

Abstract
Background
The underlying mechanisms and processes that prompt the colonisation of extreme environments, such as caves, constitute major research themes of evolutionary biology and biospeleology. The special adaptations required to survive in subterranean environments (low food availability, hypoxic waters, permanent darkness), and the geographical isolation of caves, nominate cave biodiversity as ideal subjects to answer long-standing questions concerning the interplay amongst adaptation, biogeography, and evolution. The present project aims to examine the phylogeographic patterns exhibited by two sympatric species of surface and cave-dwelling peracarid crustaceans (Asellus aquaticus and Niphargus hrabei), and in doing so elucidate the possible roles of isolation and exaptation in the colonisation and successful adaptation to the cave environment.
Results
Specimens of both species were sampled from freshwater hypogean (cave) and epigean (surface) habitats in Hungary, and additional data from neighbouring countries were sourced from Genbank. Sequencing of mitochondrial and nuclear loci revealed, through haplotype network reconstruction (TCS) and phylogenetic inference, the genetic structure, phylogeographic patterns, and divergence-time estimates of A. aquaticus and N. hrabei surface and cave populations. Contrasting phylogeographic patterns were found between species, with A. aquaticus showing strong genetic differentiation between cave and surface populations and N. hrabei lacking any evidence of genetic structure mediated by the cave environment. Furthermore, N. hrabei populations show very low levels of genetic differentiation throughout their range, which suggests the possibility of recent expansion events over the last few thousand years.
Conclusions
Isolation by cave environment, rather than distance, is likely to drive the genetic structuring observed between immediately adjacent cave and surface populations of A. aquaticus, a predominantly surface species with only moderate exaptations to subterranean life. For N. hrabei, in which populations exhibit a fully ‘cave-adapted’ (troglomorphic) phenotype, the lack of genetic structure suggests that subterranean environments do not pose a dispersal barrier for this surface-cave species.
Electronic supplementary material
The online version of this article (10.1186/s12862-017-1094-9) contains supplementary material, which is available to authorized users.
Keywords: Adaptation, Biospeleology, Exaptation, Evolution, Phylogenetics, Subterranean, Troglomorphy
Background
One of the major recurring themes in evolutionary biology and ecology is discerning the drivers of genetic differentiation and diversity among populations, and their interplay with the environment. These patterns can often be associated to a wide array of geographic and environmental factors that influence population differentiation by means of both adaptive (i.e. selection) and non-adaptive (i.e. genetic drift) processes. As it is often observed in natural systems, reduced gene flow due to geographic distance can often result in distinct patterns of genetic differentiation across a spatial continuum [1]. However, other factors besides geographic distance often affect gene-flow between populations. Biotic and abiotic interactions can impact gene-flow through a variety of mechanisms (such as local adaptation, selection against immigrants, and biased dispersal), which result in populations being isolated by environmental differences [2–4]. Isolation by environment can be identified as a driver for observed patterns of genetic differentiation when there is a positive correlation between genetic and environmental differentiation, but no correlation between genetic differentiation and spatial distances between populations (the latter being an indication of isolation by distance) [4, 5]. It is important to note, however, that isolation by distance and isolation by environment are not mutually exclusive and their effects might be particularly challenging to disentangle when environmental variables and geographic distances co-vary [3]. Caves and other subterranean habitats show marked environmental differences with adjacent surface ecosystems with a sharp boundary, most notably the absence of light and associated ecological and biogeochemical conditions. Such habitat differences can constitute significant barriers to gene flow and population connectivity, which in turn lead to high levels of genetic differentiation even at relatively small spatial scales [6, 7].
In addition to how genetic diversity is distributed across distributional space, the underlying mechanisms and processes that prompt the colonisation of extreme environments, and more specifically caves, constitute one of the major research themes of evolutionary biology [8–10]. There are two major hypotheses generally regarded to explain the transition from surface to subterranean habitats. The adaptive shift hypothesis suggests that the colonisation of subterranean habitats is a result of founder populations actively expanding into and colonising new niches [11], rather than by accidental stranding and persistence in the aphotic zones [12, 13]. The ability of a species to successfully colonise these extreme environments, however, might be mediated by ecological filtering and thus requires specific exaptations to life in darkness [9, 14–16]. Possible exaptations to cave life include morphological (e.g. reduced dependence on vision, elongation of body and appendages) and physiological (e.g. tolerance to oligoxic conditions) characteristics that are already present in numerous species inhabiting benthic and interstitial ecosystems [17–19]. On the other hand, the climatic relict hypothesis states that a species may be forced to adapt to cave life as a result of environmental change that results in uninhabitable conditions on the surface (e.g. glaciation events) [18, 20, 21]. The actual mechanisms that gave rise to contemporary cave populations are likely to be a combination of both processes, and continue to be a subject of investigation. The estimation of phylogenetic relationships from genetic data of cave-dwellers offers the possibility of elucidating the mechanisms and processes that eventually lead to cave colonisations and the persistence of cave populations. This is especially the case when genetic data of both surface and cave-dwelling organisms are coupled with their present-day geographic distributions to infer ancient events (e.g. [22]). However, to fully understand the mechanisms behind cave colonisation events through present-day phylogeographic patterns it is imperative to incorporate approaches that consider the environment and ecology of the species under study, and therefore the underlying factors that ultimately drive their evolution.
The isopod Asellus aquaticus and the amphipod Niphargus hrabei are two aquatic crustacean species that serve as ideal models to explore questions regarding the colonisation, barriers to gene-flow, and evolution of cave fauna. Asellus aquaticus is a widespread species of freshwater isopod commonly found in surface waters throughout Europe [23]. This species is also known to occasionally colonise caves where its populations exhibit “troglomorphic” phenotypes [23, 24]. Troglomorphy can be defined as the set of morphological, physiological, and behavioural characteristics associated with a species transition to life in caves (e.g. enlarged sensory and ambulatory appendages, lack of pigmentation, loss of vision, etc. [25–28]. Contrastingly, the amphipod species N. hrabei is an atypical representative of an almost exclusively cave-dwelling genus that has escaped the confines of the subterranean environment to colonise surface habitats. Its distribution spans an extensive area of central and eastern Europe with geographical ranges of up to 1300 km [29], where it lives in sympatry with A. aquaticus (e.g. in the Danube River and its floodplains). Observations suggests that N. hrabei populations are troglomorphic throughout its distribution in both caves and surface waters (Fig. 1; [28]), perhaps due to the ancient cave-origin of the genus Niphargus. The disposition to inhabit both surface and cave environments, geographical distributions, and life-history characteristics of these two crustacean species make them ideal study organisms to disentangle the effects of isolation by distance and/or isolation by environment and to reveal the mechanisms and processes at play during cave colonisation.
Fig. 1.

Asellus aquaticus displays contrasting phenotypes in and out of the cave, while Niphargus hrabei exhibits the same phenotype in both environments
The present study examines the phylogeographic patterns exhibited by sympatric surface and cave-dwelling populations of A. aquaticus and N. hrabei. We aim to test the hypothesis that isolation by environment drives the patterns of genetic differentiation of surface-exapted A. aquaticus, but not those of cave-exapted N. hrabei, for which isolation by distance is the expected mechanism. The Molnár János thermal cave system and the immediately adjacent Malom Lake (Budapest, Hungary) provide a perfect natural experiment to address questions of cave colonisation, adaptation, and population differentiation. Despite marked environmental differences between hypogean and epigean habitats, both localities are inhabited by the species under study and are hydrologically interconnected with the Danube River Basin, one of the largest and most species-rich natural floodplains in Europe [30, 31]. Therefore, the patterns of genetic differentiation that emerge from this system will allow for a better understanding of the effects of isolation (distance and/or environment) and possible roles of exaptations in the evolution of these cave and surface populations.
