

Abstract
Previous studies have found that preferential accumulation of regulatory T (Treg) cells in liver allografts during acute cellular rejection (ACR) is associated with less severe rejection, suggesting a role of Treg cells in preventing excessive progress of ACR. We investigated the impact of single nucleotide polymorphisms (SNPs) in the Forkhead box P3 (FOXP3) gene, a master regulator gene of Treg cells, on ACR severity in liver transplant (LT) recipients. In total, 102 living donor LT patients were enrolled in this study and categorized into no rejection (n = 86), steroid‐sensitive acute rejection (SSAR; n = 11), and steroid‐resistant acute rejection (SRAR; n = 5). FOXP3 SNPs –3499 A/G (rs3761547), –3279 A/C (rs3761548), and –924 A/G (rs2232365) were genotyped using the polymerase chain reaction restriction fragment length polymorphism technique. T‐cell responses to allostimulation were evaluated by the mixed lymphocyte reaction assay. We found no statistical association between the FOXP3 SNP genotype frequencies and ACR incidence. However, significantly higher incidence of SRAR was observed in LT patients with the FOXP3 rs3761548 A/C+A/A genotype than in those with the C/C genotype (A/C+A/A versus C/C; no rejection, SSAR, SRAR, 85.71%, 0%, 14.29% versus 83.58%, 16.42%, 0%, respectively; P = 0.0005). The mixed lymphocyte reaction assay performed at the time of ACR diagnosis showed higher anti‐donor CD4+ T‐cell responses in patients carrying rs3761548 A/C+A/A than in those with the C/C genotype (P = 0.019). No significant association was observed between the incidence of SRAR and either rs3761547A/G or rs2232365 A/G. Infectious complications and overall survival were not related to FOXP3 SNPs.
Conclusion: Our findings indicate that FOXP3 SNP rs3761548 A/C might be a predisposing factor for SRAR after liver transplantation. (Hepatology Communications 2017;1:406–420)
Abbreviations
- ACR
acute cellular rejection
- BSI
bloodstream infection
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- CMV
cytomegalovirus
- DSA
donor specific anti‐human leukocyte antigen antibody
- EAD
early allograft dysfunction
- FOXP3
forkhead box P3
- GCR
glucocorticoid receptor
- HLA
human leukocyte antigen
- IFN
interferon
- IL
interleukin
- iTreg
induced regulatory T cell
- LDLT
living donor liver transplantation
- LT
liver transplant
- MELD
model for end‐stage liver disease
- MLR
mixed lymphocyte reaction
- MP
methylprednisolone
- nTreg
regulatory T cell suppressing self‐antigens
- OKT3
anti‐human CD3 antibody
- PBMC
peripheral blood mononuclear cells
- PE
phycoerythrin
- PI
proliferation index
- rATG
rabbit ATG
- SI
stimulation index
- SNP
single nucleotide polymorphism
- SRAR
steroid‐resistant acute rejection
- SSAR
steroid‐sensitive acute rejection
- TAC
tacrolimus
- Treg
regulatory T cell
Introduction
Regulatory T cells (Tregs) expressing the transcription factor forkhead box P3 (Foxp3) are essential for immune homeostasis.1, 2 They naturally arise in the thymus as a lineage separate from conventional CD4+ CD25– T cells and function in the periphery, primarily to suppress responses to self‐antigens (nTregs).3 In addition, CD4+ T cells that encounter antigens in a certain microenvironment in the periphery can differentiate into “adaptive” or “induced” Treg cells (iTregs) that also express high levels of Foxp3.4 Both types of Tregs likely control allograft rejection.5 As the frequency of alloantigen‐reactive T cells in the immune repertoire of organ transplant recipients is usually higher compared with the relatively small frequency of nTreg cells, the induction of immunosuppressive therapy is indispensable to prevent allograft destruction. Interestingly, the allograft itself can induce/expand Treg populations that can protect it from rejection,6 and even when the primary allograft has been rejected, cells with characteristics of Tregs are found in the recipients.7, 8 This indicates that exposure to alloantigens can lead to the generation of iTregs and/or expansion of nTreg populations even in the progress of alloimmune responses, suggesting that the unforeseeable consequence of a rejection reaction toward allografts might be attributed to individual differences in iTreg/nTreg functions among recipients.
The development and function of Tregs is controlled by the FOXP3 gene encoding Foxp3 protein, which regulates T‐cell activation and functions as a transcriptional repressor to down‐regulate cytokine production in T cells.9 Polymorphisms in the promoter region of FOXP3 may potentially alter gene expression by changing the binding specificity of transcription factors to their binding sites and by modifying the kinetics of transcription initiation, causing Treg dysfunction and consequently the development of autoimmune diseases.10, 11 Considering the critical role of Tregs in immune responses to alloantigens after organ transplantation and previous results suggesting associations between genetic polymorphisms in the FOXP3 gene and autoimmune diseases, FOXP3 polymorphisms presumably might be associated with the severity of alloimmune responses after organ transplantation. Consistent with this, a possible association between the genotype at the FOXP3 rs3761548 locus and rejection‐free allograft survival in renal transplantation has been demonstrated, although the impact of FOXP3 polymorphisms on the severity of the rejection reaction toward kidney allografts remains to be elucidated.12
Because preferential accumulation of Tregs in liver allografts during acute cellular rejection (ACR) is reportedly associated with reduced severity of rejection,13, 14 suggesting a role of Tregs in preventing excessive progress of ACR, the present study investigated the impact of FOXP3 single nucleotide polymorphisms (SNPs) on the severity of ACR in liver transplant (LT) recipients.
Patients and Methods
PATIENT POPULATION
The study population consisted of 102 consecutive patients who received a primary living donor LT (LDLT) at Hiroshima University Hospital from 2006 to 2015. The study was performed in accordance with the declaration of Helsinki and its amendments. The study and protocol were approved by the Institutional Review Board of Hiroshima University (No. Hi‐77), and written consent was obtained from all subjects.
IMMUNOSUPPRESSIVE REGIMEN
The basic immunosuppressive regimen used following LDLT has been described.15, 16, 17 In brief, the immunosuppressive regimen consisted of tacrolimus (TAC) and methylprednisolone (MP) with gradual tapering of the dose. The whole‐blood trough level of TAC was maintained between 8 and 15 ng/mL in the first few postoperative weeks and between 5 and 10 ng/mL thereafter. For ABO‐incompatible patients, preoperative desensitization was performed. Rituximab (500 mg) was intravenously administered 2 weeks before the operation. TAC (trough, 5‐10 ng/mL) and mycophenolate mofetil were orally administered 2 weeks before the operation. Plasma exchange or double‐filtration plasmapheresis was performed when the antibody titer remained elevated. An immunosuppressive regimen similar to that for ABO‐compatible cases was administered after LDLT.
DEFINITION AND TREATMENT OF ACR
ACR was defined as graft dysfunction, evidenced by elevated transaminase and/or bilirubin, with their persistent initial elevation at least 3 times the upper normal limit in the absence of vascular or biliary complication or infection. Vascular and biliary complications were identified with the help of Doppler ultrasound. The clinical suspicion of ACR was supported by the protocoled mixed lymphocyte reaction (MLR) assay, which can rigorously monitor rejection.18 Episodes of rejection were initially treated with either mini pulse (125‐250 mg intravenous MP for 2‐3 days or more) or with steroid pulse (500 mg intravenous MP for 3 days or more), according to the clinical severity of ACR, with a gradual tapering of the dose and return to the previous oral double‐drug regimen. Rejection was considered steroid‐resistance acute rejection (SRAR) when liver function tests improved by <50% of the highest values after three steroid boluses. Most cases of SRAR were treated with anti‐human CD3 antibody (OKT3; Janssen‐Kyowa Co. Ltd., Tokyo, Japan) or rabbit ATG (rATG) (Thymoglobulin; Sanofi K.K., Shinjuku, Japan). Regardless of the response to steroids, recovery from acute rejection was considered when liver function tests returned to normal within a week after treatment. For transplants performed after 2010, biopsies of the liver allografts were taken from patients diagnosed with ACR after steroid pulse treatment to evaluate the effects of the steroid treatment and/or to distinguish between patients showing rejection and recurrence of the original disease. The histologic examination of liver biopsies was performed according to the Banff classification.19
HUMAN LEUKOCYTE ANTIGEN ANTIBODY DETERMINATION
The donor‐specific anti‐human leukocyte antigen (HLA) antibody (DSA) levels were analyzed at 1 month posttransplantation in patients with ACR. Anti‐HLA single antigen reactivity was detected on a Luminex platform (LABScan 100 flow analyzer; Luminex Corporation, Austin, TX) according to the manufacturer's protocol using LABScreen Single Antigen assays. The results were recorded as the mean fluorescence intensity, and values greater than 1,000 were considered positive as previously described.20 De novo DSAs were defined as HLA‐A, HLA‐B, HLA‐C, DRB1, or DQB1 antibodies detected against the donor HLA that were not present at the pretransplant assessment.
