

Abstract
Cytochrome P450 2E1 (CYP2E1) plays an important role in alcohol and toxin metabolism by catalyzing the conversion of substrates into more polar metabolites and producing reactive oxygen species. Reactive oxygen species‐induced oxidative stress promotes hepatocyte injury and death, which in turn induces inflammation, activation of hepatic stellate cells, and liver fibrosis. Here, we analyzed mice expressing only the human CYP2E1 gene (hCYP2E1) to determine differences in hCYP2E1 versus endogenous mouse Cyp2e1 function with different liver injuries. After intragastric alcohol feeding, CYP2E1 expression was induced in both hCYP2E1 and wild‐type (Wt) mice. hCYP2E1 mice had greater inflammation, fibrosis, and lipid peroxidation but less hepatic steatosis. In addition, hCYP2E1 mice demonstrated increased expression of fibrogenic and proinflammatory genes but decreased expression of de novo lipogenic genes compared to Wt mice. Lipidomics of free fatty acid, triacylglycerol, diacylglycerol, and cholesterol ester species and proinflammatory prostaglandins support these conclusions. Carbon tetrachloride‐induced injury suppressed expression of both mouse and human CYP2E1, but again hCYP2E1 mice exhibited greater hepatic stellate cell activation and fibrosis than Wt controls with comparable expression of proinflammatory genes. By contrast, 14‐day bile duct ligation induced comparable cholestatic injury and fibrosis in both genotypes. Conclusion: Alcohol‐induced liver fibrosis but not hepatic steatosis is more severe in the hCYP2E1 mouse than in the Wt mouse, demonstrating the use of this model to provide insight into the pathogenesis of alcoholic liver disease. (Hepatology Communications 2017;1:1043–1057)
Abbreviations
- 4‐HNE
4‐hydroxynonenal
- α‐SMA
alpha smooth muscle actin
- AA
arachidonic acid
- Acc
acetyl coenzyme A carboxylase
- ALT
alanine aminotransferase
- AMPK
adenosine monophosphate–activated protein kinase
- BDL
bile duct ligation
- CE
cholesterol ester
- CoA
coenzyme A
- col1α1
collagen type 1 alpha 1
- CYP2E1
cytochrome P450 2E1
- DAG
diacylglycerol
- DHA
docosahexaenoic acid
- FFA
free fatty acid
- hCYP2E1
humanized CYP2E1
- HSC
hepatic stellate cell
- IL
interleukin
- Ly6g
lymphocyte antigen 6 complex locus G
- mRNA
messenger RNA
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- Nox
reduced nicotinamide adenine dinucleotide phosphate oxidase
- Nrf2
nuclear erythroid 2 p45‐related factor 2
- pAccα
phosphorylated acetyl coenzyme A carboxylase alpha
- PG
prostaglandin
- PPARα
peroxisome proliferator‐activated receptor alpha
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
- SREBP‐1c
sterol regulatory element binding protein 1c
- TAG
triacylglycerol
- TGF
transforming growth factor
- TIMP1
tissue inhibitor of metalloproteinase 1
- Wt
wild‐type
Introduction
Cytochrome P450 2E1 (CYP2E1) is a member of the P450 enzyme family that plays a vital role in alcohol, drug, toxin, lipid, and carcinogen metabolism.1, 2 CYP2E1, mainly expressed in hepatocytes, catalyzes the conversion of its substrates into more polar metabolites for secretion or use as substrates for other microsomal phase II enzymes.3 CYP2E1 also transfers active electrons from reduced nicotinamide adenine dinucleotide phosphate (NADPH) or reduced nicotinamide adenine dinucleotide to oxygen to produce reactive oxygen species (ROS).3 CYP2E1‐induced toxic metabolites coupled with oxidative stress from ROS are proposed to be important mediators of liver injury by promoting an inflammatory and fibrogenic milieu to facilitate recruitment of leukocytes and activation of hepatic stellate cells (HSCs).4
Chronic alcohol consumption induces the expression of CYP2E1 protein.5 During the pathogenesis of alcoholic liver disease, metabolism of ethanol in hepatocytes by CYP2E1 generates ROS,4 which in turn facilitates lipid peroxidation, protein carbonylation, and formation of 1‐hydroxyethyl radical and lipid radical formation.6 CYP2E1 also facilitates metabolism of endobiotic chemicals, including steroids, fatty acids, and prostaglandins (PGs), through ω‐1 hydroxylation.7 ω‐1 hydroxylation is an alternative oxidation pathway for long chain fatty acids within microsomes instead of the classic β‐oxidation within mitochondria. Of particular interest is ω‐1 hydroxylation of arachidonic acid (AA) because AA‐derived eicosanoids promote proinflammatory responses, such as vasodilation, inhibition of Na+/K+‐adenosine triphosphatase, and activation of nuclear factor kappa B transactivation.8, 9 The hydroxylation of AA and other polyunsaturated fatty acids (PUFAs) within microsomes is increased during alcohol consumption.7 Decreased AA content within the liver is observed in both murine and human alcoholic liver disease,10 and administration of CYP2E1 inhibitor restores the AA concentration in the liver.11
Steatohepatitis, a major pathology for alcohol‐induced liver injury, is characterized by hepatocyte ballooning and lymphocyte infiltration. Accumulation of lipid droplets within hepatocytes results from disruption of homeostasis between de novo lipid synthesis and lipid β‐oxidation during alcohol consumption.12 During ethanol metabolism, up‐regulation of sterol regulatory element binding protein 1c (SREBP‐1c) and down‐regulation of peroxisome proliferator‐activated receptor alpha (PPARα) expression lead to the induction of fatty acid synthesis and inhibition of β‐oxidation.13 Mice subjected to alcohol feeding develop more steatosis than pair‐fed mice receiving the same caloric intake, indicating that an alcohol‐induced metabolic disorder drives steatosis. Besides CYP2E1, alcohol is also catalyzed by cytosolic alcohol dehydrogenase and subsequently by mitochondrial aldehyde dehydrogenase 2 to create reducing equivalents (reduced nicotinamide adenine dinucleotide and NADPH) and acetyl‐coenzyme A (CoA) equivalents (acetaldehyde and acetate). The ethanol metabolites NADPH and acetate are the products of lipid β‐oxidation but are the reactants of de novo lipogenesis. Therefore, during alcohol metabolism, lipid homeostasis is unbalanced and favors the direction of lipogenesis to accelerate alcoholic steatosis. The rate‐limiting enzyme during lipogenesis is acetyl‐CoA carboxylase (Acc), and the transcription factor Srebp‐1c directly up‐regulates Acc transcription.14 The de novo lipogenesis machinery uses NADPH and acetyl‐CoA to synthesize free fatty acids (FFAs). The excess FFAs are incorporated into diacylglycerol (DAG) and triacylglycerol (TAG) to form lipid droplets within hepatocytes. Excessive lipid droplet accumulation and ROS production cause hepatocyte ballooning and apoptosis. Dead hepatocytes will induce inflammatory response within the liver, triggering the recruitment of inflammatory cells to the fatty liver, activation of liver macrophage, and release of proinflammatory cytokines (tumor necrosis factor alpha, interleukin [IL]‐1β, IL‐6, and transforming growth factor [TGF]‐β1).15, 16, 17 Infiltration of neutrophils, which kill sensitized hepatocytes by introducing massive ROS and further exacerbating alcohol‐induced liver injury, is a prominent feature of alcoholic hepatitis.18 Long‐term alcohol consumption may induce liver fibrosis in which HSCs are the major source of myofibroblasts.19