Methods
Sample collection
Specimens were sampled from three main sites in Budapest, Hungary: The Molnár János thermal cave system, the adjacent thermal Malom Lake, and the Soroksár branch of the Danube River (Fig. 2). The three sites are interconnected hydrologically and the two study species (Asellus aquaticus and Niphargus hrabei) inhabit all the sites. Additional specimens of A. aquaticus were sourced from other locations in Hungary (Table 1) and sequence data for N. hrabei from neighbouring countries were obtained from GenBank to aid in the analyses [29]. All of the samples were collected using a “Sket bottle” [32] and preserved individually in 99% ethanol for subsequent molecular analyses. All of the samples employed by this study are housed in the Florida International Crustacean Collection (FICC; North Miami, FL, USA). Additional metadata associated with each specimen is securely stored in the collection’s curated electronic database.
Fig. 2.
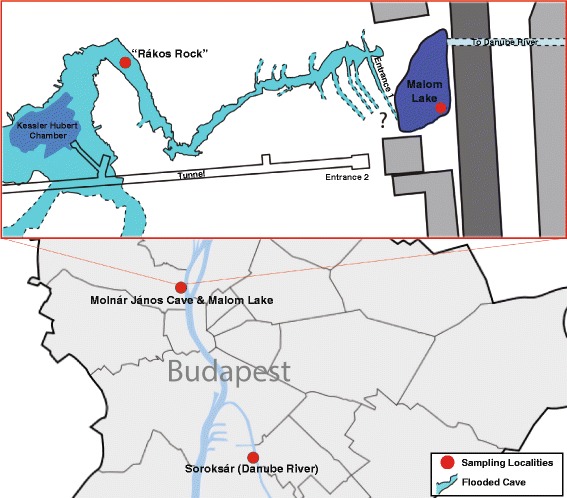
Schematic illustration of our thee main sampling localities within Budapest, Hungary. Red circles indicate exact sites within Molnár János Cave (Rákos Rock) and surface environments (Malom Lake and Danube River [Soroksár])
Table 1.
Specimens, locations, and type of habitat in which the crustacean populations were sampled
| Species | N | Locality | Coordinates | Habitat |
|---|---|---|---|---|
| Asellus aquaticus | 20 | Soroksár, Budapest, Hungary | 47.4360697 N, 19.0878143 E | Epigean |
| 20 | Molnár János Cave, Budapest, Hungary | 47.5181846 N, 19.036064 E | Hypogean | |
| 20 | Malom Lake, Budapest, Hungary | 47.5181167 N, 19.036075 E | Epigean | |
| 4 | Lipót, Hungary | 47.86316 N, 17.458875 E | Epigean | |
| 6 | Polgár, Hungary | 47.869443 N, 21.200598 E | Epigean | |
| 6 | Balatonfenyves, Hungary | 46.65515 N, 17.498538 E | Epigean | |
| 10 | Cserdi, Hungary | 46.06575 N, 17.991012 E | Epigean | |
| Niphargus hrabei | 20 | Soroksár, Budapest, Hungary | 47.4360697 N, 19.0878143 E | Epigean |
| 18 | Molnár János, Budapest Hungary | 47.5181846 N, 19.036064 E | Hypogean | |
| 20 | Malom Lake, Budapest Hungary | 47.5181167 N, 19.036075 E | Epigean | |
| a Niphargus sp. nov. | 13 | Molnár János, Budapest, Hungary | 47.5181846 N, 19.036064 E | Hypogean |
| a Niphargus forroi | 2 | Diabáz Cave, Nagyvisnyo, Hungary | 48.08809 N, 20.46627 E | Hypogean |
aUsed as outgroups for the analyses
DNA extraction and amplification
Genomic DNA was extracted from each specimen’s pereiopods and/or antennae using the commercially available QIAGEN DNeasy Blood and Tissue Kit (Cat. No. 69506). Several mitochondrial and nuclear loci were selected in order to maximise the resolution at the scale of interest (population level). Specifically, for A. aquaticus the loci chosen were: two mitochondrial ribosomal genes (12S and 16S), a mitochondrial protein-coding gene (cytochrome c oxidase subunit I, COI), and a ‘Numt’ (nuclear mitochondrial DNA segment) [33, 34] of NADH dehydrogenase 2 (hereby referred to as PseudoND2). For N. hrabei the sequenced loci included: a mitochondrial ribosomal gene (16S), a mitochondrial protein-coding gene (cytochrome c oxidase subunit I, COI), a nuclear ribosomal gene (internal transcribed spacer, ITS), and a nuclear protein-coding gene (NaK). These loci have proved to be useful in inferring intra- and interspecific relationships across the subphylum Crustacea (12S, 16S, COI, ITS, NaK) [35–39] or were specifically targeted to increase population-level resolution (PseudoND2) [34]. Polymerase Chain Reaction (PCR) amplifications were performed in reactions containing DNA template, sterile non-DEPC treated water, forward and reverse primers, and of GoTaq® Green Master Mix (Promega, M712). The thermal cycling profiles consisted of an initial denaturing step of 1 min at 94 °C, followed by 35–40 cycles of 30 s at 94 °C, annealing for 30s at 48°-62 °C (depending on primer set and species, Additional file 1), 1 min extension at 72 °C, and then a final extension of 7 min at 72 °C. PCR products were sent to Beckman Coulter Genomics (Danvers, MA, USA) for amplicon purification using solid-phase reversible immobilisation (SPRI) beads, and subsequent sequencing reactions using BigDye Terminator v3.1. Post reaction dye terminator removal was done using Agencourt CleanSEQ, after which both forward and reverse strands were sequenced on an Applied Biosystems PRISM 3730xl DNA Analyzer.
Data preparation and analyses of selection
The obtained sequences were visually inspected, quality trimmed, and cleaned manually with Geneious 8.0 [40]. Sequences from specimens heterozygous at nuclear loci were phased with PHASE v2.1 [41, 42] and SeqPHASE [43]. In instances where haplotype reconstruction during phasing resulted in more than one pair of possible sequences, pairs with the highest posterior probabilities were retained for subsequent analyses. To further verify the appropriateness of the chosen loci for phylogenetic inference, tests for selection in protein-coding loci were conducted with MEGA 7 [44]. Due to the small number of substitutions in most of the dataset, Fisher’s exact test of neutrality was chosen as the preferred option to detect evidence of selection [44, 45]. For this purpose, synonymous and non-synonymous substitutions were estimated using the Nei-Gojobori method [46].
Haplotype network reconstruction
Haplotype networks were subsequently built in PopArt 1.7 [47] using TCS (Templeton-Crandall-Sing) Networks [48]. Haplotype networks allow for informative visualisation of genealogical information at shallow divergence levels. Although several methodologies exist for constructing these networks, TCS was chosen for its effectiveness at recovering accurate population-scale phylogeographic patterns even when genetic differentiation is low (e.g. [49–52]). Subsequent to haplotype network reconstruction, the relative frequencies of the mitochondrial haplotypes identified were plotted on maps to visualise their geographic distributions in an intuitive manner.
