

Abstract
Hepatitis B virus (HBV) reactivation (HBVr) in recipients of allogeneic hematopoetic stem cells (aHSCs) appears heterogeneously with respect to its frequency, manifestation, and outcome. The aim of this study was to present data from a large German cohort of recipients of aHSC transplantation (aHSCT), focusing on the incidence of HBVr in antibody to hepatitis B core antigen (anti‐HBc)‐positive aHSCT recipients, its clinical outcome, and the role of mutations in HBV. Between 2005 and 2015, 1,871 patients received aHSCT at University Hospital Essen. A follow‐up of at least 6 months after transplant was available in 55 patients who were anti‐HBc‐positive; clinical and virologic data were analyzed. The HBV genome was sequenced with next generation technology from serum samples of 8 patients with HBVr. Thirteen out of 55 (23.6%) patients developed HBVr at a median of 26 months after aHSCT. After initiation of antiviral treatment, complete HBV DNA suppression was achieved in 7/10 (70%) patients 1 to 40 months after HBVr. Nine of 13 patients had increased alanine aminotransferase; 3 patients had compromised coagulation and model for end‐stage liver disease scores of 18‐27, and 1 of these patients died due to liver failure 5 weeks after HBVr. As a risk factor for HBVr, we identified anti‐HBc signal to cut‐off ration (S/CO) ≥7.5 before transplantation. Complete HBV DNA suppression was achieved in 7/10 patients; therapy‐relevant mutations were found in 1 patient. In 4/8 patients, immune escape mutations were detected either as majority or minority variants. Conclusion: HBVr is common in anti‐HBc‐positive aHRCT recipients and can lead to severe hepatitis with compromised coagulation. The level of anti‐HBc S/CO before transplantation is a risk factor for HBVr. Complete virologic response under adequate antiviral treatment could not be achieved in all patients. (Hepatology Communications 2017;1:1014–1023)
Abbreviations
- aHSCT
allogeneic hematopoetic stem cell transplantation
- anti‐HBc
antibody to hepatitis B core antigen
- anti‐HBe
antibody to hepatitis B e antigen
- anti‐HBs
antibody to hepatitis B surface antigen
- GVHD
graft‐versus‐host‐disease
- HBc
hepatitis B core antigen
- HBeAg
hepatitis B e antigen
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HBVr
hepatitis B virus reactivation
- HCV
hepatitis C virus
- NGS
next generation sequencing
- ROC
receiver operating characteristic
- RT
reverse transcriptase
- SHB
small hepatitis B surface antigen
- S/CO
signal to cut‐off ratio
Introduction
Reactivation of hepatitis B infection (HBVr) has been defined as the reappearance or rise of hepatitis B virus (HBV) DNA in patients with inactive or resolved HBV infection. Although it can occur spontaneously, it is often triggered by immunosuppression, for example, due to chemotherapy, rituximab treatment, or following solid organ transplantation. Clinical manifestations range from asymptomatic to clinical hepatitis with acute liver failure and may lead to immunologic control or persistence of HBV infection.1
HBVr after allogeneic hematopoetic stem cell transplantation (aHSCT) shows a heterogeneous picture concerning its frequency, manifestation, and outcome. Its incidence varies greatly among different studies, ranging from 2.6% to 86% in patients with resolved hepatitis B infection.2, 3 The time point of the reactivation varies as well, from an average of 10 to 48 months.4, 5 Clinical manifestation includes patients who are asymptomatic with no or mild biochemical hepatitis and who manage to clear the infection,5 patients that develop persistent hepatitis and maintain HBV replication even under adequate antiviral treatment,6 and patients with fulminant acute hepatitis B.7 Recently, Seto et al.5 published a prospective study investigating the course of 62 recipients of antibody to hepatitis B core antigen (anti‐HBc)‐positive/hepatitis B surface antigen (HBsAg)‐negative aHSCT. HBVr occurred at a median of 44 weeks after aHSCT. In contrast to other mainly retrospective studies, HBV DNA was measured to detect HBVr. Interestingly, HBsAg remained undetectable in nearly all patients and none of them developed severe hepatitis.5 Therefore, it might be possible that detection of HBV DNA might lead to earlier induction of antiviral therapy and might avoid hepatitis and/or liver failure. However, it remains unclear if these results from Asian patients can be transferred to Caucasian patients because HBV incidence and the time point of infection differ. To date, only one aHSCT patient cohort from Germany has been evaluated for the risk of HBVr2; however, the number of patients in that study was low, and therefore no representative study is available.
The aim of our study was to investigate the frequency and time point of HBVr as well as clinical and therapeutic outcomes in anti‐HBc‐positive patients undergoing aHSCT in a large Caucasian cohort. In addition, we tried to identify therapy‐relevant mutations in these patients by genome sequencing using next generation sequencing (NGS) technology to investigate if these mutations occur with a higher frequency compared to patients with chronic HBV.
