mendizabal, Brendan Gaut, Kateryna Makova, and an anonymous reviewer for the comments on the article and Lavanya Rishishwar for the help with the illustrations. This work was supported by grants from the National Science Foundation [SBE-131719 and MCB-1615664] and the National Institutes of Health [1R01MH103517-02].
Literature Cited
- Allis CD, Jenuwein T.. 2016. The molecular hallmarks of epigenetic control. Nat Rev Genet. 178:487–500. [DOI] [PubMed] [Google Scholar]
- Alonso C, Pérez R, Bazaga P, Herrera CM.. 2015. Global DNA cytosine methylation as an evolving trait: phylogenetic signal and correlated evolution with genome size in angiosperms. Front Genet. 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amborella Genome Project. 2013. The Amborella genome and the evolution of flowering plants. Science 342 426165:1241089. [DOI] [PubMed] [Google Scholar]
- Antequera F, Bird AP.. 1988. Unmethylated CpG islands associated with genes in higher plant DNA. Embo J. 78:2295–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Hagmann J, Müller J, Koenig D, Stegle O, Borgwardt K.. 2011. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 4807376:245.. [DOI] [PubMed] [Google Scholar]
- Bell J, et al. . 2011. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 121:R10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JT, et al. . 2012. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 84:e1002629.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick AJ, et al. . 2016. On the origin and evolutionary consequences of gene body DNA methylation. Proc Natl Acad Sci U S A. 113:9111–9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick AJ, Vogel KJ, Moore AJ, Schmitz RJ.. 2017. Evolution of DNA methylation across insects. Mol Biol Evol. 343:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhasin M, Zhang H, Reinherz EL, Reche PA.. 2005. Prediction of methylated CpGs in DNA sequences using a support vector machine. FEBS Lett. 57920:4302–4308. [DOI] [PubMed] [Google Scholar]
- Bird A. 1986. CpG-rich islands and the function of DNA methylation. Nature 3216067:209–213. [DOI] [PubMed] [Google Scholar]
- Bird A. 1995. Gene number, noise reduction and biological complexity. Trends Genet. 113:94–100. [DOI] [PubMed] [Google Scholar]
- Birdsell JA. 2002. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol Biol Evol. 197:1181–1197. [DOI] [PubMed] [Google Scholar]
- Blow MJ, et al. . 2016. The epigenomic landscape of prokaryotes. PLoS Genet. 122:e1005854.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, et al. . 2006. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. PLoS Genet. 23:e26.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Walter J, Paulsen M, Lengauer T.. 2007. CpG island mapping by epigenome prediction. PLoS Comput Biol. 36:e110.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, et al. . 2007. Divergence of transcription factor binding sites across related yeast species. Science 3175839:815.. [DOI] [PubMed] [Google Scholar]
- Busche S, et al. . 2015. Population whole-genome bisulfite sequencing across two tissues highlights the environment as the principal source of human methylome variation. Genome Biol. 16:290.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Barton N.. 2004. Genome size: does bigger mean worse? Curr Biol. 146:R233–R235. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Jain K.. 2014. Purifying selection, drift, and reversible mutation with arbitrarily high mutation rates. Genetics 1984:1587.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Barton NH, Charlesworth B.. 2017. The sources of adaptive variation. Proc R Soc B Biol Sci. 2841855:20162864.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino L, Aifantis I.. 2017. Alternative roles for oxidized mCs and TETs. Curr Opin Genet Dev. 42:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NM, Kenigsberg E, Tanay A.. 2011. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell 1455:773–786. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Taggart MH, Bird AP.. 1983. Unmethylated domains in vertebrate DNA. Nucleic Acids Res. 113:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabe EC, Sanford RS, Kohn AB, Bobkova Y, Moroz LL.. 2015. DNA methylation in basal metazoans: insights from Ctenophores. Integr Comp Biol. 556:1096–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L, Whitelaw E.. 2012. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet. 133:153–162. [DOI] [PubMed] [Google Scholar]
- Derks MFL, et al. . 2016. Gene and transposable element methylation in great tit (Parus major) brain and blood. BMC Genomics 17:332.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Clark AG.. 2002. Evolution of transcription factor binding sites in mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol. 197:1114–1121. [DOI] [PubMed] [Google Scholar]
- Doi A, Park I-H, Wen B, Murakami P, Aryee MJ, Irizarry R.. 2009. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 4112:1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, et al. . 2015. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 5287583:575–579. [DOI] [PubMed] [Google Scholar]
- Dorman CJ, Deighan P.. 2003. Regulation of gene expression by histone-like proteins in bacteria. Curr Opin Genet Dev. 132:179–184. [DOI] [PubMed] [Google Scholar]