CCl4, an environmental toxin, is metabolized by CYP2E1 and produces the toxic metabolite trichloromethyl. Trichloromethyl is a free radical and reacts with macromolecules to form DNA, protein, and lipid adducts.20 The hepatotoxicity of CCl4 is mediated by CYP2E1, and mice lacking CYP2E1 exhibit resistance to CCl4‐induced liver injury,21 but CYP2E1 itself is destabilized by active CCl4 metabolites in a phosphorylation‐dependent manner.22 The CYP2E1 inhibitor diethyldithiocarbamate or antioxidants attenuate CCl4‐induced lipid peroxidation and hepatocyte injury.23 CCl4‐induced hepatocyte necrosis, liver inflammation, activation of HSCs, and bridging fibrosis is more extensive than with alcohol.24 CCl4‐induced liver fibrosis is mainly mediated by activated HSCs.25
The excessive accumulation of bile acids is toxic to hepatocytes, resulting in cholestatic liver injury, which is manifested by proliferating bile ducts, biliary fibrosis, and jaundice.26 Activated portal fibroblasts are a major source of myofibroblasts during early biliary fibrosis.27 There are no reports establishing a correlation between hepatic cholestasis and CYP2E1. Some cases of drug‐induced liver injury are reported to cause cholestatic liver diseases, which suggests that the combination of hepatotoxin and cholestatic liver disease might be due to toxic metabolites by the CYP enzyme family.28
Although ethanol and xenobiotic metabolic pathways of microsomal enzymes are conserved between species, CYP2E1 protein has interspecies variations in the Km, Vmax, and the scope of substrates.29, 30, 31 Therefore, comparing the human CYP2E1 and mouse Cyp2e1 in several pathologic conditions may provide new insights into human liver diseases. Gonzalez and colleagues30 generated a humanized CYP2E1 (hCYP2E1) transgenic mouse by introducing a bacterial artificial chromosome containing the human CYP2E1 gene under the 2.3‐Kb upstream promoter region into a mouse with a knockout of the endogenous CYP2E1 gene. The human transgene is responsive to CYP2E1 inducers, such as acetone and acetaminophen, in the mouse.30, 32 Here, we used this hCYP2E1 mouse and wild‐type (Wt) littermates to compare the human and mouse CYP2E1 enzyme under three liver fibrosis models: alcoholic liver fibrosis by intragastric alcohol feeding, hepatotoxin‐induced liver fibrosis by CCl4 administration, and biliary fibrosis by bile duct ligation (BDL).
Materials and Methods
MICE
All mouse protocols were approved by the Institutional Animal Care and Use Committee of the University of California San Diego and the University of Southern California. Littermate male mice (Wt and hCYP2E1) were used within current study.
EXPERIMENTAL MODELS OF LIVER FIBROSIS
Alcoholic Liver Fibrosis
We used 2‐month‐old male Wt (n = 6) and hCYP2E1 (n = 12) littermates. Alcoholic steatohepatitis accompanied with liver fibrosis was achieved by intragastric alcohol feeding with a weekly binge model.33
Hepatotoxin‐Induced Liver Fibrosis
Liver fibrosis was achieved by oral administration of 25% CCl4 at 8 μL per gram body weight twice a week for 6 weeks. We used 2‐month‐old male Wt (n = 8) and hCYP2E1 (n = 8) littermates for CCl4 administration and 2‐month‐old male Wt (n = 4) and hCYP2E1 (n = 5) mice for the corn oil control. The mice were killed 48 hours after the last CCl4 administration to achieve the maximum fibrosis.
Cholestatic Liver Fibrosis
Cholestatic liver fibrosis was achieved by BDL for 14 days, as described.34 We used 2‐month‐old male Wt (n = 6) and hCYP2E1 (n = 6) littermates for BDL surgery and 2‐month‐old male Wt (n = 3) and hCYP2E1 (n = 4) mice for sham surgery control.
HISTOLOGY AND IMMUNOHISTOCHEMISTRY
Formalin‐fixed paraffin‐embedded liver tissues were sectioned to 5 μm thickness. The sections were stained with hematoxylin and eosin or Sirius Red. Immunohistochemistry was performed on formalin‐fixed livers with anti‐mouse Desmin (RB‐9014‐P0; Fisher Neuromarkers), alpha smooth muscle actin (α‐SMA) (ab5694; Abcam), F4/80 (14‐4801‐82e; Bioscience), lymphocyte antigen 6 complex locus G (Ly6g) (9668; eBioscience), 4‐hydroxynonenal (4‐HNE) (HNE11‐S; Alpha Diagnostic International Inc.), and nuclear erythroid 2 p45‐related factor 2 (Nrf2) (SC‐722; Santa Cruz Biotechnology) antibodies followed by 3,3′‐diaminobenzidine tetrahydrochloride staining (Vector Laboratories) and counterstaining with hematoxylin. The images were analyzed using Image J (National Institutes of Health, Bethesda, MD).
TARGETED HEPATIC LIPIDOMIC ANALYSIS
Fresh liver samples (∼7 mg protein) were homogenized in 1 mL phosphate‐buffered saline containing 10% methanol. For the various lipidomic analyses, aliquots were withdrawn and extracted using protocols that were optimized for each lipid category.35 Detailed analysis procedures for eicosanoids, FFAs, and TAG, DAG, and cholesteryl ester (CE) are described in the Supporting Materials and Methods.
Results
HEPATIC FIBROSIS WAS INCREASED IN hCYP2E1 MICE AFTER INTRAGASTRIC ALCOHOL FEEDING
Intragastric alcohol feeding was performed in hCYP2E1 and Wt littermates to induce alcoholic liver fibrosis. Liver weight and the liver/body ratio increased in mice fed alcohol, but there was no difference between Wt and hCYP2E1 mice (Supporting Fig. S1). Liver injury, assessed by serum alanine aminotransferase (ALT), was increased in hCYP2E1 mice versus Wt controls (Fig. 1A). The ethanol concentration in the serum was lower in hCYP2E1 mice (Fig. 1B), suggesting increased alcohol metabolism in these mice. The protein expression of both human and mouse Cyp2e1 was investigated, with antibody detecting both species. Expression of hCyp2e1 protein was higher than that of mouse Cyp2e1 from Wt liver fed normal chow. In response to alcohol feeding, expression of both human and mouse Cyp2e1 was increased, but the human protein was higher than the mouse Cyp2e1 (Fig. 1C). Messenger RNA (mRNA) levels of mouse or human Cyp2e1 were not induced by alcohol, suggesting that the induction of Cyp2e1 in protein levels was due to posttranslational stabilization (Fig. 1D), as described.5 In addition, CYP1A2, another CYP member of the mouse microsomal ethanol oxidation system, was the same in Wt and hCYP2E1 livers (Supporting Fig. S1).
Figure 1.