Patients and Methods
STUDY DESIGN
Between 2005 and 2015, 1,871 patients underwent aHSCT at University Hospital Essen. Before transplantation, all patients were tested for anti‐HBc, HBsAg, and in 1,458 cases for antibody to hepatitis B surface antigen (anti‐HBs). Detection of HBV DNA was performed in patients that were HBsAg‐positive. Of the 1,871 patients, 119 (6.4%) were anti‐HBc‐positive/HBsAg‐negative before transplantation. A posttransplant follow‐up of at least 6 months was available for 55 patients, and these patients were included in this analysis; none received prophylactic antiviral therapy. Deceased patients or patients that were lost to follow‐up were excluded from this study. Of the 55 patients, 13 (23.6%) anti‐HBc‐positive HBsAg‐negative HSCT recipients developed HBVr, defined as HBsAg positivity in patients that were previously HBsAg‐negative. HBsAg was considered positive if signal to cut‐off ratio (S/CO) was >0.05 IU/mL (quantitative assay) or >1 (qualitative assay). All patients had tested negative for HIV antibodies. One patient had a chronic hepatitis C virus (HCV) infection with detection of HCV RNA, while another patient had a post‐HCV infection with antibodies against HCV but no detection of HCV RNA. The study protocol was approved by the Ethics Committee of the University of Duisburg‐Essen. A flowchart of the study design is shown in Fig. 1.
Figure 1.
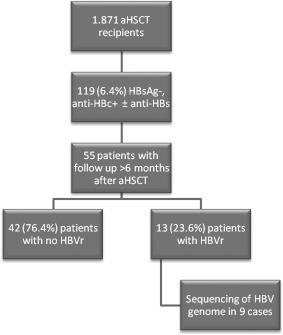
Flowchart of the study design.
LABORATORY AND SEROLOGIC PARAMETERS
Routine laboratory parameters, such as blood count, transaminases, and liver function parameters, were measured in the central laboratory of the University Hospital Essen. Clinical and virologic data were taken from patient charts, including presence of acute/chronic graft‐versus‐host disease (GVHD), comorbidities or reactivation of other viruses (cytomegalovirus, Epstein‐Barr virus, herpes simplex virus). Serologic parameters were measured at the Institute of Virology; HBsAg, anti‐HBs, and anti‐HBc were measured with Architect Abbott from 2006 to 2016 and AxSYM before 2005, according to the manufacturer's instructions (Abbott, Germany). Anti‐HBc was considered negative when the S/CO of the assay was less than 1.
SEQUENCING OF THE HBV GENOME
Archived plasma and/or serum samples obtained for diagnostic purposes were retrospectively used for DNA extraction. DNA extraction, polymerase chain reaction, and sequencing were performed as described.8 NGS of patient samples was possible in eight HBVr cases (due to serum availability or low viral load). In 1 additional patient, Sanger sequencing for genotyping and exclusion of therapy‐relevant mutation in the reverse transcriptase (RT) region had been performed. For genotype assessment and the presence of therapy‐relevant mutations in the RT region or immune escape mutations in the small hepatitis B surface antigen (SHB) region, the geno2pheno 2.0 platform was used.9
STATISTICAL ANALYSIS
Data were presented as median (interquartile range) for continuous variables and as n (percentage) for categorical variables, unless otherwise noted. We used the chi‐square/Fisher's exact test for comparison between categorical variables and the Mann‐Whitney U test for comparison between continuous variables. The Kaplan‐Meier method was used to calculate the cumulative rate of HBV reactivation at 2 years after transplantation. Data were censored if HBVr or death occurred. Receiver operating characteristic (ROC) curve calculations were also performed, and the Youden Index was applied to calculate the optimal cutoff point. The Kaplan‐Meier method was applied to present time‐to‐event variables, and the groups were formed according to the optimal cutoff point of the ROC curve and contrasted by the log‐rank test. Cox regression analysis was used to build a model to predict the outcome. Tests were performed with SPSS 21.0.
Results
ANTI‐HBc TITER AT THE TIME OF AN aHSCT IS A PREDICTIVE MARKER FOR HBVr
Clinical and biochemical data of our patient cohort are shown in Table 1. Thirteen of 55 patients developed HBVr. The cumulative risk of HBVr within 2 years was 15.1%. There were no significant differences in sex, age, body mass index, the type of underlying disease, or overall survival between patients with HBVr and patients without reactivation. In addition, donor‐specific characteristics, use of anti‐thymocyte globulin, and development of acute or chronic GVHD seem to have no impact on HBVr. Other viral reactivations, including the frequency of cytomegalovirus, Epstein‐Barr virus, or herpes simplex virus reactivation did not significantly differ between the two groups.
Table 1.
