

Abstract
The significance of the clinical impact of direct‐acting antiviral (DAA) resistance‐associated substitutions (RASs) in hepatitis C virus (HCV) on treatment failure is unclear. No standardized methods or guidelines for detection of DAA RASs in HCV exist. To facilitate further evaluations of the impact of DAA RASs in HCV, we conducted a systematic review of RAS sequencing protocols, compiled a comprehensive public library of sequencing primers, and provided expert guidance on the most appropriate methods to screen and identify RASs. The development of standardized RAS sequencing protocols is complicated due to a high genetic variability and the need for genotype‐ and subtype‐specific protocols for multiple regions. We have identified several limitations of the available methods and have highlighted areas requiring further research and development. The development, validation, and sharing of standardized methods for all genotypes and subtypes should be a priority. (Hepatology Communications 2017;1:379–390)
Abbreviations
- DAA
direct‐acting antiviral
- HCV
hepatitis C virus
- NGS
next generation sequencing
- NS
nonstructural
- PCR
polymerase chain reaction
- PRISMA
Preferred Reporting Items for Systematic Reviews and Meta‐Analyses
- RAS
resistance‐associated substitution
Introduction
The availability of tolerable and effective interferon‐free direct‐acting antiviral (DAA) therapies has revolutionized the management of hepatitis C virus (HCV) infection. However, treatment failure is often associated with antiviral HCV DAA resistance.1 Reduced responses to HCV therapy have also been observed in certain subgroups of patients with preexisting resistance‐associated substitutions (RASs).2, 3, 4, 5, 6, 7, 8, 9 Although there are new DAA therapies that are effective for people with previous treatment failure (including nonstructural [NS] 5A inhibitors) and preexisting RASs,10, 11, 12 these therapies are still in clinical development. Further, little is known about the prevalence of preexisting RASs in many regions with a high burden of HCV infection, such as Africa and Asia. As such, there is an immediate clinical need for information on available assays for the screening and detection of RASs, particularly for patients with treatment failure and those with advanced disease who require more immediate salvage therapy.
There is a growing body of literature documenting HCV sequencing assays for the detection of DAA RASs. However, there is a lack of standardization across methods, and many countries do not have commercial services available for the detection of RASs, particularly, but not exclusively, in low‐ and middle‐income countries. As such, there is a growing demand for guidance on available methods for sequencing and the detection of RASs to guide both research and clinical HCV management.
The primary objectives of this review are to systematically evaluate available HCV sequencing methods for RAS detection and develop a comprehensive public library of published sequencing primers. Additionally, this review seeks to provide expert guidance on the selection of the most appropriate methods.
Systematic Review of Available Methods
A systematic review of the literature was conducted to determine what methods currently exist for sequencing HCV to detect DAA RASs. Sequencing methods were evaluated according to several methodological parameters. This systematic review was performed according to the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) statement.13 As per a previously published systematic review on the sequencing of HCV,14 the PRISMA guidelines were customized (Supporting Table S2) to fit the scope of this study. This protocol has been registered and uploaded to the PROSPERO database (https://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42016045548) (Supporting Table S3).
A systematic search of MEDLINE (PubMed) and SCOPUS was performed on August 15, 2016, covering all studies published from January 1, 2011, to that date. The time period was restricted to capture methods related to current DAA therapy. Backward citation checks were carried out to identify further potentially relevant studies that could have included studies published before 2011. In addition, an invitation was made to researchers and laboratories with unpublished methodologies to contribute their primer sets in consultation with the HCV Resistance Working Group led by the HCV Forum.
Medline Search terms were used to search for studies relating to HCV (“HCV” OR “Hepatitis C”) and resistance (“antiviral resistance” OR “resistance mutation*” OR “resistance*associated variant*” OR “resistance*associated substitution*” OR “resistance* associated amino acid variant*” OR “polymorphism*”) from these two databases. To identify studies that used sequencing as a method, the search term ‘‘primer*’’ was also applied because it is related to polymerase chain reaction (PCR) and sequencing. The lists of studies from each search database were combined, and ineligible articles were removed after title and abstract review based on the following criteria: duplicate reference, review article, abstract or title accessible only, DAA RASs not reported, sequencing not performed, or irrelevant/insufficient method description. Studies were deemed eligible for this analysis if they were published in English, HCV DAA RASs were reported in clinical samples, HCV sequencing (population sequencing, next generation sequencing [NGS], or template‐independent amplification) was performed, and PCR/sequencing primer design or location was described. Two reviewers independently determined the eligibility of records to be included in this study (S.R.B. and A.A.E.), and disagreements were resolved by consensus.
Methodological and publication details from records deemed eligible for inclusion were recorded in a primer library (Supporting Table S1). The genome location of primers was recorded in the primer library as reported in the original record. If original records did not describe location, it was recorded in the primer library in reference to the H77 sequence (genotype 1a genome; Genbank accession number AF009606). Primers for which location could not be determined were included in the primer library but not in subsequent amplicon analyses. For studies that described a nested PCR protocol, the innermost primer was recorded in the primer library and used for the amplicon analysis. Where studies described sequencing primers in addition to PCR primers, all sequencing primers were included in the primer library as individual amplicons but only PCR primers were included in the amplicon analysis. Where multiple genotype‐specific primer pairs were described, all pairs of primers were included in the primer library as individual sequencing protocols and included in the amplicon analysis. Where a sequencing region was derived from multiple overlapping sequencing amplicons, all pairs of primers were included in the primer library as individual sequencing amplicons. For reporting of depth of coverage of NGS, the total number of reads reported for each run was divided by the number of samples that were pooled in the run, if this was described in the original record. If multiple runs were performed, the average was calculated among the runs and then divided by the number of pooled samples.