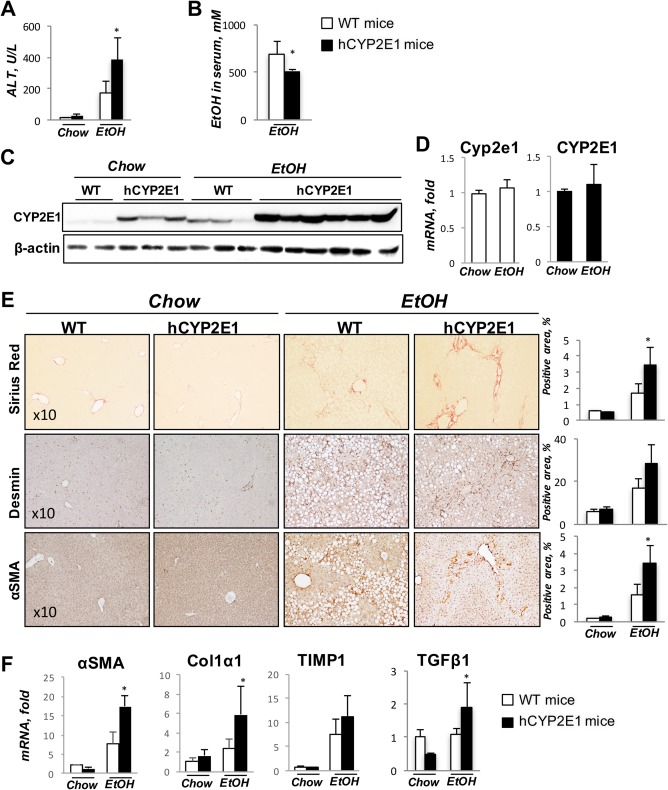
Hepatic fibrosis is increased in hCYP2E1 mice after iG alcohol feeding. Wt and hCYP2E1 mice were subjected to iG alcohol feeding with weekly binges for 8 weeks. Wt and hCYP2E1 littermates fed with normal chow were used as controls. (A) Liver function was assessed by ALT. (B) Ethanol concentration in serum was measured in Wt and hCYP2E1 mice after iG alcohol feeding. (C) Immunoblotting of CYP2E1 in liver. (D) CYP2E1 mRNA expression in both Wt and hCYP2E1 liver. (E) Representative images of Sirius Red staining, immunohistochemistry staining of Desmin, and α‐SMA. The stained area was shown with quantification of morphometric analysis of each staining. (F) Hepatic expression of fibrogenic genes was measured by mRNA‐fold induction level in Wt and hCYP2E1 livers. The P value was measured between Wt and hCYP2E1 with iG alcohol feeding; the data were shown as mean ± SEM; *P < 0.05. Abbreviations: EtOH, ethanol; iG, intragastric.
Liver fibrosis, assessed by morphometric quantification of Sirius Red staining, was increased 2‐fold in hCYP2E1 mice compared with Wt mice (Fig. 1E). There were more Desmin+ and α‐SMA+ activated HSCs in hCYP2E1 livers compared with Wt mice (Fig. 1E). In addition, hepatic mRNA levels of fibrotic genes, such as α‐SMA, collagen type 1 alpha 1 (col1α1), tissue inhibitor of metalloproteinase 1 (TIMP1), and TGF‐β1, were increased in hCYP2E1 mice compared to Wt mice after intragastric alcohol feeding (Fig. 1F).
LIVER INFLAMMATION WAS INCREASED IN hCYP2E1 MICE AFTER INTRAGASTRIC ALCOHOL FEEDING
After intragastric alcohol feeding, more Ly6g+ cells (neutrophils) were observed in hCYP2E1 mice, but there was no difference in the number of F4/80+ cells (macrophages) between Wt and hCYP2E1 mice after alcohol consumption (Fig. 2A). Hepatic mRNA expression was more elevated for inflammatory genes (IL‐6, tumor necrosis factor alpha, and IL‐1β), chemokines (monocyte chemoattractant protein 1, macrophage inflammatory protein 1, and macrophage inflammatory protein 2), and neutrophil markers (myeloperoxidase and Ly6g) in hCYP2E1 mice compared to Wt littermates. mRNA expression of the anti‐inflammatory gene IL‐10 was lower in hCYP2E1 mice (Fig. 2B). The proinflammatory PGs derived from AA, such as PGD2, PGE2, and thromboxane B2, were elevated in the hCYP2E1 liver, while protectin DX, a metabolite derived from docosahexaenoic acid (DHA) with strong anti‐inflammatory activity, was decreased in the same animals after alcohol consumption (Fig. 2C; Supporting Fig. S3).
Figure 2.
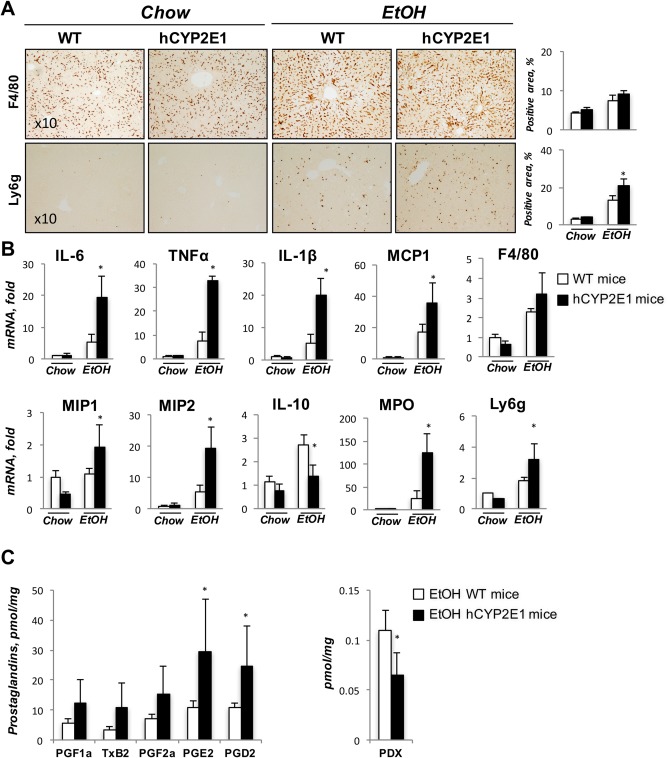
Liver inflammation is greater in hCYP2E1 than Wt mice. Alcoholic steatohepatitis is characterized by infiltration of neutrophils and up‐regulation of proinflammatory cytokines. (A) Representative images of immunohistochemistry staining of F4/80 and Ly6g are shown with quantification of stained area. (B) Hepatic mRNAs of inflammatory genes and neutrophil markers were measured in Wt and hCYP2E1 mice after iG alcohol feeding. The data are shown as the fold change of mRNA induction compared with Wt chow mice. (C) PGs, metabolites of AA, and protectin DX were measured by mass spectrometry. The P value was measured between Wt and hCYP2E1 under iG alcohol feeding; the data were shown as mean ± SEM; *P < 0.05. Abbreviations: EtOH, ethanol; iG, intragastric; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; MPO, myeloperoxidase; PDX, protectin DX; TNFα, tumor necrosis factor alpha; TxB, thromboxane B.
OXIDATIVE STRESS WAS INCREASED IN hCYP2E1 MICE AFTER INTRAGASTRIC ALCOHOL FEEDING
We measured the lipid peroxidation products 4‐HNE and malondialdehyde as parameters of oxidative stress in the liver as well as hepatic mRNA expression level of ROS production genes in Wt and hCYP2E1 mice under alcohol feeding. Immunohistochemistry showed increased 4‐HNE in hCYP2E1 livers (Fig. 3A), and malondialdehyde levels were higher (∼2‐fold) in hCYP2E1 liver (Fig. 3C). In response to alcohol feeding, hepatic mRNA levels of NADPH oxidase (Nox)2 and p67 were elevated ∼6‐fold and ∼3‐fold, respectively, in hCYP2E1 mice, but no significant differences were found in other ROS production genes (Nox1, Nox4, p47, p22) between hCYP2E1 and Wt mice (Fig. 3B).
Figure 3.