DEFINITION OF EARLY ALLOGRAFT DYSFUNCTION
Early allograft dysfunction (EAD) was defined based on previous studies21, 22 as the presence of one of the following variables after LDLT: serum bilirubin level ≥ 10 mg/dL, international normalized ratio ≥1.6 on postoperative day 7, and alanine aminotransferase or aspartate aminotransferase >2,000 IU/L within the first 7 postoperative days.
MLR ASSAY
To monitor the recipient immune status, the MLR assay using a carboxyfluorescein diacetate succinimidyl ester (CFSE) labeling technique was performed before LT as a baseline and at 1, 2, and 4 weeks after LT, with consent from the recipients, donors, and healthy volunteers.16, 17 In brief, peripheral blood mononuclear cells (PBMCs) obtained from the recipients (autologous control), donors, and healthy volunteers (third‐party control) were irradiated with 30 Gy and used as a stimulator. Responder PBMCs from the recipients were labeled with 5‐(and 6)‐CFSE (Molecular Probes, Inc., Eugene, OR). Both the stimulator and responder cells were adjusted to 2 × 106 cells/mL and cocultured in AIM‐V medium (Invitrogen, Grand Island, NY) at 37 °C in a 5% CO2 incubator in the dark for 5 days. After MLR culture, the nonadherent cells were harvested and stained using either phycoerythrin (PE)‐conjugated anti‐CD4 (RPA‐T4) or anti‐CD8 (RPA‐T8) monoclonal antibody (BD Pharmingen, San Diego, CA). Flow cytometry was carried out on a FACSCalibur dual‐laser cytometer (Becton Dickinson, Mountain View, CA) using standard Cell Quest acquisition/analysis, and fluorescence compensation was achieved using an appropriate single fluorochrome‐labeled sample. Dead cells, identified by light scatter and propidium iodide staining, were excluded from the analysis.
QUANTIFICATION OF CD4+ AND CD8+ T‐CELL PROLIFERATION
Precursor frequency (PF), proliferation index (PI), and stimulation index (SI) were quantitatively estimated as described.16, 17 In brief, divisions of reactive T cells, which were identified by their CFSE intensities, were labeled from 0 to n based on dividing time. A single cell dividing n times generates 2n daughter cells. Using this mathematic relationship, the number of division precursors was extrapolated from the number of daughter cells of each division and from proliferation events and PF in CD4+ and CD8+ T‐cell subsets. Using these values, proliferation events and PIs were calculated. The SI was calculated by dividing the PIs of allogeneic combinations by those of autologous controls.
DEFINITION OF INFECTIONS
Cytomegalovirus (CMV) infection, fungal infection, and bloodstream infection (BSI) were defined as described.23 Briefly, CMV infection was defined as detection of more than 3/50,000 CMV pp65‐positive cells along with a preemptive treatment history with valganciclovir or as CMV disease with treatment with ganciclovir. Fungal infection was confirmed by positive fungal culture in blood or abdominal cavity or evidence of infectious lesions in any organ system as demonstrated by radiographic or histologic evaluation. Positive episodes of fungal infections were both suspected and confirmed along with the treatment history of an antifungal drug. Lastly, BSI was defined according to criteria proposed by the Center for Disease Control.24 With the rise of fever higher than 38 °C, a blood culture was done, and isolation of bacteria or fungus (other than common skin contaminants) from one or more cultures in the presence of clinical symptoms was considered proof of BSI.
GENOTYPING
Foxp3 is a member of the forkhead/winged helix protein family of transcription factors. The FOXP3 gene is located on chromosome Xp11.23q13.3 in humans and is composed of 11 coding and 3 noncoding exons.25 Three SNPs in the promoter region, –3499 A/G (rs3761547), –3279 A/C (rs3761548), and –924 A/G (rs2232365), were analyzed as follows: Genomic DNA was extracted from LT‐recipient PBMCs by using the Wizard SV Genomic DNA Purification System (Promega Corporation, Madison, WI) according to the manufacturer's protocol. The three SNPs were detected by polymerase chain reaction restriction fragment length polymorphism as described.26, 27, 28 The primers and enzymes used in this technique are listed in Supporting Table S1. The thermal cycling condition for rs3761548 was initial denaturation at 98 °C for 1 minute, followed by 35 cycles of 98 °C for 30 seconds, annealing 67 °C for 30 seconds, extension at 72 °C for 1 minute, and final extension at 72 °C for 7 minutes. For rs3761547 and rs2232365, the cycling condition was initial denaturation at 95 °C for 5 minutes, followed by 35 cycles of 95 °C for 30 seconds, annealing 62 °C for 30 seconds, extension at 72 °C for 30 seconds, and final extension at 72 °C for 7 minutes.
CYTOKINE ASSAY
Serum of LDLT recipients was prepared at pretransplant and 2 weeks and 1 month after LT. Interleukin (IL)‐2, IL‐10, interferon (IFN)‐γ, and IL‐17 A were measured by using a BD Cytometric Bead Array kit (BD Bioscience, Mountain View, CA) on a BD FACS Canto II (BD Biosciences). IL‐35 was measured by using a commercial enzyme‐linked immunosorbent assay kit (Cloud‐Clone Corp., Wuhan, China) and read on a Microplate Reader‐300 (Corona Electric, Ibaraki, Japan). Both the cytometric bead array assay and enzyme‐linked immunosorbent assay were performed according to the manufacturers' instructions.
FLOW CYTOMETRY
Freshly isolated PBMCs of healthy volunteers and recipients at a pretransplant time point were used for phenotypic analysis of Tregs. As routine Treg analysis had only recently commenced, we analyzed Treg frequency in the patients from our cohort. Tregs were surface stained with the following fluorochrome‐conjugated monoclonal antibodies from BD Pharmingen: peridinin‐chlorophyll protein‐Cyanine5.5 anti‐human CD3 (SK715, 16, 17, 18), allophycocyanin‐conjugated Cyanine7 anti‐human CD4 (RPA‐T4), fluorescein isothiocyanate‐conjugated anti‐human CD25 (M‐A251), PE‐Cyanine7 anti‐human CD45RA (HI100). Intracellular Foxp3 was stained using PE‐conjugated anti‐human Foxp3 (PCH101; eBioscience). Treg phenotypes were analyzed on a BD FACS Canto II (BD Biosciences), and data were analyzed with FlowJo 7.6.5 (Tree Star Inc., Ashland, OR).
STATISTICAL ANALYSIS
Data were reported as the mean ± SD or mean ± SEM for continuous variables and as frequencies (%) for categorical variables. Clinical characteristics of the study population were compared in genotypes of each FOXP3 SNP (rs3761547, rs3761548, and rs2232365) using the Wilcoxon rank sum test for continuous variables and Pearson's chi‐squared test for categorical variables. Relationships between FOXP3 SNPs in LDLT patients and the severity of ACR, infectious complications, DSA, and EAD were analyzed using Pearson's chi‐squared test or Fisher's exact test whenever it was appropriate. To assess associations between the alloimmune response (anti‐donor and anti‐third party), concentrations of several cytokines, and FOXP3 SNPs, the Wilcoxon rank sum test was used. The Kaplan‐Meier method was used for overall survival analysis; comparisons among different groups were carried out using a log‐rank test. Statistical analyses were performed using JMP statistical software, version Genomics 8 (SAS Institute, Cary, NC). All P values presented are two‐sided. P values of < 0.05 were considered statistically significant.