Amplicons analyzed were classified by the individual protein coding region amplified and sequenced, the HCV genotype specificity of the primers used, the amplification or sequencing method they were generated by, and the year the study was published. Amplicons that were subjected to NGS were distinguished as either being generated by standard PCR‐based amplification methods (classified as an NGS method) or being generated by template‐independent amplification methods and then subjected to NGS (classified as a template‐independent method). Due to the number of primers identified that were specific for only HCV subtypes 1a or 1b, these primers and associated amplicons were analyzed in separate categories. Primers that were specific to both HCV subtype 1a and 1b or were specific for all HCV genotype 1 viruses were categorized as “Genotype 1” primers and associated amplicons. Primers and associated amplicons that were specific for non‐1 HCV genotypes were classified as specific for either HCV genotype 2, 3, or genotypes 4, 5, and 6 combined. Any primers that were specific for more than one HCV genotype were classified as “multi‐genotypic” primers and associated amplicons.
It was determined a priori that the features of an ideal candidate method to standardize HCV sequencing to detect DAA RASs would be the following: covering all key DAA RASs in all three protein coding regions of interest (NS3, NS5A, and NS5B); successfully amplifying samples from all genotypes (pan‐genotypic); and having a low limit of detection comparable to commercially available HCV resistance‐testing sequencing assays (1,000 IU/mL). The key DAA RASs were identified from the literature2, 9, 15 (Supporting Table S4). To increase relevancy, ideal candidate methods were also identified for the alternative scenario where RASs in only a single region of interest were to be determined. The ideal method for this alternative scenario would cover all key DAA RASs in that region, be pan‐genotypic, and have a limit of detection >1,000 IU/mL. These criteria were used to identify potential ideal candidate methods from the methods reviewed.
Summary of Available Literature
This study reports the first systematic review of published and unpublished HCV sequencing protocols used to detect DAA RASs in clinical samples. It identifies a panel of protocols that cover all relevant RASs and may be suitable for those looking to establish RAS testing. Guidance on the protocol assessment and validation, along with minimum reporting requirements to improve publication standards, have also been provided. We found that a large proportion of the reported protocols did not cover all clinically relevant DAA RASs, and very few studies described sufficient detail to assess validity of the method or results. Further, methods identified were dominated largely by HCV genotype 1‐specific primers. This in turn impacted the ability to identify protocols suitable for standardization. This review highlights the need for greater sharing and standardization of simple well‐validated DAA RAS sequencing methods to assess the potential impact of HCV resistance on global efforts toward HCV elimination.
A total of 910 publications were identified, and 183 were included in the full text assessment (Fig. 1). Overall, 727 records were not suitable for full text analysis after abstract and title review due to duplication of records (n = 202), being a review article (n = 112), not being in English (n = 26), being an in vitro/nonclinical study (n = 53), or irrelevant or insufficient methodological description (n = 334). Of the 183 publications eligible for full text assessment, 126 records were excluded due to unclear or insufficient methodology, 27 publications were included through back referencing, and 11 records were included for review from unpublished methods contributed by external investigators. The total number of records eligible for study inclusion was 95, from which 407 HCV PCR amplification and sequencing primer pairs were identified that had been used to detect DAA RASs in clinical samples. From these, 357 primer pairs were able to be mapped onto the HCV H77 (genotype 1a) reference genome. Primer pairs for which binding location was not reported in the original publication and could not be mapped onto the HCV H77 reference genome (n = 50) were included in the primer library but not in the subsequent amplicon analysis because the binding location was unable to be determined.
Figure 1.
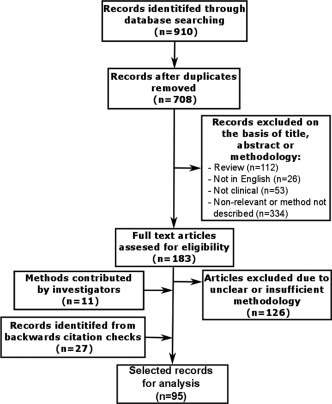
Flow diagram describing the review process and study selection.
STUDY CHARACTERISTICS
Sanger population sequencing was the most commonly performed sequencing method (71% of all amplicons), followed by NGS (28%), and template‐independent amplification (1%) (Table 1). Overall, the mean amplicon length was 1,049 nucleotides (SD, 1,403; median, 646). For population sequencing and NGS methods, the mean amplicon length was 981 nucleotides (SD, 1,053; median, 732) and 948 nucleotides (SD, 1,408; median, 468), respectively. All identified template‐independent amplification methods were full‐length (whole genome) protocols. Sequencing amplicons were identified that covered the entire HCV genome, entire protein coding region, or partial protein coding regions. The most commonly sequenced region overall was the NS3 region (n = 180), followed by NS5A (n = 95), NS5B (n = 75), and full length (n = 6) (Table 2). Methods identified from eligible records were used to assess DAA RASs in 20,411 clinical HCV samples, and 91% (n = 18,567) of these were tested using population sequencing methods.
