

Abstract
Lipotoxicity associated with insulin resistance is central to nonalcoholic steatohepatitis (NASH) pathogenesis. To date, only weight loss fully reverses NASH pathology, but mixed peroxisome proliferator–activated receptor‐alpha/delta (PPAR‐α/δ) agonists show some efficacy. Seladelpar (MBX‐8025), a selective PPAR‐δ agonist, improves atherogenic dyslipidemia. We therefore used this agent to test whether selective PPAR‐δ activation can reverse hepatic lipotoxicity and NASH in an obese, dyslipidemic, and diabetic mouse model. From weaning, female Alms1 mutant (foz/foz) mice and wild‐type littermates were fed an atherogenic diet for 16 weeks; groups (n = 8‐12) were then randomized to receive MBX‐8025 (10 mg/kg) or vehicle (1% methylcellulose) by gavage for 8 weeks. Despite minimally altering body weight, MBX‐8025 normalized hyperglycemia, hyperinsulinemia, and glucose disposal in foz/foz mice. Serum alanine aminotransferase ranged 300‐600 U/L in vehicle‐treated foz/foz mice; MBX‐8025 reduced alanine aminotransferase by 50%. In addition, MBX‐8025 normalized serum lipids and hepatic levels of free cholesterol and other lipotoxic lipids that were increased in vehicle‐treated foz/foz versus wild‐type mice. This abolished hepatocyte ballooning and apoptosis, substantially reduced steatosis and liver inflammation, and improved liver fibrosis. In vehicle‐treated foz/foz mice, the mean nonalcoholic fatty liver disease activity score was 6.9, indicating NASH; MBX‐8025 reversed NASH in all foz/foz mice (nonalcoholic fatty liver disease activity score 3.13). Conclusion: Seladelpar improves insulin sensitivity and reverses dyslipidemia and hepatic storage of lipotoxic lipids to improve NASH pathology in atherogenic diet–fed obese diabetic mice. Selective PPAR‐δ agonists act independently of weight reduction, but counter lipotoxicity related to insulin resistance, thereby providing a novel therapy for NASH. (Hepatology Communications 2017;1:663–674)
Abbreviations
- ALT
alanine transaminase
- CLS
crown‐like structure
- FA
fatty acid
- FC
free cholesterol
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NL
neutral lipid
- PPAR
peroxisome proliferator–activated receptor
- T2D
type 2 diabetes
- Wt
wild‐type
Nonalcoholic fatty liver disease (NAFLD) results from overnutrition, particularly in those with a family history of or established type 2 diabetes (T2D) and in people with metabolic syndrome and cardiovascular risk.1, 2 Thus, in genetically predisposed individuals, constant energy surplus and bodily insulin resistance increase hepatic lipid partitioning to result in steatosis (total lipids 5% of liver weight).1, 2 When hepatic lipid is comprised principally of triglyceride, liver pathology is confined to simple steatosis; but the presence of other specific lipid fractions (free fatty acids [FAs], diacylglycerides, free cholesterol [FC]) can injure hepatocytes in a process termed lipotoxicity.3, 4, 5, 6 Pathological responses to hepatic lipotoxicity include a mixed cellular inflammatory infiltrate focused on injured hepatocytes, hepatocyte ballooning and cell death, and liver fibrosis. These are the hallmarks of steatohepatitis, referred to as nonalcoholic steatohepatitis (NASH) when alcohol intake is <70 g/week in women and <140 g/week in men.2
Ten percent to 25% of NAFLD patients develop NASH. It is more likely among those with a more severe metabolic phenotype (established T2D, metabolic syndrome) and/or a family history of T2D or NAFLD/cirrhosis.1, 7 Childhood obesity, typically genetic and environmental in origin, also predicts later onset of NASH.2, 4, 8 The distinction between NASH and simple steatosis is clinically important because NASH is often associated with fibrosis, the component of NAFLD pathology predictive of progression to cirrhosis.9 Although NASH is a highly prevalent, serious disease, there is no currently approved drug therapy.1, 2, 6, 7 While reversing consequences of metabolic disorder has been attempted to target NASH, the only highly effective strategies have been bariatric surgery and lifestyle changes; reducing body weight 10% reverses NASH pathology, including liver fibrosis, but is difficult to achieve.10, 11, 12, 13, 14, 15 Pharmacologically, pathways targeted to improve NASH pathology are aimed at “movers” of metabolic obesity: glucagon‐like peptide 1 receptor agonists, adiponectin receptor agonists, glucokinase activity modulators, and selective peroxisome proliferator–activated receptor (PPAR) agonists.14, 15, 16, 17
PPARs are a nuclear receptor superfamily of ligand‐inducible transcription factors18 that induce gene expression involved in energy homeostasis, as well as inflammation pathways. PPAR‐α plays a pivotal role in hepatic lipid and lipoprotein metabolism, particularly in male rodents. Targeting this receptor is used to treat mixed dyslipidemia and hypertriglyceridemia16, 18 but is not effective against NASH. PPAR‐γ is the master regulator of adipogenesis and lipogenic pathways in adipocytes; the thiazolidinedione PPAR‐γ agonists improve peripheral and hepatic insulin sensitivity16, 18, 19 and have some efficacy in NASH14 but without reversal of NASH pathology. PPAR‐δ is expressed by multiple tissues including liver, skeletal muscle, intestine, and macrophages.20 PPAR‐δ agonists, such as GW501516, may exert beneficial effects on hyperglycemia, hyperinsulinemia, and NAFLD.20, 21 Thus, GW501516 decreased serum triglycerides, increased high‐density lipoprotein, and reduced hepatic fat.22, 23 Recent attention has turned to GFT‐505 (elafibranor), a dual‐activator of PPAR‐α/δ with a bias toward PPAR‐α (50% effective concentration = 45 nM versus 175 nM for PPAR‐δ).15, 24 The relative risk and benefits of targeting PPAR‐α versus PPAR‐δ have not been elucidated in long‐term studies, and GFT‐505 failed to activate PPAR‐δ in peripheral tissues in obese insulin‐resistant patients.24