Results
PATIENTS' DEMOGRAPHIC CHARACTERISTICS
The basic demographic and preoperative characteristics of 102 LT recipients are presented in Tables 1‐3. Besides sex, none of the patient demographics and preoperative characteristics differed among LT patients with different FOXP3 genotypes. The frequency of the rs3761548 A/C+A/A genotype was higher in female than in male patients (P = 0.001; Table 2). Similar sex leaning was observed in 82 healthy LT donors who agreed to the genotyping, possibly reflecting the FOXP3 location on chromosome X (Supporting Table S2). Contradictory results with regard to sex bias have been reported, presumably influenced by the target disease of the subjects in each study.29, 30, 31, 32, 33 On the other hand, higher frequencies of rs3761547 A/A and rs2232365 A/A genotypes in male patients were observed in this study, consistent with a previous report (Table 1, 3).34 This sex bias of FOXP3 SNPs currently remains inexplicable.
Table 1.
DEMOGRAPHICS AND PREOPERATIVE CHARACTERISTICS OF RECIPIENTS CORRELATED WITH RS3761547 GENOTYPE
| Variables | A/A (n = 66) | A/G+G/G (n = 36) | P Valuea |
|---|---|---|---|
| Gender | 0.34 | ||
| Male | 43 (65.15) | 20 (55.56) | |
| Female | 23 (34.85) | 16 (44.44) | |
| Age (year) | 53.15 ± 10.94 | 54 ± 10.97 | 0.85 |
| ABO Compatibility | 0.84 | ||
| Compatible | 56 (84.85) | 30 (83.33) | |
| Incompatible | 10 (15.15) | 6 (16.67) | |
| Original disease | 0.69 | ||
| HCV | 22 (33.33) | 15 (41.67) | |
| HBV | 13 (19.70) | 8 (22.22) | |
| Autoimmune disease | 7 (10.61) | 4 (11.11) | |
| Others | 24 (36.36) | 9 (25) | |
| MELD | 19.24 ± 8.83 | 16.25 ± 7.66 | 0.07 |
| Child‐Turcotte‐Pugh | 0.80 | ||
| A | 5 (7.58) | 3 (8.33) | |
| B | 20 (30.30) | 13 (36.11) | |
| C | 41 (62.12) | 20 (55.56) | |
| Donor | 0.42 | ||
| Parent | 6 (9.09) | 0 (0) | |
| Child | 36 (54.55) | 23 (63.89) | |
| Sibling | 9 (13.64) | 5 (13.89) | |
| Spouse | 14 (21.21) | 7 (19.44) | |
| Others | 1 (1.52) | 1 (2.78) | |
| HLA total mismatches | 3.42 ± 1.60 | 3.66 ± 1.51 | 0.53 |
| HLA Class I mismatch | 2.31 ± 1.15 | 2.47 ± 1.10 | 0.51 |
| HLA Class II mismatch | 1.10 ± 0.69 | 1.19 ± 0.66 | 0.56 |
| GRWR | 0.90 ± 0.18 | 0.89 ± 0.24 | 0.97 |
GRWR, graft‐to‐recipient weight ratio; HBV, hepatitis B virus; HCV, hepatitis C virus; LDLT, Living donor liver transplantation; MELD, model for end‐stage liver disease; SD, standard deviation
Categorical variables are presented as number(%) while continuous variables are presented as mean±SD
Pearson's chi‐squared test/Wilcoxon rank sum tests, A difference was considered significant if P‐value was < 0.05.
Typed HLA antigens: HLA‐A1, HLA‐A2, HLA‐B1, HLA‐B2, HLA‐DR1, and HLA‐DR2.
Table 3.
DEMOGRAPHICS AND PREOPERATIVE CHARACTERISTICS OF RECIPIENTS CORRELATED WITH RS2232365 GENOTYPE
| Variables | A/A (n = 85) | A/G+G/G (n = 17) | P Valuea |
|---|---|---|---|
| Gender | 0.05 | ||
| Male | 56 (65.88) | 7 (41.18) | |
| Female | 29 (34.12) | 10 (58.82) | |
| Age (year) | 53.12 ± 11.09 | 55.05 ± 10.07 | 0.71 |
| ABO Compatibility | 0.22 | ||
| Compatible | 70 (82.35) | 16 (94.12) | |
| Incompatible | 15 (17.65) | 1 (5.88) | |
| Original disease | 0.74 | ||
| HCV | 29 (34.12) | 8 (47.06) | |
| HBV | 18 (21.18) | 3 (17.65) | |
| Autoimmune disease | 10 (11.76) | 1 (5.88) | |
| Others | 28 (32.94) | 5 (29.41) | |
| MELD | 18.08 ± 8.68 | 18.70 ± 7.94 | 0.47 |
| Child‐Turcotte‐Pugh | 0.61 | ||
| A | 6 (7.06) | 2 (11.76) | |
| B | 30 (34.12) | 4 (23.53) | |
| C | 50 (58.82) | 11 (64.71) | |
| Donor | 0.74 | ||
| Parent | 5 (5.88) | 1 (5.88) | |
| Child | 47 (55.29) | 12 (70.59) | |
| Sibling | 13 (15.29) | 1 (5.88) | |
| Spouse | 18 (21.18) | 3 (17.65) | |
| Others | 2 (2.35) | 0 (0) | |
| HLA total mismatches | 3.49 ± 1.60 | 3.58 ± 1.41 | 0.97 |
| HLA Class I mismatches | 2.41 ± 1.06 | 2.41 ± 1.06 | 0.90 |
| HLA Class II mismatch | 1.13 ± 0.69 | 1.17 ± 0.63 | 0.84 |
| GRWR | 0.90 ± 0.21 | 0.85 ± 0.17 | 0.26 |
GRWR, graft‐to‐recipient weight ratio; HBV, hepatitis B virus; HCV, hepatitis C virus; LDLT, Living donor liver transplantation; MELD, model for end‐stage liver disease; SD, standard deviation
Categorical variables are presented as number (%), while continuous variables are presented as mean ± SD.
Pearson's chi‐squared test/ Wilcoxon rank sum tests, a difference was considered significant if P‐value was < 0.05.
Typed HLA antigens: HLA‐A1, HLA‐A2, HLA‐B1, HLA‐B2, HLA‐DR1, and HLA‐DR2.
Table 2.
DEMOGRAPHICS AND PREOPERATIVE CHARACTERISTICS OF RECIPIENTS CORRELATED WITH RS3761548 GENOTYPE
| Variables | C/C (n = 67) | A/C+A/A (n = 35) | P Valuea |
|---|---|---|---|
| Gender | 0.0011a | ||
| Male | 49 (73.13) | 14 (40) | |
| Female | 18 (26.87) | 21 (60) | |
| Age (year) | 53.62 ± 9.46 | 53.11 ± 13.39 | 0.53 |
| ABO Compatibility | 0.15 | ||
| Compatible | 54 (80.60) | 32 (91.43) | |
| Incompatible | 13 (19.40) | 3 (8.57) | |
| Original disease | 0.72 | ||
| HCV | 23 (34.33) | 14 (40) | |
| HBV | 16 (23.88) | 5 (14.29) | |
| Autoimmune disease | 7 (10.45) | 4 (11.43) | |
| Others | 21 (31.34) | 12 (34.29) | |
| MELD | 18.34 ± 8.99 | 17.88 ± 7.66 | 0.71 |
| Child‐Turcotte‐Pugh | 0.58 | ||
| A | 5 (7.46) | 3 (8.57) | |
| B | 24 (35.82) | 9 (25.71) | |
| C | 38 (56.72) | 23 (65.71) | |
| Donor | 0.74 | ||
| Parent | 3 (4.48) | 3 (8.57) | |
| Child | 38 (56.72) | 21 (60) | |
| Sibling | 10(14.93) | 4 (11.43) | |
| Spouse | 14 (22.90) | 7 (20) | |
| Others | 2 (2.99) | 0 (0) | |
| HLA total mismatches | 3.47 ± 1.60 | 3.57 ± 1.52 | 0.75 |
| HLA Class I mismatch | 2.36 ± 1.18 | 2.37 ± 1.05 | 0.93 |
| HLA Class II mismatch | 1.10 ± 0.66 | 1.20 ± 0.71 | 0.48 |
| GRWR | 0.89 ± 0.20 | 0.91 ± 0.20 | 0.86 |
GRWR, graft‐to‐recipient weight ratio; HBV, hepatitis B virus; HCV, hepatitis C virus; LDLT, Living donor liver transplantation; MELD, model for end‐stage liver disease; SD, standard deviation.
