

Abstract
The emergence of resistance mutations in the reverse transcriptase gene of hepatitis B virus (HBV) is associated with treatment failure. Entecavir (ETV) is one of the most potent anti‐HBV reagents; it has a very low resistance rate and is used as the first‐line treatment for chronic hepatitis B. In this study, we isolated HBVs in 4 ETV‐refractory patients (2 with viral breakthrough, 1 with partial virological response, and 1 with flare‐up) and assessed ETV resistance using replication‐competent 1.38‐fold HBV genome‐length molecular clones. The full genome sequences of infected HBVs in ETV‐refractory patients were determined. The HBV molecular clones were generated with the patient‐derived sequences. After transfection of these molecular clones into HepG2 cells, viral replications and ETV susceptibilities were evaluated by measuring the amount of intracellular core‐particle‐associated HBV DNA using Southern blotting and real‐time polymerase chain reaction. Among these cases, ETV‐resistant variants were detected in 2 patients with viral breakthrough and responsible amino acid mutations in reverse transcriptase were successfully identified in these variants. No ETV‐resistant mutation was detected in the other cases. The identified ETV‐resistant mutations did not confer resistance to tenofovir disoproxil fumarate. Conclusion: The HBV replication model with patient‐derived sequences is useful for assessing replication efficiency, susceptibility to anti‐HBV reagents, and responsible resistance mutations and can aid in choosing the appropriate treatment strategy for treatment‐failure cases of chronic hepatitis B. (Hepatology Communications 2017;1:110‐121)
Abbreviations
- CI
confidence interval
- EC50
effective concentrations required to inhibit 50%
- ETV
entecavir
- HBeAg
hepatitis B e antigen
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HLA
human leucocyte antigen
- LAM
lamivudine
- NA
nucleos(t)ide analogue
- PCR
polymerase chain reaction
- RT
reverse transcriptase
- TDF
tenofovir disoproxil fumarate
- VBT
viral breakthrough
Introduction
Hepatitis B virus (HBV), a member of the Hepadnaviridae family, has a partially double‐stranded 3.2‐kb DNA genome and comprises four open reading frames coding for hepatitis B surface antigen (HBsAg), core protein/hepatitis B e antigen (HBeAg), viral polymerase, and X protein.1, 2 Infection with this virus causes severe liver diseases, including acute, chronic, and fulminant hepatitis; cirrhosis; and hepatocellular carcinoma (HCC).3 Although effective vaccines against HBV are available in many countries, chronic hepatitis B (CHB) infection remains a significant global health problem. The World Health Organization estimates that approximately 240 million people are afflicted with CHB infections, and these people are at risk for cirrhosis and HCC.4, 5 Currently, two classes of anti‐HBV reagents are clinically available: interferon‐α and nucleos(t)ide analogues (NAs). Interferon treatment can reduce HBeAg and HBsAg through a combination of direct antiviral and immunomodulatory effects,6 but it is associated with unfavorable side effects. NAs target the step of reverse transcription and inhibit the production of HBV DNA from pregenomic RNA. Although these reagents ameliorate liver diseases and prevent disease progression to HCC, such long‐term treatment often causes the emergence of resistance mutations. In Japan, NAs of lamivudine (LAM), adefovir dipivoxil (ADV), entecavir (ETV), and tenofovir disoproxil fumarate (TDF) have been approved. Because these NAs share a common target for the viral reverse transcriptase (RT), resistance mutations to these reagents have been reported only in RT domains. Among these NAs, ETV is one of the most potent anti‐HBV reagents; it has a very low resistance rate in treatment‐naive patients7, 8, 9, 10, 11 and has long been used as a first‐line reagent. To date, a limited number of ETV‐resistant mutations have been reported. They include the LAM‐resistant‐associated amino acid mutations at rt180 and rt204 and require additional mutations at rt169, rt184, rt202, or rt250.8, 12 Mutations of rtI163V and rtA186T in addition to the LAM‐resistant mutations have been reported to confer ETV resistance.13
In this study, we isolated HBVs from four ETV‐refractory cases (2 viral breakthrough [VBT] patients, 1 partial virological response patient, and 1 flare‐up patient) and generated replication‐competent 1.38‐fold HBV genome‐length molecular clones with patient‐derived sequences. After transfection of these molecular clones into HepG2 cells, viral replication and ETV susceptibility were evaluated by measuring the amount of intracellular core‐particle‐associated HBV DNA using Southern blotting and real‐time polymerase chain reaction (PCR).