Table 1.
OVERALL RESULTS OF DATA INCLUDED IN THE PRIMER LIBRARY
| Total | Population | NGS | TIA | |
|---|---|---|---|---|
| Number of records (%)a | 95 (100) | 65 (68) | 27 (28) | 3 (3) |
| Number of sequencing amplicons (%)a | 403 (100) | 286 (71) | 114 (28) | 3 (1) |
| Mean amplicon length (median; range) | 1,049 (646;118‐9,646) | 981 (732; 181‐8,991) | 948 (468; 118‐8,964) | 9,646 (9,646; 9,646‐9,646) |
| Most commonly sequenced region | NS3 | NS3 | NS3 | Full length |
| Number of clinical samples tested (%)a | 20,411 (100) | 18,567 (91) | 1,778 (9) | 66 (0.3) |
Percentages are row percentage. Abbreviation: TIA, template‐independent amplification.
Table 2.
NUMBER OF FULL AND PARTIAL SEQUENCING AMPLICONS ANALYZED BY PROTEIN CODING REGIONS OF THE HCV GENOME THAT WAS SEQUENCED AND THE HCV SUBTYPE/GENOTYPE SPECIFICITY OF THE PRIMERS USED TO GENERATE AMPLICONS
| NS3 (n = 180)a | NS5A (n = 95)a | NS5B (n = 75)a | Full Genome | Other | ||||
|---|---|---|---|---|---|---|---|---|
| Fullb | Partialc | Fullb | Partialc | Fullb | Partialc | Fullb | Partialc | |
| Total | 15 | 165 | 20 | 75 | 9 | 66 | 7 | 4 |
| Gt1a | 3 | 42 | 2 | 22 | 2 | 24 | 2 | 0 |
| Gt1b | 4 | 53 | 9 | 37 | 3 | 24 | 2 | 2 |
| Gt1 | 4 | 32 | 1 | 0 | 0 | 1 | 0 | 0 |
| Gt2 | 2 | 8 | 1 | 0 | 0 | 1 | 0 | 0 |
| Gt3 | 2 | 11 | 6 | 10 | 0 | 3 | 0 | 0 |
| Gt4/5/6 | 0 | 13 | 1 | 5 | 4 | 4 | 0 | 0 |
| Multi‐genotypicd | 0 | 6 | 0 | 1 | 0 | 9 | 3 | 2 |
357 individual amplicons were analyzed. If an amplicon covered more than one region, it was counted in both regions it covered; therefore, row total adds up to 360.
Full refers to whether the full protein coding region is covered by the amplicon (i.e., entire NS3 region).
Partial refers to whether only a partial protein coding region is covered by the amplicon (i.e., only part of the NS3 region).
Multi‐genotypic refers to primers that were designed for one or more HCV genotypes. Abbreviation: Gt, genotype.
AMPLICONS COVERING PARTIAL OR FULL PROTEIN CODING REGIONS
Of all amplicons in NS3, NS5A, or NS5B, 13% (n = 44) covered the full protein coding region and 88% (n = 306) partially covered the protein coding region (Table 2; Figs. 2, 3, 4). In all three regions (NS3, NS5A, and NS5B), the majority of sequencing amplicons were specific to either HCV subtype 1a or 1b (Table 2; Figs. 2, 3, 4). Amplicons specific to HCV genotypes 2, 3, 4, 5, and 6 accounted for only 20% of NS3 methods and 24% of both NS5A and NS5B methods. While 66% and 88% of all methods identified in NS3 and NS5A, respectively, covered all key DAA RASs, only 36% of identified methods in NS5B covered all key DAA RASs (Figs. 3 and 4).
Figure 2.
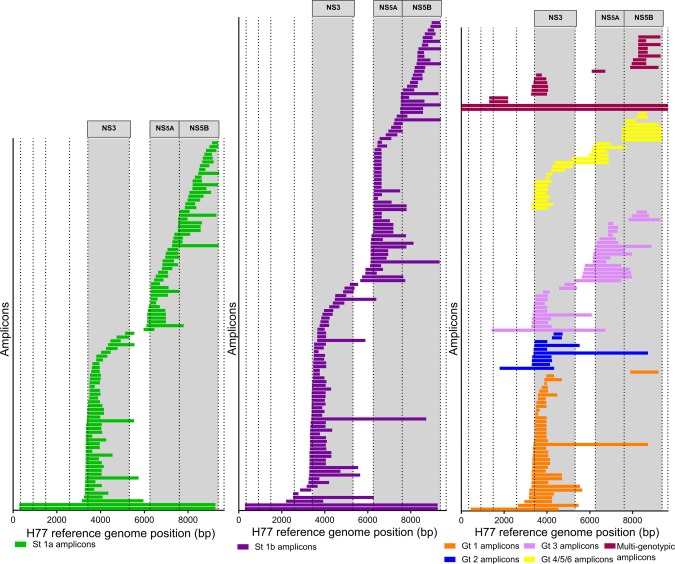
Location of amplicons identified grouped by HCV genotype specificity. Length and location of amplicons identified for HCV subtype 1a (green bars), 1b (purple bars), HCV genotype 1 (additional to subtype‐specific amplicons, orange bars), 2 (blue bars), 3 (pink bars), 4/5/6 combined (yellow bars), and multi‐genotypic amplicons (primers that are universal for two or more genotypes, maroon bars). Each bar represents one amplicon. Dotted lines represent the beginning/end of individual protein coding regions of the HCV genome. Gray‐shaded columns indicate the NS3, NS5A, and NS5B regions of the HCV genome, respectively. Abbreviations: bp, base pair; Gt, genotype; St, subtype.
