

Abstract
Nonalcoholic fatty liver disease (NAFLD) plays a crucial role in type 2 diabetes and hepatocellular carcinoma. The major underlying pathogenesis is hepatic insulin resistance. The aim of the present study was to characterize patients with NAFLD with paradoxically normal hepatic insulin sensitivity relative to patients with NAFLD with hepatic insulin resistance. We recruited 26 patients with NAFLD and divided them into three groups ranked by the level of hepatic insulin sensitivity (HIS; high‐HIS, mid‐HIS, low‐HIS), as assessed by the hyperinsulinemic‐euglycemic clamp studies using stable isotope. Hepatic insulin sensitivity of the high‐HIS group was identical to that of the non‐NAFLD lean control (clamped percent suppression of endogenous glucose production, 91.1% ± 5.2% versus 91.0% ± 8.5%, respectively) and was significantly higher than that of the low‐HIS group (66.6% ± 7.5%; P < 0.01). Adiposity (subcutaneous, visceral, intrahepatic, and muscular lipid content), hepatic histopathology, and expression levels of various genes by using liver biopsies, muscle, and adipose tissue insulin sensitivity, plasma metabolites by metabolomics analysis, putative biomarkers, and lifestyles were assessed and compared between the high‐HIS and low‐HIS groups. Among these, adipose tissue insulin sensitivity assessed by clamped percent suppression of free fatty acid, serum high molecular weight adiponectin, and plasma tricarboxylic acid cycle metabolites, such as citric acid and cis‐aconitic acid, were significantly higher in the high‐HIS group compared to the low‐HIS group. In contrast, there were no differences in adiposity, including intrahepatic lipid content assessed by proton magnetic resonance spectroscopy (28.3% ± 16.1% versus 20.4% ± 9.9%, respectively), hepatic histopathology, other putative biomarkers, and lifestyles. Conclusion: High levels of adipose tissue insulin sensitivity, serum high molecular weight adiponectin, and plasma tricarboxylic acid cycle metabolites are unique characteristics that define patients with hepatic insulin‐sensitive NAFLD regardless of intrahepatic lipid content. (Hepatology Communications 2017;1:634–647)
Abbreviations
- CYP2C19
cytochrome P450, family 2, subfamily C, polypeptide 19
- HCC
hepatocellular carcinoma
- HIS
hepatic insulin sensitivity
- 1H‐MRS
proton magnetic resonance spectroscopy
- HMW
high molecular weight
- mRNA
messenger RNA
- MSEA
metabolite set enrichment analysis
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PC
pyruvate carboxylase
- RT‐PCR
real‐time polymerase chain reaction
- T2DM
type 2 diabetes mellitus
- TCA
tricarboxylic acid
Introduction
With the current high rate of obesity among the general population, we are confronted with an epidemic of nonalcoholic fatty liver disease (NAFLD) that has an estimated incidence of 25% worldwide.1 NAFLD is also associated with type 2 diabetes mellitus (T2DM) and hepatocellular carcinoma (HCC), which are serious problems for both individuals and societies. The major underlying pathophysiological mechanism linking NAFLD to T2DM or HCC is hepatic insulin resistance.2, 3
NAFLD‐associated hepatic insulin resistance has been investigated extensively in rodent models of NAFLD and in patients with NAFLD.3, 4 To date, several potential molecular mechanisms have been recognized, together with the identification of several potential therapeutic targets for NAFLD‐associated hepatic insulin resistance, such as activation of diacylglycerol‐mediated protein kinase C epsilon,5, 6 endoplasmic reticulum stress,7 inflammation,8 and oxidative stress.9 However, in clinical practice, NAFLD‐associated hepatic insulin resistance remains unresolved in humans. Interestingly, recent studies have reported the existence of hepatic steatosis dissociated from insulin resistance in humans,10, 11, 12 but its clinical or cellular features remain poorly understood. It is important, therefore, to determine the clinical characteristics that can help distinguish hepatic insulin‐sensitive NAFLD from hepatic insulin‐resistant NAFLD.
We recently encountered a cohort of patients with NAFLD and with normal hepatic insulin sensitivity. The goal of the present study was to define the characteristics of these individuals compared to patients with NAFLD with hepatic insulin resistance through a comprehensive analysis of various histopathologic and biochemical parameters. We recruited 26 Japanese patients with NAFLD (mean body mass index, 30 kg/m2) and assessed various background parameters, including adiposity and lipid distribution, tissue‐specific insulin sensitivity (liver, muscle, and adipose tissue) assessed by hyperinsulinemic‐euglycemic clamp studies using stable isotope, hepatic histopathology and gene expression using liver biopsies, plasma metabolomics and putative biomarkers, and lifestyle patterns. The unique characteristics may allow the identification of diagnostic markers and/or therapeutic targets to preserve hepatic insulin sensitivity and prevent the development of T2DM and HCC in patients with NAFLD.