Materials and Methods
PATIENTS
We performed a retrospective analysis of 70 patients with CHB (male/female, 53/17; mean age, 46.6 ± 8.9 years) who received ETV treatment at Seizankai Kiyokawa Hospital, Tokyo, Japan, from December 2006 to May 2016. All patients were clinically diagnosed with chronic hepatitis and were HBsAg‐positive and HBV DNA‐positive for at least 6 months prior to the start of treatment. None were diagnosed with cirrhosis or HCC. All patients received 0.5 mg ETV for more than 1 year, and no patient had received other NAs. This study was approved by the Ethics Committees of our institutions (approval numbers 246 from Seizankai Kiyokawa Hospital and 377 from the National Institute of Infectious Diseases), and written informed consent was obtained from each patient. Of the 70 patients with CHB treated with ETV, four cases appeared to be refractory (Table 1). These four subjects were male, positive for HBsAg and HBeAg, and infected with HBV genotype C. Their ages ranged from 40 to 59 years, and all were negative for serological markers of hepatitis C virus and human immunodeficiency virus infections. Profiles of the 4 ETV‐refractory patients are as follows: Case A (40‐year‐old male; Fig. 1A): The administration of ETV (0.5 mg daily) was started in August 2007. Serum HBV DNA became undetectable after 3 years of ETV treatment but re‐emerged after 3.3 years of treatment. The HBV DNA titer was 8.0 log copies/mL at that time. The increased dosage of ETV (1.0 mg daily) was not effective. This case was diagnosed as a VBT case. HBV was isolated from serum samples collected at two time points, at the beginning of treatment (time point 1) and at VBT (time point 2) (Fig. 1A). The isolated HBVs were named clones A1 and A2, respectively. The entire genomes of the isolated HBVs were sequenced, and the deduced amino acids in the RT region were aligned and compared with the genotype C consensus sequence (Supporting Fig. S1). The known resistance mutation to NAs was not detected in clone A1. However, reported LAM‐resistant mutations of rtV173L/L180M/M204V emerged in clone A2. Case B (43‐year‐old male; Fig. 1B): The administration of ETV (0.5 mg daily) was started in April 2007. Serum HBV DNA was undetectable after 2.5 years of ETV treatment and remained undetectable until 6.8 years from the start of ETV when HBV DNA re‐emerged and reached 8.3 log copies/mL. This case was also diagnosed as a VBT case. HBVs isolated at the beginning of treatment (time point 1) and at VBT (time point 2) were sequenced (Fig. 1B). As in the HBVs isolated in case A, no resistance mutation was detected in clone B1. However, mutations of rtN238H/L269I accompanied by LAM‐resistant mutations of rtL180Q/M204V emerged in clone B2. Case C (59‐year‐old male; Fig. 1C): The administration of ETV (0.5 mg daily) was started in June 2010. Serum HBV DNA was 9.1 log copies/mL before treatment and decreased to 5.0 log copies/mL soon after starting ETV; however, HBV DNA remained positive for 1.5 years of ETV treatment. This case was considered to be a partial virological response case. HBV isolated from serum samples at multiple time points (time points 1‐3 in Fig. 1C) were sequenced. Of these HBV clones, the deduced amino acid sequences in the RT region were identical but had several differences compared with the genotype C consensus sequence. Case D (53‐year‐old male; Fig. 1D): The administration of ETV (0.5 mg daily) was started in June 2007. After 2 years of treatment with ETV, serum HBV DNA became undetectable. After 3 years from the start of ETV treatment, HBV DNA suddenly flared up. This flare‐up of HBV DNA was transient and decreased to an undetectable level without additional treatment. Therefore, this case was defined as a flare‐up case. HBV was isolated at three time points: the beginning of treatment (time point 1), the flare‐up point (time point 2), and the subsided point (time point 3) (Fig. 1D). The deduced amino acid sequences in the RT region were identical in clones D1 and D3 and an amino acid substitution of V191I was detected in clone D2. All four patients were treatment naive and confirmed their medication adherence.
Table 1.
PROFILES OF ETV‐REFRACTORY CASES
| Patients | Case A | Case B | Case C | Case D |
|---|---|---|---|---|
| Sex | male | male | male | male |
| Agea (year) | 40 | 43 | 59 | 53 |
| ALTa (IU/mL) | 312 | 211 | 291 | 309 |
| HBV genotype | C | C | C | C |
| HBeAg | positive | positive | positive | positive |
| Core promoter (nt 1762/1764) | mutant/mutant | mutant/mutant | wild/wild | mutant/mutant |
| Precore(nt 1896) | wild | wild | wild | wild |
| HBV DNA*(log copy/mL) | 7.6 | 8.6 | 9.1 | 8.7 |
Values at the beginning of ETV treatment.
Figure 1.
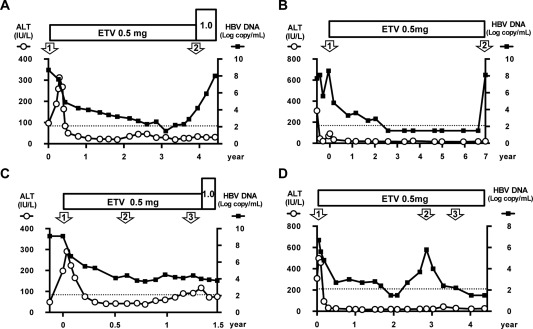
Clinical courses of 4 ETV‐refractory patients (A: Case A with VBT, B: Case B with VBT, C: Case C with partial virological response, D: Case D with flare‐up). Serum HBV DNA titers (solid squares) and ALT levels (open circles) are indicated. The lower detection limit of HBV DNA is indicated by the dotted line. The serum samples were collected at the time points indicated by open arrows with numbers, and the sequences of infected HBV were analyzed. Abbreviation: ALT, alanine aminotransferase.
VIROLOGICAL MARKERS FOR HBV
HBsAg and HBeAg were measured by chemiluminescent immunoassay using a commercial assay kit (Abbott Japan Co., Ltd., Tokyo, Japan). The HBV genotype was determined by enzyme immunoassay (IMMUNIS HBV Genotype EIA; Institute of Immunology Co., Ltd., Tokyo, Japan). HBV DNA titer was determined by the COBAS TaqMan HBV test ver. 2.0 (Roche Diagnostics K.K., Tokyo, Japan) or the Amplicor HBV Monitor assay (Roche Diagnostics K.K.); the lower detection limits of these assays were 2.1 and 2.6 log copies/mL, respectively.
SEQUENCE ANALYSIS OF HBV
Total DNA was isolated from 200 µL of patient serum collected at the indicated time points in Fig. 1 using the QIAamp Blood Mini Kit (Qiagen K.K., Tokyo, Japan). The entire genome of HBV was amplified by PCR using primers as described.14 Amplified fragments were sequenced directly with an automated DNA sequencer (3500 Genetic Analyzer; Applied Biosystems, Foster City, CA).