Figure 3.
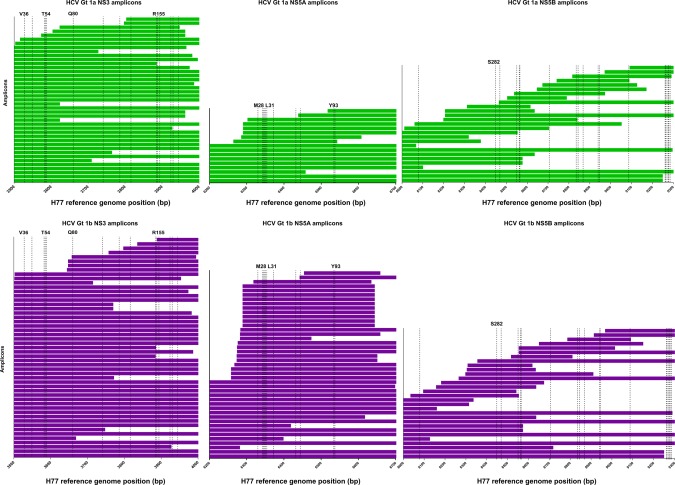
HCV sequencing protocols identified from the review in relation to clinically relevant DAA RASs specific for HCV subtypes 1a and 1b. Only HCV protein coding regions that contain clinically relevant DAA RASs are depicted (NS3, NS5A, and NS5B). DAA RAS positions are indicated by dashed lines and are listed in full in Supporting Table S4.9, 15 Each bar represents one amplicon. Amplicons specific for HCV subtype 1a are depcited by green bars, and amplicons specific for HCV subtype 1b are depicted by purple bars. Abbreviations: bp, base pair; Gt, genotype.
Figure 4.
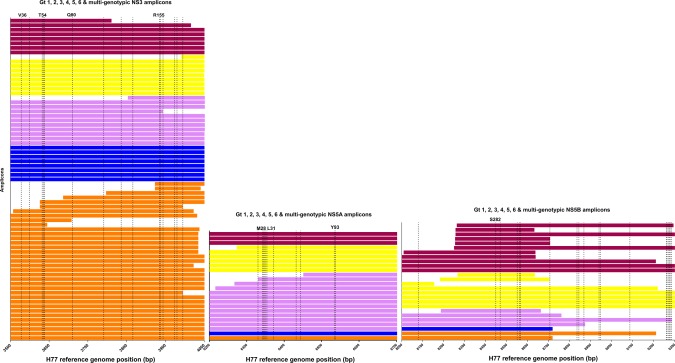
HCV sequencing protocols identified from the review in relation to clinically relevant DAA RASs specific for HCV genotypes 1‐6. Only HCV protein coding regions that contain clinically relevant DAA RASs are depicted (NS3, NS5A, and NS5B). DAA RAS positions are indicated by dashed lines and are listed in full in Supporting Table S4.9, 15 Each bar represents one amplicon. Amplicons specific for HCV genotype 1 (additional to subtype‐specific amplicons) are depicted by orange bars, amplicons specific to HCV genotype 2 are depicted by blue bars, amplicons specific to HCV genotype 3 are depicted by pink bars, amplicons specific to HCV genotypes 4, 5, or 6 are depicted by yellow bars, and amplicons that are specific for two or more HCV genotypes are depicted by maroon bars. Abbreviations: bp, base pair; Gt, genotype.
Table 3.
CANDIDATE METHODS FOR STANDARDIZATION OF HCV SEQUENCING TO DETECT DAA RASS
| HCV Region Targeted | Study Author | Year of Publication | LoD | Number Tested | Genotype/Subtype Specificity | Amplicon Length | Method | Primer Library Ref (Supporting Table S1) |
|---|---|---|---|---|---|---|---|---|
| Full length | Hedskog et al. | 2015 | 40,000 IU/mL | 17 | Multi‐genotypic [1‐6] | ∼9,000 bp | TIA | Ref01817 |
| NS3 | Besse et al. | 2012 | 40 IU/mL | 140 | Multi‐genotypic [1‐6] | ∼700 bp | Population sequencing | Ref02719 |
| NS5A | Lindstrom et al. | 2015 | Not reported | 127 | Multi‐genotypic [1a/1b/2b/3a] | ∼636 bp | Population sequencing | Ref04320 |
| NS5A | McCormick et al. | 2015 | Not reported | 138 | 4 | ∼810 bp | Population sequencing | Ref08421 |
| NS5A | Ku et al. | 2016 | Not reported | 7 | 5a | ∼1,500 bp | NGS | Ref03522 |
| NS5B | Tian et al. | 2012 | Not reported | 200 | Multi‐genotypic [1‐6] | ∼1,030 bp | Population sequencing | Ref07623 |
| NS5B | Garcia et al. | * | Not reported | ‐ | Multi‐genotypic [1‐6] | ∼1,300 bp | Population sequencing | Ref093a |
| NS5B | Bergfors et al. | * | 1000 IU/mL | 54 | Multi‐genotypic [1a/1b/2b/3a] | ∼1,030 bp | Population sequencing | Ref091a |
Unpublished method contributed by an external investigator. Abbreviations: bp, base pairs; LoD, lower limit of detection; Ref, reference; TIA, template‐independent sequencing.