Phylogenetic analyses
Individual and concatenated gene trees were estimated using Maximum Likelihood (GARLI 2.01 [53]) and Bayesian inference (MrBayes 3.2.6 [54]) methods as implemented in the CIPRES portal [55], after using PartitionFinder v1.1.1 [56] to identify the best-fit models of molecular evolution and partitioning schemes for the dataset (Additional file 1). Maximum Likelihood phylogenetic trees were reconstructed with an initial search for the best tree, using 10 parallel runs via GARLI 2.01. Additionally, 10,000 bootstrap replicates were generated in 40 independent runs to assess nodal support of the best tree. All ML trees were then summarised with a 95% consensus rule and annotated using the SumTrees.py python script from the DendroPy library [57]. Bayesian phylogenetic trees were inferred with the same partitioning scheme as in the ML analyses. MrBayes 3.2.6 was executed with two independent runs, each consisting of 4 chains running for 10 M generations. The MCMC run was sampled every 1000 generations, and a relative burn-in frequency of 25% was set for accurate posterior sampling. After assessing for convergence (Tracer v1.6 [58], the SumTrees.py script was again invoked to extract the Maximum Clade Credibility Tree (MCCT) and to annotate the phylogenetic trees’ nodal support as posterior probabilities. Further, population trees were estimated under a multi-locus coalescent model using *BEAST (BEAST 2.4.0 [59]). Intraspecific divergence times of A. aquaticus and N. hrabei populations were concurrently estimated using molecular clock rate calibrations for peracarid crustaceans’ COI (1.25% of substitutions per site per million years [60]; and between 0.34% and 0.76% of substitutions per site per Myr [61]), which were previously estimated for taxa closely related to our species of study (Stenasellidae and Niphargiidae). All of *BEAST and BEAST analyses were run in triplicate at Florida International University’s High Performance Computing cluster (Panther) for 200 M generations after which they were assessed for convergence using Tracer v1.6. The *BEAST speciation models for which there was no evidence for convergence were discarded. The runs using a Yule model of speciation as a tree prior with a strict molecular clock calibration were retained for subsequent analyses. After discarding 25% of the sampled trees as burn-in, nodal support was annotated as posterior probabilities on the MCCT of each population tree analysis (TreeAnnotator; [59]. For each species, population trees that included divergence time estimates were plotted using the geoscale.phylo function of the R package ‘strap’ [62]. These were then georeferenced on precomputed maps with custom scripts that made use of the R package ‘phytools’ [63].
Genealogical Sorting Indices (gsi) were calculated using the R package ‘genealogicalSorting’ to quantify intraspecific lineage divergence, which allowed for the evaluation of monophyly in each population [64, 65]. Lastly, a modified approach to calculate gsi (pairwise-gsi or pwgsi) was used to independently quantify the divergence of every population-pair [66]. This modified approach thus prevented possible bias and false-positives that could have arisen as by-products of the trees’ topologies outside the main groups of interest [66]. Outgroup populations of N. hrabei were excluded from this final analysis due to sample size requirements of the pwgsi approach. Nonetheless, this exclusion bears no impact on the comparisons amongst the target populations (Molnár János Cave, Malom Lake, and the Danube River’s Soroksár).
Demographic history
Additionally, we sought to better understand the demographic histories of A. aquaticus and N. hrabei, as reflected in their sequence data, by estimating changes in their population sizes over time. For this purpose, we conducted Extended Bayesian skyline (EBS) analyses [67]) as implemented in BEAST [59]. Extended Bayesian skyline analyses allow for the incorporation of multi-locus datasets to estimate population history over time along with an assessment of the estimations’ uncertainty [67]. The parameters employed for these analyses were maintained as in the previous successful BEAST runs, with the exception of the priors associated to the species tree, which was set to “Coalescent Extended Bayesian Skyline”. These analyses were also run in triplicate at Florida International University’s High Performance Computing cluster (Panther) for 200 M generations after which they were assessed for convergence using Tracer v1.6. EBS run logs were subsequently combined, after discarding 25% as burn-in, and the demographic histories of both species were plotted using custom R scripts for ease of visualisation and further inferences.
Results
DNA sequences and data deposition
A total of ~1690 and ~2757 base pairs (bp) of nucleotide sequence data were recovered for A. aquaticus (81 sequences for 12S, 83 for 16S, 76 for COI, and 84 for PseudoND2) and N. hrabei (55 sequences for 16S, 54 for COI, 51 for ITS, and 58 for NaK) respectively. All sequence data from this project were curated, annotated with their respective metadata, and deposited in the NCBI’s Genbank database to allow for their dissemination and future use by other researchers (See Additional file 1 for accession numbers).
Testing for neutrality of selected loci
Fisher’s exact test of neutrality was employed to determine the suitability of the selected loci for phylogeographic inference by ensuring that they are not being subject to selective pressures. The probability (P) of rejecting the null hypothesis of strict-neutrality in favor of the alternative hypothesis of positive selection was larger than 0.05 for all loci in both species, and therefore not considered significant at an alpha value (α) of 5%. The chosen loci were therefore deemed suitable for subsequent analyses.
Haplotype network reconstruction
Analyses of haplotype networks display evident genetic structuring in surface and cave A. aquaticus, with particularly distinct haplotypes differentiating Molnár János Cave’s population from Malom Lake’s despite their spatial proximity (Fig. 3). The haplotypes found in Molnár János Cave and Malom Lake are exclusive to each locality, with the exception of a single “surface-phenotype” cave specimen that shared a haplotype found in both Malom Lake and the western population of Lipót (Figs. 3, 4). This strong differentiation and lack of shared haplotypes between the cave’s and adjacent surface populations is not the case for N. hrabei, where Malom Lake’s and Molnár János Cave’s mtDNA haplotypes are the same and no genetic structuring is evident (Fig. 5). Mitochondrial haplotypes (COI and 16S) of N. hrabei are not only closely related, but also found widely distributed throughout its range (Figs. 5, 6).
Fig. 3.
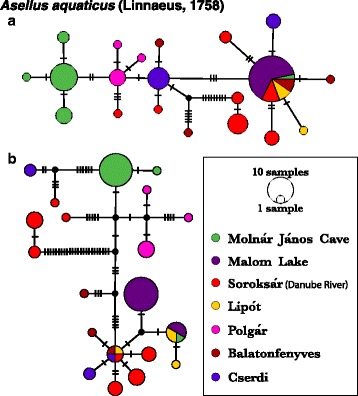
Haplotype networks of Asellus aquaticus: nuclear (a- PseudoND2) and mitochondrial (b- 16S, 12S, and COI) loci. Node diameter and annotation denote sample sizes, while hatch marks represent mutational steps between haplotypes. Colours represent sampling locality, as illustrated in the legend
Fig. 4.
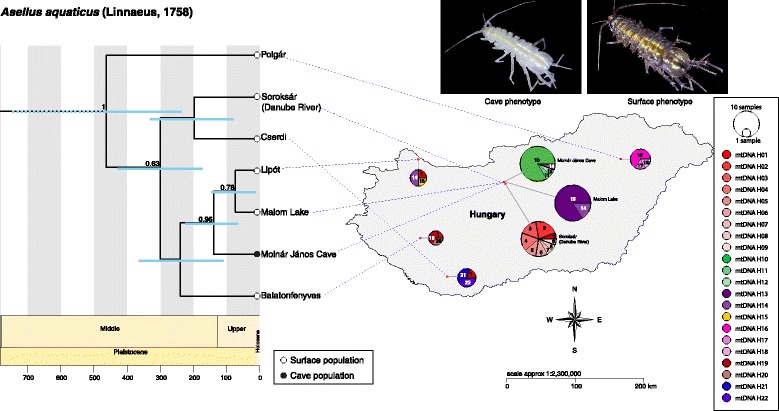
Divergence time estimates (x axis in thousands of years) of Asellus aquaticus populations (calculated with a multi-locus coalescent model in *BEAST; outgroups not shown) and the distribution of its populations with relative mtDNA haplotype frequencies throughout Hungary. Phylogenetic and population tree analyses support the inclusion of the cave phenotype as part of the species, but with evident population structuring as a result of the cave environment
Fig. 5.
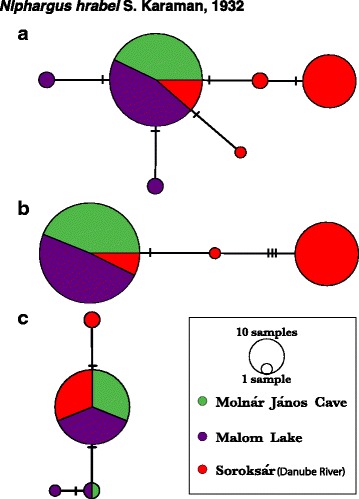
Haplotype networks of Niphargus hrabei: nuclear (a- ITS; b- NaK) and mitochondrial (c- 16S and COI) loci. Node diameter and annotation denote sample sizes (alleles in the case of nuclear genes), while hatch marks represent mutational steps between haplotypes. Colours represent sampling locality, as illustrated in the legend
Fig. 6.