CONSTRUCTION OF REPLICATION‐COMPETENT MOLECULAR CLONES
PCR‐amplified fragments were inserted into the pGEM‐T Easy Vector system (Promega, Madison, WI). At least five clones of each fragment were sequenced, and the consensus sequence was determined. Using the obtained clones as templates, two material fragments (fragment 1, nucleotide [nt] 970‐3215/1‐178 and fragment 2, nt 178‐2186; the number of nucleotides was referred from the genotype C prototype clone [accession number AB246344, https://www.ncbi.nlm.nih.gov/nuccore/AB246344]) with consensus sequences for construction generated using the fusion PCR technique. The replication‐competent HBV molecules with 1.38‐fold genome length were constructed by ligation of two material fragments (Fig. 2). The intended mutations were introduced by site‐directed PCR and fragment swap. Established molecular clones were sequenced and confirmed to be correctly constructed.
Figure 2.
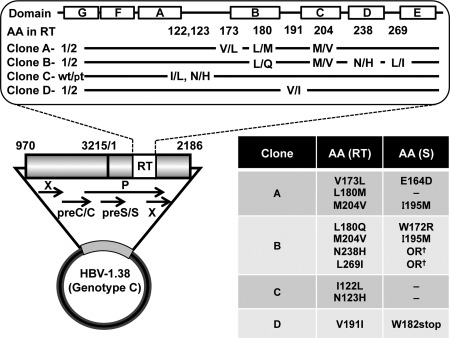
Construction of replication‐competent 1.38‐fold HBV genome length molecular clone. The schema of the constructed replication‐competent 1.38‐fold HBV genome is indicated. This construct contains 4,432 bp of HBV genome and covers all four open reading frames. The upper panel shows the domain of RT and the introduced amino acid mutations in each clone. The lower panel indicates amino acid mutations in the RT region and the corresponding surface gene. Abbreviations: OR, out of region of the surface gene; AA, amino acid; preC/C, precore/core region; X, X gene of HBV; P, polymerase gene of HBV; preS/S, preS/S region; S, surface gene of HBV.
TRANSFECTION INTO HepG2 CELLS
HepG2 cells were obtained from the European Collection of Authenticated Cell Cultures (Salisbury, UK, https://www.phe-culturecollections.org.uk/collections/ecacc.aspx) and cultured at 37°C in a 5% CO2 environment in complete growth medium. HepG2 cells at 80%‐90% confluence in 100‐mm dishes were transfected with 2.5 µg of HBV molecular clones using Lipofectamine 3000 Reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Twenty‐four hours after transfection, cells were washed and the medium was changed to fresh medium with or without reagents. The transfected cells were harvested at 3 days after transfection. The anti‐HBV reagents of ETV and TDF were purchased from LKT Laboratories, Inc. (St. Paul, MN) and Selleck Chemicals (Houston, TX), respectively.
ISOLATION OF CORE‐PARTICLE‐ASSOCIATED HBV DNA
Isolation of intracellular core‐particle‐associated HBV DNA was accomplished as described by Günther et al. with some modifications.15 Briefly, cells were suspended in 500 μL of lysis buffer (100 mM Tris‐HCl pH 8.0, 0.2% Nonidet P40), and nuclei were pelleted by centrifugation. The supernatant was transferred and treated with DNase (RQ1 RNase‐free DNase; Promega) and RNase (RNase A; Qiagen K.K.). Then, proteinase K, 500 mM ethylene diamine tetraacetic acid, 5 M NaCl, and 10% sodium dodecyl sulfate were added and the samples were incubated at 55°C for 1 hour. HBV DNA was then recovered by phenol–chloroform (1:1) extraction and 2‐propanol precipitation.
SOUTHERN BLOT HYBRIDIZATION OF HBV DNA
The isolated core‐particle‐associated HBV DNA was separated on a 1.0% agarose gel and transferred to a nylon membrane (positively charged; Roche Diagnostics, Penzberg, Germany). The transferred membrane was immobilized by an ultraviolet crosslinker and hybridized by an alkaline‐ phosphatase‐labeled probe of a full‐length HBV DNA fragment (accession number AB246344, https://www.ncbi.nlm.nih.gov/nuccore/AB246344) generated with the Gene Images AlkPhos Direct labeling system (DIG‐High Prime DNA Labeling and Detection Starter Kit II; Roche Diagnostics). Chemiluminescent detection was performed with CDP‐Star (DIG‐High Prime DNA Labeling and Detection Starter Kit II) and analyzed using an LAS4000 image analyzer (Fuji Photo Film, Tokyo, Japan).
QUANTIFICATION OF HBV DNA BY REAL‐TIME PCR
The titer of core‐particle‐associated HBV DNA was also evaluated by real‐time PCR with the TaqMan Fast Universal PCR Master Mix (Applied Biosystems, Foster City, CA). The sequences of primers and probe used have been described.16 Amplification and detection were performed with StepOnePlus Real‐Time PCR Systems (Applied Biosystems).
CALCULATION OF 50% EFFECTIVE CONCENTRATION VALUE AND FIT MODEL
Dose‐response curves were drawn using GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA). The effective concentrations required to inhibit 50% (EC50) values were calculated with data obtained by real‐time PCR. The following model was used to draw the dose‐response curve: Y = Bottom + (Top – Bottom)/(1 + 10∧((X – logEC50))), where X is the concentration of the reagent, Y is the response expressed as the percentage of core‐particle‐associated HBV DNA related to the corresponding control, logEC50 represents the log EC50 control, and Top and Bottom represent the Y values at the top and bottom plateau of the fitted curve, respectively.
STATISTICAL ANALYSIS
Statistical analysis was performed using an unpaired two‐tailed Student t test with Welch's correction. P values < 0.05 were considered statistically significant.