MULTI‐GENOTYPIC AND FULL‐LENGTH METHODS
For amplicons identified in the NS3 region, six were generated with multi‐genotypic (designed to target two or more HCV genotypes) primers (Figs. 2, 3, 4). One amplicon generated by multi‐genotypic primers was identified in the NS5A region, and nine amplicons were identified in the NS5B region generated by multi‐genotypic primers (Figs. 2, 3, 4). Only a small number of multi‐genotypic primers were identified that produced amplicons covering all key DAA RASs in the regions sequenced (NS3, n = 3; NS5A, n = 1; NS5B, n = 2; Fig. 4; Table 2). In total, seven full‐length amplicons were identified, with four derived from primers specific for genotype 1a (n = 2) and 1b (n = 2). Three full‐length amplicons were identified generated from template‐independent amplification, which, due to not using traditional PCR primer‐based amplification, were classified as multi‐genotypic methods of amplification.
DESCRIPTION OF LEVEL OF VALIDATION REPORTED
For population sequencing methods, 6% (n = 4) of studies reported sensitivity of minor variant detection, 11% (n = 7) reported amplification sensitivity, and 38% (n = 25) reported the success rate. For NGS methods, 48% (n = 13) of studies reported sensitivity of minor variant detection and 7% (n = 2) reported amplification sensitivity. For NGS studies, 26% (n = 7) reported the success rate. In the three studies that reported using template‐independent amplification methods, one reported sensitivity of minor variant detection, amplification sensitivity (reported to be 40,000 IU/mL), and the diversity or geographic origin of samples tested.16 While all template‐independent amplification methods reported being multi‐genotypic, one study reported being tested on clinical samples from all six genotypes17; the other two studies reported being tested in clinical samples from two genotypes (genotype 6 and genotype 1)16 and four genotypes (genotypes 1‐4).18
IDEAL CANDIDATE METHODS
No methods were identified that matched all the features of an ideal candidate to standardize HCV sequencing that we determined a priori. Of all methods reviewed, the closest match to the ideal features was Ref018.17 This method was reported to be full‐length, pan‐genotypic, and validated successfully in a large panel of diverse samples from all genotypes and many subtypes; however, the limit of detection was not reported for this study. With an unreported and potentially low limit of detection, this method would not be appropriate for determining the presence of DAA RASs in samples with low HCV RNA levels and may not adequately sample underlying viral subpopulations. For the alternative scenario that was considered where DAA RASs are to be determined only in single protein coding regions of HCV (NS3, NS5A, or NS5B), one method matching all features of an ideal candidate was identified in the NS3 region, two in the NS5B region, and none in the NS5A region. As no methods were identified in the NS5A region, the next closest matches were identified.
For NS3, Ref02719 was the only method identified that covered all key DAA RASs and was pan‐genotypic. It was reported to have been successfully validated in a panel of diverse samples from all genotypes and multiple subtypes and had a limit of detection of 40 IU/mL. Therefore, it matched all the features of an ideal candidate to standardize sequencing of HCV to detect DAA RASs in the NS3 region.
No pan‐genotypic methods were identified for NS5A covering all key DAA RASs. However, one multi‐genotypic method, Ref043,20 was identified covering all key DAA RASs and was validated for subtypes 1a/1b/2b/3a. This method may be suitable to standardize sequencing of HCV to detect DAA RASs in NS5A as it has been demonstrated to amplify the most globally prevalent genotypes and all genotypes for which NS5A RASs have been demonstrated to be clinically relevant. To standardize sequencing of samples of genotypes 4, 5, and 6, genotype‐specific methods covering all key DAA RASs in NS5A specific to these genotypes were identified from the methods reviewed. The HCV genotype 4‐specific method, Ref084,21 covered all key DAA RASs and reported the most in‐depth validation of all genotype 4‐specific methods identified. The HCV subtype 5a‐specific method, Ref03522, covered all key DAA RASs and was the only method specific for genotype 5 that was identified. No methods covering all DAA RASs in NS5A designed to amplify genotype 6 were identified.
For NS5B, two methods were identified that were pan‐genotypic and covered all key DAA RASs in this region, Ref 07623 and Ref093 (Garcia et al., unpublished data); however the limit of detection was not reported for either method. A multi‐genotypic method, Ref091 (Bergfors et al., unpublished data), was also identified as a possible candidate as it covered all key DAA RASs in this region, had a reported limit of detection of 1000 IU/mL, and was specific for the most prevalent HCV subtypes globally (genotypes 1a, 1b, 2b, and 3a).