Patients and Methods
STUDY DESIGN AND POPULATION
The study subjects were recruited between November 2014 and September 2015 at Toho University Hospital, Japan. For the patients with NAFLD, we set the following criteria for inclusion in the study: 1) clinical suspicion of NAFLD based on ultrasonography and a serum alanine aminotransferase level of >30 IU/L; 2) age between 20 and 70 years; and 3) hemoglobin A1c <9%. We also used the following exclusion criteria: 1) liver disease other than NAFLD (e.g., hepatitis B or C, autoimmune hepatitis, drug‐induced hepatitis, and alcoholic hepatitis); 2) any acute or chronic disease other than NAFLD, T2DM, hypertension, or dyslipidemia; 3) type 1 or secondary diabetes mellitus. Based on these criteria, 26 patients with NAFLD were analyzed in the present study. NAFLD was defined as an intrahepatic lipid content of >5% as determined by proton magnetic resonance spectroscopy (1H‐MRS). We also recruited 5 non‐NAFLD subjects for the control. Of the 26 patients with NAFLD, 16 had T2DM, 8 of whom were being treated with glucose‐lowering agents. After providing a signed consent form, each patient was advised to switch all glucose‐lowering medications to α‐glucosidase inhibitors and/or rapid‐acting insulin secretagogues to avoid the effects of such medications on hepatic steatosis and insulin sensitivity. Thus, 1 patient was switched to α‐glucosidase inhibitors while the remaining 7 were switched to a combination of α‐glucosidase inhibitors and rapid‐acting insulin secretagogues. Patients continued dietary and exercise therapy and other medications, and a comprehensive set of clinical and laboratory measures were obtained at 3 months after the consent. All measurements were performed after an overnight fast.
This study was approved by the medical ethics committee of Toho and Juntendo universities and was conducted according to the Declaration of Helsinki and current legal regulations in Japan. Written informed consent was obtained from all patients. This study was registered on the University Hospital Medical Information Network Clinical Trials Registry (UMIN000017735), a nonprofit organization in Japan that meets the requirements of the International Committee of Medical Journal Editors.
HYPERINSULINEMIC‐EUGLYCEMIC CLAMP STUDIES
Hyperinsulinemic‐euglycemic clamp studies were performed using an artificial endocrine pancreas (STG55; Nikkiso, Tokyo, Japan) as described.13, 14 An intravenous catheter was placed in the forearm for tracer infusion; another catheter was placed in the contralateral hand vein for blood withdrawal. The hand was kept warm by a heating device for arterial blood sampling. Over a 3‐hour baseline period, 6,6‐[2H2] glucose (99% atom percent enrichment; Cambridge Isotope Laboratories, Inc., Andover, MA) was infused intravenously by a priming dose (200 × fasting blood glucose [mg/dL] / 100 mg/m2 body surface area), followed by continuous infusion of 2 mg/m2 body surface area/minute. After the basal equilibration period, the hyperinsulinemic‐euglycemic clamp was started with a primed‐constant infusion of insulin (40 mU/m2/minute; U‐100 Humulin R; Eli Lilly, Indianapolis, IN). Although a dose of 10 mU/m2/minute is usually used to evaluate hepatic insulin sensitivity while avoiding complete suppression of hepatic glucose production specifically in normal individuals,15, 16 20 or 40 mU/m2/minute is sometimes used to evaluate hepatic insulin sensitivity.17, 18, 19 We chose 40 mU/m2/minute because we focused on 26 patients with NAFLD, including 14 patients with T2DM and NAFLD in the main analysis. Eventually, 40 mU/m2/minute did not exceed the threshold of maximum suppression of glucose production. To maintain a plasma glucose concentration at approximately 95 mg/dL, a variable infusion of 20% glucose containing 2.5% 6,6‐[2H2]‐glucose was administered.20 Blood samples were obtained at –180, –30, –15, 0, 15, 30, 60, 120, 160, 170, and 180 minutes. The atom percent enrichment of glucosem+2 in plasma was measured by high‐performance liquid chromatography with an LTQ Orbitrap XL mass spectrometer (Thermo Scientific, Fremont, CA). Glucosem+2 enrichment was determined from the m/z ratio 332.2:330.2. Basal and clamped endogenous glucose production and the clamped glucose disposal rate were calculated using a steady‐state equation as described.21 Clamped percent suppression of serum free fatty acid by insulin, which represents insulin sensitivity of adipose tissue, was calculated as (serum free fatty acid concentration at baseline – mean free fatty acid concentration at time 160‐180 minutes)/free fatty acid concentration at baseline.