Results
CONSTRUCTION OF HBV MOLECULAR CLONES
Using the full genome sequence of HBV isolated at the beginning of treatment in each case, we constructed replication‐competent 1.38‐fold HBV genome‐length molecular clones. These molecular clones contain the 4,432 bp of HBV genome covering all four open reading frames (Fig. 2). In cases A, B, and D, mutations that emerged during ETV treatment were introduced into the prototype clone (clones A1, B1, and D1) of each case and generated clones A2, B2, and D2, respectively (upper panel of Fig. 2; Supporting Fig. S1). Some of these mutations in RT were accompanied by amino acid mutations of overlapping surface genes, as indicated in the lower panel of Fig. 2. In case C, all isolated clones have identical amino acids in the RT region but have several differences in comparison with the genotype C consensus sequence. We selected the amino acid substitution of I122L/N123H detected between the RT domains of B and C. We introduced these mutations into a patient‐isolated clone, clone C‐pt, and generated clone C‐wt. These mutations do not affect amino acids in the surface gene.
ANALYSIS FOR SUSCEPTIBILITIES TO ETV
Using these generated HBV molecular clones, we assessed the replication efficiencies and susceptibilities to ETV in culture cells. Plasmids of HBV clones were transfected into HepG2 cells, and ETV was administered to the transfected cells at concentrations of 500, 50, 5, and 0.5 nM. After 3 days of treatment with ETV, the cells were harvested and intracellular core‐particle‐associated HBV DNA was extracted. The amounts of core‐particle‐associated HBV DNA were assessed by Southern blotting and real‐time PCR. All patient‐derived HBV molecules were replicable in HepG2 cells, and by administration of ETV, HBV replication was reduced in a dose‐dependent manner (Fig. 3). These reductions of HBV replication were similarly observed by both Southern blotting and real‐time PCR. To assess the susceptibility to ETV, the EC50 values were determined by drawing the dose‐response curves with data obtained by real‐time PCR (Fig. 4; Table 2).
Figure 3.
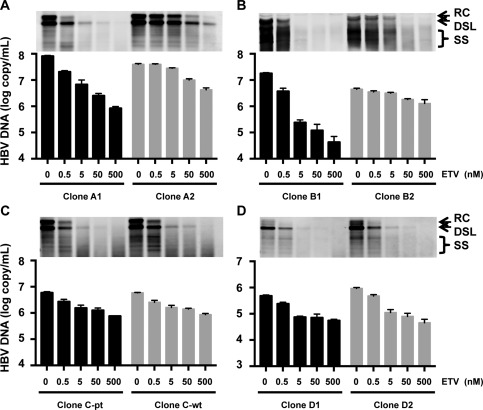
Replication of patient‐derived HBV molecular clones and susceptibility to ETV (A: Case A with VBT, B: Case B with VBT, C: Case C with partial virological response, D: Case D with flare‐up). HBV molecular clones were transfected into HepG2 cells, and ETV was administered to the transfected cells at the indicated concentrations. HBV replication and its reduction by ETV administration were assessed by measuring the intracellular core‐particle‐associated HBV DNA using Southern blotting (upper panel) and real‐time PCR (lower panel). The results are shown as the means ± SD. Abbreviations: DSL, double‐stranded liner HBV DNA; RC, relaxed circular HBV DNA; SS, single‐stranded HBV DNA.
Figure 4.
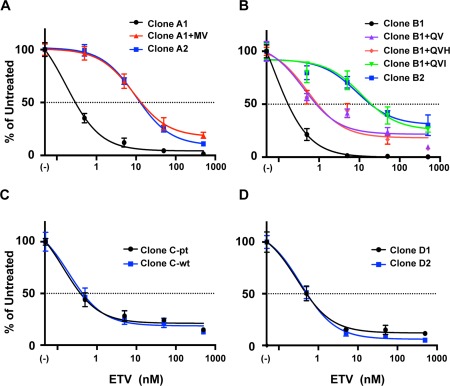
Dose‐response curve analysis of susceptibilities of patient‐derived and identified mutation‐introduced HBV clones (A: Case A with VBT, B: Case B with VBT, C: Case C with partial virological response, D: Case D with flare‐up). The dose‐response curves were drawn with data obtained by real‐time PCR described in Fig. 3 and Supporting Fig. S2. The amount of core‐particle‐associated HBV DNA without ETV treatment was defined as the untreated control, and percentages of the amount of core‐particle‐associated HBV DNA at the indicated concentration of ETV were calculated. The results are shown as the means ± SD.
Table 2.
REPLICATION EFFICIENCY AND EC50 VALUES FOR ETV OF HBV CLONES
| Replication Efficiency | ETV Resistance | |||
|---|---|---|---|---|
| Clones | HBV DNA (copy/mL) | Fold | EC50 in nM (95% CI) | Fold Resistance |
| Clone A1 | 8.06 × 107 ± 5.06 × 106 | 1 | 0.0865 (0.0533‐0.141) | 1 |
| Clone A2 | 3.98 × 107 ± 2.32 × 106 a | 0.494 | 8.60 (5.68‐13.0) | 99.3 |
| Clone A1+MV | 5.24 × 106 ± 2.78 × 105 a | 0.0651 | 9.95 (7.39‐13.4) | 115 |
| Clone B1 | 1.80 × 107 ± 1.10 × 106 | 1 | 0.135 (0.0999‐0.182) | 1 |
| Clone B2 | 4.44 × 106 ± 3.89 × 105 a | 0.247 | 8.63 (3.26‐22.8) | 63.9 |
| Clone B1+QV | 1.22 × 106 ± 1.14 × 105 a | 0.0675 | 0.530 (0.244‐1.15) | 3.92 |
| Clone B1+QVH | 8.37 × 105 ± 3.17 × 104 a | 0.0465 | 0.670 (0.286‐1.57) | 4.96 |
| Clone B1+QVI | 1.54 × 106 ± 1.15 × 105 a | 0.0855 | 11.4 (4.30‐30.4) | 84.6 |
| Clone C‐pt | 5.86 × 106 ± 1.93 × 105 | 1 | 0.271 (0.153‐0.480) | 1 |
| Clone C‐wt | 5.88 × 106 ± 5.40 × 105 | 1.00 | 0.215 (0.132‐0.350) | 0.794 |
| Clone D1 | 4.84 × 105 ± 4.74 × 104 | 1 | 0.377 (0.257‐0.552) | 1 |
| Clone D2 | 9.37 × 105 ± 5.83 × 104 a | 1.94 | 0.454 (0.351‐0.587) | 1.21 |
P < 0.05 in comparison with the prototype of each clone.