Seladelpar (also known as MBX‐8025) is a novel, potent, and selective PPAR‐δ agonist (50% effect concentration human PPAR‐δ = 2 nM, PPAR‐α = 1,600 nM) that demonstrates favorable effects on insulin resistance, diabetes, and atherogenic dyslipidemia.23, 25, 26, 27 In an 8‐week placebo‐controlled, active comparator study of obese patients with mixed dyslipidemia, MBX‐8025 reduced low‐density lipoprotein cholesterol and triglyceride, accompanied by reduced concentrations of small, dense low‐density lipoprotein particles.25, 26 In the present study, we addressed the effects of MBX‐8025 treatment on insulin resistance, glucose tolerance, hepatic lipid fractions, and liver pathology in wild‐type (Wt) and appetite‐defective (Alms1 mutant foz/foz) mice fed an atherogenic diet. foz/foz mice develop obesity complicated by T2D and resemble humans with NASH by exhibiting high blood pressure, atherogenic dyslipidemia, hypoadiponectinemia, and steatohepatitis complicated by pericellular fibrosis.3, 28, 29, 30
Materials and Methods
ANIMAL PROCEDURES
Experimental procedures were approved by the Australian National University Animal Ethics Committee (pA2012/20). From weaning (week 4), Alms1 mutant (foz/foz) NOD.B10 mice or Wt littermates (female mice in both groups) were fed an atherogenic diet (23% fat, 0.2% cholesterol and 45% simple carbohydrate; 4.78 kcal/g digestible energy; SpecialtyFeeds, Glen Forrest, Australia) ad libitum for 16 weeks, after which groups were randomized (n = 8‐12 mice/group) to once‐a‐day oral administration (by gavage) for 8 weeks of MBX‐8025 (10 mg/kg in 1% methylcellulose) or vehicle (controls). Animals were housed under 12‐hour light/dark cycle and constant temperature of 22°C and received maximal humane care.
EXPERIMENTAL PROCEDURES
Seladelpar (MBX‐8025; Supporting Fig. S1) was synthesized and characterized as described.31 We measured body weight each week to calculate amounts of MBX‐8025 or vehicle for gavage. One week before sacrifice (week 27), we performed an intraperitoneal glucose tolerance test after a 4‐hour fast; following glucose injection (2 g/kg lean body mass), blood glucose was measured at 0, 15, 30, 60, and 120 minutes by glucometer (Accu‐Chek Advantage; Roche Diagnostics, Mannheim, Germany). One week later, mice were fasted for 4 hours and anesthetized (100 mg/kg ketamine, 16 mg/kg xylazine), and blood and liver were harvested. Circulating blood insulin was assessed using an enzyme‐linked immunosorbent assay kit (EMD Millipore, Darmstadt, Germany). The homeostatic model assessment of insulin resistance was calculated as [fasting insulin (ng/mL) × 25 × fasting glucose (mmol/L)]/22.5. Serum alanine aminotransferase (ALT), total triglyceride, and cholesterol were analyzed by ACT Pathology (The Canberra Hospital) using multichannel autoanalyzer.
ASSESSMENT OF HEPATIC LIPIDS AND LIVER HISTOLOGY
Total neutral lipid (NL) content was assessed by staining of homogenized frozen livers (50 mg) with oil red O (3.67 mM), then measuring the absorbance (at 450 nm) of eluted dye. For lipidomic analyses, liver total lipids were extracted by a modified Folch extraction protocol. Total FAs were analyzed as methyl esters by gas chromatography on a 60‐m HP‐INNOWax column programmed from 80°C to 220°C (Agilent Technologies, Santa Clara, CA) in a GC‐17A (Shimadzu Scientific, Kyoto, Japan). The NL fraction was obtained from the total lipid extract by solid‐phase separation on an Agilent Bond Elut Si cartridge (500 mg), eluting with 1% acetic acid in chloroform. The eluate was evaporated to dryness and the residue redissolved in 1% acetic acid in hexane for high‐pressure liquid chromatography on a LiChrospher DIOL column (Alltech, Deerfield, IL) with detection by an LT‐II light‐scattering detector (Shimadzu). Hepatic macrosteatosis, necroinflammation, ballooning scores, and their sum (overall NAFLD activity score) were determined by an expert (blinded) liver pathologist on hematoxylin and eosin–stained liver sections, according to the system devised for human NAFLD.30 To quantify liver fibrosis, sirius red–stained sections were analyzed by collagen densitometry.3, 29 Hepatocyte apoptosis and proliferation were visualized by immunostaining of caspase‐cleaved cytokeratin‐18 fragment (M30) and proliferating cell nuclear antigen (PCNA), respectively. Epidermal growth factor–like module–containing mucin‐like hormone receptor‐like 1 (F4/80) was used to detect hepatic macrophage crown‐like structures (CLSs).28
QUANTIFICATION OF HEPATIC GENE EXPRESSION
Liver homogenates were prepared in TRI reagent, total RNA was isolated,29 and complementary DNA was obtained from 1 μg total RNA.13 mRNA levels were quantified by real‐time polymerase chain‐reaction using SYBR green primers (Sigma‐Aldrich, St. Louis, MO), normalizing data to the geometric mean of β‐actin and Gapdh. Liver proteins were analyzed as described.13
STATISTICAL ANALYSES
Parametric data are presented as mean ± SEM. Most experimental procedures were performed in duplicate, with a minimum of eight samples/group. Data significance was assessed by one‐way or two‐way analysis of variance, followed by Bonferroni's post hoc analysis. Group differences were considered significant when P < 0.05. Other than for assessment of liver pathology, the investigators involved in this research were not blinded during the experimental procedures.