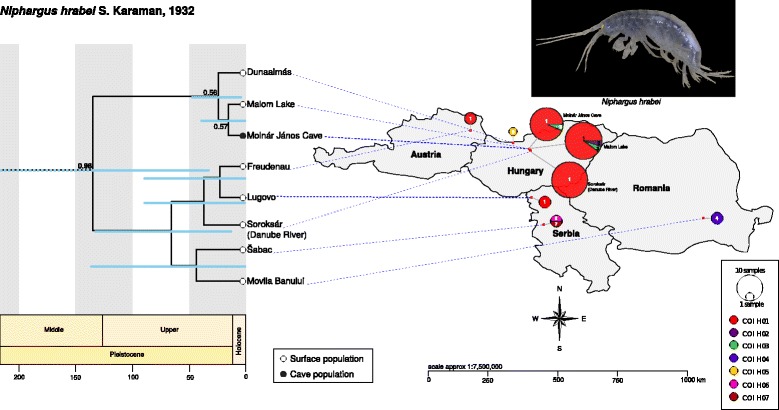
Divergence time estimates (x axis in thousands of years) of Niphargus hrabei populations (calculated with a multi-locus coalescent model in *BEAST; outgroups not shown) and the distribution of its populations with relative COI haplotype frequencies in our three main Hungarian sites and neighbouring populations. Phylogenetic and population tree analyses do not support any evident genetic structuring between cave and surface populations
Phylogeographic and genealogical sorting analyses
The patterns observed in the haplotype network reconstructions are reflected in and confirmed by the concatenated phylogenetic trees (tree-files are available in the figshare respository at: https://doi.org/10.6084/m9.figshare.5660542.v1). The final Maximum Likelihood and Bayesian trees for each species are nearly identical, with the exception of a few unsupported nodes, and are concordant with the *BEAST population trees estimated with all the sequenced loci (A. aquaticus, Fig. 4; N. hrabei, Fig. 6; see Additional file 1 for evolutionary model selection details). Furthermore, these population trees show that cave and surface populations of A. aquaticus diverged from each other at least 60 k years ago (Table 2). There is no evidence for genetic structuring between cave and surface populations of N. hrabei, and the phylogenetic split between these is not supported. The Pairwise Genealogical Sorting Index (pwgsi) estimates follow patterns similar to those of the population tree topologies recovered (A. aquaticus, Table 3; N. hrabei, Table 4). In A. aquaticus, the distinction between the Molnár János Cave population and the adjacent Malom Lake is clear despite its proximity (pwgsi = 1.00, p < 0.001), with the former having higher affinities to the south-western population of Balatonfenyves (pwgsi = 0.22, p < 0.001). The pwgsi between Molnár János Cave’s and Malom Lake’s N. hrabei populations on the other hand, provides no evidence of genealogical differentiation between these (pwgsi = 0.14, p = 0.52). Nevertheless, both populations display a statistically significant but modest degree of reciprocal monophyly when compared to the next geographically closest population, the Soroksár branch of the Danube River (pwgsi = 0.46 and 0.48 respectively, both p < 0.001).
Table 2.
Divergence time estimations between Molnár János Cave and phylogenetically closest surface populations of the peracarid crustaceans under study (thousands of years [95% H.P.D.])
| Molecular Clock Rate | ||
|---|---|---|
| Species | Ketmaier et al. (2003) | Lefébure et al. (2006) |
| Asellus aquaticus | 60.81 (28.68, 96.53) | 139.21 (67.07, 222.26) |
| Niphargus hrabei | Surface/cave split not supported | Surface/cave split not supported |
Table 3.
Pairwise Genealogical Sorting Index estimates for each population pair of Asellus aquaticus
| Bayesian Phylogeny | Maximum Likelihood Phylogeny | ||||
|---|---|---|---|---|---|
| Population 1 | Population 2 | pwgsi | p-value | pwgsi | p-value |
| Molnár János Cave | Soroksár (Danube) | 0.71 | < 0.001 | 0.68 | < 0.001 |
| Molnár János Cave | Malom Lake | 1.00 | < 0.001 | 1.00 | < 0.001 |
| Molnár János Cave | Lipót | 0.46 | < 0.001 | 0.35 | < 0.001 |
| Molnár János Cave | Polgár | 0.89 | < 0.001 | 0.89 | < 0.001 |
| Molnár János Cave | Balatonfenyves | 0.22 | < 0.001 | 0.22 | < 0.001 |
| Molnár János Cave | Cserdi | 0.56 | < 0.001 | 0.57 | < 0.001 |
| Soroksár (Danube) | Malom Lake | 1.00 | < 0.001 | 1.00 | < 0.001 |
| Soroksár (Danube) | Lipót | 0.49 | < 0.001 | 0.43 | < 0.001 |
| Soroksár (Danube) | Polgár | 0.89 | < 0.001 | 0.89 | < 0.001 |
| Soroksár (Danube) | Balatonfenyves | 0.17 | 0.003 | 0.13 | 0.001 |
| Soroksár (Danube) | Cserdi | 0.46 | < 0.001 | 0.43 | < 0.001 |
| Malom Lake | Lipót | 1.00 | < 0.001 | 1.0 | < 0.001 |
| Malom Lake | Polgár | 1.00 | < 0.001 | 1.0 | < 0.001 |
| Malom Lake | Balatonfenyves | 1.00 | < 0.001 | 1.0 | < 0.001 |
| Malom Lake | Cserdi | 1.00 | < 0.001 | 1.0 | < 0.001 |
| Lipót | Polgár | 1.00 | < 0.001 | 1.0 | < 0.001 |
| Lipót | Balatonfenyves | 0.38 | < 0.001 | 0.23 | < 0.001 |
| Lipót | Cserdi | 0.47 | < 0.001 | 0.47 | < 0.001 |
| Polgár | Balatonfenyves | 0.78 | < 0.001 | 0.78 | < 0.001 |
| Polgár | Cserdi | 0.83 | < 0.001 | 0.83 | < 0.001 |
| Balatonfenyves | Cserdi | 0.15 | 0.012 | 0.15 | < 0.001 |
P-values assess significance that exclusive ancestry observed for every population pair is greater than that which would be observed at random
Table 4.
Pairwise Genealogical Sorting Index estimates for each population pair of Niphargus hrabei
| Bayesian Phylogeny | Maximum Likelihood Phylogeny | ||||
|---|---|---|---|---|---|
| Population 1 | Population 2 | pwgsi | p-value | pwgsi | p-value |
| Soroksár (Danube) | Molnár János Cave | 0.46 | <0.001 | 0.15 | <0.001 |
| Soroksár (Danube) | Malom Lake | 0.48 | <0.001 | 0.18 | <0.001 |
| Molnár János Cave | Malom Lake | 0.14 | 0.521 | 0.06 | 0.005 |
P-values assess significance that exclusive ancestry observed for every population pair is greater than that which would be observed at random
Demographic history
We conducted Extended Bayesian Skyline (EBS) analyses [67]), as implemented in BEAST [59], to evaluate the demographic history of our two study species and investigate if there is any evidence of possible climate-associated population changes. The EBS plot for A. aquaticus shows a gradual population contraction reaching a minimum approximately between 100 and 200 thousand years ago and a gradual recovery thereafter (Fig. 7). Contrastingly, EBS analyses for N. hrabei point to a sharper decline beginning at a later date (~ 60 thousand years ago). The 95% H.P.D. interval suggests that an evident population bottleneck followed by a rapid expansion occurred approximately 10 thousand years before present (Fig. 8), roughly corresponding to the end of the Würm glaciation (~ 11,700 years ago).
Fig. 7.
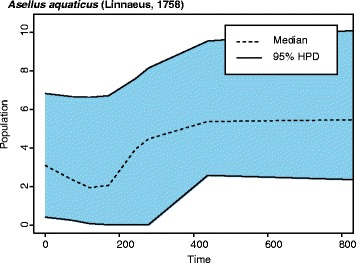
An Extended Bayesian Skyline Plot illustrates the demographic history of the sampled Hungarian Asellus aquaticus populations over the last 800,000 years. The x-axis represents time before present in thousands of years, while the y-axis denotes the estimated population size (θ) assuming a generation time of one year
Fig. 8.