In assessments of HBV clones isolated from patients A and B, HBV DNA titers without ETV treatment were significantly lower (P < 0.05) in clones of A2 and B2 in comparison with the prototype clones of A1 and B1 (0.494‐fold and 0.247‐fold, respectively), suggesting lower replication efficiencies of emerged clones during ETV treatment (Table 2). The mutation‐introduced clones of A2 and B2 exhibited significantly lower susceptibility to ETV in comparison with the prototype clones (clones A1 and B1). The calculated EC50 values of clones A2 and B2 were 99.3‐fold and 63.9‐fold higher than that of clones A1 and B1, respectively. In assessments of HBV clones isolated from patient C, HBV DNA titers of clone C‐pt without ETV treatment were similar to clone C‐wt. However, in assessments of HBV clones isolated from patient D, HBV DNA titers of clone D2 without ETV treatment were significantly higher (1.94‐fold) than that of clone D1. The EC50 values of clone C‐pt and D2 were comparable to those of clones C‐wt and D1, respectively.
IDENTIFICATION OF RESPONSIBLE MUTATIONS OF ETV RESISTANCE
In our assessment with patient‐derived HBV molecular clones, the clones of A2 and B2 were revealed to be resistant to ETV. Both clones have key LAM‐resistant mutations plus one or two additional mutations (Fig. 2). To identify the mutation responsible for ETV resistance, we performed a mapping study by generating additional clones as follows: Clone A2 has the mutation rtV173L with the key LAM‐resistant mutations rtL180M/M204V. We generated a clone that has only the LAM‐resistant mutations rtL180M/M204V and named the clone A1+MV. Clone B2 has the mutations rtN238H/L269I accompanied by the LAM‐resistant mutations rtL180Q/M204V. Thus, we generated three clones as follows: clone B1+QV (introduced only the LAM‐resistant mutations rtL180Q/M204V to clone A1), clone B1+QVH (introduced rtL180Q/M204V plus rtN238H to clone A1), and clone B1+QVI (introduced rtL180Q/M204V plus rtL269I to clone A1).
Using these additional clones, we compared the replication efficiencies and susceptibilities to ETV with the prototype of each clone (Fig. 4; Supporting Fig. S2). In comparison with clone A1, clone A1+MV exhibited resistance to ETV. The EC50 value of clone A1+MV was 115‐fold higher than that of clone A1 and was comparable to clone A2 (Table 2). On the other hand, the replication efficiency of clone A1+MV was substantially lower. The HBV DNA titer of clone A1+MV without ETV treatment was 0.0651‐fold lower than that of clone A1. These data indicate that the introduction of the additional mutation of rtV173L rescued the replication efficiency to 0.494‐fold of clone A1. In the case of clone B, the clone with the LAM‐resistant mutations rtL180Q/M204V (clone B1+QV) exhibited slightly reduced susceptibility to ETV. The EC50 value of this clone was 3.92‐fold higher than that of clone B1. The addition of rtN238H to this clone (clone B1+QVH) did not enhance the ETV resistance. However, the addition of the mutation rtL269I (clone B1+QVI) caused severe deterioration of ETV susceptibility. The EC50 value of clone B1+QVI was 84.6‐fold higher than that of clone B1 and was comparable to clone B2. The replication levels of clones B1+QV, B1+QVH, and B1+QVI were almost identical; however, when these mutations were combined (clone B2), the replication efficiency was enhanced to 0.247‐fold of clone B1. We also analyzed the effect of mutation rtL269I alone. The EC50 value of clone B1+I was 0.472 nM (95% confidence interval [CI], 0.318‐0.699) and was 3.50‐fold higher than that of clone B1.
ANALYSIS FOR SUSCEPTIBILITIES TO TDF OF ETV‐RESISTANT CLONES
To assess the susceptibility to TDF of these ETV‐resistant clones, the clones were transfected into HepG2 cells and treated with TDF at concentrations of 500, 50, 5, and 0.5 nM. The replication of these HBV clones was similarly inhibited by TDF treatment in a dose‐dependent manner (Fig. 5). The EC50 values of clones A1 and A2 were 10.5 nM (95% CI, 6.65‐16.6) and 10.7 nM (95% CI, 6.41‐17.8), respectively. The EC50 values of clones B1 and B2 were 11.4 nM (95% CI, 5.45‐23.9) and 10.5 nM (95% CI, 3.46‐32.1), respectively. These data indicate that the ETV‐resistant mutations identified in this study do not confer resistance to TDF.
Figure 5.
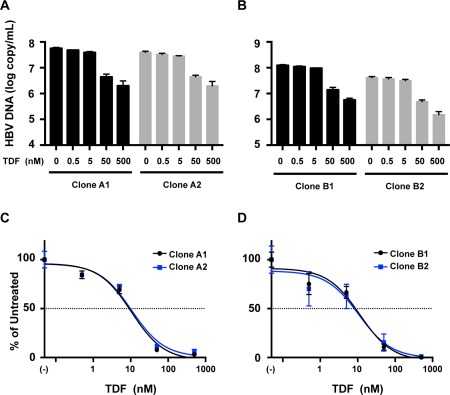
Susceptibilities to TDF of ETV‐resistant clones identified in this study. HBV molecular clones derived from patients A and B were transfected into HepG2 cells, and TDF was administered to the transfected cells at the indicated concentrations. (A, B) HBV replication and its reduction by TDF administration were assessed by measuring the intracellular core‐particle‐associated HBV DNA using real‐time PCR. (C, D) Dose‐response curves were drawn with the obtained data. The amount of core‐particle‐associated HBV DNA without TDF treatment was defined as the untreated control, and percentages of the amount of core‐particle‐associated HBV DNA at the indicated concentrations of TDF were calculated. The results are shown as the means ± SD.