DATA OF PATIENTS POSITIVE FOR ANTI‐HBc
| Variable |
HBVr Patients n = 13 |
Non‐HBVr Patients n = 42 |
P |
|---|---|---|---|
| Age in years | 54 (19) | 52 (20) | 0.992 |
| Sex (male/female) | 8 / 5 | 23 / 19 | 0.756 |
| BMI (kg/m2) | 25.51 (4.61) | 24.72 (5.22) | 0.725 |
| Primary disease, n (%) (AML/ALL vs lymphoma vs MDS vs other) | 7 (53.8%) / 3 (23.1) / 1 (7.7%) / 2 (15.4%) | 20 (47.6%) / 5 (11.9%) / 7 (16.7%) / 10 (23.8%) | 0.614 |
| Donor (related/unrelated), n | 5 / 8 | 17 / 24 | 1 |
| HLA (matched/mismatched), n | 10 / 3 | 36 / 3 | 0.157 |
| Presence of acute GVHD, n (%) | 7 (53.8%) | 29 (69%) | 0.336 |
| Presence of chronic GVHD, n (%) | 11 (84.6%) | 34 (81%) | 1 |
| Use of ATG in conditioning protocol, n (%) | 5/13 (38.5%) | 15/41 (36.6%) | 1 |
| Follow‐up since aHSCT (months) | 52 (70) | 58.5 (63.5) | 0.968 |
| Survival 2 years posttransplant, n (%) | 11/12 (91.7%) | 36/40 (90%) | 1 |
| Survival 1 year posttransplant, n (%) | 13/13 (100%) | 41/42 (97.6%) | 1 |
| Survival 6 months posttransplant, n (%) | 13/13 (100%) | 42/42 (100%) | 1 |
| anti‐HBs ≥10 mIU/mL before aHSCT | 10 (76.9%) | 37 (88.1%) | 0.376 |
| anti‐HBc titer (S/CO) before aHSCT | 8.93 (3.32) | 6.69 (7.05) | 0.022 |
| ALT before aHSCT | 27.5 (29.75) | 24 (22.5) | 0.799 |
| INR before aHSCT | 1.11 (0.17) | 1.08 (0.12) | 0.639 |
| anti‐HBc loss after aHSCT | 6 (46.2%) | 19 (52.8%)‡ | 0.754 |
| CMV reactivation | 8 (61.5%) | 21 (50%) | 0.537 |
| EBV reactivation | 8 (61.5%) | 34 (81%) | 0.260 |
| HSV reactivation | 1 (7.7%) | 6 (16.7%)‡ | 0.658 |
| Time to HBVr (months) | 26 (25) | ‐ | ‐ |
| ALT (U/L) at HBVr | 75 (355) | ‐ | ‐ |
| anti‐HBs ≥10 mIU/mL at HBVr* | 3 (25%) | ‐ | ‐ |
| anti‐HBe positivity/HBeAg negativity at HBVr diagnosis, n (%) | 5 (38.5%) | ‐ | ‐ |
| HBV DNA (IU/mL) at HBVr† | 25.630 (217.144) | ‐ | ‐ |
Data are presented as median (interquartile range) for continuous variables and as n (percentage) for categorical variables. We used chi‐square/Fisher's exact test for comparison between categorical variables and Mann‐Whitney U test for comparison between continuous variables. Tests were performed with SPSS 21.0. Bold script indicates a statistically significant result.
*Data available for 12 patients. †Data available for 11 patients. ‡Data available for 36 patients.
Abbreviations: ALL, acute lymphoblastic leukemia; ALT, alanine transaminase; AML, acute myeloid leukemia; ATG, anti‐thymocyte globulin; BMI, body mass index; CMV, cytomegalovirus; EBV, Epstein‐Barr Virus; HLA, human leukocyte antigen; HSV, Herpes simplex virus; INR, international normalized ratio; MDS, myelodysplastic syndrome.
Interestingly, anti‐HBc titers (in S/CO) before transplantation differed between the two cohorts; patients with HBVr had significantly higher anti‐HBc titers than patients who did not have reactivation of the HBV infection. We performed an ROC curve analysis to evaluate different continuous parameters as potential predictive factors for HBVr. The only parameter with predictive value was anti‐HBc S/CO before transplantation; the ROC curve analysis indicated that it could be used to predict HBVr after aHRST with an area under the curve of 0.712 and an optimal cutoff point of 7.5, corresponding to a sensitivity of 76.9% and a specificity of 61.9% (Fig. 2). In addition, we performed a Kaplan‐Meier analysis to further evaluate the prognostic accuracy of anti‐HBc S/CO. An anti‐HBc S/CO more than 7.5 pretransplantation was significantly associated with a higher HBVr rate in our patient cohort (Fig. 3). Univariate Cox regression analysis was applied to produce a model predicting HBVr; with anti‐HBc S/CO ≥7.5 as a predictor for HBVr, we produced a model with a hazard ratio of 4.608 (1.265‐16.777) and a regression coefficient of 1.528 (0.659) at P = 0.021.
Figure 2.
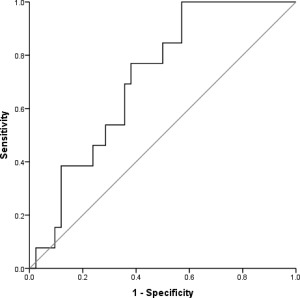
Receiver operating characteristic curve for anti‐HBc S/CO before aHRST exhibited a reasonable overall performance to discriminate patients with HBVr from those without HBVr. Diagonal segments are produced by ties.
Figure 3.