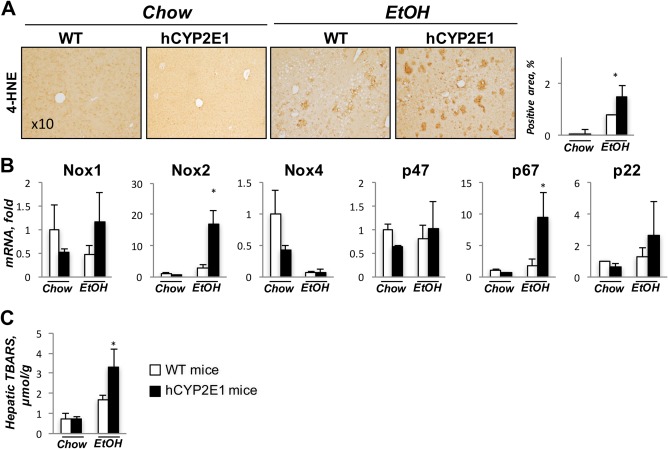
Alcohol‐induced oxidative stress is increased in hCYP2E1 compared to Wt mice. CYP2E1‐induced oxidative stress during alcohol consumption is a major pathology during alcoholic liver disease. (A) Representative images of immunohistochemistry staining of 4‐HNE are shown with quantification of the positive area. (B) Liver mRNA of ROS production genes was measured in Wt and hCYP2E1 mice after iG alcohol feeding. (C) Hepatic lipid peroxidation was assessed by measuring TBARS formation. The data are shown as the fold change of mRNA induction compared with Wt chow mice. The P value was measured between Wt and hCYP2E1 under iG alcohol feeding; the data were shown as mean ± SEM; *P < 0.05. Abbreviations: EtOH, ethanol; iG, intragastric; TBARS, thiobarbituric acid reactive substances.
HEPATIC STEATOSIS WAS ATTENUATED AND LIPID PROFILES WERE CHANGED IN hCYP2E1 MICE AFTER INTRAGASTRIC ALCOHOL FEEDING
After intragastric alcohol feeding, lipid droplets accumulated within hepatocytes as both macrovesicular and microvesicular steatosis in both Wt and hCYP2E1 mice (Fig. 4A). Steatosis was ∼35% less in hCYP2E1 mice compared to Wt mice (Fig. 4A). Hepatic mRNA expression of Srebp‐1c, fatty acid synthase, and Accα were up‐regulated in both genotypes after alcohol consumption but less in hCYP2E1 livers (Fig. 4B). PPARα, a surrogate for the key transcription factor involved in fatty acid β‐oxidation, was more highly expressed in hCYP2E1 liver compared to Wt mice (Fig. 4B). This gene expression profile is consistent with increased lipogenesis and decreased β‐oxidation, which would produce increased FFA content in Wt versus hCYP2E1 mice. Indeed, we observed higher hepatic FFAs in the Wt mice fed alcohol than in the hCYP2E1 mice (Fig. 4C).
Figure 4.
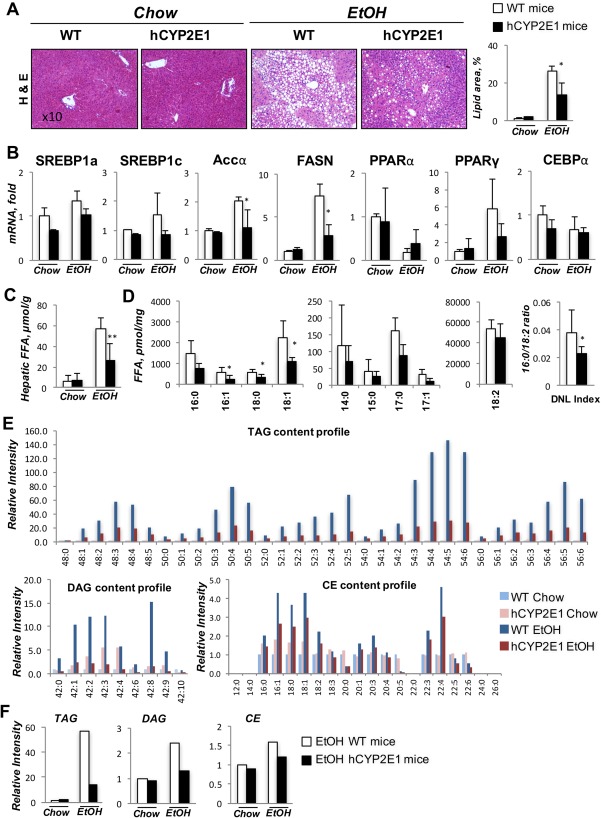
Alcoholic steatosis and de novo lipogenesis are greater in Wt than hCYP2E1 mice. Alcoholic steatosis is characterized by lipid droplet accumulation and up‐regulation of de novo lipogenesis genes. (A) Representative images of H&E staining of liver sections with quantification of lipid area. (B) Level of hepatic mRNAs of lipogenesis genes were measured in Wt and hCYP2E1 mice after iG alcohol feeding. (C) Hepatic content of total FFAs was evaluated by enzymatic reactions in the liver from chow and iG feeding mice. (D) Individual FFA species were measured by mass spectrometry. The index of de novo lipogenesis was calculated according to the ratio of 16:0 and 18:2. (E) Individual TAG, DAG, and CE species were measured in the liver from Wt and hCYP2E1 mice subjected to normal chow or iG alcohol feeding. (F) The total TAG, DAG, and CE were calculated by adding individual lipid species. The P value was measured between Wt and hCYP2E1 under iG alcohol feeding; the data were shown as mean ± SEM; *P < 0.05. Abbreviations: CEBPα, CCAAT‐enhancer‐binding protein alpha; DNL, de novo lipogenesis; EtOH, ethanol; FASN, fatty acid synthase; H&E, hematoxylin and eosin; iG, intragastric.
We also undertook targeted lipidomic profiling of individual FFA species (Fig. 4D). Diet‐derived FFAs (C14 and C15) and essential unsaturated fatty acids (C18:2, C18:3, C18:4, C18:6, C20:3 n3, C20:3 n9) were slightly decreased between Wt and hCYP2E1 groups (Fig. 4D); however, FFAs derived from de novo lipogenesis (C16:0, C18:0, and C18:1), were decreased significantly in the hCYP2E1 compared to the Wt liver. Moreover, the de novo lipogenesis index (16:0/18:2 ratio)36 was decreased in the hCYP2E1 liver (Fig. 4D).
Excess FFAs will be incorporated into TAG, DAG, and CE species to form lipid droplets within hepatocytes during steatohepatitis. All detected TAG species were reduced in hCYP2E1 livers compared to Wt livers (Fig. 4E,F). The most striking up‐regulations of TAG species were those containing unsaturated fatty acids, like 50:4, 52:5, 54:4, 54:5, 54:6, 56:4, and 56:5 (a full version of TAG and DAG content profiles is shown in Supporting Fig. S2). Although all DAG and most CE lipid species were increased in Wt livers after alcohol consumption, the extent of the increase of TAG, DAG, and CE (Fig. 4E,F) suggested that TAG lipid species were the major lipids contributing to the difference in steatosis between Wt and hCYP2E1 livers.
hCYP2E1 INDUCED Nrf2, AN Accα INHIBITOR
Up‐regulation of hepatic mRNA expression of Accα and Srebp‐1c (Fig. 4B) along with increased Accα protein (Fig. 5A) in Wt mice were greater than in hCYP2E1 mice with intragastric ethanol (Figs. 4B, 5A). Expression of phosphorylated Accα (pAccα) and its upstream kinase adenosine monophosphate–activated protein kinase (AMPK) was similar in the two genotypes (Fig. 5A). Nrf2 mRNA after alcohol feeding was increased to similar levels in Wt and hCYP2E1 liver; however, Nrf2 target genes Metallothionein 1, heme oxygenase 1, NAD(P) dehydrogenase, quinone 1 and glutamate‐cysteine ligase, catalytic subunit were higher in hCYP2E1 mice (Fig. 5C). In agreement, immunohistochemistry showed more abundant Nrf2 nuclear staining within hCYP2E1 hepatocytes than within Wt hepatocytes (Fig. 5B). These results imply that higher ROS induced by human CYP2E1 promoted Nrf2 nuclear localization and sequentially inhibited the transcription of Accα.