Discussion
HBV is a significant human pathogen that is spread worldwide. Approximately 240 million people globally have CHB infection.4 Although advances have been made in anti‐HBV therapy with NAs, patients infected with this virus still need many years of treatment or lifelong treatment.17 Even in treatment with ETV, one of the most potent anti‐HBV reagents with a very low resistance rate, long‐term administration of this reagent is required, and it often leads to emergence of resistance‐associated variants in refractory cases. Therefore, the analysis of resistance‐associated variants in such cases is indispensable. In this study, to assess ETV‐resistant mutations, we generated replication‐competent HBV molecular clones using sequences from four ETV‐refractory cases and assessed the resistance to NAs, ETV, and TDF after transient transfection into HepG2 cells. We evaluated the replication abilities and susceptibilities of these clones by Southern blotting and found that they could be quantified by real‐time PCR for core‐particle‐associated HBV DNA. We used the 1.38‐fold‐genome length molecular clone, which contains two copies of the X region (Fig. 2) and therefore has some advantages in replication efficiencies in comparison with the minimum‐length molecular clones.18 We considered it preferable to obtain sufficient replication efficiency of the HBV molecular clones with patient‐derived sequences. Among the four ETV‐refractory cases, we detected the ETV‐resistant variants in two VBT patients (cases A and B). The HBV clones isolated from other cases exhibited susceptibilities to ETV.
ETV‐resistant variants were detected in VBT cases, and the responsible mutations were identified in both clones. In case A, emerged HBV at VBT (clone A2) had three mutations compared with the prototype clone A1 that was isolated at the beginning of treatment. The identified mutations, rtV173L/L180M/M204V, are known to be LAM resistant.19 Among these, rtL180M/M204V have been reported previously to confer LAM resistance associated with a decline in replication efficiency.20 The rtV173L mutation did not affect susceptibility to LAM but instead enhanced viral replication.19 Similarly, the introduction of the rtL180M/M204V mutations in clone A1 could confer ETV resistance. The EC50 value of the clone with these mutations was 99.3‐fold higher than that of the prototype clone and was not affected by the introduction of the additional mutation of rtV173L. On the other hand, the replication efficiency was strongly reduced by mutations of rtL180M/M204V but rescued by rtV173L. These observations of compensation by rtV173L were analogous with the case of LAM resistance,19 and we could clarify that these mutations are also associated with ETV resistance. The ETV‐resistant level of clone A1+MV seemed to be high, although the essential role of LAM‐resistant mutations to ETV is well known. Moreover, rtL180M/M204V‐associated high‐level resistance to ETV has been reported in patient‐derived strains but not laboratory strains.21 Such high‐level ETV resistance attributable to these mutations may be peculiar to the ETV refractory patient‐derived clone, and other amino acid residues in the RT region or other regions may be associated with this ETV resistance. These data suggest that the switch to or add‐on of ETV to LAM treatment‐failure patients described in reports was not recommended.22, 23
In case B, clone B2 with the four mutations rtL180Q/M204V/N238H/L269I emerged during ETV treatment. The mapping study of these mutations revealed that the key LAM‐resistant mutations rtL180Q/M204V could confer ETV resistance, similar to the HBV isolated in case A, but the extent of resistance was not high. The EC50 value was 3.92‐fold higher than that of the prototype clone. This lower contribution of these mutations to ETV resistance may be attributed to the difference of the mutations at rt180; M or Q, or to the genetic backgrounds of strains in other regions. By the addition of rtL269I to clone B1+QV, ETV resistance was enhanced and EC50 was increased to a level similar to clone B2, although the addition of the rtN238H mutation had minimal effects on ETV resistance. The rtN238H mutation was sometimes detected in NA‐treated patients and has been reported not to influence the susceptibilities to LAM, ADV, or other NAs.24, 25, 26 The rtL269I mutation was also detected in NA treatment‐failure patients. This mutation has been reported to enhance the replication efficiency and to be associated with enhanced replication efficiency when combined with the rtM204I mutation, although rtL269I alone did not alter susceptibilities to NAs.25, 27 In our experiment, rtL269I but not rtN238H could enhance ETV resistance when combined with rtL180Q/M204V, which is consistent with previous data.27, 28
In this study, we detected two ETV‐resistant variants, clones A2 and B2. Clone A2 comprised LAM‐resistant mutations, and clone B2 harbored novel ETV‐resistant mutations. Both clones were isolated at VBT and exhibited 99.3‐fold and 63.9‐fold resistance, respectively, compared with the prototype. In such clinical situations, selection and administration of other appropriate NAs will be required. We assessed the susceptibilities to TDF of these ETV‐resistant variants and found that the replication of these variants was efficiently suppressed. EC50 values were similar to those of prototype clones. In fact, the patient of case B was treated with 300 mg TDF daily after VBT and achieved sufficient reduction of HBV DNA and HBsAg levels (data not shown). Thus, the HBV replication system will be useful to determine effective reagents for treatment‐failure cases.
On the other hand, we did not detect ETV‐resistant variants in cases of partial virological response (case C) and flare‐up (case D). We found that the HBV strain in case C was constant. We selected the amino acid substitutions in comparison with the genotype C consensus sequence and assessed the effects on susceptibility to ETV by introducing these mutations. However, no remarkable difference in replication efficiency or susceptibility was detected. Thus, in this case, other factors for resistance, such as host factors associated with antiviral responses, were considerable. Genome‐wide association studies have indicated that genetic variants in the human leucocyte antigen (HLA)‐DP and HLA‐DQ are associated with the risk of persistent infection with HBV.29, 30 The polymorphisms in HLA‐DP were also reported to be associated with clearance of HBV by LAM treatment.31 Such genetic variations might be related to the partial virological response to ETV administration observed in case C, although these variations have not been investigated.