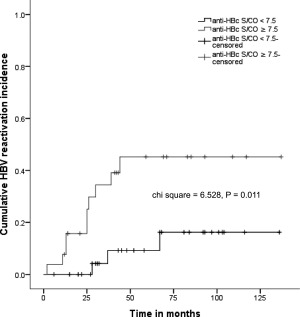
HBV reactivation rate was significantly lower in patients with anti‐HBc S/CO <7.5 (as shown in the Kaplan‐Meier analysis curve). Patients were grouped by anti‐HBc S/CO ≥7.5 or <7.5 based on the optimal cutoff point generated by the operator curve analysis. Chi‐square and P values from the log‐rank test are shown.
NOT ALL PATIENTS WITH HBVr ACHIEVED COMPLETE HBV DNA SUPPRESSION UNDER ANTIVIRAL TREATMENT
We next investigated the clinical outcome of patients with HBVr. As shown in Tables 1 and 2, patients developed HBVr after 2‐67 months (median 26 months). Most patients with HBVr were hepatitis B e antigen (HBeAg)‐positive and antibody to HBeAg (anti‐HBe)‐negative at the time of HBVr, while about half the patients in both groups lost anti‐HBc after aHCST. Twelve of 13 patients were treated either with lamivudine (n = 4), entecavir (n = 5), or tenofovir (n = 1). One patient was treated with adefovir, and 1 patient was switched from lamivudine to tenofovir due to lamivudine resistance. One patient did not receive any antiviral therapy due to recurrence of the underlying disease and death.
Table 2.
CHARACTERISTICS OF PATIENTS WITH HBVr
| Age | Sex | Disease | Time of HBVr (Months) | Outcome | Time of HBVr to Last Follow‐Up | Antiviral Treatment | HBV DNA Suppression (Months) | HBsAg Loss After HBVr (Months) | ALT (IU/mL) at HBVr | INR at HBVr | MELD Score at HBVr | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 28 | M | T‐NHL | 13 | survival | 18 | Lamivudin | 9 | ‐ | 31 | 0.99 | 7 |
| 2 | 36 | M | AML | 13 | death (sepsis) | 14 | Entecavir | ‐ | ‐ | 157 | 1.06 | 6 |
| 3 | 54 | M | AML | 2 | survival | 7 | Entecavir | NA (lost to follow‐up) | ‐ | 20 | 0.91 | 6 |
| 4 | 54 | F | AML | 44 | survival | 83 | Lamivudin | 5 | 5 | 88 | 0.90 | 6 |
| 5 | 41 | M | MDS | 30 | survival | 65 | Lamivudin, Tenofovir | 40 | 45 | 384 | 0.83 | 7 |
| 6 | 61 | F | AML | 39 | survival | 54 | Entecavir | ‐ | ‐ | 26 | 0.95 | 6 |
| 7 | 61 | F | AML | 67 | survival | 6 | Tenofovir | 6 | ‐ | 159 | NA | |
| 8 | 57 | F | sNHL | 37 | survival | 27 | Entecavir | ‐ | ‐ | 34 | 0.87 | 8 |
| 9 | 48 | M | AML | 25 | death | 22 | Entecavir | 4 | 11 | 617 | 1.66 | 27 |
| 10 | 55 | M | CML | 28 | survival | 11 | ‐ | 4 | 4 | 53 | 1.01 | 11 |
| 11 | 40 | M | HL | 26 | death (recurrence) | 0 | ‐ | NA (death after 1 week) | ‐ | 68 | 1.18 | 18 |
| 12 | 57 | M | IMF | 11 | death (liver failure) | 1 | Lamivudin | NA (death after 5 weeks) | ‐ | 229 | 1.44 | 19 |
| 13 | 61 | F | AML | 25 | survival | 16 | Adefovir | 1 | 2 | 1125 | 1.27 | 18 |
Abbreviations: ALT, alanine transaminase; AML, acute myeloid leukemia; CML, chronic myeloid leukemia; HL, Hodgkin lymphoma; IMF, idiopathic osteomyelofibrosis; INR, international normalized ratio; MDS, myelodysplastic syndrome; MELD, model for end‐stage liver disease; NA, unknown; sNHL, secondary non‐Hodgkin lymphoma; T‐NHL, T‐cell non‐Hodgkin lymphoma.
Complete HBV DNA suppression was achieved in 7/10 (70%) of patients 1 to 40 months after HBVr, while in 3 patients a follow‐up of HBV DNA was not performed. Two patients died 1 and 5 weeks after HBVr due to recurrence of the underlying disease or liver failure. One patient was lost to follow‐up 16 months after aHSCT. In 3 patients, low viral loads remained detectable 7 months to 4.5 years after initiating a specific antiviral therapy. Nine of 13 patients showed clinical signs of hepatitis with elevated alanine transaminase. Three patients even had compromised coagulation and model for end‐stage liver disease scores of 18‐27, while 1 patient died due to liver failure. In addition, HBsAg loss as the ultimate goal of HBV treatment was only achieved in 5 patients.
We compared our data with 11 published studies investigating HBVr after aHSCT. HBVr incidence, time point, and patient outcome compared to our cohort are presented in Supporting Tables S1 and S2. Overall, we found similar rates of HBVr; however, the clinical course and outcome appear to be worse when compared to the study by Seto et al.5 Importantly and in line with several other studies, we could confirm that patients with HBVr after aHSCT had no higher rates of mutations associated with resistance to antiviral treatment, as shown in Table 3.