Figure 5.
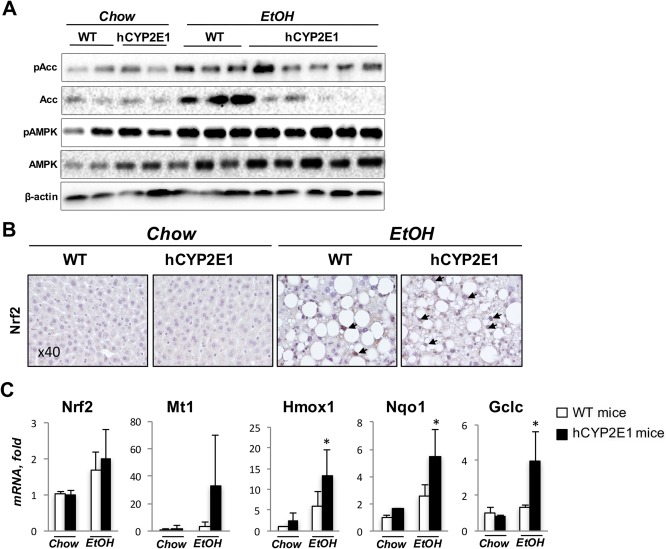
Transcriptional regulation of Accα by Nrf2. Acc is the speed‐limiting enzyme for de novo lipogenesis. Acc is regulated by AMPK at the posttranslational level and Nrf2 at the transcriptional level. (A) Western blotting of pAcc, Acc, pAMPK, and AMPK from the liver of Wt or hCYP2E1 mice. (B) Immunohistochemistry staining of Nrf2 on the liver sections of Wt and hCYP2E1 mice. The nuclear staining of Nrf2 is indicated by arrows. (C) mRNA level of Nrf2 and its transcriptional targets in the liver. The P value was measured between Wt and hCYP2E1 under iG alcohol feeding; the data were shown as mean ± SEM; *P < 0.05. Abbreviations: EtOH, ethanol; iG, intragastric; pAMPK, phosphorylated AMPK. Abbreviations: EtOH, ethanol; Gclc, glutamate‐cysteine ligase, catalytic subunit; Hmox1, heme oxygenase 1; Mt1, metallothionein 1; Nrf2, nuclear factor, erythroid derived 2, like 2; Nqo, NAD(P)H dehydrogenase, quinone 1.
HEPATIC FIBROSIS WAS INCREASED IN hCYP2E1 MICE AFTER CCl4 INJURY
We next investigated the role of human CYP2E1 in response to toxic hepatic injury induced by CCl4 administration in Wt and hCYP2E1 mice. Serum ALT levels were elevated in hCYP2E1 mice compared with Wt mice after CCl4 administration (Fig. 6A). Protein expressions of mouse and human Cyp2e1 were down‐regulated after CCl4 injury (Fig. 6B), reportedly due to phosphorylation‐dependent protein degradation.22 The mRNAs of mouse and human Cyp2e1 were also suppressed in response to CCl4 (Fig. 6B). Compared to Wt mice after CCl4 injury, hCYP2E1 mice had more fibrosis and more Desmin+ and α‐SMA+ HSCs (Fig. 6C). Hepatic lipid peroxidation was increased in the hCYP2E1 mice under CCl4 administration (Fig. 6C). No difference was observed in F4/80+ cells in Wt and hCYP2E1 livers after CCl4 administration (Fig. 6C). Levels of hepatic mRNA of fibrogenic genes, such as α‐SMA and TIMP1, were more elevated in hCYP2E1 mice in response to CCl4 compared to Wt mice, while hepatic mRNA levels of Col1α1 and TGF‐β1 genes were elevated equally (Fig. 6D).
Figure 6.
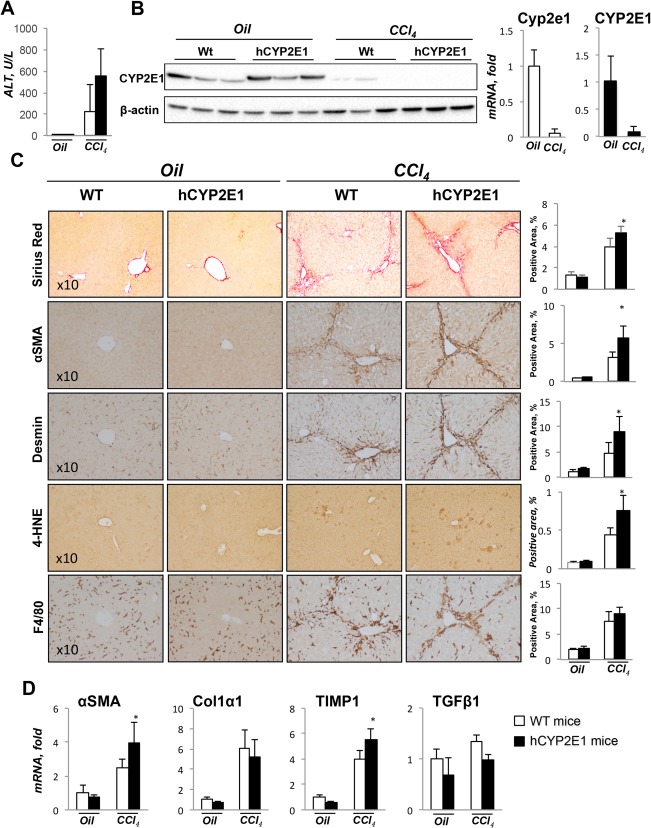
Hepatic fibrosis is increased in hCYP2E1 mice after CCl4 injury. Liver fibrosis induced by CCl4 was assessed in Wt and hCYP2E1 mice. Mice receiving corn oil were used as controls. (A) CCl4‐induced liver injury was evaluated by serum ALT level. (B) Immunoblotting and mRNA expression level of CYP2E1 in liver from both corn oil and CCl4 groups. (C) Representative images of Sirius Red and immunohistochemistry staining of Desmin, α‐SMA, 4‐HNE, and F4/80 are presented with quantification. (D) mRNA levels of fibrogenic genes were measured in Wt and hCYP2E1 mice after CCl4 injury or oil application. Data are shown as the fold change of mRNA induction compared with Wt mice receiving corn oil. The P value was measured between Wt and hCYP2E1 mice after CCl4 injury; the data were shown as mean ± SEM; *P < 0.05.