- Dowen RH, et al. . 2012. Widespread dynamic DNA methylation in response to biotic stress. Proc Natl Acad Sci U S A. 10932:E2183–E2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin MJ, et al. . 2015. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 4:e05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichten SR, Briskine R, Song J, Li Q, Swanson-Wagner R, Hermanson PJ.. 2013. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell 258:2783.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango N, Hunt BG, Goodisman MA, Yi SV.. 2009. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc Natl Acad Sci U S A. 10627:11206–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango N, Kim S-H, Program NCS, Vigoda E, Yi SV.. 2008. Mutations of different molecular origins exhibit contrasting patterns of regional substitution rate variation. PLoS Comput Biol. 4:e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango N, Yi SV.. 2008. DNA methylation and structural and functional bimodality of vertebrate promoters. Mol Biol Evol. 258:1602–1608. [DOI] [PubMed] [Google Scholar]
- Fagny M, et al. . 2015. The epigenomic landscape of African rainforest hunter-gatherers and farmers. Nat Commun. 6:10047.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltus FA, Lee EK, Costello JF, Plass C, Vertino PM.. 2003. Predicting aberrant CpG island methylation. Proc Natl Acad Sci U S A. 10021:12253–12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, et al. . 2005. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 10230:10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, et al. . 2017. Evolution of the sperm methylome of primates is associated with retrotransposon insertions and genome instability. Hum Mol Genet. 26: 3508–3519. [DOI] [PubMed] [Google Scholar]
- Gaidatzis D, et al. . 2014. DNA sequence explains seemingly disordered methylation levels in partially methylated domains of mammalian genomes. PLoS Genet. 102:e1004143.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M.. 1987. CpG islands in vertebrate genomes. J Mol Biol. 1962:261–282. [DOI] [PubMed] [Google Scholar]
- Gaunt TR, et al. . 2016. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 17:61.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavery MR, Roberts SB.. 2013. Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 1:e215.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JR, et al. . 2010. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 65:e1000952.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, et al. . 2015. DNA methylation on N(6)-adenine in C. elegans. Cell 1614:868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoretti I, Lee Y-M, Goodson HV.. 2004. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 3381:17–31. [DOI] [PubMed] [Google Scholar]
- Hagmann J, Becker C, Müller J, Stegle O, Meyer RC, Wang G.. 2015. Century-scale methylome stability in a recently diverged Arabidopsis thaliana lineage. PLoS Genet. 111:e1004920.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon E, et al. . 2015. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 191:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Martienssen RA.. 2014. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 1571:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando-Herraez I, et al. . 2015. The interplay between DNA methylation and sequence divergence in recent human evolution. Nucleic Acids Res. 4317:8204–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, et al. . 2013. DNA methylation presents distinct binding sites for human transcription factors. eLife 2:e00726.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh I, Wu X, Park T, Yi SV.. 2017. Detecting differential DNA methylation from sequencing of bisulfite converted DNA of diverse species. Brief Bioinformatics bbx077. 10.1093/bib/bbx077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt BG, Glastad KM, Yi SV, Goodisman MA.. 2013a. The function of intragenic DNA methylation: insights from insect epigenomes. Integr Comp Biol. 532:319–328. [DOI] [PubMed] [Google Scholar]
- Hunt BG, Glastad K, Yi SV, Goodisman MAD.. 2013b. Patterning and regulatory associations of DNA methylation are mirrored by histone modifications in insects. Genome Biol Evol. 53:591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huska M, Vingron M.. 2016. Improved prediction of non-methylated islands in vertebrates highlights different characteristic sequence patterns. PLoS Comput Biol. 1212:e1005249.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke J, Collignon E, Fuks F.. 2016. Portraits of TET-mediated DNA hydroxymethylation in cancer. Curr Opin Genet Dev. 36:16–26. [DOI] [PubMed] [Google Scholar]
- Illingworth R, et al. . 2008. A movel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 6:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth RS, Bird AP.. 2009. CpG islands-’A rough guide’. FEBS Lett. 58311:1713–1720. [DOI] [PubMed] [Google Scholar]
- Illingworth RS, et al. . 2010. Orphan CpG islands identify mumerous conserved promoters in the mammalian genome. PLoS Genet. 69:e1001134.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller TE, Han P, Yi SV.. 2016a. Evolutionary transition of promoter and gene body DNA methylation across invertebrate-vertebrate boundary. Mol Biol Evol. 334:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller TE, Lasky JR, Yi SV.. 2016b. The multivariate association between genomewide DNA methylation and climate across the range of Arabidopsis thaliana. Mol Ecol. 258:1823–1837. [DOI] [PubMed] [Google Scholar]
- Krebs AR, Dessus-Babus S, Burger L, Schübeler D.. 2014. High-throughput engineering of a mammalian genome reveals building principles of methylation states at CG rich regions. eLife 3:e04094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen F, Solheim J, Prydz H.. 1993. A methylated CpG island 3’ in the apolipoprotein-E gene does not repress its transcription. Hum Mol Genet. 26:775–780. [DOI] [PubMed] [Google Scholar]