Table 3.
HBV SEQUENCE CHARACTERISTICS OF aHSCT PATIENTS AT HBVr
| Number | GT | RT Drug Resistance Mutations | SHB Escape Mutations | SHB Mutation Compared To Genotype‐Specific Reference Sequence (Geno2pheno HBV) |
|---|---|---|---|---|
| 1 | D | no known drug resistance mutations | 120S, 105L | F8FV, F19FV, L21FL, L22FL, T23AST, I25IN, T27PT, S31RS, W36GW, T37AST, G43GW, V47GV, G50CG, Q51PQ, S61PS, C65CR, D99DGNS, Y100HY, G102GS, M103IM, L104FLV, P105LP, V106LV, C107CST, P108ILPT, I110HIKLNQ, P111*PQS, G112*G, P120S, T127P, Y134K, I150T, S154P, W163*W, T189I, V190AV, W191CW |
| 2 | E | no known drug resistance mutations | no known escape mutations in SHB | G10EG, F19FV, L21FL, I25IN, Q51QR, P66PS, T148AT, E164V, W191GW |
| 3 | D | no known drug resistance mutations, 80F (minority mutation of unknown relevance at a known mutation amino acid) | 122K, 131N | V14AV, F19FV, L21FL, L22FL, T23ST, I25IN, T27PT, I28IK, S31RS, W36GW, T37AT, N40NS, T45AST, T46PT, V47GV, S64PS, T68IT, Y72FSY, S114ST, R122KQR, T125MT, T127PT, T131NT, Y134FY, S143ST, S154PS, G159AG, F161FY, A168AV, V194AV, Y200FY, L209LV, L213IL |
| 4 | NA | NA | NA | NA |
| 5 | D | resistance to lamivudin and telbivudine, partly resistant against entecavir: V193L, L180M, M204V | no known escape mutations in SHB | F19FV, L21FL, T118A, T126AT, T131AT, W163GW, E164V, P188AP, I195M, L213FL, L216LS |
| 6 | A2 | no known drug resistance mutations | 120T, 120S, 144A | F8FV, F19FV, L21FL, S64FS, T114PT,P120PST, D144AD, A166AG, L209V |
| 7 | D | no known drug resistance mutations | no known escape mutations in SHB | F19FV, L21FL, T23AT, T27KT, C69CR, T125M |
| 8 | A2 | no known drug resistance mutations | no known escape mutations in SHB | F19FV, L21FL, S193L, L209V |
| 9 | A | no known drug resistance mutationsa | NA | NA |
| 10 | NA | NA | NA | NA |
| 11 | NA | NA | NA | NA |
| 12 | NA | NA | NA | NA |
| 13 | A2 | no known drug resistance mutations, 80F (minority mutation of unknown relevance at a known mutation amino acid) | 122K, 131N | G7EGKR, F19FV, L21FL, L22FL, T23ST, L26LP, I28IK, S31RS, T37AT, S45ST, P46PT, P56LP, N59NS, S64PS, I68IT, Y72FSY, T114ST, K122KQR, T125MT, P127PT, N131NT, F134FY, T143ST, A159AG, Y161FY, E164EG, V168AV, A194AV, L209LV, I213IL |
Mutations seen in >10% of the reads (NGS) are depicted in bold characters, mutations detected in <10% in normal characters.
Sequencing for genotype and drug‐relevant mutations in RT was performed with Sanger sequencing.
Abbreviations: GT, genotype; NA, unknown; RT, reverse transcriptase.
NO INCREASE IN THERAPY‐RELEVANT MUTATIONS DETECTED IN PATIENTS WITH HBVr
To investigate if resistance‐mediating mutations occur more frequently in patients after aHSCT, which might explain why not all of these patients responded to antiviral therapy, we performed NGS in 8 patients with HBVr. In 1 additional patient, Sanger sequencing was already available. All individual mutations compared to the genotype‐specific reference sequence (Geno2pheno HBV) are summarized in Table 3. Overall, we could detect therapy‐relevant mutations (173L, 180M, 204V) in the RT region of HBV in only 1/9 patients. Importantly, this patient achieved complete DNA suppression and even HBsAg clearance after switching to tenofovir from lamivudine. In the remaining 8 patients, no therapy‐relevant mutations were detected; however, complete virologic response was observed in 4/7 patients. One patient was lost to follow‐up. Interestingly, we were able to detect escape mutations in the SHB region as majority (120P, 120S, 120T, 122K, 131N) or minority (105L, 120P, 102S, 120T, 122K, 131N, 144A) variants in 4/8 (50%) patients. Two of these patients achieved complete HBV DNA suppression under antiviral treatment, 1 patient did not, and 1 patient was lost on follow‐up.