Categorical variables are presented as number (%) while continuous variables are presented as mean ± SD.
Pearson's chi‐squared test/ Wilcoxon rank sum tests, a difference was considered significant if P‐value was < 0.05.
Typed HLA antigens: HLA‐A1, HLA‐A2, HLA‐B1, HLA‐B2, HLA‐DR1, and HLA‐DR2.
RS3761548 A/C IS ASSOCIATED WITH ACR SEVERITY
We investigated whether the incidence and severity of ACR differed among patients with different FOXP3 genotypes. Sixteen out of the 102 (15.69%) patients experienced ACR within 6 weeks after LDLT (rejection patients). The median time to ACR was 13.5 days (range, 7‐32 days). The median dose of total intravenous MP used was 937.5 mg (range, 500‐3,525 mg), and the median duration of treatment was 5 days (range, 2‐8 days). Consistent with the incidence of SRAR in previous reports,35, 36, 37 5 out of 16 (31.25%) patients suffering from ACR were diagnosed as having SRAR. One of these patients was treated with OKT3, 3 with rATG, and 1 without OKT3/rATG. Since 2010, 6 patients have been diagnosed with ACR and have received steroid bolus for rejection therapy. Liver biopsies of these patients after treatment showed that 3 patients responded well to steroid boluses and were thus clinically diagnosed with steroid‐sensitive acute rejection (SSAR); indeed, the histologic findings showed no evidence of rejection in the 3 patients. The other 3 patients, whose liver function tests improved by <50% of the highest values, were diagnosed with SRAR, and the histology revealed the remaining mild/moderate cellular rejection (Supporting Table S3).
Table 4 shows the relationship between the FOXP3 genotypes in LDLT patients and ACR types. The baseline demographics and preoperative variables, such as age, sex, ABO incompatibility, primary disease, donor's relation, model for end‐stage liver disease score, Child‐Turcotte‐Pugh, and HLA mismatches did not differ between the patients who had never suffered ACR (no‐rejection patients) and rejection patients (data not shown). The proportion of rs3761548 (C/C versus A/C+A/A) was significantly different among patients with and without an episode of SSAR or SRAR (P = 0.0005). Of note, all 5 patients suffering from SRAR exhibited the rs3761548 A/C+A/A genotype, while all 11 patients suffering from SSAR exhibited the rs3761548 C/C genotype. There was no statistically significant association between the type of ACR and either rs3761547 or rs2232365. When comparing patients with an episode of SSAR and those with an episode of SRAR, no difference in the baseline demographics and preoperative variables described above was found. Consistent with previous reports,35, 38 peak values of aspartate aminotransferase and alkaline phosphatase did not differ between SSAR and SRAR patients. SRAR patients in our cohort showed higher serum bilirubin at the start of rejection, although the difference did not reach statistical significance. This was consistent with a previous report that patients with elevated serum bilirubin at the start of rejection were at greater risk of developing SRAR.35 The total dose of the systemic steroid used as first‐line therapy for ACR was lower in SSAR patients (P = 0.016), while treatment duration was not different between SSAR and SRAR patients (Supporting Table S4). These findings indicated that the rs3761548 A/C+A/A genotype is a solitary risk factor for an SRAR episode, while other conventional markers were not significantly associated.
Table 4.
ASSOCIATION BETWEEN THE FOXP3 GENOTYPES OF RECIPIENT AND ACRa
| rs3761547 | rs3761548 | rs2232365 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| A/A (n = 66) | A/G+G/G (n = 36) | P Valueb | C/C (n = 67) | A/C+A/A (n = 35) | P Valueb | A/A (n = 85) | A/G+G/G (n = 17) | P Valueb | |
| No rejection | 57 (86.36) | 29 (80.56) | 0.30 | 56 (83.58) | 30 (85.71) | 0.0005 | 73 (85.88) | 13 (76.47) | 0.37 |
| SSAR | 5 (7.58) | 6 (16.67) | 11 (16.42) | 0 (0) | 9 (10.59) | 2 (11.76) | |||
| SRAR | 4 (6.06) | 1 (2.78) | 0 (0) | 5 (14.29) | 3 (3.53) | 2 (11.76) | |||
Data presented as number (%).
Pearson's chi‐squared test; a difference was considered significant if P < 0.05.
ASSOCIATION BETWEEN ACR SEVERITY AND DE NOVO DSA FORMATION
A previous study demonstrated that early ACR in LDLT was significantly associated with DSA formation within the first month after transplantation,39 raising a query as to whether the incidence of de novo DSA formation differs between SSAR and SRAR patients. To address this issue, DSA detection was carried out in the sera of 15 ACR patients (the sample of 1 ACR patient was unavailable) pretransplant and 1‐month posttransplant. None of the patients had preformed DSAs. Two of the 10 (20%) SSAR patients with the rs3761548 C/C genotype developed de novo DSAs, whereas 4 of the 5 (80%) SRAR patients with the rs3761548 A/C+A/A genotype developed de novo DSAs (Table 5). Although the difference between the two groups did not reach statistical significance (P = 0.08), these results suggest that ACR severity associated with de novo DSA formation is potentially influenced by the rs3761548 SNP.
Table 5.
DETAIL OF DSA ANALYSIS OF ACR PATIENT
| HLA Class I | HLA Class II | ||||||
|---|---|---|---|---|---|---|---|
| Patient No. | A | B | C | DR | DQ | De Novo DSA | |
| SSAR | 9 | — | — | — | — | — | Negative |
| 19 | — | — | — | — | — | Negative | |
| 27 | 33:03 (2854.72) | 58:01(2498.17) | 03:02 (1215.76) 03:03 (1453.31) | 52 (3326.78) | — | Positive | |
| 39 | — | — | — | — | — | Negative | |
| 43 | — | — | — | — | — | Negative | |
| 47 | — | — | — | — | — | Negative | |
| 53 | — | — | — | — | — | Negative | |
| 60 | — | — | — | — | — | Negative | |
| 61 | — | — | — | — | 4 (7927.88) | Positive | |
| 99 | — | — | — | — | — | Negative | |
| SRAR | 57 | — | — | — | 04:01 (1377.20) | — | Positive |
| 59 | 03:01 (1014) | — | — | 01:01 (20978.80) 15:02 (20148.47) | — | Positive | |
| 74 | — | — | — | — | 4 (6333.69) | Positive | |
| 82 | — | — | — | — | 3 (16686.90) | Positive | |
| 83 | — | — | — | — | — | Negative | |
DSA was analyzed by using LAB Screen Single Antigen assay.
Data presented as type of DSA with mean immunofluorescence intensity (MFI) in parentheses and hyphen indicates HLA allele negative i.e. (MFI < 1000) for DSA.
DSA, donor specific anti‐HLA antibody; SSAR, steroid sensitive acute rejection; SRAR, steroid resistant acute rejection.
FREQUENCY OF Tregs IN PERIPHERAL BLOOD IS NOT ASSOCIATED WITH FOXP3 RS3761548 SNPs
We next analyzed whether the frequency of Tregs in peripheral blood was different among patients with different FOXP3 genotypes. To define the Tregs, PBMCs from 11 irrelevant healthy volunteers and the last 14 LT recipients at a pretransplant time point were analyzed. In both the healthy volunteers and the LT recipients, there was no significant association between the proportion of CD25highFoxp3+Tregs between either CD4+ T cells or whole PBMCs and the rs3761548 SNP (Supporting Fig. S1). Further phenotypic analyses also revealed no association of the frequency of either the CD4+CD45RA+Foxp3high effector Treg subset or the CD4+CD45RA–Foxp3low non‐Treg subset with the rs3761548 SNP (Supporting Fig. S2).