HEPATIC INJURY WAS NOT AFFECTED IN hCYP2E1 MICE AFTER BDL INJURY
To investigate whether human Cyp2e1 affects cholestatic hepatic injury, 14‐day BDL and sham surgery were performed in Wt and hCYP2E1 mice. Hepatic injury was equal in Wt and hCYP2E1 after BDL, as reflected by increased serum ALT and alkaline phosphatase levels (Fig. 7A). Expressions of human and mouse Cyp2e1 were down‐regulated at both protein and mRNA levels after BDL (Fig. 7B). Liver fibrosis as assessed by Sirius Red staining showed no difference between Wt and hCYP2E1 mice (Fig. 7C). Additionally, no changes in hepatic mRNA level of fibrogenic genes (α‐SMA, Col1α1, and TIMP1) were found in hCYP2E1 compared to Wt mice (Fig. 7D).
Figure 7.
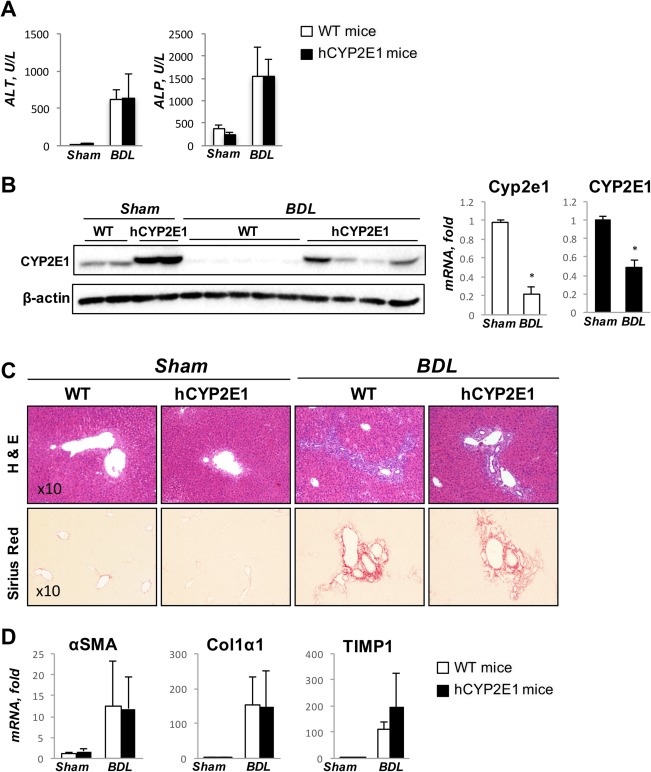
Biliary fibrosis shows no difference between Wt and hCYP2E1 mice. Wt and hCYP2E1 mice underwent BDL for 14 days. Wt and hCYP2E1 littermates receiving sham surgery were used as the control. (A) Liver function was assessed by serum ALT and ALP for cholestatic liver injury. (B) Immunoblotting and mRNA levels of CYP2E1 in livers from sham or BDL mice. (C) Representative images of H&E and Sirius Red staining of liver sections from sham or BDL mice. (D) mRNA level of fibrogenic genes was measured in Wt and hCYP2E1 mice. The data shown are the fold change of mRNA induction compared with Wt chow mice. Abbreviations: ALP, alkaline phosphatase; H&E, hematoxylin and eosin.
Discussion
We compared the contribution of mouse and human CYP2E1 in three models of liver fibrosis: alcoholic liver fibrosis by intragastric alcohol feeding plus multiple binges, hepatotoxin‐induced liver fibrosis by CCl4 administration, and biliary fibrosis after 14 days of BDL. The CYP2E1 enzyme played different roles in the different models. Both mouse and human CYP2E1 were induced by alcohol consumption, but the hCYP2E1 mice had increased CYP2E1 protein levels, lipid peroxidation, oxidative stress, inflammation, and development of liver fibrosis. Although CCl4 administration inhibited hepatic CYP2E1 expression as reported,21 greater fibrosis was generated in the hCYP2E1 mice than in the Wt littermates but with equal levels of hepatic inflammation. During BDL‐induced cholestatic liver injury, the mouse and human CYP2E1s did not change the development of fibrosis.
Previous liver fibrosis studies in mice relied on the murine CYP2E1 enzyme, but human CYP2E1 has distinctive enzymatic activity on several substrates, such as chlorzoxazone,29, 30 p‐nitrophenol,30 and FFAs.31 During alcohol consumption, CYP2E1 expression is induced by acetone, which is a toxic metabolite from ethanol and stabilizes the CYP2E1 protein.37 Indeed, we observed up‐regulation of the CYP2E1 protein but not mRNA from the alcohol‐consuming mice (Fig. 1C,D). Furthermore, hCYP2E1 mice had greater CYP2E1 induction by ethanol than their Wt littermates at the protein level (Fig. 1C), although the mRNA expression level of human CYP2E1 was higher than mouse Cyp2e1 at the basal level (Supporting Fig. S1). Human CYP2E1 has been reported with higher enzymatic activity to alcohol than mice Cyp2e1.30, 38 In agreement with a previous report, we observed a higher level of liver injury in the alcohol‐treated hCYP2E1 mice indicated by higher serum ALT level (Fig. 1A), higher ethanol metabolism rate (Fig. 1B), and increased lipid peroxidation within hepatocytes (Fig. 3A). Furthermore, increased ethanol metabolism by hCYP2E1 did not affect the expression of Adh genes, which are enzymes that also oxidize ethanol to acetaldehyde.
The correlation between CYP2E1 activity and alcoholic steatohepatitis has been controversial. This might be due to different alcoholic mouse models applied by different groups or by a compensatory increase of other cytochrome P450s in the CYP2E1 knockout mouse. For example, Kono et al.39 reported that CYP2E1 did not contribute to alcohol‐induced early hepatic steatosis, inflammation, and necrosis after 4 weeks of intragastric alcohol feeding by comparing Wt and Cyp2e1 knockout mice. In contrast, Cederbaum and colleagues38 demonstrated decreased liver injury in Cyp2e1 knockout mice during alcohol consumption by using the Lieber–Dicarli alcohol feeding method for 3 weeks. Here, we used intragastric alcohol feeding plus weekly binges for 8 weeks to directly compare the role of murine and human CYP2E1, and we observed more severe inflammation and fibrosis in hCYP2E1 mice. Hepatic injury correlated with the level of CYP2E1 induction and ROS production (Figs. 1, 3), but CYP2E1 expression and ROS correlated inversely with hepatic steatosis (Fig. 4). The reduced hepatic steatosis was associated with reduced expression of de novo lipogenesis genes, such as Srebp‐1c, Accα, and PPARγ (Fig. 4B).