Results
MBX‐8025 REDUCED GLUCOSE INTOLERANCE AND HYPERINSULINEMIA INDEPENDENT OF WEIGHT CHANGE IN ATHEROGENIC DIET–FED foz/foz AND Wt MICE
In atherogenic diet‐fed Wt mice, administration of MBX‐8025 reduced body weight by ∼18% (P < 0.05; Fig. 1A). In contrast, MBX‐8025 produced minimal effect on body weight in atherogenic diet–fed foz/foz mice (Fig. 1A). As expected,3, 28 these animals developed severe hyperglycemia, hyperinsulinemia, and whole‐body insulin resistance after 16 weeks (P < 0.05; Fig. 1B); MBX‐8025 strikingly improved these indices (P < 0.05). After intraperitoneal glucose injection, blood glucose reached ∼32 mmol/L in vehicle‐treated versus ∼14 mmol/L in MBX‐8025‐treated foz/foz mice (P < 0.05; Fig. 1C); the area under the blood glucose disappearance curve was correspondingly lower in MBX‐8025‐treated foz/foz mice (P < 0.05; Fig. 1C). MBX‐8025 produced a proportionally similar effect on glucose handling in atherogenic diet–fed Wt mice (P < 0.05; Fig. 1C).
Figure 1.
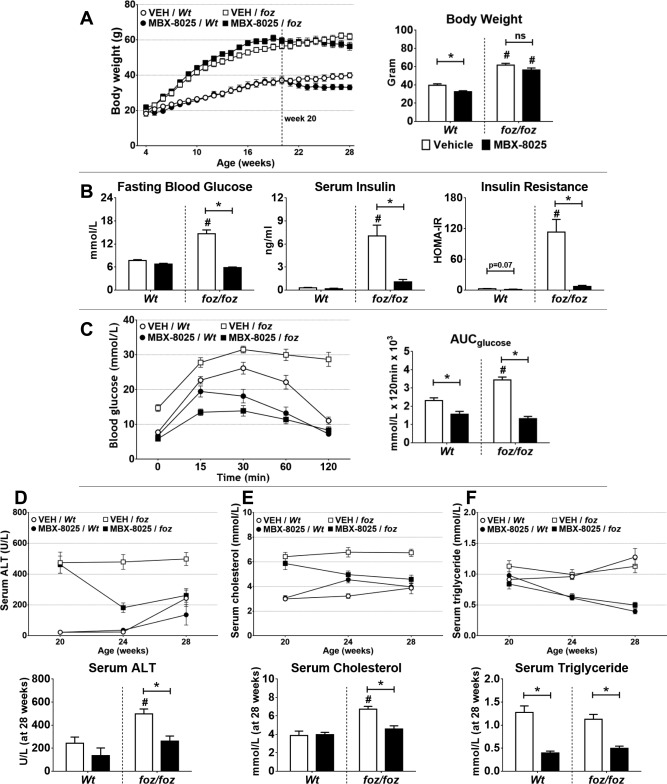
Effects of MBX‐8025 on body weight, hyperglycemia, hyperinsulinemia, serum ALT, cholesterol, and triglyceride in atherogenic diet–fed foz/foz and Wt mice. (A) MBX‐8025 caused significant weight reduction in Wt, but not foz/foz, mice. Compared to Wt, atherogenic diet–fed foz/foz mice developed (B) hyperglycemia, hyperinsulinemia, and insulin resistance; MBX‐8025 improved, or fully corrected, all of these values in these mice. (C) By intraperitoneal glucose tolerance testing, MBX‐8025 improved glucose tolerance in both foz/foz and Wt mice. At 20 weeks (D) serum ALT was markedly increased in foz/foz compared to Wt mice; MBX‐8025 significantly lowered serum ALT in foz/foz mice, with similar (but not significant) effects in Wt mice. (E) Serum total cholesterol was higher in atherogenic diet–fed foz/foz than in Wt mice; after 8 weeks of treatment with MBX‐8025, values returned to those of Wt. (F) Serum triglyceride concentrations were similar in vehicle‐treated foz/foz and Wt mice and reduced by MBX‐8025 in both genotypes. Data are mean ± SEM (n = 8‐12 per group). * P < 0.05 compared with genotype‐matched control (MBX‐8025 effect), # P < 0.05 compared with treatment‐matched control (genotype effect). Abbreviations: ALT, alanine transaminase; AUC, area under the curve; HOMA‐IR, homeostatic model assessment of insulin resistance; ns, not significant; VEH, vehicle.
After 16 weeks of atherogenic dietary intake, serum ALT was higher in foz/foz than Wt mice (P < 0.05; Fig. 1D). MBX‐8025 lowered serum ALT levels in foz/foz mice (P < 0.05; Fig. 1D) and similarly (but not significantly) in Wt mice (Fig. 1D). Serum cholesterol was markedly increased in foz/foz compared to Wt mice (P < 0.05; Fig. 1E); similar to the effects seen in patients with hypercholesterolemia,25, 26 MBX‐8025 normalized serum cholesterol. Serum triglyceride concentrations were similar between foz/foz and Wt mice at 20 weeks of age, and again similar to patients with elevated triglycerides.25, 26 MBX‐8025 decreased triglycerides in both genotypes (P < 0.05; Fig. 1F).
MBX‐8025 NORMALIZED HEPATIC LIPID PARTITIONING AND REVERSED NASH PATHOLOGY IN ATHEROGENIC DIET–FED foz/foz MICE
Hepatic total NLs (oil red O incorporation) were higher in atherogenic diet–fed foz/foz versus Wt mice (P < 0.05); MBX‐8025 profoundly reduced total NLs in foz/foz as well as Wt mice (P < 0.05; Fig. 2A). This was reflected by chromatographic analyses of hepatic triglyceride, diacylglycerol, and total FA content (P < 0.05; Fig. 2B‐D). Hepatic FC increases in human NASH,5 and as reported,3, 32 hepatic FC and cholesteryl esters were strikingly increased in atherogenic diet–fed foz/foz mice compared to Wt (P < 0.05; Fig. 2E); MBX‐8025 reduced both FC and cholesteryl ester content. In addition, we measured total amounts of individual hepatic FA levels such as palmitic, palmitoleic, oleic, stearic, linoleic, and docosahexaenoic acid levels; MBX‐8025 consistently reduced hepatic levels of these molecules in foz/foz mice (P < 0.05; Supporting Fig. S2). As indicated by hematoxylin and eosin staining, macrovesicular steatosis was mild in Wt and severe in foz/foz mouse livers (Fig. 2F and Table 1). MBX‐8025 appeared to reduce macrovesicular steatosis in both lines (Fig. 2F), nearly halving the steatosis score in foz/foz mouse livers (Table 1). MBX‐8025 abolished hepatocyte ballooning (P < 0.05; Fig. 2F and Table 1). The overall NAFLD activity score was ∼6.9 in vehicle‐treated foz/foz mice, indicating “definite NASH” in all foz/foz mice (Table 1). MBX‐8025 decreased the NAFLD activity score by ∼50% and reversed NASH in 8/8 mice. Thus, all PPAR‐δ agonist–treated, atherogenic diet–fed foz/foz mice showed simple steatosis (Fig. 2F and Table 1).