ALLOIMMUNE RESPONSES DURING ACR IN FOXP3 RS3761548 A/C+A/A INDIVIDUALS ARE MORE VIGOROUS THAN THOSE IN C/C INDIVIDUALS
We monitored anti‐donor alloreactivity at regular intervals by using an MLR assay employing an intracellular CFSE labeling technique. This allowed us to examine the relationship between alloimmune responses and the FOXP3 rs3761548 SNP. Among patients who had never experienced ACR, the average anti‐donor CD4+/CD8+ T‐cell SI in patients with the rs3761548 A/C+A/A genotype was higher at all time points during the observation period than that in patients with the C/C genotype, although the difference did not reach statistical significance (Fig. 1).
Figure 1.
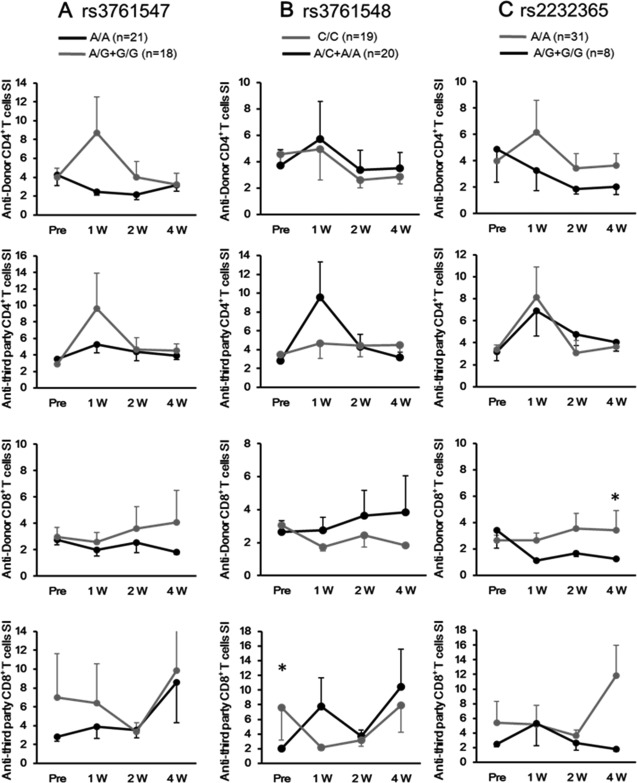
LT patients with FOXP3 rs3761548 A/C+A/A genotype show higher anti‐donor T‐cell response than those with the C/C genotype. Kinetics of the SI of anti‐donor or anti‐third party reactive CD4+ or CD8+ T cells in no‐rejection patients were compared between (A) FOXP3 rs3761547 A/A (n = 21) and A/G+G/G (n = 18), (B) FOXP3 rs3761548 C/C (n = 20) and A/C+A/A (n = 19), (C) or FOXP3 rs2232365 A/A (n = 31) and A/G+G/G (n = 8) within 4 weeks after liver transplantation. Data are presented as the mean ± SEM. *P < 0.05
When patients developed significant disorders in liver function as determined by laboratory tests after LT, MLR was performed for diagnosing ACR immediately after liver dysfunction had occurred. The anti‐donor CD4+ T‐cell SI determined by MLR assay at the time of ACR diagnosis was higher in patients with the rs3761548 A/C+A/A genotype than in patients with the C/C genotype, whereas the difference in anti‐third party CD4+ T‐cell SI between the genetically disparate two did not reach statistical significance (Fig. 2B). Similarly, the anti‐donor CD4+ T‐cell SI in patients with the rs3761547 A/A genotype was higher than that in patients with the A/G+G/G genotype, probably reflecting that 80% of FOXP3 rs3761548 A/C+A/A individuals suffering from SRAR displayed the rs3761547 A/A genotype (Table 4). We observed no association between the SI and rs2232365 SNPs (Fig. 2A,C) nor between the anti‐donor CD8+ T‐cell SI and FOXP3 SNPs. Thus, alloimmune responses in CD4+ T cells during ACR after LT were more vigorous in FOXP3 rs3761548 A/C+A/A individuals than in C/C individuals.
Figure 2.
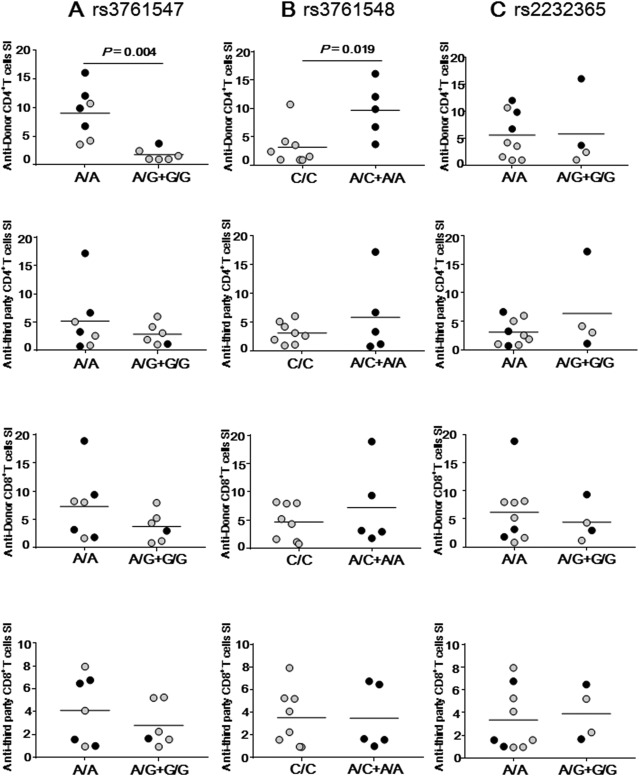
LT patients with the FOXP3 rs3761548 A/C+A/A genotype show higher anti‐donor CD4+T‐cell response than those with the C/C genotype at the time of ACR. SI of anti‐donor or anti‐third party reactive CD4+ or CD8+ T cells by using CFSE MLR in patients with SSAR and SRAR were compared between (A) FOXP3 rs3761547 A/A (n = 7) and A/G+G/G (n = 6), (B) FOXP3 rs3761548 C/C (n = 8) and A/C+A/A (n = 5), or (C) FOXP3 rs2232365 A/A (n = 9) and A/G+G/G (n = 4). Data not shown (n = 3). Gray circles represent SSAR patients (n = 8) and black circles represent SRAR patients (n = 5) in each genotype group. Horizontal bars represent grand mean of each group.
RELATIONSHIP BETWEEN SERUM LEVELS OF IMMUNE‐REGULATORY CYTOKINES AND FOXP3 SNPs
We further investigated whether serum levels of immune‐regulatory cytokines were influenced by the FOXP3 genotype in LT patients. We found no significant association between serum levels of IL‐2, IL‐10, IFN‐γ, IL‐17A, or IL‐35 and rs3761548 at any time point (Fig. 3). Of note, among LT patients who had experienced ACR, serum IFN‐γ levels at 2 weeks were markedly higher in patients with the rs3761548 A/C+A/A genotype than in patients with the C/C genotype. Such association was not observed for rs3761547 and rs2232365 SNPs (Supporting Figs. S3 and S4). Statistical differences in preoperative serum IL‐2 levels among the rs3761547 SNP disparate patients and in serum IL‐35 levels at 2 weeks among the rs2232365 SNP disparate patients were observed, although these findings are not clinically well interpreted.
Figure 3.