Discussion
Universal prophylaxis with antivirals for anti‐HBc‐positive/HBsAg‐negative aHSCT recipients remains controversial. It is recommended in several10, 11, 12 but not all guidelines,13, 14 and doubt about its cost effectiveness in HBV‐endemic regions has been expressed.5 It remains unclear whether prophylactic administration of antivirals is necessary or cost effective, which patients might benefit most, which antiviral agent is most efficient, and for how long it should be given. Furthermore, there is no standardized accepted screening method for HBVr after aHSCT.12, 13 The aim of this study is to present data of recipients of aHSCT from a large German center, focusing on the incidence, time of occurrence, and outcome of HBVr in anti‐HBc‐positive cases to identify risk factors associated with HBVr after aHSCT.
Overall, the incidence of HBVr varies among published retrospective studies, ranging from 2.6% to 86%. Reasons for the large variations might be the different screening methods that were used, the different populations, and possible bias due to the retrospective nature of the studies. The cumulative rate of HBVr within the first 2 years after aHSCT ranged from 6.3% to 21.7%6, 15, 16 in previous studies and is comparable to the rate in our cohort, which was 15.1% 2 years after aHSCT. In contrast, the cumulative rate of HBVr in the Seto et al. study,5 which was performed prospectively, was much higher, reaching 40.8%. The disparity in the results is even more intriguing when considering the differences in GVHD prevalence in patients with HBVr in our cohort compared to those of Seto et al. The prevalence of chronic GVHD in that study's cohort was 54% compared to 81% in our patients, although anti‐thymocyte globulin was used less frequently. Considering that the level of immunosuppression influenced the chronic GVHD prevalence as well as the HBVr rate, we would have expected a higher HBVr rate in our population.
Additionally, it is of particular interest that Seto et al.5 reported HBVr on an average of 10 months after aHSCT. The median time of the reactivation in published retrospective studies varied from 12 to 48 months. HBVr was most common approximately 19 months after aHSCT (Supporting Table S1). In our cohort, HBVr was diagnosed even later, at a median of 26 months after aHSCT. In fact, only in 4/13 (31%) patients, HBVr was diagnosed within the first 2 years after aHSCT. One reason for the observed disparity in the onset and incidence of HBVr in different cohorts, apart from the different populations, might be the different screening methods that were used. In most retrospective studies, including our own, HBsAg detection was used to diagnose HBVr as opposed to HBV DNA in the prospective study performed by Seto et al.5 There is evidence that HBV DNA detection usually precedes HBsAg detection in an HBVr setting with a median of 4 months (range 0 to 39 months),2 although reverse seroconversion preceding HBV DNA detection has been described.17 The use of HBsAg instead of HBV DNA for HBVr detection could limit the number of patients diagnosed with HBVr and/or delay the time point of diagnosis for HBVr. It is interesting that all but 1 patient in the study by Seto et al.5 were HBsAg‐negative at the time of HBVr diagnosis and showed no signs of hepatitis, such as elevated liver enzymes. HBV DNA could be an earlier and better screening marker than HBsAg for HBV reactivation in recipients of aHSCT, and early treatment of HBV reactivation in recipients of HSCT might be crucial for a favorable outcome. However, the clinical significance of HBVr without HBsAg reverse seroconversion or signs of hepatitis and the possible time point of therapy cessation remain unclear; further prospective studies are needed to answer this question. Using HBV DNA as a screening method to diagnose HBVr in immunosuppressed patients not receiving antiviral prophylaxis is recommended in several11, 14 but not all guidelines.13 An additional aspect to consider when comparing HBsAg versus HBV DNA as a detection method for HBVr is the availability and cost of polymerase chain reaction versus measurement of HBsAg and the cost of both screening methods versus the prophylactic administration of antivirals.
An anti‐HBc titer of more than 7.5 S/CO has been identified as a risk factor for HBVr in our cohort. This is in line with the results of a study by Bae et al.18 in which the authors concluded that an anti‐HBc S/CO ≥8 is associated with increased risk for HBVr in patients positive for anti‐HBc. In general, HBV DNA could be detected in patients with chronic HBV at very low levels after HBsAg seroclearance, and disappeared with an incidence of 10‐20% 5 and 10 years after HBsAg seroclearance.19 Also anti‐HBc titers decreased after HBV clearance.19 Another study by Kobyashi et al.20 demonstrated that lower anti‐HBc S/CO levels were associated with HBV DNA disappearance after HBsAg clearance. Thus, high anti‐HBc titers might correlate with a larger hepatic reservoir of HBV, which slowly decreases after seroclearance and could potentially explain the association of a high anti‐HBc titer with HBVr.