We detected one amino acid mutation, V191I, in the RT region of HBV isolated at the time point of flare‐up in case D. In our in vitro assessment with this mutation‐introduced clone, we could not detect any effect on susceptibility to ETV. rtA181T mutation‐associated ADV resistance has been reported.28, 32, 33, 34 This mutation is often associated with a stop codon at the overlapping surface gene, and the sW172stop mutation impairs HBsAg secretion by generating a truncated protein and also causes the susceptibility to ADV to deteriorate. The rtV191I mutation is also associated with a stop codon of the surface gene (sW182stop), but its influence on ETV resistance was not confirmed. Instead, we found that this mutation had an effect on replication efficiency. The introduction of this mutation into the prototype clone enhanced replication 1.94‐fold. This enhanced replication might be associated with HBV DNA‐titer flare‐up in this case. This mutation is also detected in ADV‐resistant cases,35, 36 although in vitro analysis revealed that this mutation is not associated with ADV resistance.
In this study, to evaluate the resistance to NAs, we used replication‐competent HBV molecular clones. The full genome sequence of infected HBV in each patient was determined and exploited to make molecular clones. Using this strategy, we found that the contribution of mutations to ETV resistance was clone dependent. The introduction of similar mutations at positions 180 and 204 in the RT region into the A1 and B1 clones resulted in different resistance levels. Therefore, the use of patient‐derived HBV clones is important to determine the correct resistance level of infected HBV in patients.
In conclusion, using the HBV replication model with patient‐derived sequences, we identified ETV‐resistant mutations. The system used in this study is useful for assessing replication efficiency, susceptibility to anti‐HBV reagents, and responsible resistance mutations. Such evaluation can provide worthwhile information when choosing the appropriate treatment strategy for treatment‐failure cases of CHB.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1022/suppinfo.
Supporting Information
Acknowledgment
We thank Ms. Nao Sugiyama for laboratory work and support.
Supported by grants for Research Programs on Hepatitis (16fk0310503j0005 and 16fk0310509j0105) from the Japan Agency for Medical Research and Development. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
The nucleotide sequences in this paper will appear in the DDBJ/EMBL/GenBank with accession numbers LC170474‐LC170477.
Potential conflict of interest: K.K. received honoraria for lectures from Bristol‐Meyers Squibb. The other authors have nothing to disclose.
REFERENCES
- 1. Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology 2007;132:1574–1585. [DOI] [PubMed] [Google Scholar]
- 2. Michailidis E, Kirby KA, Hachiya A, Yoo W, Hong SP, Kim SO, et al. Antiviral therapies: focus on hepatitis B reverse transcriptase. Int J Biochem Cell Biol 2012;44:1060–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet 1981;2:1129–1133. [DOI] [PubMed] [Google Scholar]
- 4. Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age‐specific HBsAg seroprevalence and endemicity. Vaccine 2012;30:2212–2219. [DOI] [PubMed] [Google Scholar]
- 5. Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, et al. Present and future therapies of hepatitis B: from discovery to cure. Hepatology 2015;62:1893–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suzuki F, Arase Y, Suzuki Y, Akuta N, Sezaki H, Seko Y, et al. Long‐term efficacy of interferon therapy in patients with chronic hepatitis B virus infection in Japan. J Gastroenterol 2012;47:814–822. [DOI] [PubMed] [Google Scholar]
- 7. Colonno RJ, Rose R, Baldick CJ, Levine S, Pokornowski K, Yu CF, et al. Entecavir resistance is rare in nucleoside naive patients with hepatitis B. Hepatology 2006;44:1656–1665. [DOI] [PubMed] [Google Scholar]
- 8. Baldick CJ, Tenney DJ, Mazzucco CE, Eggers BJ, Rose RE, Pokornowski KA, et al. Comprehensive evaluation of hepatitis B virus reverse transcriptase substitutions associated with entecavir resistance. Hepatology 2008;47:1473–1482. [DOI] [PubMed] [Google Scholar]
- 9. Tenney DJ, Rose RE, Baldick CJ, Pokornowski KA, Eggers BJ, Fang J, et al. Long‐term monitoring shows hepatitis B virus resistance to entecavir in nucleoside‐naive patients is rare through 5 years of therapy. Hepatology 2009;49:1503–1514. [DOI] [PubMed] [Google Scholar]
- 10. Yuen MF, Seto WK, Fung J, Wong DK, Yuen JC, Lai CL. Three years of continuous entecavir therapy in treatment‐naive chronic hepatitis B patients: VIRAL suppression, viral resistance, and clinical safety. Am J Gastroenterol 2011;106:1264–1271. [DOI] [PubMed] [Google Scholar]
- 11. Zoutendijk R, Reijnders JG, Brown A, Zoulim F, Mutimer D, Deterding K, et al. Entecavir treatment for chronic hepatitis B: adaptation is not needed for the majority of naive patients with a partial virological response. Hepatology 2011;54:443–451. [DOI] [PubMed] [Google Scholar]
- 12. Tenney DJ, Levine SM, Rose RE, Walsh AW, Weinheimer SP, Discotto L, et al. Clinical emergence of entecavir‐resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob Agents Chemother 2004;48:3498–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayashi S, Murakami S, Omagari K, Matsui T, Iio E, Isogawa M, et al. Characterization of novel entecavir resistance mutations. J Hepatol 2015;63:546–553. [DOI] [PubMed] [Google Scholar]