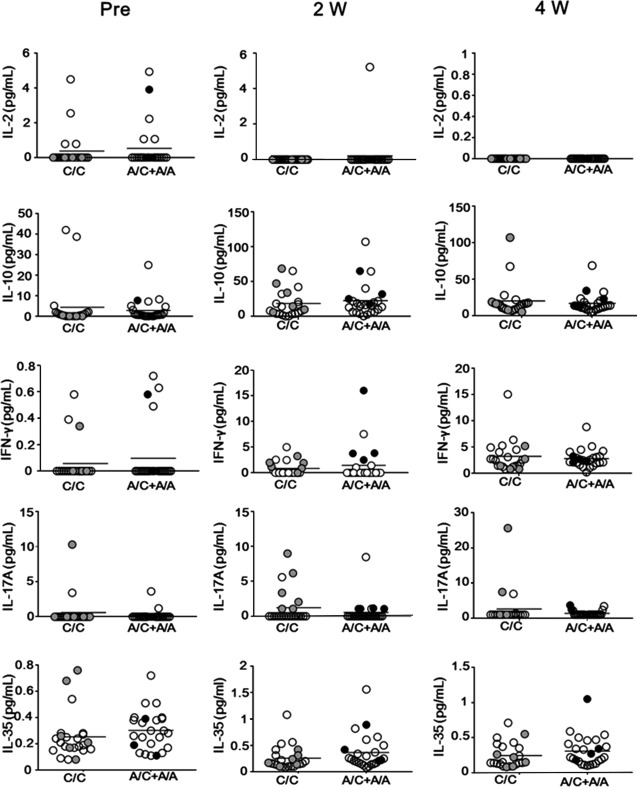
rs3761548 A/C+A/A recipients show a higher level of IFN‐γ than patients with the C/C genotype. Serum levels of IL‐2, IL‐10, IFN‐γ, IL‐17A, and IL‐35 of LT recipients as quantified by cytometric bead assay or enzyme‐linked immunosorbent assay at pretransplant and 2 weeks and 4 weeks after transplantation among FOXP3 rs3761548 A/C genotypes. Open white circles represent no‐rejection patients (n = 38), gray round circles represent SSAR patients (n = 6), and black round circles represent SRAR patients (n = 4). Horizontal bars represent grand mean of each group.
RELATIONSHIPS BETWEEN THE INCIDENCE OF EAD AND FOXP3 SNPs
EAD is considered to be an important complication after LDLT. Therefore, we next evaluated the relationship between FOXP3 SNP genotypes and EAD. Fifteen of the total 102 patients (14.7%) developed EAD, which is consistent with the incidence of EAD in previous reports.21, 22 The recipient and donor characteristics, surgical details of the patients, as well as FOXP3 SNPs and incidence of EAD are summarized in Supporting Table S5. The model for end‐stage liver disease score was significantly higher in the EAD group than the non‐EAD group (P = 0.0002). Similarly, the total bilirubin level and the international normalized ratio on posttransplant day 7 were also significantly higher in the EAD group. There was no significant association between any of the FOXP3 SNPs and the incidence of EAD among the whole cohort, and even among patients without an ACR episode, the incidence of EAD was 17.85% (10 of 56) for the C/C genotype and 6.67% (2 of 30) for the rs3761548 A/C+A/A genotype; P = 0.20).
RELATIONSHIPS BETWEEN THE INCIDENCE OF INFECTIOUS COMPLICATIONS/PATIENT SURVIVAL AND FOXP3 SNPs
The overall incidences of fungal infection, CMV infection, and BSI within 6 weeks after LT were 17.6%, 33.3%, and 31.4%, respectively. None of these infection episodes in LT recipients differed among any of the FOXP3 genotypes (Table 6). The overall survival did not differ among patients with different FOXP3 SNPs of rs3761547, rs3761548, or rs2232365 (Fig. 4). The cause of mortality was either recurrence of original disease (24%), surgical complications (16%), BSI (20%), rejection (12%), or another cause (28%). These findings indicate that FOXP3 genotypes are not associated with the incidence of infectious complications and patient survival in our cohort of LT recipients.
Table 6.
ASSOCIATION BETWEEN THE FOXP3 GENOTYPES OF RECIPIENTS AND INFECTIOUS COMPLICATIONSa
| rs3761547 | rs3761548 | rs2232365 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| A/A (n = 66) | A/G+G/G (n = 36) | P Valueb | C/C (n = 67) | A/C+A/A (n = 35) | P Valueb | A/A (n = 85) | A/G+G/G (n = 17) | P Valueb | |
| Fungal | 10 (15.15) | 8 (22.22) | 0.42 | 13 (19.40) | 5 (14.29) | 0.59 | 15 (17.65) | 3 (17.65) | 1.00 |
| CMV | 19 (28.79) | 15 (41.67) | 0.19 | 23 (34.33) | 11 (31.43) | 0.82 | 25 (29.41) | 9 (52.94) | 0.08 |
| BSI | 23 (34.85) | 9 (25) | 0.37 | 20 (29.85) | 12 (34.29) | 0.65 | 28 (32.94) | 4 (23.53) | 0.57 |
Data presented as number (%).
Fisher's exact test; a difference was considered significant if P < 0.05.
Figure 4.
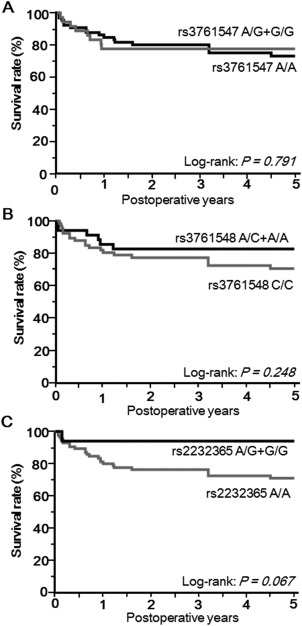
Overall survival is not related to FOXP3 SNPs. Kaplan‐Meier curve showing overall 5‐year survival (combined mortality and graft loss) according to recipient (A) FOXP3 rs3761547 A/A (n = 66) versus A/G+G/G (n = 36), (B) FOXP3 rs3761548 C/C (n = 67) versus A/C+A/A (n = 35), and (C) FOXP3 rs2232365 A/A (n = 85) versus A/G+G/G (n = 17) genotypes. P values were calculated using a log‐rank test.
Discussion
ACR after liver transplantation, which occurs in one third of all recipients, can be mild to severe and should be treated according to its severity. Although the gold standard for diagnosing the presence and severity of ACR of liver allografts is histologic evaluation based on the Banff criteria,19 histologic analysis cannot precisely predict the clinical response to first‐line rejection therapy with steroid pulse.40 Several retrospective studies have attempted to define either a predictive parameter or a risk factor for SRAR. One study could not display remarkable differences in clinical, biochemical, or immunologic parameters between patients with SSAR and SRAR.38 Conti et al.35 reported that LT patients who had a pretransplant history of steroid administration for autoimmune disease were at high risk of developing SRAR compared to recipients without such history. The same group explored the possible roles of IL‐1, IL‐1Ra, and IL‐15 in SRAR and concluded that IL‐1Ra secretion was consistently reduced in patients with SRAR.37 They also demonstrated that plasma IL‐15 was enhanced particularly during SRAR and chronic rejection.41 However, current methods to predict the risk of SRAR still display insufficient specificity and sensitivity, making implementation of targeted therapies difficult. Although treatment with rATG is an effective therapeutic option for SRAR with acceptable complications in LT recipients,36, 42, 43 emerging surrogate markers of SRAR would further improve the safety, efficacy, and outcomes of this specific remedy.
The effects of steroids depend on their capacity to bind to the glucocorticoid receptor (GCR) and form a steroid‐GCR complex, which acts as a transcription factor enhancing the expression of numerous genes.44 GCR has been described as an enhancer of FOXP3 gene expression.45 Several studies in both transplant and nontransplant fields have reported the capability of steroids to enhance Foxp3 expression in vitro and in vivo to generate Tregs.46, 47, 48, 49 Mathian et al.49 and Karagiannidis et al.46 found that systemic administration of MP induced dramatic and rapid but transient increases in circulatory Tregs in patients with asthma and systemic lupus erythematosus, respectively. On the other hand, Prado et al.47 found that in vitro dexamethasone up‐regulated messenger RNA, enhanced FOXP3 expression, and generated CD25high cells with phenotypic characteristics attributable to natural Tregs. Despite their hyporesponsiveness and enhanced Foxp3 expression, these Tregs did not exert suppressive activity. In another study done in renal transplant patients treated with MP for acute kidney rejection, Seissler et al.48 observed an increase in a specific subset of Tregs that displayed the CD4+Foxp3+CD127lowCD45RA−HLA–DR+ phenotype; however, they did not observe an increase in other Treg subsets. Such contradictory findings among studies likely reflect individual variation in Treg activity, suggesting that a predisposing genetic factor in FOXP3 might play an important role in this scenario.
Several gene disease association studies suggested that FOXP3 SNP rs3761548 may be the potent regulatory site of transcription of FOXP3.10, 28, 50 In accordance with the TRANSFAC6.0 public database, the rs3761548 C/A polymorphism is located in the core of ‘GGGCGG’ sequence of the putative binding site for the transcription factor specificity protein 1 (Sp1); this C>A allele mutation might therefore interfere with the interaction of Sp1 with the FOXP3 promoter. Thus, the rs3761548 A allele may affect Foxp3 expression and the Treg function.50 These molecular facts may explain significantly higher incidence of SRAR observed in LT patients with the FOXP3 rs3761548 A/C+A/A genotype than those with the C/C genotype in the current study.