Strengths of Current Methods
Despite its breadth and scope, this review did not identify any methods matching the ideal candidate features determined a priori; however, this is not unexpected given the extensive genetic diversity of HCV. Therefore, in lieu of an ideal candidate method, several methods were identified as alternatives to standardize sequencing for particular scenarios. Template‐independent amplification methods can produce full‐length sequences and are pan‐genotypic due to their nonreliance on traditional PCR primers. This methodology has not been widely used for detection of HCV DAA RASs; however, method Ref01817 could be a candidate for determining the presence of all key DAA RASs across all regions of interest in samples of any genotype. The strengths of this method are that it is full length and pan‐genotypic; however, as current template‐independent amplification methods have a high limit of detection and NGS is only cost effective for high throughput applications, this method may not be universally suitable for standardization of HCV sequencing to detect DAA RASs.
Where NGS is suitable, it may be preferable for its numerous advantages, such as the possibility to sequence the whole HCV genome pan‐genotypically and potentially having the sensitivity to detect substitutions in minority variants if sufficient numbers of viral genome templates are sampled. However, despite a potential sensitivity for NGS of ∼1%, it should be noted that a 15% cutoff for minor variant frequency, comparable to population sequencing, better predicts DAA treatment failure due to the selection of resistant viruses.2, 8 Therefore, it has been proposed that a 15% cutoff should be used in clinical trials, real‐life studies, and in clinical practice to report the presence of resistant variants assessed by NGS2, 24 in patients naïve to treatment with a particular DAA class. This would also allow for a comparison of results generated from population sequencing with NGS or template‐independent amplification assays.
For the alternative scenario that was considered, where DAA RASs are to be determined only in a single region of HCV (NS3, NS5A, or NS5B), only one method was identified that matched all the criteria for an ideal candidate method in one of the regions of interest. This was Ref027,19 which would be an ideal candidate to standardize sequencing to detect DAA RASs in NS3. In the NS5A and NS5B regions, no methods matched all the criteria for an ideal candidate; therefore, the methods identified compromised on either being pan‐genotypic (only multi‐genotypic or genotype‐specific methods were identified) or having a limit of detection of 1,000 IU/mL. While these methods may not match the ideal criteria, they may still be adequate for most scenarios.
Limitations of Current Methods
Due to the limited number of methods identified that covered all key DAA RASs in each region of interest and the poor reporting of performance characteristics, it was not possible to rank methods over each other. Therefore, all candidate methods identified in this review only have a conditional recommendation. It is, however, possible to put forward recommendations for minimum performance characteristics and validation that should be reported and performed for all publications reporting sequencing of HCV to detect DAA RASs. Reporting of success rate (both numerator and denominator), sensitivity of minor variant detection, PCR sensitivity, and limit of detection in publications should be improved. This would facilitate selecting and implementing appropriate standardized methods in new laboratories in the future. It was observed that the majority of samples tested were HCV genotype 1, and there is considerable uniformity in geographic origin of samples tested. Given the extreme level of genetic variation across HCV genotypes and subtypes, particularly in HCV genotype 4 and 6 viruses, recommending protocols is fraught where proof of robustness across diverse subtypes does not exist.
To report methods as pan‐genotypic, demonstrated success in many clinical samples from all genotypes and many subtypes should be mandatory. Development and validation of protocols for genotypes 4‐6, particularly in the NS3 and NS5A regions, should be explored further, particularly to enhance surveillance in low‐ and middle‐income countries. Given estimates of HCV burden in low‐ and middle‐income countries are considerable,25, 26 it is necessary to ensure that suitable, robust methods to screen and detect DAA RASs in clinical samples from genotypes 2, 4, 5, and 6 are developed. Public databases with sequence data from diverse subtypes would facilitate the design of robust methods for these genotypes. Additionally, the availability of a standardized panel of HCV samples of diverse genotypic and global origin for method development, particularly for genotypes 4‐6, would greatly assist in this endeavor and should be a priority. Most studies reported the binding location of all primers in addition to the primer sequence; however, some did not report the primer binding location. Due to the genetic variation of HCV, it was not possible to determine the binding location of all primer sequences that were not reported in the original publication. This issue was particularly exacerbated for primers specific to non‐genotype 1 or 3 and highlights the need for authors to properly identify the binding location of primers in their associated publications.
Key clinically relevant DAA RASs were identified, and amplicons covering these positions in each protein coding region of HCV were reviewed. In NS3, 467 base pairs are required to be sequenced to cover all key DAA RASs, while only 197 are required in NS5A. However in NS5B, 1,256 base pairs are required to be covered to include all clinically relevant DAA RASs. This may have resulted in a large number of protocols being developed to cover this region with multiple overlapping amplicons. As our primary criteria for identifying candidate protocols for standardization was to cover all key DAA RASs in a single amplicon, this may have reduced the number of candidate protocols identified in NS5B. However, it is important that a candidate method for standardization of sequencing cover all key DAA RASs in a single amplicon to maximize the applicability of the method and to simplify reporting of resistance for both clinical and research purposes. It would be preferable for assays to cover the entire protein coding region that is targeted by a particular drug class in the event that new inhibitors have clinically relevant RASs outside of the current subregions of interest. However, until methodological improvements are made to facilitate this, targeting subregions of interest will continue to be recommended. Going forward, to enhance knowledge of the prevalence of DAA RASs globally and to improve access to screening and detection, standardization of HCV regions sequenced to detect DAA RASs should be implemented
This study is the first review of methods used for sequencing HCV to detect DAA RASs in clinical samples. The study was considerably strengthened by the expertise of several expert investigators and the inclusion of unpublished methods submitted by invited researchers. While external contributions of unpublished methods were invited, no service laboratory methods were contributed because of patent or commercial interests. Therefore, it is possible that well‐validated robust methods routinely used in clinical samples to sequence HCV for detection of DAA RASs have not been included in this study. However, some of these methods are impossible to assess externally without sufficient validation parameters and might not be as robust, which impacts whether recommendations can be made for the broader use of these assays. An inherent limitation of reviews conducted on topics that are particularly pertinent or have a rapid pace of discovery is the volume of literature that may become available between when the review is carried out and when it is published. In this instance, we attempted to overcome this limitation by inviting researchers to contribute methods that were unpublished or under review. Furthermore, it should be noted that the purpose of this study was to improve and standardize the technical aspects of RAS detection. This study was not meant to address issues related to the interpretation of the impact of the presence of RASs on clinical outcomes, which may vary greatly in different HCV genotypes/subtypes and patient populations. The reporting, clinical interpretation, and standardization of RAS detection in HCV should be a future priority.