In addition, the clinical presentation of HBVr varied greatly among the different cohorts. In some cohorts, the patients had a favorable outcome; in the study by Seto et al.5 no patient developed elevated liver transaminases and all patients achieved complete HBV DNA suppression after 4‐10 weeks under antiviral therapy. In two other surveys, most patients managed to clear the reinfection and lost HBsAg.4, 21 However, in other cohorts, including our own, the clinical course and outcome were less favorable. Hepatitis, clinically manifested or not, has often been documented in patients with HBVr after aHSCT, as shown in Table 2. Even fulminant hepatitis in patients with HBVr after aHSCT has been reported.7, 22 In our cohort, 9/13 of our patients had increased alanine transaminase. Three patients had compromised coagulation and model for end‐stage liver disease scores of 18‐26, while 1 patient died due to liver failure. In 1 patient, drug therapy‐relevant mutations were detected, leading to a switch in therapy from lamivudine to tenofovir. Although no relevant mutation could be detected in 8 patients of our cohort, complete virologic response was only seen in 4 patients and HBsAg clearance in 2 of the 4. Three patients failed to achieve complete suppression of HBV replication despite adequate antiviral treatment. The occurrence of an incomplete response of adequate antiviral treatment in aHSCT HBVr patients has also been described in other studies.2, 6 In the absence of therapy‐relevant mutations in the viral genome and comparable immunosuppression to other patients, a possible explanation for the phenomenon might be other unknown viral or host factors. Early treatment of HBVr in recipients of HSCT might be crucial for a favorable outcome when considering the favorable outcome of Seto et al.5 In any case, our results show that the time point of the presentation can vary widely, from 2 to 44 months, so any strategy against HBVr (HBV monitoring and therapy on demand or antiviral prophylaxis) should be extended for several years after aHSCT.
In 50% of patients, we detected known immune escape mutations, more often as minority variants but also as majority variants. Immune escape mutations have been described in the HBV genome of blood donors infected with hepatitis B with a prevalence of 14.1%23 and in patients developing HBVr upon immunosuppression,24 although to the best of our knowledge, not in the setting of HBVR following aHSCT. It has been postulated that immunosuppression could favor the production of mutated viral species with increased potential to evade immune responses.24 Some of the SHB mutations we detected, such as 120S, 120T, 131N, and 144A, have been observed in vaccine or immunoglobulin therapy escape cases.25, 26 It is impossible to know if these mutations were part of the viral template (cccDNA) in the hepatocytes prior to reactivation and have possibly aided in the reactivation of the virus or if the immunosuppressed state of the patients allowed the development of many quasi‐species, some including escape mutations. In any case, the detection of these mutations, in combination with the detection of drug therapy‐relevant mutations and the potentially unfavorable clinical course of patients with HBVr underscores the need for prophylaxis or at least careful patient monitoring and, in case of HBVr, the importance of treating the patients with an antiviral substance with a high genetic barrier to resistance.
To the limitations of our study belong its retrospective character and the relatively limited number of anti‐HBc‐positive patients in our patient cohort. HBVr can vary greatly in its clinical manifestation and outcome. HBVr is frequently diagnosed in anti‐HBc‐positive aHRCT recipients and can lead to hepatitis with compromised coagulation in some cases. Complete virologic response under adequate antiviral treatment cannot be achieved in some patients. Importantly, the time point of HBVr varies between 2 and 44 months, so any strategy against HBVr (HBV monitoring and therapy on demand or antiviral prophylaxis) should be extended for several years after aHSCT.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1118/full.
Supporting Information
Potential conflict of interest: Nothing to report.
Supported by the German Research Association DFG‐TRR60 OA 2015‐2017.
REFERENCES
- 1. Hoofnagle JH. Reactivation of hepatitis B. Hepatology 2009;49(Suppl.):S156‐S165. [DOI] [PubMed] [Google Scholar]
- 2. Knoll A, Boehm S, Hahn J, Holler E, Jilg W. Long‐term surveillance of haematopoietic stem cell recipients with resolved hepatitis B: high risk of viral reactivation even in a recipient with a vaccinated donor. J Viral Hepat 2007;14:478‐483. [DOI] [PubMed] [Google Scholar]
- 3. Park S, Kim K, Kim DH, Jang JH, Kim SJ, Kim WS, et al. Changes of hepatitis B virus serologic status after allogeneic hematopoietic stem cell transplantation and impact of donor immunity on hepatitis B virus. Biol Blood Marrow Transplant 2011;17:1630‐1637. [DOI] [PubMed] [Google Scholar]
- 4. Onozawa M, Hashino S, Izumiyama K, Kahata K, Chuma M, Mori A, et al. Progressive disappearance of anti‐hepatitis B surface antigen antibody and reverse seroconversion after allogeneic hematopoietic stem cell transplantation in patients with previous hepatitis B virus infection. Transplantation 2005;79:616‐619. [DOI] [PubMed] [Google Scholar]
- 5. Seto WK, Chan TS, Hwang YY, Wong DK, Fung J, Liu KS, et al. Hepatitis B reactivation in occult viral carriers undergoing hematopoietic stem cell transplantation: a prospective study. Hepatology 2017;65:1451‐1461. [DOI] [PubMed] [Google Scholar]
- 6. Hammond SP, Borchelt AM, Ukomadu C, Ho VT, Baden LR, Marty FM. Hepatitis B virus reactivation following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2009;15:1049‐1059. [DOI] [PubMed] [Google Scholar]
- 7. Kitano K, Kobayashi H, Hanamura M, Furuta K, Ueno M, Rokuhara A, et al. Fulminant hepatitis after allogenic bone marrow transplantation caused by reactivation of hepatitis B virus with gene mutations in the core promotor region. Eur J Haematol 2006;77:255‐258. [DOI] [PubMed] [Google Scholar]
- 8. Anastasiou OE, Widera M, Verheyen J, Korth J, Gerken G, Helfritz FA, et al. Clinical course and core variability in HBV infected patients without detectable anti‐HBc antibodies. J Clin Virol 2017;93:46‐52. [DOI] [PubMed] [Google Scholar]
- 9. Beggel B, Buch J, Däumer M, Hoffmann D, Kaiser R, Lengauer T, et al. Geno2pheno [HBV]. http://hbv.bioinf.mpi‐inf.mpg.de/index.php. 2009.