- 14. Yamada N, Shigefuku R, Sugiyama R, Kobayashi M, Ikeda H, Takahashi H, et al. Acute hepatitis B of genotype H resulting in persistent infection. World J Gastroenterol 2014;20:3044–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gunther S, Li BC, Miska S, Kruger DH, Meisel H, Will H. A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J Virol 1995;69:5437–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abe A, Inoue K, Tanaka T, Kato J, Kajiyama N, Kawaguchi R, et al. Quantitation of hepatitis B virus genomic DNA by real‐time detection PCR. J Clin Microbiol 1999;37:2899–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chotiyaputta W, Peterson C, Ditah FA, Goodwin D, Lok AS. Persistence and adherence to nucleos(t)ide analogue treatment for chronic hepatitis B. J Hepatol 2011;54:12–18. [DOI] [PubMed] [Google Scholar]
- 18. Sugiyama M, Tanaka Y, Kato T, Orito E, Ito K, Acharya SK, et al. Influence of hepatitis B virus genotypes on the intra‐ and extracellular expression of viral DNA and antigens. Hepatology 2006;44:915–924. [DOI] [PubMed] [Google Scholar]
- 19. Delaney WE, Yang H, Westland CE, Das K, Arnold E, Gibbs CS, et al. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J Virol 2003;77:11833–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allen MI, Deslauriers M, Andrews CW, Tipples GA, Walters KA, Tyrrell DL, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998;27:1670–1677. [DOI] [PubMed] [Google Scholar]
- 21. Brunelle MN, Lucifora J, Neyts J, Villet S, Holy A, Trepo C, et al. In vitro activity of 2,4‐diamino‐6‐[2‐(phosphonomethoxy) ethoxy]‐pyrimidine against multidrug‐resistant hepatitis B virus mutants. Antimicrob Agents Chemother 2007;51:2240–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang TT, Gish RG, Hadziyannis SJ, Cianciara J, Rizzetto M, Schiff ER, et al. A dose‐ranging study of the efficacy and tolerability of entecavir in Lamivudine‐refractory chronic hepatitis B patients. Gastroenterology 2005;129:1198–1209. [DOI] [PubMed] [Google Scholar]
- 23. Sherman M, Yurdaydin C, Sollano J, Silva M, Liaw YF, Cianciara J, et al. Entecavir for treatment of lamivudine‐refractory, HBeAg‐positive chronic hepatitis B. Gastroenterology 2006;130:2039–2049. [DOI] [PubMed] [Google Scholar]
- 24. Angus P, Vaughan R, Xiong S, Yang H, Delaney W, Gibbs C, et al. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology 2003;125:292–297. [DOI] [PubMed] [Google Scholar]
- 25. Kwon SY, Park YK, Ahn SH, Cho ES, Choe WH, Lee CH, et al. Identification and characterization of clevudine‐resistant mutants of hepatitis B virus isolated from chronic hepatitis B patients. J Virol 2010;84:4494–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhong Y, Lv J, Li J, Xing X, Zhu H, Su H, et al. Prevalence, virology and antiviral drugs susceptibility of hepatitis B virus rtN238H polymerase mutation from 1865 Chinese patients with chronic hepatitis B. Antiviral Res 2012;93:185–190. [DOI] [PubMed] [Google Scholar]
- 27. Ahn SH, Kim DH, Lee AR, Kim BK, Park YK, Park ES, Ahn SH, et al. Substitution at rt269 in hepatitis B virus polymerase is a compensatory mutation associated with multi‐drug resistance. PLoS One 2015;10:e0136728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahn SH, Park YK, Park ES, Kim JH, Kim DH, Lim KH, et al. The impact of the hepatitis B virus polymerase rtA181T mutation on replication and drug resistance is potentially affected by overlapping changes in surface gene. J Virol 2014;88:6805–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, et al. A genome‐wide association study identifies variants in the HLA‐DP locus associated with chronic hepatitis B in Asians. Nat Genet 2009;41:591–595. [DOI] [PubMed] [Google Scholar]
- 30. Mbarek H, Ochi H, Urabe Y, Kumar V, Kubo M, Hosono N, et al. A genome‐wide association study of chronic hepatitis B identified novel risk locus in a Japanese population. Hum Mol Genet 2011;20:3884–3892. [DOI] [PubMed] [Google Scholar]
- 31. Hosaka T, Suzuki F, Kobayashi M, Fukushima T, Kawamura Y, Sezaki H, et al. HLA‐DP genes polymorphisms associate with hepatitis B surface antigen kinetics and seroclearance during nucleot(s)ide analogue therapy. Liver Int 2015;35:1290–1302. [DOI] [PubMed] [Google Scholar]
- 32. Warner N, Locarnini S. The antiviral drug selected hepatitis B virus rtA181T/sW172* mutant has a dominant negative secretion defect and alters the typical profile of viral rebound. Hepatology 2008;48:88–98. [DOI] [PubMed] [Google Scholar]
- 33. Villet S, Pichoud C, Billioud G, Barraud L, Durantel S, Trepo C, et al. Impact of hepatitis B virus rtA181V/T mutants on hepatitis B treatment failure. J Hepatol 2008;48:747–755. [DOI] [PubMed] [Google Scholar]
- 34. Dai J, Chen EQ, Bai L, Gong DY, Zhou QL, Cheng X, Huang FJ, et al. Biological characteristics of the rtA181T/sW172* mutant strain of Hepatitis B virus in animal model. Virol J 2012;9:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang H, Westland CE, Delaney WE, Heathcote EJ, Ho V, Fry J, et al. Resistance surveillance in chronic hepatitis B patients treated with adefovir dipivoxil for up to 60 weeks. Hepatology 2002;36:464–473. [DOI] [PubMed] [Google Scholar]
- 36. Sheldon J, Rodes B, Zoulim F, Bartholomeusz A, Soriano V. Mutations affecting the replication capacity of the hepatitis B virus. J Viral Hepat 2006;13:427–434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1022/suppinfo.
Supporting Information