Expert Commentary
We describe the first review of primers used for sequencing HCV to detect DAA resistance. The development of standardized RAS sequencing protocols is complicated by high genetic variability, reflected in the lack of genotype 4‐6 protocols. Template‐independent amplification protocols may provide multi‐genotypic or pan‐genotypic methods, provided the amplification sensitivity can be enhanced. The combined development, validation, and sharing of standardized methods for all genotypes and subtypes is a priority to improve understanding of the real‐world impact of RASs, particularly in low‐ and middle‐income countries. To facilitate the sharing of standardized methods, basic validation should be carried out, and these parameters should be consistently reported in publications.
Future Perspectives
We have identified eight methods that are candidates for further evaluation and validation and may be useful for standardizing DAA RAS testing. Further method development is still required for the assessment of RASs in HCV genotypes 4‐6, particularly in the NS5A region. The HCV Resistance Working Group, led by the HCV Forum, has proposed creating a standardized panel of HCV samples of diverse genotypic and global origins to be available for HCV diagnostic and method development. This effort would be particularly useful for the development of methods to detect RASs suitable for genotypes 4‐6, and the panel could also be used for quality assurance programs, not only for RAS testing, but also for genotyping and viral load testing. Global efforts are underway to create a collaborative database of HCV sequence and clinical data for the monitoring of DAA RASs, which will further inform the clinical relevance of RAS testing into the future.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1050/suppinfo.
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Table 4
Acknowledgment
Unpublished HCV sequencing primers and methodologies were contributed to this review by Johan Lennerstrand, Nader Kameli, Federico Garcia, Ana‐Belén Pérez, Natalia Chueca, Janke Schinkel, and Francesca Ceccherini‐Silberstein. Monica Parker provided advice on the assessment of laboratory‐developed molecular tests.
Potential conflict of interest: Nothing to report.
The views expressed are those of the authors and do not necessarily represent official policy of the U.S. Food and Drug Administration.
REFERENCES
- 1. Komatsu TE, Boyd S, Sherwat A, Tracy L, Naeger LK, O'Rear JJ, et al. Regulatory analysis of effects of hepatitis C virus NS5A polymorphisms on efficacy of elbasvir and grazoprevir. Gastroenterology 2017; 152:586‐597. [DOI] [PubMed] [Google Scholar]
- 2. Pawlotsky J‐M. Hepatitis C virus resistance to direct‐acting antiviral drugs in interferon‐free regimens. Gastroenterology 2016;151:70‐86. [DOI] [PubMed] [Google Scholar]
- 3. Dore GJ, Altice F, Litwin AH, Dalgard O, Gane EJ, Shibolet O, et al. Elbasvir‐grazoprevir to treat hepatitis C virus infection in persons receiving opioid agonist therapy: a randomized trial. Ann Intern Med 2016;165:625‐634. [DOI] [PubMed] [Google Scholar]
- 4. Forns X, Gordon SC, Zuckerman E, Lawitz E, Calleja JL, Hofer H, et al. Grazoprevir and elbasvir plus ribavirin for chronic HCV genotype‐1 infection after failure of combination therapy containing a direct‐acting antiviral agent. J Hepatol 2015;63:564‐572. [DOI] [PubMed] [Google Scholar]
- 5. Zeuzem S, Ghalib R, Reddy KR, Pockros PJ, Ben Ari Z, Zhao Y, et al. Grazoprevir‐elbasvir combination therapy for treatment‐naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection a randomized trial. Ann Intern Med 2015;163:1‐13. [DOI] [PubMed] [Google Scholar]
- 6. Nelson DR, Cooper JN, Lalezari JP, Lawitz E, Pockros PJ, Gitlin N, et al. All‐oral 12‐week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY‐3 phase III study. Hepatology 2015;61:1127‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sarrazin C, Dvory‐Sobol H, Svarovskaia ES, Doehle BP, Pang PS, Chuang S‐M, et al. Prevalence of resistance‐associated substitutions in HCV NS5A, NS5B, or NS3 and outcomes of treatment with ledipasvir and sofosbuvir. Gastroenterology 2016;151:501‐512.e501. [DOI] [PubMed] [Google Scholar]
- 8. Zeuzem S, Mizokami M, Pianko S, Mangia A, Han KH, Martin R, et al. NS5A resistance‐associated substitutions in patients with genotype 1 hepatitis C virus: prevalence and effect on treatment outcome. J Hepatol 2017;66:910‐918. [DOI] [PubMed] [Google Scholar]