The contribution of ROS to the development of alcoholic steatosis is complex as increased oxidative stress during alcohol consumption regulates multiple signaling pathways, which play opposing roles in lipogenesis. Knocking out Cyp2e1 reduces alcohol‐induced hepatic oxidative stress and prevents development of alcoholic steatosis,38 and administration of antioxidants could ameliorate alcoholic steatosis.40 In contrast to previous reports, Chen et al.41 demonstrated that depletion of glutathione promotes ROS accumulation during alcohol consumption but protects the liver from alcoholic steatosis. Increased ROS promotes phosphorylation of 5′ AMPK, which becomes activated and promotes phosphorylation and inactivation of Accα (pAccα).42 In our study, up‐regulation of AMPK and phosphorylated AMPK were detected after alcohol consumption in both Wt and hCYP2E1 liver to a similar level (Fig. 5A). In agreement with this observation, the phosphorylation of Ser79 at Accα showed no significant difference between Wt and hCYP2E1 liver (Fig. 5A). Instead of pAccα, total Accα was significantly increased in the Wt alcohol group compared to the hCYP2E1 mice, and this was consistent with the higher Accα mRNA expression in the Wt mice (Fig. 4B). As a defensive mechanism, increased ROS promotes nuclear translocation of Nrf2 by degrading the cytoplasmic Keap1‐Nrf2.43 Nrf2 binds to antioxidant response elements, causing transcription of antioxidant genes, such as superoxide dismutases, peroxiredoxins, sulfiredoxin, and glutathione reductase.44 Although the Nrf2 mRNA level was increased to a similar level after alcohol consumption in Wt and hCYP2E1 (Fig. 5C), Nrf2 nuclear localization and up‐regulation of Nrf2 direct‐target genes was higher in the hCYP2E1 liver (Fig. 5B,C). Our results are consistent with studies demonstrating that Nrf2 nuclearization results in inhibition of Accα transcription and Nrf2 knockout mice or Nrf2 inhibitor results in reduced steatosis in nonalcoholic steatohepatitis rodent models.45
Higher Acc expression in the Wt mice may contribute to the excessive production of FFAs in liver (Fig. 4C). The end product of de novo lipogenesis is palmitic acid (C16:0), and elongase adds two more carbons to produce stearic acid (C18:0). Stearoyl‐CoA desaturase converts stearic acid into oleic acid (C18:1). These three fatty acids comprise the majority of FFAs that enter an active metabolic pool for DAG, TAG, and CE formation.35 Within alcoholic liver, TAG contributed more than 90% of the glycerolipids within both Wt and hCYP2E1 mice (Fig. 4F). In Wt livers, the TAG content increased ∼60‐fold, while DAG and CE were increased 2.2‐fold and 1.6‐fold, respectively (Fig. 4F). This suggests that alcoholic steatosis mainly resulted from the accumulation of TAG and that the driving force for reduced steatosis within hCYP2E1 mice was due to reduced TAG formation. Interestingly, the serum TAG was not changed between Wt and hCYP2E1 mice after alcohol consumption (Supporting Fig. S1), and the body weight and liver were not significantly affected by hCYP2E1 (Supporting Fig. S1). The TAG content profile indicated that most C48, C50, C52, and C54 species were significantly reduced within hCYP2E1 liver compared to Wt liver, and especially the TAGs containing unsaturated fatty acids, including C48:3, C50:4, C50:5, C52:5, C54:3, C54:4, C54:5, and C54:6, had the greatest reduction (Fig. 4E). These TAG species are mainly composed of essential PUFAs, like linoleic acid (18:2 linoleic acid), AA (20:4 AA), eicosapentaenoic acid (20:5 eicosapentaenoic acid), and DHA (22:6 DHA). These ω‐3 and ω‐6 fatty acids cannot be synthesized by mammalian cells, and the only source for these PUFAs is from diet. Because in the intragastric alcohol feeding model the mice passively consumed the same liquid food within both groups, we expected that there would be more PUFAs oxidized within hCYP2E1 liver in the more oxidative environment. Indeed, eicosanoids, more specifically PGD2, PGE2, and thromboxane B2, were increased in the hCYP2E1 liver (Fig. 2C). These PGs are derived from AA and are produced enzymatically involving cyclooxygenase 1‐ and cyclooxygenase 2‐dependent pathways.46, 47, 48 Higher CYP2E1 activity contributes to ω‐1 hydroxylation of AA after chronic alcohol consumption and is responsible for the decrease of AA content.10, 49, 50 Our study demonstrated higher metabolism of AA by human than mouse CYP2E1, which would contribute to the increased proinflammatory AA metabolites in hCYP2E1 liver.
Consistent with previous reports, we observed decreased CYP2E1 mRNA and protein after CCl4 administration (Fig. 6B). CYP2E1 is highly expressed within the hepatocytes located around the pericentral zone.38 Therefore, CCl4‐induced injury and cell death might occur in the hepatocytes with high expression of CYP2E1, further causing reduced CYP2E1 mRNA in the liver. Similarly, we observed reduced CYP2E1 protein and mRNA within the mouse liver after BDL (Fig. 7B), consistent with previous reports of reduced CYP2E1 mRNA expression in the liver of patients with cholestatic diseases.28 However, because CYP2E1 does not participate in biliary fibrosis, the two different CYP2E1s have no effect on the phenotype of this model.
The present study demonstrates that CYP2E1 played different roles in varied experimental liver fibrosis models. Most dramatically, alcoholic liver disease had more severe ROS, inflammation, and fibrosis but less steatosis in the hCYP2E1 mice. Future studies on different types of liver injury may benefit by using hCYP2E1 mice to more closely reflect human biochemistry.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1115/full.
Supporting Information
Acknowledgment
We thank Karin Diggle for her excellent technical support.
Potential conflict of interest: Dr. Dennis is a cofounder, director, and consultant to LipoNexus, Inc. and holds equity in the company. The other authors have nothing to report.
Supported by National Institutes of Health grants 2 P50 AA011999 (D.A.B.), 5 U01 AA021856 (D.A.B.), 5 P42 ES010337 (D.A.B.), 2P30DK064391 (E.A.D., O.Q.), HL38180, DK112378, DK56260, DK52574 (N.O.D.), and R01DK105961 (E.A.D., O.Q.).
REFERENCES
- 1. Lieber CS. Cytochrome P‐4502E1: its physiological and pathological role. Physiol Rev 1997;77:517‐544. [DOI] [PubMed] [Google Scholar]
- 2. Kessova I, Cederbaum AI. CYP2E1: biochemistry, toxicology, regulation and function in ethanol‐induced liver injury. Curr Mol Med 2003;3:509‐518. [DOI] [PubMed] [Google Scholar]
- 3. Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med 2008;44:723‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cederbaum AI. Microsomal generation of reactive oxygen species and their possible role in alcohol hepatotoxicity. Alcohol Alcohol Suppl 1991;1:291‐296. [PubMed] [Google Scholar]
- 5. Song BJ, Veech RL, Park SS, Gelboin HV, Gonzalez FJ. Induction of rat hepatic N‐nitrosodimethylamine demethylase by acetone is due to protein stabilization. J Biol Chem 1989;264:3568‐3572. [PubMed] [Google Scholar]
- 6. Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J 2001;15:1335‐1349. [DOI] [PubMed] [Google Scholar]
- 7. Adas F, Salaun JP, Berthou F, Picart D, Simon B, Amet Y. Requirement for omega and (omega;‐1)‐hydroxylations of fatty acids by human cytochromes P450 2E1 and 4A11. J Lipid Res 1999;40:1990‐1997. [PubMed] [Google Scholar]
- 8. Nieto N. Ethanol and fish oil induce NFkappaB transactivation of the collagen alpha2(I) promoter through lipid peroxidation‐driven activation of the PKC‐PI3K‐Akt pathway. Hepatology 2007;45:1433‐1445. [DOI] [PubMed] [Google Scholar]
- 9. Makita K, Falck JR, Capdevila JH. Cytochrome P450, the arachidonic acid cascade, and hypertension: new vistas for an old enzyme system. FASEB J 1996;10:1456‐1463. [DOI] [PubMed] [Google Scholar]