FOXP3 SNPs have been reported to influence genetic susceptibility to various autoimmune diseases and tumors. More precisely, the rs3761548 genotype has been associated with autoimmune thyroid disease, unexplained recurrent spontaneous abortion, rheumatoid arthritis, psoriasis, and allergic rhinitis.26, 27, 28, 32, 33 In the transplantation field, Qui et al.12 reported that the rs3761548 A/A genotype has an increased risk of allograft rejection 2, 3, and 5 years after renal transplantation, despite a lack of association with early rejection.
In this study, we serially monitored alloimmune responses in recipients by MLR assay instead of using a pathologic investigation/biopsy for diagnosing ACR. The MLR assay using the CFSE labeling technique assay has proven effective for recognizing an individual patient's posttransplant status and distinguishing rejection from relapse of the original disease.16, 17, 18 The MLR data revealed that alloimmune responses in CD4+ T cells during ACR after LT in individuals with FOXP3 rs3761548 A/C+A/A were more vigorous than those in C/C homozygous individuals. The C/C individuals, who had competent transcription of FOXP3, may well proliferate competent Tregs that can counter the cytotoxic T cells, thus leading to mild ACR or steroid‐sensitive ACR. On the other hand, A‐allele‐carrying individuals, who have a defective transcription of FOXP3, may produce incompetent Tregs that cannot suppress ongoing proliferation of cytotoxic T cells against allografts, thus leading to severe ACR, which can only be suppressed by the use of depleting antibodies targeting T cells. Thus, the rs3761548 A/C+A/A genotype can be considered a risk factor for resistance to steroid pulse therapy for treating ACR but does not appear to be a risk factor for the onset of ACR.
In conclusion, the FOXP3 gene rs3761548 SNP in LT recipients was significantly associated with susceptibility to SRAR. This fact suggests that the immunosuppression regime and/or anti‐ACR treatment regimen should be adjusted on an individual basis by identifying FOXP3 SNPs, implying a need for personalized medicine in the field. Although this study is preliminary with a relatively small cohort, it can nevertheless serve as a useful starting point for further large‐scale prospective studies with a more rigorous and controlled design, including quantitation of Foxp3+ Tregs in liver allografts of patients with various genotypes.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1052/suppinfo.
Supporting Information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information Table 4
Supporting Information Table 5
Acknowledgment
We thank Dr. Kohei Ishiyama, Dr. Kentaro Ide, Dr. Masahiro Ohira, and Dr. Hiroyuki Tahara for their advice and encouragement and Dr. Jinlian Piao, Dr. Das Lalit Kumar, and Mr. Yu Sasaki for technical assistance.
Potential conflict of interest: Nothing to report.
Partly supported by the Japan Society for the Promotion of Science (KAKENHI grant number JP15K10026) received (to Y.T.) and a Grant‐in‐Aid for Research on Hepatitis from the Japan Agency for Medical Research and Development (AMED 16fk0210107h0001) received (to H.O.).
Contributor Information
Yuka Tanaka, Email: yukasan@hiroshima-u.ac.jp.
Hideki Ohdan, Email: hohdan@hiroshima-u.ac.jp.
REFERENCES
- 1. Kim CH. FOXP3 and its role in the immune system. Adv Exp Med Biol 2009;665:17‐29. [DOI] [PubMed] [Google Scholar]
- 2. Sakaguchi S. Naturally arising Foxp3‐expressing CD25 + CD4 + regulatory T cells in immunological tolerance to self and non‐self. Nat Immunol 2005;6:345‐352. [DOI] [PubMed] [Google Scholar]
- 3. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3 + regulatory T cells in the human immune system. Nat Rev Immunol 2010;10:490‐500. [DOI] [PubMed] [Google Scholar]
- 4. Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity 2009;30:616‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol 2012;12:417‐430. [DOI] [PubMed] [Google Scholar]
- 6. Hamano K, Rawsthorne MA, Bushell AR, Morris PJ, Wood KJ. Evidence that the continued presence of the organ graft and not peripheral donor microchimerism is essential for maintenance of tolerance to alloantigen in vivo in anti‐CD4 treated recipients. Transplantation 1996;62:856‐860. [DOI] [PubMed] [Google Scholar]
- 7. Tullius SG, Nieminen M, Bechstein WO, Jonas S, Steinmüller T, Pratschke J, et al. Chronically rejected rat kidney allografts induce donor‐specific tolerance. Transplantation 1997;64:158‐161. [DOI] [PubMed] [Google Scholar]
- 8. Steger U, Kingsley CI, Karim M, Bushell AR, Wood KJ. CD25+CD4 + regulatory T cells develop in mice not only during spontaneous acceptance of liver allografts but also after acute allograft rejection. Transplantation 2006;82:1202‐1209. [DOI] [PubMed] [Google Scholar]
- 9. Hori S, Nomura T, Sakaguchi S. Control of regulatory T Cell development by the transcription factor Foxp3. Science 2003;299:1057‐1061. [PubMed] [Google Scholar]
- 10. Oda JM, Hirata BK, Guembarovski RL, Watanabe MA. Genetic polymorphism in FOXP3 gene: Imbalance in regulatory T‐cell role and development of human diseases. J Genet 2013;92:163‐171. [DOI] [PubMed] [Google Scholar]
- 11. He Y, Na H, Li Y, Qiu Z, Li W. FoxP3 rs3761548 polymorphism predicts autoimmune disease susceptibility: a meta‐analysis. Hum Immunol 2013;74:1665‐1671. [DOI] [PubMed] [Google Scholar]
- 12. Qiu XY, Jiao Z, Zhang M, Chen JP, Shi XJ, Zhong MK. Genetic association of FOXP3 gene polymorphisms with allograft rejection in renal transplant patients. Nephrology 2012;17:423‐430. [DOI] [PubMed] [Google Scholar]
- 13. Baumann AK, Schlue J, Noyan F, Hardtke‐Wolenski M, Lehner F, Barg‐Hock H, et al. Preferential accumulation of T helper cells but not cytotoxic T cells characterizes benign subclinical rejection of human liver allografts. Liver Transpl 2016;22:943‐955. [DOI] [PubMed] [Google Scholar]
- 14. Taubert R, Pischke S, Schlue J, Wedemeyer H, Noyan F, Heim A, et al. Enrichment of regulatory T Cells in acutely rejected human liver allografts. Am J Transplant 2012;12:3425‐3436. [DOI] [PubMed] [Google Scholar]
- 15. Ide K, Tanaka Y, Onoe T, Banshodani M, Tazawa H, Igarashi Y, et al. Evidence for the immunosuppressive potential of calcineurin inhibitor‐sparing regimens in liver transplant recipients with impaired renal function. J Transpl 2011;2011:483728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka Y, Tashiro H, Onoe T, Ide K, Ishiyama K, Ohdan H. Optimization of immunosuppressive therapy based on a multiparametric mixed lymphocyte reaction assay reduces infectious complications and mortality in living donor liver transplant recipients. Transplant Proc 2012;44:555‐559. [DOI] [PubMed] [Google Scholar]
- 17. Tanaka Y, Ohira M, Tashiro H, Imamura M, Chayama K, Ohdan H. Impact of alloimmune T cell responses on hepatitis C virus replication in liver transplant recipients. Hum Immunol 2014;75:1259‐1267. [DOI] [PubMed] [Google Scholar]
- 18. Tanaka Y, Ohdan H, Onoe T, Mitsuta H, Tashiro H, Itamoto T, et al. Low incidence of acute rejection after living‐donor liver transplantation: immunologic analyses by mixed lymphocyte reaction using a carboxyfluorescein diacetate succinimidyl ester labeling technique. Transplantation 2005;79:1262‐1267. [DOI] [PubMed] [Google Scholar]
- 19. Ormonde DG, de Boer WB, Kierath A, Bell R, Shilkin KB, House AK, et al. Banff schema for grading liver allograft rejection: utility in clinical practice. Liver Transpl Surg 1999;261‐268. [DOI] [PubMed] [Google Scholar]