Figure 2.
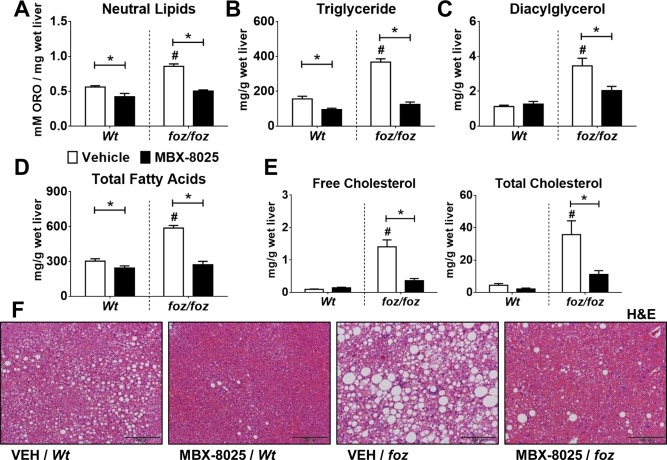
Effects of MBX‐8025 on hepatic lipid stores and steatosis in atherogenic diet–fed foz/foz and Wt mice. (A) As determined by elution of oil red O from liver lysates, hepatic total lipids were increased in vehicle‐treated foz/foz compared to Wt mice; in both genotypes, MBX‐8025 lowered these values substantially. Hepatic (B) triglyceride, (C) diacylglycerol, and (D) total FAs were all increased in vehicle‐treated foz/foz versus Wt mice; MBX‐8025 lowered these levels significantly. (E) Vehicle‐treated foz/foz mice showed increased hepatic FC and cholesteryl ester contents compared to Wt counterparts; MBX‐8025 treatment reduced the amount of both lipid fractions. (F) Liver histology in vehicle‐treated foz/foz mouse livers displayed macrovesicular steatosis, hepatocyte ballooning, and inflammatory cell recruitment; MBX‐8025 reversed NASH histology in foz/foz mice, shown in representative hematoxylin & eosin–stained liver sections (Table 1 summarizes group data). Data are mean ± SEM (n = 8‐12 per group). * P < 0.05 compared with genotype‐matched control (MBX‐8025 effect), # P < 0.05 compared with treatment‐matched control (genotype effect). Abbreviations: H&E, hematoxylin and eosin; ORO, oil red O; VEH, vehicle.
Table 1.
Effects of MBX‐8025 on Liver Weight and Histology in foz/foz and Wt Mice Fed an Atherogenic Diet
| n | Weight (g) | Steatosis Score | Inflammation Score | Ballooning Score | NAS | ||
|---|---|---|---|---|---|---|---|
| foz/foz | Vehicle | 8 | 6.77 ± 0.57b | 3.00 ± 0a, b | 1.86 ± 0.14b | 2 ± 0a, b | 6.86 ± 0.14a, b |
| MBX‐8025 | 8 | 6.41 ± 0.40b | 1.88 ± 0.13b | 1.25 ± 0.31 | 0 ± 0 | 3.13 ± 0.40b | |
| Wt | Vehicle | 12 | 1.64 ± 0.07 | 0.46 ± 0.23 | 0.38 ± 0.19 | 0.15 ± 0.11 | 1 ± 0.48 |
| MBX‐8025 | 10 | 2.53 ± 0.11 | 0.50 ± 0.22 | 0.50 ± 0.23 | 0 ± 0 | 1 ± 0.30 | |
Data are mean ± SEM; scores were generated by assessment of blinded liver slides by an expert pathologist (M.M.Y.).30
P < 0.05 (versus genotype‐matched, treatment comparison).
P < 0.05 (versus treatment‐matched, genotype comparison).
Abbreviation: NAS, NAFLD activity score.
MBX‐8025 REDUCED HEPATOCELLULAR APOPTOSIS AND LIVER INFLAMMATION IN ATHEROGENIC DIET–FED foz/foz MICE
foz/foz mice developed hepatomegaly; consistent with the known effects of PPAR‐δ agonists on liver, MBX‐8025 did not affect this increase (Table 1). M30–positive hepatocytes, reflecting apoptosis,33 were more abundant in foz/foz mouse livers than Wt (P < 0.05; Fig. 3A); MBX‐8025 normalized apoptosis levels to those of Wt controls (P < 0.05). Liver injury provokes an adaptive hepatocellular proliferative response. Thus, the number of PCNA–positive hepatocyte nuclei was higher in foz/foz mouse livers compared to Wt; MBX‐8025 suppressed this to Wt levels (P < 0.05; Fig. 3B). While NASH pathology is of clinical significance in NAFLD, the histological factor that correlates best with progression to cirrhosis is liver fibrosis.9 As determined by the sirius red–positive (collagen‐stained) area, liver fibrosis was barely evident after 24 weeks of atherogenic dietary intake in Wt mice (Fig. 3C), but by this time foz/foz mice exhibited substantial fibrosis (Fig. 3C), as noted28, 29; MBX‐8025 significantly reduced sirius red–positive areas in foz/foz mice (P < 0.05; Fig. 3C). This change was supported by a trend (not significant) for a decrease in hepatic alpha‐smooth muscle actin mRNA levels in MBX‐8025‐treated foz/foz mice and reductions in Col1‐α1, Ctgf, and Timp1 transcripts (P < 0.05; Supporting Fig. S3).