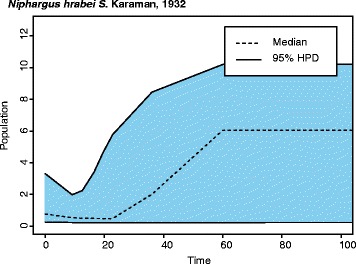
An Extended Bayesian Skyline Plot of Niphargus hrabei’s demographic history, from 100,000 years ago to today, depicts a possible genetic bottleneck at approximately 10,000 years ago. The x-axis represents time before present in thousands of years, while the y-axis denotes the estimated population size (θ) assuming a generation time of one year
Discussion
The most intriguing finding of the present study is the population differentiation between cave and surface populations of Asellus aquaticus, a pattern which is not reflected in Niphargus hrabei. Here, we will discuss the phylogeographic patterns in the light of alternate isolation mechanisms (geographic distance vs. environment). Second, we focus on the Molnár János Cave system, and discuss its potential role as a climatic refugium together with the role of exaptation in successful cave colonisation. Lastly, we conclude by illustrating future possibilities and directions for research in this emergent study system.
Contrasting phylogeographies: Isolation by distance, environment or both?
Our first objective was to resolve the phylogeographic patterns between sympatric surface and cave populations of two crustaceans in order to investigate if the cave environment is acting as a barrier for dispersal and connectivity of populations. The A. aquaticus populations throughout Hungary are genetically diverse with each population being comprised mostly by distinct, but closely related, mitochondrial haplotypes exclusive to their respective localities. Phylogenetic and population tree analyses do provide strong statistical support for the genetic differentiation between Molnár János Cave’s subterranean population and its epigean counterparts. Our results further suggest that the observed genetic and phenotypic differentiation between cave and surface isopods result from the cave acting as an isolating environment. Nevertheless, despite this differentiation, the Molnár János Cave A. aquaticus population still falls well within the species range for A. aquaticus in a phylogenetic context (Fig. 4). Furthermore, the presence of a single haplotype (mtDNA H01) in all of the western Hungarian populations sampled for this study suggests that movement over large distances does occur, suggesting a smaller role for geographical distance (vs. environment) as a driver of genetic differentiation (Fig. 4). The relatively high haplotypic diversity found in the Danube River (Soroksár, in comparison to the other localities examined) further supports its role as a dispersal avenue for isopods inhabiting surface waters. Effective dispersal in surface environments, but not into the cave, is further evident in our phylogenetic analyses by the lack of nodal support for the differentiation of the surface populations.
Interestingly, movement of individuals from Malom Lake into the cave was detected by the sampling of a single isopod with both surface haplotype (mtDNA H14) and phenotype (Figs. 3 & 4). Personal observations confirm that, in rare instances, surface isopods can be found in the aphotic zone well within Molnár János Cave. However, surface isopods do not persist in the cave habitat and the lack of shared mitochondrial and nuclear haplotypes suggests infrequent or non-existent admixture between these two genetically close but phenotypically distinct populations (Figs. 3 & 4). A similar pattern was observed in Slovenian and Romanian A. aquaticus where no population connectivity was found between troglomorphic cave isopods and other nearby populations from surface waters [24]. This recurrent pattern could be explained by a variety of mechanisms ultimately driven by the environmental differences between subterranean and epigean habitats [2–4]. It is thus feasible that surface individuals who wander into the cave are outcompeted by the troglomorphic resident population (i.e. competitive exclusion) before successful breeding takes place, and/or that hybrid individuals are at a significant fitness disadvantage that prevents their genes from persisting in the cave population [3]. Investigating and understanding which exact mechanisms might be at play in the Molnár János Cave system undoubtedly constitutes an important question to address in future studies.
Unlike that of A. aquaticus, haplotype network reconstruction and population tree analyses of N. hrabei show no evidence of genetic structuring between surface and cave populations (Figs. 5 & 6). In fact, haplotypic diversity seems to be relatively low throughout its range (Fig. 6). The only phylogeographic pattern recovered is the segregation of Hungarian populations as a distinct clade (Fig. 6). However, geography does not explain the inclusion of Danube River (Soroksár) individuals within the clade comprised by the Austrian, Serbian, and Romanian specimens. Low genetic diversity, weak genetic structuring despite large geographical distances, and lack of statistical support for any of the populations sampled, suggest that this species’ modern populations resulted from a recent expansion event and do not provide any clear evidence for isolation by distance nor environment. This is further evidenced by the results of our EBS analyses that suggest a population bottleneck for N. hrabei at approximately 10 thousand years ago followed by a rapid expansion (Fig. 8). It is important to note that sampling for some of the localities examined was limited and this could have a negative effect on statistical support for those populations. Nevertheless, the three main target populations of the present study (Molnár János cave, Malom Lake and Danube River [Soroksár]) were sufficiently sampled to adequately evaluate the population structure of surface vs. adjacent cave populations. Therefore, for N. hrabei, whose populations exhibit a fully ‘troglomorphic’ phenotype in all environments, the lack of genetic structure at this scale suggests that subterranean environments do not pose a barrier for this species. Further analyses employing high-resolution data (e.g. genome-wide SNPs) from next-generation sequencing methodologies would undoubtedly be of advantage to clarify whether this lack of genetic structure is truly due to unimpeded movement in and out of the cave or a by-product of N. hrabei’s recent colonisation of the habitats under investigation [10].
The Molnár János thermal cave system: A climatic refugium and a possible role for exaptation
Divergence-time estimates, calibrated with peracarid COI molecular clock rates [60, 61], place the divergence of A. aquaticus populations from Molnár János Cave and Malom Lake at approximately 60,000 to 140,000 years ago (Fig. 4). This relatively recent split falls within the Pleistocene, a period of time during which severe climatic changes associated with glaciation events impacted the geographical distributions of numerous taxa across the globe [21, 68]. Even though Hungary was not directly glaciated, hydrological changes in the region would have altered the viability of this crustacean’s surface populations and shifted their latitudinal distributions [21, 68].
Caves have been known to act as refuges during rapid environmental changes on the surface [21, 23, 69–71]. Molnár János Cave’s waters are exclusively fed by thermomineral springs with a constant water temperature (~ 24 °C), which increases its suitability as a refugium for organisms with exaptations that help them subsist in cave environments. Being a moderately exapted species, it is possible that A. aquaticus would have been able to take refuge in the cave where it remained isolated from surface populations. This isolation eventually resulted in the emergence of troglomorphic phenotypes via adaptive and neutral processes. Upon cessation of this isolation, it is possible that competitive exclusion prevented new and/or returning surface populations from successfully invading the cave and vice versa. This mechanism would be in accordance with the observed phylogeographic patterns. Extended Bayesian Skyline Plot analyses illustrate a population decline for A. aquaticus and the possibility of the aforementioned events occurring approximately 100–200 thousand years ago (Fig. 7). It is also possible that A. aquaticus may have had a constant food source independent from the surrounding surface environment, as bacterial communities in Molnár János Cave, upon which A. aquaticus feeds (pers. obs.), have been shown to thrive via chemoautotrophic processes [72].