- 10. Nanji AA, Sadrzadeh SM, Dannenberg AJ. Liver microsomal fatty acid composition in ethanol‐fed rats: effect of different dietary fats and relationship to liver injury. Alcohol Clin Exp Res 1994;18:1024‐1028. [DOI] [PubMed] [Google Scholar]
- 11. Morimoto M, Reitz RC, Morin RJ, Nguyen K, Ingelman‐Sundberg M, French SW. CYP‐2E1 inhibitors partially ameliorate the changes in hepatic fatty acid composition induced in rats by chronic administration of ethanol and a high fat diet. J Nutr 1995;125:2953‐2964. [DOI] [PubMed] [Google Scholar]
- 12. Purohit V, Gao B, Song BJ. Molecular mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res 2009;33:191‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oh SY, Park SK, Kim JW, Ahn YH, Park SW, Kim KS. Acetyl‐CoA carboxylase beta gene is regulated by sterol regulatory element‐binding protein‐1 in liver. J Biol Chem 2003;278:28410‐28417. [DOI] [PubMed] [Google Scholar]
- 15. McClain CJ, Barve S, Barve S, Deaciuc I, Hill DB. Tumor necrosis factor and alcoholic liver disease. Alcohol Clin Exp Res 1998;22(Suppl.):248S‐252S. [DOI] [PubMed] [Google Scholar]
- 16. Hong F, Kim WH, Tian Z, Jaruga B, Ishac E, Shen X, et al. Elevated interleukin‐6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol‐induced apoptosis in the liver: involvement of induction of Bcl‐2 and Bcl‐x(L) proteins. Oncogene 2002;21:32‐43. [DOI] [PubMed] [Google Scholar]
- 17. Mathews S, Gao B. Therapeutic potential of interleukin 1 inhibitors in the treatment of alcoholic liver disease. Hepatology 2013;57:2078‐2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu R, Huang H, Zhang Z, Wang FS. The role of neutrophils in the development of liver diseases. Cell Mol Immunol 2014;11:224‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Friedman SL. Stellate cell activation in alcoholic fibrosis‐‐an overview. Alcohol Clin Exp Res 1999;23:904‐910. [PubMed] [Google Scholar]
- 20. Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol 2003;33:105‐136. [DOI] [PubMed] [Google Scholar]
- 21. Wong FW, Chan WY, Lee SS. Resistance to carbon tetrachloride‐induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol Appl Pharmacol 1998;153:109‐118. [DOI] [PubMed] [Google Scholar]
- 22. Eliasson E, Johansson I, Ingelman‐Sundberg M. Substrate‐, hormone‐, and cAMP‐regulated cytochrome P450 degradation. Proc Natl Acad Sci U S A 1990;87:3225‐3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knockaert L, Berson A, Ribault C, Prost PE, Fautrel A, Pajaud J, et al. Carbon tetrachloride‐mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab Invest 2012;92:396‐410. [DOI] [PubMed] [Google Scholar]
- 24. Lan T, Kisseleva T, Brenner DA. Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis in mice through inhibition of hepatic stellate cell activation. PLoS One 2015;10:e0129743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dyson JK, Hirschfield GM, Adams DH, Beuers U, Mann DA, Lindor KD, et al. Novel therapeutic targets in primary biliary cirrhosis. Nat Rev Gastroenterol Hepatol 2015;12:147‐158. [DOI] [PubMed] [Google Scholar]
- 27. Fausther M, Goree JR, Lavoie EG, Graham AL, Sevigny J, Dranoff JA. Establishment and characterization of rat portal myofibroblast cell lines. PLoS One 2015;10:e0121161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Padda MS, Sanchez M, Akhtar AJ, Boyer JL. Drug‐induced cholestasis. Hepatology 2011;53:1377‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bogaards JJ, Bertrand M, Jackson P, Oudshoorn MJ, Weaver RJ, van Bladeren PJ, et al. Determining the best animal model for human cytochrome P450 activities: a comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica 2000;30:1131‐1152. [DOI] [PubMed] [Google Scholar]
- 30. Cheung C, Yu AM, Ward JM, Krausz KW, Akiyama TE, Feigenbaum L, et al. The cyp2e1‐humanized transgenic mouse: role of cyp2e1 in acetaminophen hepatotoxicity. Drug Metab Dispos 2005;33:449‐457. [DOI] [PubMed] [Google Scholar]
- 31. Adas F, Berthou F, Salaun JP, Dreano Y, Amet Y. Interspecies variations in fatty acid hydroxylations involving cytochromes P450 2E1 and 4A. Toxicol Lett 1999;110:43‐55. [DOI] [PubMed] [Google Scholar]
- 32. Cheung C, Gonzalez FJ. Humanized mouse lines and their application for prediction of human drug metabolism and toxicological risk assessment. J Pharmacol Exp Ther 2008;327:288‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ueno A, Lazaro R, Wang PY, Higashiyama R, Machida K, Tsukamoto H. Mouse intragastric infusion (iG) model. Nat Protoc 2012;7:771‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iwaisako K, Jiang C, Zhang M, Cong M, Moore‐Morris TJ, Park TJ, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci USA 2014;111:E3297‐E3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J Lipid Res 2010;51:3299‐3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Newberry EP, Xie Y, Kennedy SM, Graham MJ, Crooke RM, Jiang H, et al. Prevention of hepatic fibrosis with liver microsomal triglyceride transfer protein deletion in liver fatty acid binding protein null mice. Hepatology 2017;65:836‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song BJ, Veech RL, Park SS, Gelboin HV, Gonzalez FJ. Induction of rat hepatic N‐nitrosodimethylamine demethylase by acetone is due to protein stabilization. J Biol Chem 1989;264:3568‐3572. [PubMed] [Google Scholar]
- 38. Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol‐induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock‐in mice. Free Radic Biol Med 2010;49:1406‐1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kono H, Bradford BU, Yin M, Sulik KK, Koop DR, Peters JM, et al. CYP2E1 is not involved in early alcohol‐induced liver injury. Am J Physiol 1999;277:G1259‐G1267. [DOI] [PubMed] [Google Scholar]
- 40. Wheeler MD, Nakagami M, Bradford BU, Uesugi T, Mason RP, Connor HD, et al. Overexpression of manganese superoxide dismutase prevents alcohol‐induced liver injury in the rat. J Biol Chem 2001;276:36664‐36672. [DOI] [PubMed] [Google Scholar]
- 41. Chen Y, Singh S, Matsumoto A, Manna SK, Abdelmegeed MA, Golla S, et al. Chronic glutathione depletion confers protection against alcohol‐induced steatosis: implication for redox activation of AMP‐activated protein kinase pathway. Sci Rep 2016;6:29743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fediuc S, Gaidhu MP, Ceddia RB. Regulation of AMP‐activated protein kinase and acetyl‐CoA carboxylase phosphorylation by palmitate in skeletal muscle cells. J Lipid Res 2006;47:412‐420. [DOI] [PubMed] [Google Scholar]
- 43. Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1‐Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 2013;1:45‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hayes JD, Dinkova‐Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 2014;39:199‐218. [DOI] [PubMed] [Google Scholar]
- 45. Xu J, Kulkarni SR, Donepudi AC, More VR, Slitt AL. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin‐deficient mice. Diabetes 2012;61:3208‐3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 2001;294:1871‐1875. [DOI] [PubMed] [Google Scholar]
- 47. Buczynski MW, Dumlao DS, Dennis EA. Thematic review series: proteomics. An integrated omics analysis of eicosanoid biology. J Lipid Res 2009;50:1015‐1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol 2015;15:511‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. French SW, Ihrig TJ, Morin RJ. Lipid composition of RBC ghosts, liver mitochondria and microsomes of ethanol‐fed rats. Q J Stud Alcohol 1970;31:801‐809. [PubMed] [Google Scholar]
- 50. Cairns SR, Peters TJ. Biochemical analysis of hepatic lipid in alcoholic and diabetic and control subjects. Clin Sci (Lond) 1983;65:645‐652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1115/full.
Supporting Information