Figure 3.
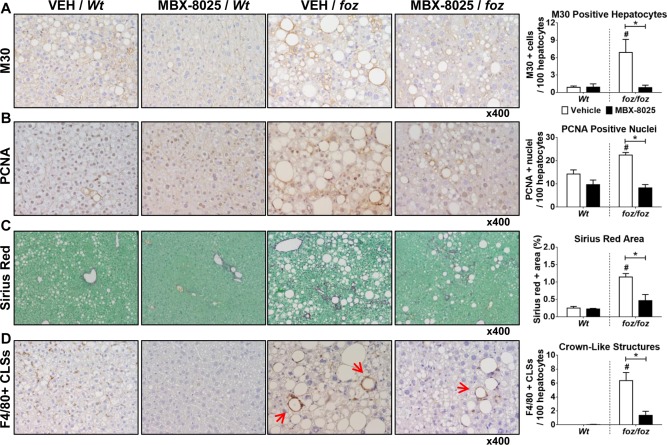
Effects of MBX‐8025 on hepatocyte apoptosis, proliferation, fibrosis, and liver inflammation in atherogenic diet–fed foz/foz and Wt mice. (A) As quantified by cytokeratin‐18 fragmentation (M30) immunohistochemistry, vehicle‐treated foz/foz mouse livers showed more hepatocytes undergoing apoptosis than Wt; MBX‐8025 normalized this to Wt levels. (B) The number of PCNA–positive liver cells, reflecting hepatocytes in the cell cycle, was higher in vehicle‐treated foz/foz mice than Wt; and MBX‐8025 lowered this value after 8 weeks of treatment. (C) Densitometry of collagen+ areas by sirius red staining showed that Wt mice developed very mild fibrosis after 24 weeks of atherogenic dietary feeding, so MBX‐8025 could not affect liver fibrosis in these mice. foz/foz mice developed significant fibrosis, and MBX‐8025 treatment reduced collagen density in these mice. (D) Macrophage CLSs around injured hepatocytes (arrows) were abundant in livers of vehicle‐treated foz/foz, but not Wt, mice; MBX‐8025 reduced the number of hepatic CLSs in these mice. Data are mean ± SEM (n = 8‐12 per group). * P < 0.05 compared with genotype‐matched control (MBX‐8025 effect). # P < 0.05 compared with treatment‐matched control (genotype effect). Abbreviations: CLS, crown‐like structure; PCNA, proliferating cell nuclear antigen; VEH, vehicle.
The number of contiguous cells positive for F4/80 and forming CLSs indicates a focal proinflammatory response to injured hepatocytes. There were no CLSs in Wt mouse livers (P < 0.05; Fig. 3D), but the number was very high in livers of vehicle‐treated foz/foz mice with NASH. MBX‐8025 diminished the hepatic CLS number in foz/foz mouse livers compared to vehicle‐treated controls (P < 0.05; Fig. 3D). Despite MBX‐8025 treatment resulting in a substantial reduction in steatosis and ballooning scores and an impressive decrease in CLSs around injured hepatocytes, there was only a modest decline in the inflammation score (Table 1). We considered it possible that macrophages accumulating during tissue repair could confound interpretation of the inflammatory score in this context. We therefore sought other evidence for anti‐inflammatory effects of MBX‐8025 by measuring gene expression for chemokines and chemokine receptors pertinent to hepatic inflammation. Mcp1 mRNA was higher in foz/foz than Wt mouse liver (P < 0.05; Supporting Fig. S4A); MBX‐8025 suppressed hepatic expression of this gene but did not affect expression of its receptor Ccr2 (Supporting Fig. S4B). Transcripts for Ccl5 and Cx3Cr1 were also higher in livers of foz/foz versus Wt mice (P < 0.05); MBX‐8025 reduced Cx3Cr1 mRNA in foz/foz and Ccl5 mRNA in Wt mouse livers (P < 0.05; Supporting Fig. S4C,D). Cd11b and Cd11c mRNA levels were also increased in foz/foz mouse livers (versus Wt) (Supporting Fig. S4E,F); MBX‐8025 lowered Cd11c, but not Cd11b, mRNA.
TRANSCRIPT EXPRESSION OF GENES INVOLVED IN HEPATIC LIPID METABOLISM WAS INCREASED BY MBX‐8025 IN ATHEROGENIC DIET–FED foz/foz MICE
We considered it likely that lower hepatic content of lipotoxic and proinflammatory lipid species such as FC, diacylglycerol, and FAs could influence NAFLD pathology in atherogenic diet–fed foz/foz mice. We therefore studied genes whose expression is regulated by PPAR‐α, PPAR‐δ, or both, in some cases at the protein as well as mRNA level. Pnpla2 promotes mitochondrial function, so as to increase FA oxidation in the cell.34 In the present study, MBX‐8025 increased hepatic expression of Pnpla2 gene (mRNA and protein) in both genotypes (P < 0.05; Fig. 4A). The genes Cidea and Plin2 regulate lipid droplet formation and lipid storage35; MBX‐8025 treatment increased hepatic Cidea and Plin2 mRNA levels in both genotypes (P < 0.05; Fig. 4B). mRNA levels of the FA oxidation factors Acadl and Cpt2 were also increased (P < 0.05) in livers of MBX‐8025‐treated versus vehicle‐treated foz/foz mice (Fig. 4C; no treatment effect on Cpt1 mRNA [data not shown]). Peroxisomal Acox1 and microsomal Cyp4a10 mRNA expression was induced by MBX‐8025 in both genotypes (P < 0.05; Fig. 4C), with no differences between vehicle‐treated foz/foz and Wt mice (Fig. 4C). In addition, hepatic mRNA levels of Ucp2 and Pdk4, which are involved in mitochondrial metabolism, were substantially increased by MBX‐8025 in both genotypes (P < 0.05; Fig. 4D). Hepatic mRNA expression of Abca1 was increased in MBX‐8025‐treated Wt mice but not influenced by MBX‐8025 in foz/foz mice (Fig. 4E), and any change in Angptl4 in both lines did not reach statistical significance (Fig. 4E).