Niphargus hrabei is likely to have colonised Molnár János Cave thousands of years later, as suggested by the divergence-time estimates (Fig. 6). Niphargus hrabei cave and surface populations appear to be panmictic and show no evidence of isolation by the cave environment or of competitive exclusion within the cave. They have successfully colonised the cave from surface populations and appear to have no limitations with dispersing from and into cave environments. Niphargus hrabei’s facility for dispersal and its exceptional adaptability to markedly different habitats is reflected by an atypical large distributional range [29]. This adaptability is also evidenced by its unimpeded presence despite putative competitors in Molnár János Cave: the isopod A. aquaticus and two other species of Niphargus that are yet to be described (pers. obs.). However, an alternative explanation would be that older populations of N. hrabei once inhabited the Molnár János Cave, but were outcompeted by the obligate cave-dwellers in absence of migrants from surface waters. In fact, our EBS analyses show an abrupt population decline with a bottleneck and subsequent expansion at approximately 10 thousand years ago (Fig. 8) that roughly coincides with the end of the Würm glaciation, event which took place approximately between 115,000 and 11,700 years ago [73]. The end of the Würm glaciation is known for great climatic variability [74] with major temperature fluctuations that played a significant role in shaping the modern distributional patterns of other European species ([73, 75, 76]). Nevertheless, niphargiid coexistence in other caves has also been previously explained by evolutionary and ecological processes such as niche differentiation [77]. Understanding the exact mechanisms by which these processes take place continues to be an important research theme in evolutionary biology and biospeleology.
Conclusions
The Molnár János Cave system and its inhabitants serve as ideal models for phylogeographic and biospeleological studies in an evolutionary context. While the present study has provided significant insights into the phylogeographic histories of two species and their transition into and out of caves, important questions remain to be answered. Further analyses will greatly aid in the understanding of the exact causes of the observed patterns, as well as in the elucidation of the mechanisms by which exaptations have helped them thrive in such extreme environments. An integrative approach incorporating different sources of molecular data (e.g. genomic, transcriptomic, epigenetic, etc.) has been initiated and will be of definitive advantage to address these outstanding questions [10]. Advances in modern molecular methodologies will undoubtedly enable future high-resolution studies of the adaptive processes that underlie the contrasting phylogeographic patterns revealed by this study.
Acknowledgments
We would like to thank Dr. Péter Borza for his assistance in the collection of specimens of Asellus aquaticus from Hungarian localities. We also extend our gratitude to Attila Hosszú, Szabolcs Storozynski, Péter Zsoldos, and Zoltán Bauer for their help as support divers during the collection of specimens in Molnár János Cave. The authors would like to acknowledge the Instructional & Research Computing Center (IRCC) at Florida International University for providing High Performance Computing resources that have contributed to the research results reported within this article. This is contribution #69 of the Marine Education and Research Center of the Institute for Water and the Environment at Florida International University.
Funding
J.P.M. was supported by the Philip M. Smith Graduate Research Grant for Cave and Karst Research from the Cave Research Foundation and The Crustacean Society Scholarship in Graduate Studies. This work was partially funded by the National Science Foundation (Doctoral Dissertation Improvement Grant #1701835) awarded to J.P.M and H.B.G. G.B. and G.H. were supported by the Hungarian Scientific Research Fund (#K-105517), the Hungarian Ministry of Human Capacities (#ÚNKP-17-3), and by the National Research, Development and Innovation Fund forinternational cooperation (#SNN 125627).
Availability of data and materials
The dataset supporting the conclusions of this article is available in the NCBI’s Genbank database. Accession numbers are detailed in the Additional file 1 included with this publication. Phylogenetic trees resulting from the study are available in the figshare respository at: https://doi.org/10.6084/m9.figshare.5660542.v1.
Abbreviations
- DEPC
Diethylpyrocarbonate
- DNA
Deoxyribonucleic acid
- EBS
Extended Bayesian Skyline
- FICC
Florida International Crustacean Collection
- gsi
Genealogical Sorting Index
- HPD
Highest Posterior Density
- MCCT
Maximum Clade Credibility Topology
- MCMC
Markov chain Monte Carlo
- ML
Maximum Likelihood
- mtDNA
Mitochondrial DNA
- NCBI
National Center for Biotechnology Information
- Numt
Nuclear mitochondrial DNA segment
- PCR
Polymerase Chain Reaction
- pwgsi
Pairwise Genealogical Sorting Index
- SPRI
Solid Phase Reversible Immobilization
- TCS
Templeton-Crandall-Sing
Additional file
Additional tables with information regarding evolutionary models selected for phylogenetic analyses, the genetic loci sequence and primers employed, accession numbers for data deposited in GenBank, and Genealogical Sorting Index estimates for all populations of Asellus aquaticus and Niphargus hrabei. (DOCX 56 kb)
Authors’ contributions
JPM and GB conceived the project and contributed equally towards its completion; JPM, GB, GH, and HBG designed the experiments; GB collected the samples; JPM, GB, and BW produced the data; JPM analysed the data and led the writing; GH and HBG funded the project and provided conceptual advice. JPM, GB, BW, GH, and HBG discussed the results and implications, interpreted the data, participated in the writing, and provided critical contributions towards the final version of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12862-017-1094-9) contains supplementary material, which is available to authorized users.
Contributor Information
Jorge L. Pérez-Moreno, Email: jpere645@fiu.edu
Gergely Balázs, Email: balazsgrg@gmail.com.
Blake Wilkins, Email: bwilk010@fiu.edu.
Gábor Herczeg, Email: gaborherczeg@caesar.elte.hu.
Heather D. Bracken-Grissom, Email: hbracken@fiu.edu
References
- 1.Wright S. Isolation by distance. Genetics. 1943;28:114–138. doi: 10.1093/genetics/28.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nosil P, Egan SP, Funk DJ. Heterogeneous genomic differentiation between walking-stick ecotypes: “isolation by adaptation” and multiple roles for divergent selection. Evolution. 2008;62:316–336. doi: 10.1111/j.1558-5646.2007.00299.x. [DOI] [PubMed] [Google Scholar]
- 3.Wang I, Bradburd G. Isolation by environment. Ecol Lett. 2014:5649–62. [DOI] [PubMed]
- 4.Manthey JD, Moyle RG. Isolation by environment in white-breasted nuthatches (Sitta carolinensis) of the Madrean archipelago sky islands: a landscape genomics approach. Mol Ecol. 2015;24:3628–3638. doi: 10.1111/mec.13258. [DOI] [PubMed] [Google Scholar]
- 5.Orsini L, Vanoverbeke J, Swillen I, Mergeay J, De Meester L. Drivers of population genetic differentiation in the wild: isolation by dispersal limitation, isolation by adaptation and isolation by colonization. Mol Ecol. 2013;22:5983–5999. doi: 10.1111/mec.12561. [DOI] [PubMed] [Google Scholar]
- 6.Mathieu J, Jeannerod F, Hervant F, Kane TC. Genetic differentiation of Niphargus rhenorhodanensis (Amphipoda) from interstitial and karst environments. Aquat Sci. 1997;59:39–47. doi: 10.1007/BF02522549. [DOI] [Google Scholar]