- 10. Cornberg M, Protzer U, Petersen J, Wedemeyer H, Berg T, Jilg W, et al. Prophylaxis, diagnosis and therapy of hepatitis B virus infection ‐ the German guideline. [in German] Z Gastroenterol 2011;49:871‐930. 21748700 [Google Scholar]
- 11. European Association for the Study of the Liver . EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J Hepatol 2012;57:167‐185. [DOI] [PubMed] [Google Scholar]
- 12. Sarin SK, Kumar M, Lau GK, Abbas Z, Chan HL, Chen CJ, et al. Asian‐Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hepatol Int 2016;10:1‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reddy KR, Beavers KL, Hammond SP, Lim JK, Falck‐Ytter YT. 2015. American Gastroenterological Association Institute guideline on the prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology 2015;148:215‐219. [DOI] [PubMed] [Google Scholar]
- 14. Hwang JP, Somerfield MR, Alston‐Johnson DE, Cryer DR, Feld JJ, Kramer BS, et al. Hepatitis B virus screening for patients with cancer before therapy: American Society of Clinical Oncology provisional clinical opinion update. J Clin Oncol 2015;33:2212‐2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mikulska M, Nicolini L, Signori A, Rivoli G, Del Bono V, Raiola AM, et al. Hepatitis B reactivation in HBsAg‐negative/HBcAb‐positive allogeneic haematopoietic stem cell transplant recipients: risk factors and outcome. Clin Microbiol Infect 2014;20:O694‐O701. [DOI] [PubMed] [Google Scholar]
- 16. Vigano M, Vener C, Lampertico P, Annaloro C, Pichoud C, Zoulim F, et al. Risk of hepatitis B surface antigen seroreversion after allogeneic hematopoietic SCT. Bone Marrow Transplant 2011;46:125‐131. [DOI] [PubMed] [Google Scholar]
- 17. Kanaan N, Kabamba B, Marechal C, Pirson Y, Beguin C, Goffin E, et al. Significant rate of hepatitis B reactivation following kidney transplantation in patients with resolved infection. J Clin Virol 2012;55:233‐238. [DOI] [PubMed] [Google Scholar]
- 18. Bae SK, Gushima T, Saito N, Yamanaka I, Shimokawa T, Matsuo Y, et al. The impact of hepatitis B core antibody levels on HBV reactivation after allogeneic hematopoietic SCT: an 11‐year experience at a single center. Bone Marrow Transplant 2016;51:1496‐1498. [DOI] [PubMed] [Google Scholar]
- 19. Arase Y, Suzuki F, Suzuki Y, Saitoh S, Kobayashi M, Akuta N, et al. Long‐term presence of HBV in the sera of chronic hepatitis B patients with HBsAg seroclearance. Intervirology 2007;50:161‐165. [DOI] [PubMed] [Google Scholar]
- 20. Kobyashi M, Chayama K, Arase Y, Tsubota A, Saitoh S, Suzuki Y, et al. Progressive and sufficient decrease of hepatitis B core antibody can predict the disappearance of hepatitis B virus DNA in Japanese patients with hepatitis B surface antigen clearance. J Gastroenterol 2000;35:753‐757. [DOI] [PubMed] [Google Scholar]
- 21. Dhedin N, Douvin C, Kuentz M, Saint Marc MF, Reman O, Rieux C, et al. Reverse seroconversion of hepatitis B after allogeneic bone marrow transplantation: a retrospective study of 37 patients with pretransplant anti‐HBs and anti‐HBc. Transplantation 1998;66:616‐619. [DOI] [PubMed] [Google Scholar]
- 22. Schubert A, Michel D, Mertens T. Late HBsAg seroreversion of mutated hepatitis B virus after bone marrow transplantation. BMC Infect Dis 2013;13:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harris BJ, Holzmayer V, Qureshi H, Khan MA, Khan SA, Salamat N, et al. Hepatitis B genotypes and surface antigen mutants present in Pakistani blood donors. PloS One 2017;12:e0178988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salpini R, Colagrossi L, Bellocchi MC, Surdo M, Becker C, Alteri C, et al. Hepatitis B surface antigen genetic elements critical for immune escape correlate with hepatitis B virus reactivation upon immunosuppression. Hepatology 2015;61:823‐833. [DOI] [PubMed] [Google Scholar]
- 25. Echevarria JM, Avellon A. Hepatitis B virus genetic diversity. J Med Virol 2006;78(Suppl. 1):S36‐S42. [DOI] [PubMed] [Google Scholar]
- 26. Chen WN, Oon CJ. Hepatitis B virus surface antigen (HBsAg) mutants in Singapore adults and vaccinated children with high anti‐hepatitis B virus antibody levels but negative for HBsAg. J Clin Microbiol 2000;38:2793‐2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1118/full.
Supporting Information