Figure 4.
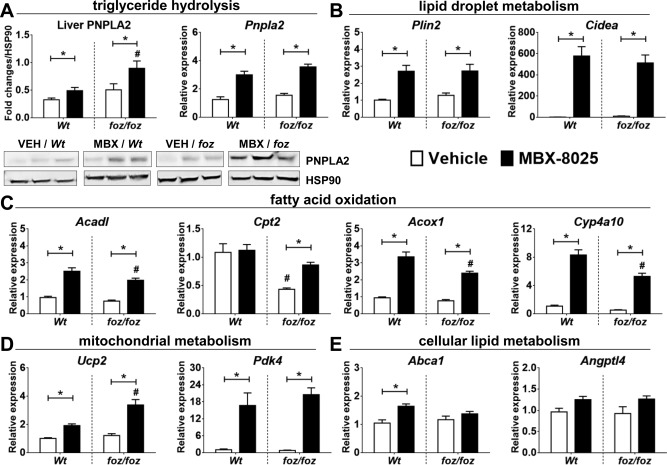
Effects of MBX‐8025 on hepatocellular lipid metabolism in atherogenic diet–fed foz/foz and Wt mice. We compared markers for which expression is controlled by PPAR‐α, PPAR‐δ, or both and that are relevant to healthy hepatocellular function in terms of storage and turnover of lipid molecules. Data are mean ± SEM (n = 8‐12 per group). * P < 0.05 compared with genotype‐matched control (MBX‐8025 effect), # P < 0.05 compared with treatment‐matched control (genotype effect).
Discussion
In individuals with genetic predisposition, especially those with a family history of T2D,1, 7 excess calorie intake is complicated by insulin resistance. The resultant hyperinsulinemia up‐regulates hepatic FA uptake and synthesis in the liver.36 In addition, hyperinsulinemia programs the hepatic uptake of FC and down‐regulates its biotransformation into bile acids.3, 5 Such dysregulation of hepatic cholesterol turnover was first documented in the appetite‐dysregulated Alms1 mutant (foz/foz) mice used here,3, 28 and as later confirmed in human NASH,5 it results in accumulation of FC. FC and possibly other lipid and bile acid–related molecules have been shown to cause lipotoxicity in primary hepatocytes.32, 37 Physical activity and weight loss improve insulin sensitivity and so correct the hepatic accumulation of lipotoxic lipids.3, 10, 29 However, attempts to combat hepatic lipid partitioning and promote insulin sensitization pharmacologically have been only partially effective, in the case of PPAR‐γ and farnesoid X receptor agonists,11, 12, 13 or ineffective (metformin and PPAR‐α agonists).11
The most important finding of the present study is that a selective PPAR‐δ agonist, seladelpar (MBX‐8025), exerts powerful effects on hepatic lipid partitioning in the face of morbid obesity, complicated by diabetes and metabolic syndrome in mice. MBX‐8025 returns hepatic levels of candidate lipotoxic lipids (FC, diacylglycerol, and FAs) to normal, and this alteration in liver lipid profile was achieved with minimal or no reduction in body weight. Further, restitution of physiological hepatic lipid levels was associated with improvement of steatosis, resolution of hepatocyte ballooning and apoptosis, and reduction in macrophage recruitment and Kupffer cell activation to form CLSs. This culminated in the reversal of NASH pathology, with significant improvement in liver fibrosis.
A key question is how does a selective PPAR‐δ agonist act independently of weight loss to reduce circulating and liver tissue cholesterol and triglyceride levels in obese diabetic mice? We attribute these changes to an MBX‐8025‐regulated increase in insulin sensitivity and enhanced energy use, changes that have been demonstrated by PPAR‐δ stimulation with tool agents that have not progressed to clinical studies.38, 39 Thus, MBX‐8025 lowered fasting blood glucose, decreased serum insulin concentrations and whole‐body insulin resistance (homeostatic model assessment of insulin resistance), and normalized glucose disposition after an intraperitoneal load. MBX‐8025 has been shown to improve atherogenic dyslipidemia, particularly lowering serum low‐density lipoprotein cholesterol in humans and rodents23, 25, 26, 27; the present study extends this by demonstrating that enhanced insulin sensitivity is part of the mechanism. Others have shown that mixed PPAR‐β/δ agonists increase energy use by skeletal muscle38, 40; for example, GW501516, a prototypic PPAR‐δ agonist, reduces glucose use by muscle cells while increasing mitochondrial uncoupling and lipid use.41 Clinical trials of this agent were terminated because of safety concerns.42, 43 By contrast, MBX‐8025, a potent PPAR‐δ agonist with high binding affinity to its receptor, caused no side effects during this study, nor have they been a feature of clinical trials to date.25, 26, 27
In the present study, MBX‐8025 reduced the amount of hepatic NLs in atherogenic diet–fed Wt mice, which is a simple dietary model of steatosis. The atherogenic diet–fed foz/foz mouse is a multifactorial model of NASH in which both a dietary component and genetically determined drive to overeat exert interactive effects. MBX‐8025 reduced liver total NLs in this more severe model compared to levels found in Wt mice. To understand whether this reduction was attributable to a direct effect of MBX‐8025 on the liver, we assayed PPAR‐δ target molecules concerned with intracellular lipid metabolism and lipid droplet formation and function. The increase in PNPLA2 protein expression produced by MBX‐8025 is consistent with the known role of PPAR‐δ in FA oxidation and provides a rational explanation for the reduction of cellular triglyceride levels and decreased steatosis; PNPLA2 protein regulates lipolysis so as to lower triglyceride levels and provide substrate for FA oxidation.34 In addition, MBX‐8025 increased mRNA expression of lipid droplet factors such as Plin2 and Cidea. Cidea expression correlates with “browning” of adipocytes and fatty hepatocytes, a phenotypic switch in which cells become metabolically more active.35 MBX‐8025 also increased expression of FA oxidation genes Acadl, Cpt2 (mitochondrial), Acox1 (peroxisomal), and Cyp4a10 (microsomal) in foz/foz mouse livers. We interpret the profile of these changes as indicating that MBX‐8025 treatment could trigger hepatocytes to become metabolically more active. This proposal was supported by increased transcripts of the mitochondrial factors Ucp2 and Pdk4. In light of these data, it seems possible that MBX‐8025 acts on hepatocytes to reduce steatosis by activating pathways for enhancing lipid droplet storage, possibly also to promote cellular lipid metabolism and oxidation.