- Lassalle F, et al. . 2015. GC-content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLoS Genet. 112:e1004941.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M, et al. . 2013. The correlation of genome size and DNA methylation rate in metazoans. Theory Biosci. 1321:47–60. [DOI] [PubMed] [Google Scholar]
- Li J, et al. . 2015. Genome-wide DNA methylome variation in two genetically distinct chicken lines using MethylC-seq. BMC Genomics 16:851.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienert F, et al. . 2011. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet. 4311:1091–1097. [DOI] [PubMed] [Google Scholar]
- Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH.. 2008. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 1333:523.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, King HW, Patient RK, Odom DT, Klose RJ.. 2016. Protection of CpG islands from DNA methylation is DNA-encoded and evolutionarily conserved. Nucleic Acids Res. 4414:6693–6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowdon RF, Jang HS, Wang T.. 2016. Evolution of epigenetic regulation in vertebrate genomes. Trends Genet. 325:269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunnon K, et al. . 2016. Variation in 5-hydroxymethylcytosine across human cortex and cerebellum. Genome Biol. 17:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F, Foret S, Kucharski R, Wolf S, Falckenhayn C, Maleszka R.. 2010. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 811:e1000506.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS.. 2003. The origins of genome complexity. Science 3025649:1401–1404. [DOI] [PubMed] [Google Scholar]
- Marsh VL, Peak-Chew SY, Bell SD.. 2005. Sir2 and the Acetyltransferase, Pat, regulate the archaeal chromatin protein, Alba. J Biol Chem. 28022:21122–21128. [DOI] [PubMed] [Google Scholar]
- McClay JL, et al. . 2015. High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol. 161:291.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae AF, et al. . 2014. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 155:R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal I, et al. . 2016. Comparative methylome analyses identify epigenetic regulatory loci of human brain evolution. Mol Biol Evol. 3311:2947–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal I, Yi SV.. 2016. Whole-genome bisulfite sequencing maps from multiple human tissues reveal novel CpG island associated with tissue-specific regulation. Hum Mol Genet. 251:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal I, Yi SV.. 2017. Diversity of human CpG islands In: Patel PB, Preedy VR, editors. Handbook of nutrition, diet, and epigenetics. New York: Springer. DOI 10.1007/978-3-319-31143-2_67-1. [Google Scholar]
- Moses AM, et al. . 2006. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput Biol. 210:e130.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederhuth CE, et al. . 2016. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 171:194.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty A, et al. . 2005. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309:2033–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, et al. . 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 3275961:92.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette M, Jackson L, Chimileski S, Papke RT.. 2015. Genome-wide DNA methylation analysis of Haloferax volcanii H26 and identification of DNA methyltransferase related PD-(D/E)XK nuclease family protein HVO_A0006. Front Microbiol. 6:251.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia E, et al. . 2012. Evidence for widespread GC-biased gene conversion in eukaryotes. Genome Biol Evol. 47:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve JN, Sandman K, Daniels CJ.. 1997. Archaeal histones, nucleosomes, and transcription initiation. Cell 897:999–1002. [DOI] [PubMed] [Google Scholar]
- Roberts RJ, Vincze T, Posfai J, Macelis D.. 2015. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43(D1):D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler K, Takuno S, Gaut BS.. 2016. CG methylation covaries with differential gene expression between leaf and floral bud tissues of Brachypodium distachyon. PLoS One 113:e0150002.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Carlier E, Riviere G.. 2015. Regulation of Hox orthologues in the oyster Crassostrea gigas evidences a functional role for promoter DNA methylation in an invertebrate. FEBS Lett. 58913:1459–1466. [DOI] [PubMed] [Google Scholar]
- Sandman K, Reeve JN.. 2006. Archaeal histones and the origin of the histone fold. Curr Opin Microbiol. 95:520–525. [DOI] [PubMed] [Google Scholar]
- Sarda S, Zeng J, Hunt BG, Yi SV.. 2012. The evolution of invertebrate gene body methylation. Mol Biol Evol. 298:1907–1916. [DOI] [PubMed] [Google Scholar]
- Schmitz RJ, et al. . 2011. Transgenerational epigenetic instability is a source of novel methylation variants. Science 3346054:369.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, et al. . 2013. Patterns of population epigenomic diversity. Nature 4957440:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour DK, Koenig D, Hagmann J, Becker C, Weigel D.. 2014. Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization. PLoS Genet. 1011:e1004785.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmen MW, et al. . 1999. Nonmethylated transposable elements and methylated genes in a chordate genome. Science 2835405:1164–1167. [DOI] [PubMed] [Google Scholar]