- 9. Sarrazin C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J Hepatol 2016;64:486‐504. [DOI] [PubMed] [Google Scholar]
- 10. Bourlière M, Gordon SC, Ramji A, Ravendhran N, Tran TT, Hyland RH, et al. Sofosbuvir/velpatasvir/voxilaprevir for 12 weeks as a salvage regimen in NS5A inhibitor‐experienced patients with genotype 1‐6 infection: The Phase 3 POLARIS‐1 Study. Hepatology 2016;64:1‐136. [Google Scholar]
- 11. Zeuzem S, Flamm SL, Tong MJ, Vier‐ling JM, Pianko S, Buggisch P, et al. A randomized, controlled, phase 3 trial of sofosbuvir/ velpatasvir/voxilaprevir or sofosbuvir/velpatasvir for 12 weeks in direct acting antiviral‐experienced patients with genotype 1‐6 HCV infection: The POLARIS‐4 Study. Hepatology 2016;64:1‐136. [Google Scholar]
- 12. Vermehren J, Dietz J, Susser S, Hahn Tv, Petersen J, Hinrichsen H, et al. Retreatment of patients who failed direct‐acting antiviral (DAA) therapies: real world experience from a large european hepatitis C resistance database (P894). In: The 67th Annual Meeting of the American Association for the Study of Liver Diseases: The Liver Meeting 2016. Boston, MA. Hepatology; 2016;64:361‐601. [Google Scholar]
- 13. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group . Preferred reporting items for systematic reviews and meta‐analyses: The PRISMA Statement. PLoS Med 2009;6:e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jacka B, Lamoury F, Simmonds P, Dore GJ, Grebely J, Applegate T. Sequencing of the hepatitis C virus: a systematic review. PLoS One 2013;8:e67073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lontok E, Harrington P, Howe A, Kieffer T, Lennerstrand J, Lenz O, et al. Hepatitis C virus drug resistance‐associated substitutions: state of the art summary. Hepatology 2015;62:1623‐1632. [DOI] [PubMed] [Google Scholar]
- 16. Wei B, Kang J, Kibukawa M, Chen L, Qiu P, Lahser F, et al. Development and validation of a template‐independent next‐generation sequencing assay for detecting low‐level resistance‐associated variants of hepatitis C virus. J Mol Diagn 2016;18:643‐656. [DOI] [PubMed] [Google Scholar]
- 17. Hedskog C, Chodavarapu K, Ku KS, Xu S, Martin R, Miller MD, et al. Genotype‐ and subtype‐independent full‐genome sequencing assay for hepatitis C virus. J Clin Microbiol 2015;53:2049‐2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bowden R, Bonsall D, Ansari MA, Ip C, Trebes A, Brown A, et al. ve‐SEQ: robust, unbiased enrichment for streamlined detection and whole‐genome sequencing of HCV and other highly diverse pathogens. F1000Res 2015;4:1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Besse B, Coste‐Burel M, Bourgeois N, Feray C, Imbert‐Marcille BM, Andre‐Garnier E. Genotyping and resistance profile of hepatitis C (HCV) genotypes 1‐6 by sequencing the NS3 protease region using a single optimized sensitive method. J Virol Methods 2012;185:94‐100. [DOI] [PubMed] [Google Scholar]
- 20. Lindstrom I, Kjellin M, Palanisamy N, Bondeson K, Wesslen L, Lannergard A, et al. Prevalence of polymorphisms with significant resistance to NS5A inhibitors in treatment‐naive patients with hepatitis C virus genotypes 1a and 3a in Sweden. Infect Dis (Lond) 2015;47:555‐562. [DOI] [PubMed] [Google Scholar]
- 21. McCormick AL, Wang L, Garcia‐Diaz A, Macartney MJ, Webster DP, Haque T. Prevalence of baseline polymorphisms for potential resistance to NS5A inhibitors in drug‐naive individuals infected with hepatitis C genotypes 1‐4. Antivir Ther 2015;20:81‐85. [DOI] [PubMed] [Google Scholar]
- 22. Ku KS, Chodavarapu RK, Martin R, Miller MD, Mo H, Svarovskaia ES. Sequencing analysis of NS3/4A, NS5A, and NS5B genes from patients infected with hepatitis c virus genotypes 5 and 6. J Clin Microl 2016;54:1835–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tian D, Li L, Liu Y, Li H, Xu X, Li J. Different HCV genotype distributions of HIV‐infected individuals in Henan and Guangxi, China. PLoS ONE 2012;7:e50343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perales C, Domingo E. Antiviral strategies based on lethal mutagenesis and error threshold. Curr Top Microbiol Immunol 2016;392:323‐339. [DOI] [PubMed] [Google Scholar]
- 25. Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age‐specific antibody to HCV seroprevalence. Hepatology 2013;57:1333‐1342. [DOI] [PubMed] [Google Scholar]
- 26. Gower E, Estes C, Blach S, Razavi‐Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 2014;61(1 Suppl):S45‐57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1050/suppinfo.
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Table 4