The predominant inflammatory cell types in livers with NASH are F4/80–positive Kupffer cells and infiltrated macrophages. Such activated macrophages coalesce around injured hepatocytes in a crown‐like structure pattern. MBX‐8025 treatment substantially lowered the number of CLSs in liver sections of foz/foz mice. It is salient to note that PPAR‐δ is expressed in macrophages, where it promotes their polarization toward an anti‐inflammatory phenotype.21 As expected,3, 28, 29 the NASH‐related liver inflammation in foz/foz mice fed an atherogenic diet was associated with increased expression of macrophage factors such as Mcp1, Cx3Cr1, and Cd11c mRNA; and MBX‐8025 decreased their expression in foz/foz mouse livers. Inflammation‐driven fibrosis is an important aspect of NASH clinically,3, 27, 28 and MBX‐8025 showed promising effects on reducing fibrosis, as shown by collagen densitometry. However, further study is required to establish whether MBX‐8025 arrests fibrosis deposition by extinguishing NASH pathology or whether it has effects on matrix‐depositing cells, growth factors, and macrophages that can promote liver fibrosis resolution.
In summary, we provide novel evidence that the selective PPAR‐δ agonist MBX‐8025 reduces hepatic lipid partitioning, near totally correcting levels of lipotoxic lipids in mice with metabolic obesity associated with insulin resistance and diabetes. The beneficial effects of MBX‐8025 result from effects on extrahepatic metabolism that contribute to improved insulin sensitivity and glucose handling and possibly from direct effects on hepatic lipid storage and oxidation. By showing a close correlation between removal of FC and/or other lipotoxic lipids and resolution of hepatocyte injury and NASH pathology, the present data reinforce a mechanistic view that NASH is a form of liver lipotoxicity.3, 4, 5 The translational potential of the present findings is considerable as today there is still no drug licensed for use in NASH and none in clinical trial that reverses NASH in the majority of cases. Should the reversal of NASH documented in this murine model, which replicates the predisposing metabolic milieu and liver pathology of human NASH, be reproduced in patients, seladelpar (MBX‐8025) and other selective PPAR‐δ agonists could provide a mechanism‐based, novel approach to treat NASH.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1072/suppinfo.
Supporting Information Figures.
Potential conflict of interest: Nothing to report.
Supported by CymaBay Therapeutics, Inc., and the Australia National Health and Medical Research Council (1044288 and 102818).
REFERENCES
- 1. Loomba R, Abraham M, Unalp A, Wilson L, Lavine J, Doo E, et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology 2012;56:943‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Farrell GC, Wong VW‐S, Chitturi S. NAFLD in Asia—as common and important as in the West. Nat Rev Gastroenterol Hepatol 2013;10:307‐318. [DOI] [PubMed] [Google Scholar]
- 3. Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, et al. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011;141:1393‐1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 2012;142:711‐725. [DOI] [PubMed] [Google Scholar]
- 5. Min H‐K, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab 2012;15:665‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neuschwander‐Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774‐788. [DOI] [PubMed] [Google Scholar]
- 7. Chitturi S, Abeygunasekera S, Farrell GC, Holmes‐Walker J, Hui JM, Fung C, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002;35:373‐379. [DOI] [PubMed] [Google Scholar]
- 8. Ayonrinde OT, Olynyk JK, Marsh JA, Beilin LJ, Mori TA, Oddy WH, et al. Childhood adiposity trajectories and risk of nonalcoholic fatty liver disease in adolescents. J Gastroenterol Hepatol 2015;30:163‐171. [DOI] [PubMed] [Google Scholar]
- 9. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vilar‐Gomez E, Martinez‐Perez Y, Calzadilla‐Bertot L, Torres‐Gonzalez A, Gra‐Oramas B, Gonzalez‐Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015;149:367‐378. [DOI] [PubMed] [Google Scholar]
- 11. Musso G, Cassader M, Gambino R. Non‐alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov 2016;15:249‐274. [DOI] [PubMed] [Google Scholar]
- 12. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haczeyni F, Poekes L, Wang H, Mridha AR, Barn V, Geoffrey Haigh W, et al. Obeticholic acid improves adipose morphometry and inflammation and reduces steatosis in dietary but not metabolic obesity in mice. Obesity 2017;25:155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ratziu V, Goodman Z, Sanyal A. Current efforts and trends in the treatment of NASH. J Hepatol 2015;62:S65‐S75. [DOI] [PubMed] [Google Scholar]
- 15. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator−activated receptor‐α and ‐δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016;150:1147‐1159. [DOI] [PubMed] [Google Scholar]
- 16. Souza‐Mello V. Peroxisome proliferator–activated receptors as targets to treat non‐alcoholic fatty liver disease. World J Hepatol 2015;7:1012‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larrain S, Rinella ME. A myriad of pathways to NASH. Clin Liver Dis 2012;16:525‐548. [DOI] [PubMed] [Google Scholar]
- 18. Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature 2000;405:421‐424. [DOI] [PubMed] [Google Scholar]
- 19. Phielix E, Szendroedi J, Roden M. The role of metformin and thiazolidinediones in the regulation of hepatic glucose metabolism and its clinical impact. Trends Pharmacol Sci 2011;32:607‐616. [DOI] [PubMed] [Google Scholar]