- 20. Morimoto H, Ide K, Tanaka Y, Ishiyama K, Ohira M, Tahara H, et al. Different sensitivity of rituximab‐treatment to B‐cells between ABO‐incompatible kidney and liver transplantation. Hum Immunol 2016;77:456‐463. [DOI] [PubMed] [Google Scholar]
- 21. Olthoff KM, Kulik L, Samstein B, Kaminski M, Abecassis M, Emond J, et al. Validation of a current definition of early allograft dysfunction in liver transplant recipients and analysis of risk factors. Liver Transpl 2010;16:943‐949. [DOI] [PubMed] [Google Scholar]
- 22. Hong SH, Kwak JA, Chon JY, Park CS. Prediction of early allograft dysfunction using serum phosphorus level in living donor liver transplantation. Transpl Int 2013;26:402‐410. [DOI] [PubMed] [Google Scholar]
- 23. Shimizu S, Tanaka Y, Tazawa H, Verma S, Onoe T, Ishiyama K, et al. Fc‐gamma receptor polymorphisms predispose patients to infectious complications after liver transplantation. Am J Transplant 2016;16:625‐633. [DOI] [PubMed] [Google Scholar]
- 24. Horan TC, Andrus M, Dudeck MA. CDC/NHSN surveillance definition of health care‐associated infection and criteria for specific types of infections in the acute care setting. Am J Infect Control 2008;36:309‐332. [DOI] [PubMed] [Google Scholar]
- 25. Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood 2009;114:3727‐3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Inoue N, Watanabe M, Morita M, Tomizawa R, Akamizu T, Tatsumi K, et al. Association of functional polymorphisms related to the transcriptional level of FOXP3 with prognosis of autoimmune thyroid diseases. Clin Exp Immunol 2010;162:402‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. You Z, Wu Z, Zhang C, Li Z, Su X, Zhang X, et al. Association between functional polymorphisms of Foxp3 gene and the occurrence of unexplained recurrent spontaneous abortion in a Chinese Han population. Clin Dev Immunol 2012;2012:896458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paradowska‐Gorycka A, Jurkowska M, Felis‐Giemza A, Romanowska‐Prochnicka K, Manczak M, Maslinski S, et al. Genetic polymorphisms of Foxp3 in patients with rheumatoid arthritis. J Rheumatol 2015;42:170‐180. [DOI] [PubMed] [Google Scholar]
- 29. Park O, Grishina I, Leung PS, Gershwin ME, Prindiville T. Analysis of the Foxp3/scurfin gene in Crohn's disease. Ann N Y Acad Sci 2005;1051:218‐228. [DOI] [PubMed] [Google Scholar]
- 30. Hassannia H, Ghaffari J. FOXP3 and TGF‐β gene polymorphisms in allergic rhinitis. Iran J Immunol 2011;8:218‐225. [PubMed] [Google Scholar]
- 31. Jahan P, Cheruvu R, Tippisetty S, Komaravalli PL, Valluri V, Ishaq M. Association of FOXP3 (rs3761548) promoter polymorphism with nondermatomal vitiligo: a study from India. J Am Acad Dermatol 2013;69:262‐266. [DOI] [PubMed] [Google Scholar]
- 32. Gao L, Li K, Li F, Li H, Liu L, Wang L, et al. Polymorphisms in the FOXP3 gene in Han Chinese psoriasis patients. J Dermatol Sci 2010;57:51‐56. [DOI] [PubMed] [Google Scholar]
- 33. Fodor E, Garaczi E, Polyanka H, Koreck A, Kemeny L, Szell M. The rs3761548 polymorphism of FOXP3 is a protective genetic factor against allergic rhinitis in the Hungarian female population. Hum Immunol 2011;72:926‐929. [DOI] [PubMed] [Google Scholar]
- 34. Song Q‐H, Shen Z, Xing X‐J, Yin R, Wu Y‐Z, You Y, et al. An association study of single nucleotide polymorphisms of the FOXP3 intron‐1 and the risk of Psoriasis vulgaris. Indian J Biochem Biophys 2012;49:25‐35. [PubMed] [Google Scholar]
- 35. Conti F, Dousset B, Archambeua D, Louvel A, Houssin D, Calmus Y. Enhanced risk of steroid‐resistant acute rejection following pretransplant steroid therapy in liver graft recipients. Transplantation 1995;60:1104‐1108. [DOI] [PubMed] [Google Scholar]
- 36. Lee JG, Lee J, Lee JJ, Song SH, Ju MK, Choi GH, et al. Efficacy of rabbit anti‐thymocyte globulin for steroid‐resistant acute rejection after liver transplantation. Medicine (Baltimore) 2016;95:e3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Conti F, Breton S, Batteux F, Furlan V, Houssin D, Weill B, et al. Defective interleukin‐1 receptor antagonist production is associated with resistance of acute liver graft rejection to steroid therapy. Am J Pathol 2000;157:1685‐1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bijleveld CG, Klompmaker IJ, Van den Berg AP, Gouw AS, Hepkema BG, Haagsma EB, et al. Incidence, risk factors, and outcome of antithymocyte globulin treatment of steroid‐resistant rejection after liver transplantation. Transpl Int 1996;9:570‐575. [DOI] [PubMed] [Google Scholar]
- 39. Kasahara M, Kiuchi T, Takakura K, Uryuhara K, Egawa H, Asonuma K, et al. Postoperative flow cytometry crossmatch in living donor liver transplantation: clinical significance of humoral immunity in acute rejection. Transplantation 1999;67:568‐575. [DOI] [PubMed] [Google Scholar]
- 40. Horoldt BS, Burattin M, Gunson BK, Bramhall SR, Nightingale P, Hubscher SG, et al. Does the Banff rejection activity index predict outcome in patients with early acute cellular rejection following liver transplantation? Liver Transpl 2006;12:1144‐1151. [DOI] [PubMed] [Google Scholar]
- 41. Conti F, Frappier J, Dharancy S, Chereau C, Houssin D, Weill B, et al. Interleukin‐15 production during liver allograft rejection in humans. Transplantation 2003;76:210‐216. [DOI] [PubMed] [Google Scholar]
- 42. Schmitt TM, Phillips M, Sawyer RG, Northup P, Hagspiel KD, Pruett TL, et al. Anti‐thymocyte globulin for the treatment of acute cellular rejection following liver transplantation. Dig Dis Sci 2010;55:3224‐3234. [DOI] [PubMed] [Google Scholar]
- 43. Benjamin MM, Dasher KJ, Trotter JF. A comparison of outcomes between OKT3 and antithymocyte globulin for treatment of steroid‐resistant rejection in hepatitis C liver transplant recipients. Transplantation 2014;97:470‐473. [DOI] [PubMed] [Google Scholar]
- 44. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids 2010;75:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rudra D, DeRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol 2012;13:1010‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karagiannidis C, Akdis M, Holopainen P, Woolley NJ, Hense G, Ruckert B, et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol 2004;114:1425‐1433. [DOI] [PubMed] [Google Scholar]
- 47. Prado C, Gómez J, López P, de Paz B, Gutiérrez C, Suárez A. Dexamethasone upregulates FOXP3 expression without increasing regulatory activity. Immunobiology 2011;216:386‐392. [DOI] [PubMed] [Google Scholar]
- 48. Seissler N, Schmitt E, Hug F, Sommerer C, Zeier M, Schaier M, et al. Methylprednisolone treatment increases the proportion of the highly suppressive HLA‐DR+‐Treg‐cells in transplanted patients. Transpl Immunol 2012;27:157‐161. [DOI] [PubMed] [Google Scholar]
- 49. Mathian A, Jouenne R, Chader D, Cohen‐Aubart F, Haroche J, Fadlallah J, et al. Regulatory T cell responses to high‐dose methylprednisolone in active systemic lupus erythematosus. PLoS One 2015;10:e0143689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Song P, Wang XW, Li HX, Li K, Liu L, Wei C, et al. Association between FOXP3 polymorphisms and vitiligo in a Han Chinese population. British Journal of Dermatology 2013;169:571‐578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1052/suppinfo.
Supporting Information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information Table 4
Supporting Information Table 5