- 7.Tobler M, Dewitt TJ, Schlupp I. García de León FJ, Herrmann R, Feulner PGD, et al. toxic hydrogen sulfide and dark caves: phenotypic and genetic divergence across two abiotic environmental gradients in Poecilia mexicana. Evolution. 2008;62:2643–2659. doi: 10.1111/j.1558-5646.2008.00466.x. [DOI] [PubMed] [Google Scholar]
- 8.Juan C, Guzik MT, Jaume D, Cooper SJB. Evolution in caves: Darwin’s “wrecks of ancient life” in the molecular era. Mol Ecol. 2010;19:3865–3880. doi: 10.1111/j.1365-294X.2010.04759.x. [DOI] [PubMed] [Google Scholar]
- 9.Benvenuto C, Knott B, Weeks S. Crustaceans of extreme environments. In: Thiel M, Watling L, editors. The natural history of the Crustacea. New York: Oxford University Press; 2015. pp. 379–417. [Google Scholar]
- 10.Pérez-Moreno JL, Iliffe TM, Bracken-Grissom HD. Life in the underworld: Anchialine cave biology in the era of speleogenomics. Int J Speleol. 2016;45:149–170. doi: 10.5038/1827-806X.45.2.1954. [DOI] [Google Scholar]
- 11.Rouch R, Danielopol DL. origine de la fauna aquatique souterraine, entre le paradigme du refuge et le modèle de la colonization active. Stygologia. 1987;3:345–372. [Google Scholar]
- 12.Kane TC, Culver DC, Jones RT. Genetic structure of morphologically differentiated populations of the amphipod Gammarus minus. Evolution. 1992;46:272–278. doi: 10.1111/j.1558-5646.1992.tb02002.x. [DOI] [PubMed] [Google Scholar]
- 13.Kano Y, Kase T. Genetic exchange between anchialine cave populations by means of larval dispersal: the case of a new gastropod species Neritilia cavernicola. Zool Scr. 2004;33:423–437. doi: 10.1111/j.0300-3256.2004.00159.x. [DOI] [Google Scholar]
- 14.Gould SJ, Vrba ES. Exaptation—a missing term in the science of form. Paleobiology. 1982;8:4–15. doi: 10.1017/S0094837300004310. [DOI] [Google Scholar]
- 15.Gibert J, Deharveng L. Subterranean ecosystems : a truncated functional biodiversity. Bioscience. 2002;52:473–481. doi: 10.1641/0006-3568(2002)052[0473:SEATFB]2.0.CO;2. [DOI] [Google Scholar]
- 16.Gilgado JD, Ledesma E, Cuesta E, Arrechea E, de la Vega JL Z, Sánchez-Ruiz A, et al. Dima assoi Pérez Arcas 1872 (Coleoptera: Elateridae): from montane to hypogean life. An example of exaptations to the subterranean environment. Annales de la Société entomologique de France (N.S.) 2014;50:264–271. doi: 10.1080/00379271.2014.981421. [DOI] [Google Scholar]
- 17.Humphreys W, Eberhard S. Subterranean fauna of Christmas Island. Indian Ocean Helictite. 2001;37:59–74. [Google Scholar]
- 18.Leys R, Watts CH, Cooper SJ, Humphreys W. Evolution of subterranean diving beetles (Coleoptera: Dytiscidae: Hydroporini, Bidessini) in the arid zone of Australia. Evolution. 2003;57:2819–2834. doi: 10.1111/j.0014-3820.2003.tb01523.x. [DOI] [PubMed] [Google Scholar]
- 19.Suárez-Morales E, Iliffe TMA. New Exumella (Crustacea: Copepoda: Ridgewayiidae) from anchialine waters of the western Caribbean, with comments on regional biogeography. Bull Mar Sci. 2005;77:409–423. [Google Scholar]
- 20.Barr TC, Holsinger JR. Speciation in cave faunas. Annu Rev Ecol Syst. 1985;16:313–337. doi: 10.1146/annurev.es.16.110185.001525. [DOI] [Google Scholar]
- 21.Bryson RWJ, Prendini L, Savary WE, Pearman PB. Caves as microrefugia: Pleistocene phylogeography of the troglophilic north American scorpion Pseudouroctonus reddelli. BMC Evol Biol. 2014;14 [DOI] [PMC free article] [PubMed]
- 22.Villacorta C, Jaume D, Oromí P, Juan C. Under the volcano: phylogeography and evolution of the cave-dwelling Palmorchestia hypogaea (Amphipoda, Crustacea) at La Palma (Canary Islands) BMC Biol. 2008;6:7. doi: 10.1186/1741-7007-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verovnik R, Sket B, Trontelj P. The colonization of Europe by the freshwater crustacean Asellus aquaticus (Crustacea: isopoda) proceeded from ancient refugia and was directed by habitat connectivity. Mol Ecol. 2005;14:4355–4369. doi: 10.1111/j.1365-294X.2005.02745.x. [DOI] [PubMed] [Google Scholar]
- 24.Konec M, Prevorčnik S, Sarbu SM, Verovnik R, Trontelj P. Parallels between two geographically and ecologically disparate cave invasions by the same species, Asellus aquaticus (Isopoda, Crustacea) J Evolutionary Biology. 2015;28:n/a–a. doi: 10.1111/jeb.12610. [DOI] [PubMed] [Google Scholar]
- 25.Turk S, Sket B, Sarbu S. Comparison between some epigean and hypogean populations of Asellus aquaticus (Crustacea: isopoda: Asellidae) Hydrobiologia. 1996;337:161–170. doi: 10.1007/BF00028517. [DOI] [Google Scholar]
- 26.Li H, Cooper R. Spatial familiarity in the blind cave crayfish, Orconectes australis packardi. Crustaceana. 2001;74:417–433. doi: 10.1163/156854001750243018. [DOI] [Google Scholar]
- 27.Li H, Cooper RL. The effect of ambient light on blind cave crayfish: social interactions. J Crustac Biol. 2002;22:449–458. doi: 10.1163/20021975-99990252. [DOI] [Google Scholar]
- 28.Mejía-Ortíz LM, Hartnoll RA. New use for useless eyes in cave crustaceans. Crustaceana. 2006;79:593–600. doi: 10.1163/156854006777584313. [DOI] [Google Scholar]
- 29.Copilas-Ciocianu D, Fišer C, Borza P, Balázs G, Angyal D, Petrusek A. Low intraspecific genetic divergence and weak niche differentiation despite wide ranges and extensive sympatry in two epigean Niphargus species (Crustacea: Amphipoda). Zool J Linnean Soc. 2017;
- 30.Schiemer F. Conservation of biodiversity in floodplain rivers. River Systems. 1999;11:423–438. doi: 10.1127/lr/11/1999/423. [DOI] [Google Scholar]
- 31.Danielopol DL, Pospisil P. Hidden biodiversity in the groundwater of the Danube flood plain national park (Austria) Biodivers Conserv. 2001;10:1711–1721. doi: 10.1023/A:1012098706986. [DOI] [Google Scholar]
- 32.Chevaldonné P, Sket B, Marschal C, Lejeusne C, Calado R. Improvements to the “Sket bottle”: a simple manual device for sampling small crustaceans from marine caves and other cryptic habitats. J Crustac Biol. 2008;28:185–188. doi: 10.1651/07-2923R.1. [DOI] [Google Scholar]
- 33.Lopez JV, Yuhki N, Masuda R, Modi W, O’Brien SJ. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J Mol Evol. 1994;39:174–190. doi: 10.1007/BF00163806. [DOI] [PubMed] [Google Scholar]
- 34.Miraldo A, Hewitt GM. Dear PH, Paulo OS, Emerson BC. Numts help to reconstruct the demographic history of the ocellated lizard ( Lacerta lepida ) in a secondary contact zone. Mol Ecol. 2012;21:1005–1018. doi: 10.1111/j.1365-294X.2011.05422.x. [DOI] [PubMed] [Google Scholar]
- 35.Bracken HD, De Grave S, Toon A, Felder DL, Crandall KA. Phylogenetic position, systematic status, and divergence time of the Procarididea (Crustacea: Decapoda) Zool Scr. 2010;39:198–212. doi: 10.1111/j.1463-6409.2009.00410.x. [DOI] [Google Scholar]
- 36.Bracken H, Grave SD, Felder D. Phylogeny of the infraorder Caridea based on mitochondrial and nuclear genes (Crustacea: Decapoda) In: Martin J, Crandall K, Felder DL, editors. Decapod crustacean Phylogenetics. Boca Raton: Taylor and Francis/CRC Press; 2009. pp. 274–298. [Google Scholar]
- 37.Bracken-Grissom HD, Enders T, Jara CG, Crandall KA. Molecular diversity of two freshwater anomuran crab species in southern Chile (Decapoda: Anomura: Aeglidae) compared to associated morphometric differences. In: Held C, Schubart C, Koenemann S, editors. Phylogeography and population genetics in Crustacea. Boca Raton: Taylor and Francis/CRC Press; 2011. pp. 305–322. [Google Scholar]