- 20. Ehrenborg E, Skogsberg J. Peroxisome proliferator–activated receptor delta and cardiovascular disease. Atherosclerosis 2013;231:95‐106. [DOI] [PubMed] [Google Scholar]
- 21. Odegaard JI, Ricardo‐Gonzalez RR, Eagle AR, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity‐induced insulin resistance. Cell Metab 2008;7:496‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olson EJ, Pearce GL, Jones NP, Sprecher DL. Lipid effects of peroxisome proliferator–activated receptor‐Δ agonist GW501516 in subjects with low high‐density lipoprotein cholesterol characteristics of metabolic syndrome. Arterioscler Thromb Vasc Biol 2012;32:2289‐2294. [DOI] [PubMed] [Google Scholar]
- 23. Ooi EM, Watts GF, Sprecher DL, Chan DC, Barrett PHR. Mechanism of action of a peroxisome proliferator–activated receptor (PPAR)‐δ agonist on lipoprotein metabolism in dyslipidemic subjects with central obesity. J Clin Endocrinol Metab 2011;96:E1568‐E1576. [DOI] [PubMed] [Google Scholar]
- 24. Cariou B, Staels B. GFT505 for the treatment of nonalcoholic steatohepatitis and type 2 diabetes. Expert Opin Investig Drugs 2014;23:1441‐1448. [DOI] [PubMed] [Google Scholar]
- 25. Choi Y‐J, Roberts BK, Wang X, Geaney JC, Naim S, Wojnoonski K, et al. Effects of the PPAR‐δ agonist MBX‐8025 on atherogenic dyslipidemia. Atherosclerosis 2012;220:470‐476. [DOI] [PubMed] [Google Scholar]
- 26. Bays HE, Schwartz S, Littlejohn T III, Kerzner B, Krauss RM, Karpf DB, et al. MBX‐8025, a novel peroxisome proliferator receptor‐δ agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J Clin Endocrinol Metab 2011;96:2889‐2897. [DOI] [PubMed] [Google Scholar]
- 27. Risérus U, Sprecher D, Johnson T, Olson E, Hirschberg S, Liu A, et al. Activation of peroxisome proliferator–activated receptor (PPAR) δ promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008;57:332‐339. [DOI] [PubMed] [Google Scholar]
- 28. Van Rooyen DM, Gan LT, Yeh MM, Haigh WG, Larter CZ, Ioannou G, et al. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J Hepatol 2013;59:144‐152. [DOI] [PubMed] [Google Scholar]
- 29. Haczeyni F, Barn V, Mridha AR, Yeh MM, Estevez E, Febbraio MA, et al. Exercise improves adipose function and inflammation and ameliorates fatty liver disease in obese diabetic mice. Obesity 2015;23:1845‐1855. [DOI] [PubMed] [Google Scholar]
- 30. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 31. Zhang R, Wang A, DeAngelis A, Pelton P, Xu J, Zhu P, et al. Discovery of para‐alkylthiophenoxyacetic acids as a novel series of potent and selective PPARδ agonists. Bioorg Med Chem Lett 2007;17:3855‐3859. [DOI] [PubMed] [Google Scholar]
- 32. Gan LT, Van Rooyen DM, Koina ME, McCuskey RS, Teoh NC, Farrell GC. Hepatocyte free cholesterol lipotoxicity results from JNK1‐mediated mitochondrial injury and is HMGB1 and TLR4‐dependent. J Hepatol 2014;61:1376‐1384. [DOI] [PubMed] [Google Scholar]
- 33. Kramer G, Erdal H, Mertens HJ, Nap M, Mauermann J, Steiner G, et al. Differentiation between cell death modes using measurements of different soluble forms of extracellular cytokeratin 18. Cancer Res 2004;64:1751‐1756. [DOI] [PubMed] [Google Scholar]
- 34. Tang T, Abbott MJ, Ahmadian M, Lopes AB, Wang Y, Sul HS. Desnutrin/ATGL activates PPARδ to promote mitochondrial function for insulin secretion in islet β cells. Cell Metab 2013;18:883‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosell M, Kaforou M, Frontini A, Okolo A, Chan Y‐W, Nikolopoulou E, et al. Brown and white adipose tissues: intrinsic differences in gene expression and response to cold exposure in mice. Am J Physiol Endocrinol Metab 2014;306:E945‐E964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002;109:1125‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, et al. Mechanisms of lysophosphatidylcholine‐induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol 2012;302:G77‐G84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, et al. Activation of peroxisome proliferator–activated receptor δ induces fatty acid β‐oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci 2003;100:15924‐15929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krämer DK, Al‐Khalili L, Guigas B, Leng Y, Garcia‐Roves PM, Krook A. Role of AMP kinase and PPARδ in the regulation of lipid and glucose metabolism in human skeletal muscle. J Biol Chem 2007;282:19313‐19320. [DOI] [PubMed] [Google Scholar]
- 40. Coll T, Álvarez‐Guardia D, Barroso E, Gómez‐Foix AM, Palomer X, Laguna JC, et al. Activation of peroxisome proliferator–activated receptor‐δ by GW501516 prevents fatty acid–induced nuclear factor‐κB activation and insulin resistance in skeletal muscle cells. Endocrinology 2010;151:1560‐1569. [DOI] [PubMed] [Google Scholar]
- 41. Brunmair B, Staniek K, Dörig J, Szöcs Z, Stadlbauer K, Marian V, et al. Activation of PPAR‐δ in isolated rat skeletal muscle switches fuel preference from glucose to fatty acids. Diabetologia 2006;49:2713‐2722. [DOI] [PubMed] [Google Scholar]
- 42. Sahebkar A, Chew GT, Watts GF. New peroxisome proliferator–activated receptor agonists: potential treatments for atherogenic dyslipidemia and non‐alcoholic fatty liver disease. Expert Opin Pharmacother 2014;15:493‐503. [DOI] [PubMed] [Google Scholar]
- 43. Billin AN. PPAR‐β/δ agonists for type 2 diabetes and dyslipidemia: an adopted orphan still looking for a home. Expert Opin Investig Drugs 2008;17:1465‐1471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1072/suppinfo.
Supporting Information Figures.