- Slesarev AI, Belova GI, Kozyavkin SA, Lake JA.. 1998. Evidence for an early prokaryotic origin of histones H2A and H4 prior to the emergence of eukaryotes. Nucleic Acids Res. 262:427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava M, et al. . 2010. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466:720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, et al. . 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 4807378:490–495. [DOI] [PubMed] [Google Scholar]
- Suzuki MM, Bird A.. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 96:465–476. [DOI] [PubMed] [Google Scholar]
- Suzuki MM, Kerr ARW, De Sousa D, Bird A.. 2007. CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 175:625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, et al. . 2017. Integration of CpG-free DNA induces de novo methylation of CpG islands in pluripotent stem cells. Science 3566337:503.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, Jones PA.. 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 996:3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuno S, Gaut BS.. 2012. Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol Biol Evol. 291:219.. [DOI] [PubMed] [Google Scholar]
- Takuno S, Gaut BS.. 2013. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc Natl Acad Sci U S A. 1105:1797.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuno S, Ran J-H, Gaut BS.. 2016. Evolutionary patterns of genic DNA methylation vary across land plants. Nat Plants 2:15222.. [DOI] [PubMed] [Google Scholar]
- Taudt A, Colomé-Tatché M, Johannes F.. 2016. Genetic sources of population epigenomic variation. Nat Rev Genet. 176:319–332. [DOI] [PubMed] [Google Scholar]
- Teschendorff AE, West J, Beck S.. 2013. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet. 22(R1):R7–R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Honeybee Genome Sequencing Consortium. 2006. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443:931–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie S, Charlton J, Clark V, Bird A.. 1997. Methylation of genomes and genes at the invertebrate-vertebrate boundary. Mol Cell Biol. 173:1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Graaf A, et al. . 2015. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc Natl Acad Sci U S A. 112:6676–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidalis A, et al. . 2016. Methylome evolution in plants. Genome Biol. 171:264.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter E, et al. . 2014. Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife 3:e03397.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. 1957. The strategy of genes. London: George Allen & Unwin, Ltd. [Google Scholar]
- Wang Y, et al. . 2006. Functional CpG methylation system in a social insect. Science 3145799:645–647. [DOI] [PubMed] [Google Scholar]
- Weber M, et al. . 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 378:853–862. [DOI] [PubMed] [Google Scholar]
- Wu X, Zhang Y.. 2017. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 18:517–534. [DOI] [PubMed] [Google Scholar]
- Yamada Y, et al. . 2004. A comprehensive analysis of allelic methylation status of CpG islands on human chromosome 21q. Genome Res. 142:247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi SV, Goodisman MAD.. 2009. Computational approaches for understanding the evolution of DNA methylation in animals. Epigenetics 48:551–556. [DOI] [PubMed] [Google Scholar]
- Yin Y, et al. . 2017. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 3566337:eaaj2239.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH.. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 138:335–340. [DOI] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D.. 2010. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 3285980:916–919. [DOI] [PubMed] [Google Scholar]
- Zeng J, Yi SV.. 2010. DNA methylation and genome evolution in honeybee: gene length, expression, functional enrichment covary with the evolutionary signature of DNA methylation. Genome Biol Evol. 2:770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, et al. . 2012. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am J Hum Genet. 913:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Nagrajan HK, Yi SV.. 2014. Fundamental diversity of human CpG islands at multiple biological levels. Epigenetics 94:483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW-L, Chen H.. 2006. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 1266:1189.. [DOI] [PubMed] [Google Scholar]
- Zhang D, et al. . 2010. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 863:411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, et al. . 2015. Genome-wide and single-base resolution DNA methylomes of the sea lamprey (Petromyzon marinus) reveal gradual transition of the genomic methylation pattern in early vertebrates. bioRxiv. 10.1101/033233. [DOI] [Google Scholar]
- Zheng X, et al. . 2013. Transgenerational variations in DNA methylation induced by drought stress in two rice varieties with distinguished difference to drought resistance. PLoS One 811:e80253.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wang G, Qian J.. 2016. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 179:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, et al. . 2013. Charting a dynamic DNA methylation landscape of the human genome. Nature 5007463:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]