- 38.Bracken-Grissom HHD, Cannon ME, Cabezas P, Feldmann RM, Schweitzer CE, Ahyong ST, et al. A comprehensive and integrative reconstruction of evolutionary history for Anomura (Crustacea: Decapoda) BMC Evol Biol. 2013;13:128–8. doi: 10.1186/1471-2148-13-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bybee SM, Bracken-Grissom HD, Hermansen RA, Clement MJ, Crandall KA, Felder DL. Directed next generation sequencing for phylogenetics: an example using Decapoda (Crustacea) Zoologischer Anzeiger - A Journal of Comparative Zoology. 2011;250:497–506. doi: 10.1016/j.jcz.2011.05.010. [DOI] [Google Scholar]
- 40.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stephens M, Smith NJ, Donnelly PA. New statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stephens M, Donnelly PA. Comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flot J-FSEQPHASE. A web tool for interconverting PHASE input/output files and FASTA sequence alignments. Mol Ecol Resour. 2010;10:162–166. doi: 10.1111/j.1755-0998.2009.02732.x. [DOI] [PubMed] [Google Scholar]
- 44.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Kumar S, Nei M. Small-sample tests of episodic adaptive evolution: a case study of primate lysozymes. Mol Biol Evol. 1997;14:1335–1338. doi: 10.1093/oxfordjournals.molbev.a025743. [DOI] [PubMed] [Google Scholar]
- 46.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 47.Leigh JW, Bryant D. popart : full-feature software for haplotype network construction. Nakagawa S. Methods Ecol Evol. 2015;6:1110–1116. doi: 10.1111/2041-210X.12410. [DOI] [Google Scholar]
- 48.Templeton AR, Crandall KA, Sing CFA. Cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992;132:619–633. doi: 10.1093/genetics/132.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerber AS, Templeton AR. Population sizes and within-deme movement of Trimerotropis saxatilis (Acrididae), a grasshopper with a fragmented distribution. Oecologia. 1996;105:343–350. doi: 10.1007/BF00328737. [DOI] [PubMed] [Google Scholar]
- 50.Gomez-Zurita J, Petitpierre E, Juan C. Nested cladistic analysis, phylogeography and speciation in the Timarcha goettingensis complex (Coleoptera, chrysomelidae) Mol Ecol. 2000;9:557–570. doi: 10.1046/j.1365-294x.2000.00900.x. [DOI] [PubMed] [Google Scholar]
- 51.Johnson JB, Jordan S. Phylogenetic divergence in leatherside chub (Gila copei) inferred from mitochondrial cytochrome b sequences. Mol Ecol. 2000;9:1029–1035. doi: 10.1046/j.1365-294x.2000.00960.x. [DOI] [PubMed] [Google Scholar]
- 52.Turner TF, Trexler JC, Harris JL, Haynes JL. Nested cladistic analysis indicates population fragmentation shapes genetic diversity in a freshwater mussel. Genetics. 2000;154:777–785. doi: 10.1093/genetics/154.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion: The University of Texas at Austin; 2006. Available from: http://hdl.handle.net/2152/2666?.
- 54.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 55.Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. Gateway computing environments workshop (GCE), 2010. IEEE. 2010:1–8.
- 56.Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29:1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- 57.Sukumaran J, Holder MT. DendroPy: a python library for phylogenetic computing. Bioinformatics. 2010;26:1569–1571. doi: 10.1093/bioinformatics/btq228. [DOI] [PubMed] [Google Scholar]
- 58.Rambaut A, Suchard M, Drummond A. Tracer v1.6 [Internet]. 2014. Available from: http://tree.bio.ed.ac.uk/software/tracer/?
- 59.Drummond AJ, Rambaut ABEAST. Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ketmaier V, Argano R. Caccone a. Phylogeography and molecular rates of subterranean aquatic Stenasellid isopods with a peri-Tyrrhenian distribution. Mol Ecol. 2003;12:547–555. doi: 10.1046/j.1365-294X.2003.01734.x. [DOI] [PubMed] [Google Scholar]
- 61.Lefébure T, Douady CJ, Gouy M, Trontelj P, Briolay J, Gibert J. Phylogeography of a subterranean amphipod reveals cryptic diversity and dynamic evolution in extreme environments. Mol Ecol. 2006;15:1797–1806. doi: 10.1111/j.1365-294X.2006.02888.x. [DOI] [PubMed] [Google Scholar]
- 62.Bell MA, Lloyd GT. Strap: an R package for plotting phylogenies against stratigraphy and assessing their stratigraphic congruence. Palaeontology. 2015;58:379–389. doi: 10.1111/pala.12142. [DOI] [Google Scholar]
- 63.Revell LJ. Phytools: an R package for phylogenetic comparative biology (and other things): phytools: R package. Methods Ecol Evol. 2012;3:217–223. doi: 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- 64.Cummings MP, Neel MC, Shaw KLA. Genealogical approach to quantifying lineage divergence. Evolution; international journal of organic evolution. 2008;62:2411–2422. doi: 10.1111/j.1558-5646.2008.00442.x. [DOI] [PubMed] [Google Scholar]
- 65.Development Core R, Team R, Language A. Environment for statistical computing [internet]. Vienna, Austria: R Foundation for Statistical. Computing. 2008; Available from: http://www.R-project.org
- 66.Winter DJ, Trewick SA, Waters JM, Spencer HG. The genealogical sorting index and species delimitation [Internet]. 2016 Jan. Report No.: biorxiv;036525v1. Available from: http://biorxiv.org/lookup/doi/10.1101/036525
- 67.Ho SYW, Shapiro B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol Ecol Resour. 2011;11:423–434. doi: 10.1111/j.1755-0998.2011.02988.x. [DOI] [PubMed] [Google Scholar]
- 68.Hewitt GM. The structure of biodiversity – insights from molecular phylogeography. Front Zool. 2004;1 [DOI] [PMC free article] [PubMed]
- 69.Howarth FG. Ecology of cave arthropods. Annu Rev Entomol. 1983;28:365–389. doi: 10.1146/annurev.en.28.010183.002053. [DOI] [Google Scholar]
- 70.Prendini L, Francke OF. Vignoli V. Troglomorphism, trichobothriotaxy and typhlochactid phylogeny (Scorpiones, Chactoidea): more evidence that troglobitism is not an evolutionary dead-end. Cladistics. 2010;26:117–142. doi: 10.1111/j.1096-0031.2009.00277.x. [DOI] [PubMed] [Google Scholar]
- 71.Allegrucci G, Todisco V, Sbordoni V. Molecular phylogeography of Dolichopoda cave crickets (Orthoptera, Rhaphidophoridae): a scenario suggested by mitochondrial DNA. Mol Phylogenet Evol. 2005;37:153–164. doi: 10.1016/j.ympev.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 72.Borsodi AK, Knáb M, Krett G, Makk J, Márialigeti K, Erőss A, et al. Biofilm bacterial communities inhabiting the cave walls of the Buda thermal karst system, Hungary. Geomicrobiol J. 2012;29:611–627. doi: 10.1080/01490451.2011.602801. [DOI] [Google Scholar]
- 73.Schmitt T, Seitz A. Intraspecific allozymatic differentiation reveals the glacial refugia and the postglacial expansions of European Erebia medusa (Lepidoptera: Nymphalidae) Biol J Linn Soc. 2001;74:429–458. [Google Scholar]
- 74.Ashworth A. A late-glacial insect fauna from red Moss, Lancashire. England Insect Systematics & Evolution. 1992;3:211–224. doi: 10.1163/187631272X00300. [DOI] [Google Scholar]
- 75.Coope GR. The response of insect faunas to glacial-interglacial climatic fluctuations. Philosophical Transactions: Biological Sciences. 1994;344:19–26. doi: 10.1098/rstb.1994.0046. [DOI] [Google Scholar]
- 76.Hewitt GM. Post-glacial re-colonization of European biota. Biol J Linn Soc. 1999;68:87–112. doi: 10.1111/j.1095-8312.1999.tb01160.x. [DOI] [Google Scholar]
- 77.Trontelj P, Blejec A, Fišer C. Ecomorphological convergence of cave communities. Evolution. 2012;66:3852–3865. doi: 10.1111/j.1558-5646.2012.01734.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset supporting the conclusions of this article is available in the NCBI’s Genbank database. Accession numbers are detailed in the Additional file 1 included with this publication. Phylogenetic trees resulting from the study are available in the figshare respository at: https://doi.org/10.6084/m9.figshare.5660542.v1.