1H‐MRS
Intrahepatic lipid and intramyocellular lipid content were measured by 1H‐MRS after an overnight fast as described.22 Details are provided in the Supporting Methods.
LABORATORY TESTS
Measurements were performed at the central laboratory of the hospital or were outsourced to a private laboratory (SRL Laboratory, Tokyo, Japan). Details of the methods used for the measurements are provided in the Supporting Methods.
MEASUREMENT OF PLASMA METABOLITES
Metabolome analysis was conducted with the Dual Scan package of Human Metabolome Technologies Inc. (Tsuruoka, Japan) using capillary electrophoresis time‐of‐flight mass spectrometry and liquid chromatography time‐of‐flight mass spectrometry, based on the methods described.23, 24 Details of the methods are provided in the Supporting Methods.
METABOLITE SET ENRICHMENT ANALYSIS
To identify significant metabolic pathways, metabolite set enrichment analysis (MSEA) was performed as described.25 First, Welch's t test was applied to the entire 356 detected metabolites between hepatic insulin‐sensitive and insulin‐resistant patients. Significantly different metabolites (P < 0.05) were selected for MSEA. For technical reasons, MSEA could not be applied to fatty acids and acylcarnitine. In order to control the false discovery rate associated with multiple testing, the Benjamin and Hochberg method was applied to adjust the q value.
ASSESSMENT OF BODY COMPOSITION
Whole body fat and muscle mass were measured by dual‐energy x‐ray absorptiometry. The whole body skeletal muscle mass index was calculated by whole body skeletal muscle mass (kg) / body height (m)2. The amounts of subcutaneous and visceral fat were computed by measuring the area at the umbilical level on abdominal computed tomography and Synapse Vincent software (ver 4.3.0001; Fujifilm Medical, Japan).
LIFESTYLE ASSESSMENT
Validated questionnaires were used to assess well‐being (Well‐Being Questionnaire 12 and Beck Depression Inventory II), sleep quantity and quality (Morning–Evening Questionnaire and Pittsburgh Sleep Quality Index), and food intake (Brief Self‐Administered Diet‐History Questionnaire). Physical activity and caloric consumption were assessed by the Lifecorder GS (Suzuken Co., Aichi, Japan).26 Total daily energy expenditure and physical activity were corrected by body weight. Patients were asked to keep their physical activity and food intake during the study at levels similar to their daily routines.
LIVER BIOPSY
An ultrasound‐guided liver needle biopsy was obtained from patients with suspected nonalcoholic steatohepatitis (NASH). Liver needle biopsies were performed at unit V based on the Couinaud classification, avoiding vessels under sonography with a 16‐gauge liver biopsy needle (Core IITM semiautomatic biopsy instrument; InterV Clinical Products, Dartmouth, MA). One punch (∼10 mg) was flash frozen in liquid nitrogen for gene expression analysis, and another punch was used for routine histopathologic examination. A liver biopsy was not obtained from non‐NAFLD subjects.
HISTOPATHOLOGIC ASSESSMENT
The liver biopsy specimen was fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin along with azan for evaluation of the NAFLD activity score and fibrosis, respectively. Liver biopsy specimens were evaluated by an experienced pathologist blinded to the identity of subjects and clinical information. The NAFLD activity score and fibrosis were evaluated using standard histologic criteria.27 Details of the scoring are described in the Supporting Methods.
REAL‐TIME POLYMERASE CHAIN REACTION
Total RNA was extracted from ∼10 mg of flash‐frozen liver biopsy using the RNeasy mini kit (Qiagen, Tokyo). RNA was reverse‐transcribed into complementary DNA using the QuantiTect Reverse Transcription kit (Qiagen, Hilden, Germany). The abundance of transcripts was assessed by real‐time polymerase chain reaction (RT‐PCR) on an Applied Biosystems 7500 Fast Real‐Time PCR System (Thermo Fisher Diagnostics, Tokyo, Japan) with a Fast SYBR Green Master Mix (Thermo Fisher Diagnostics). The expression level of each gene of interest was normalized for the efficiency of amplification with β‐actin messenger RNA (mRNA) as the invariant control as determined by a standard curve.28
MICROARRAY AND GENE ONTOLOGY ANALYSES
Microarray analysis was performed using the obtained RNA from liver biopsies. Briefly, robust multichip array normalization was performed using the Affymetrix Expression console. Statistical analyses were performed using R software (http://www.R‐project.org). Differences between the two groups were computed using the significance analysis of microarrays t test; genes with more than 1.5‐fold differences in expression between the groups on average (P < 0.05) were selected as differently expressed genes. A complete set of microarray data was deposited at the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) repository under accession number GSE92746. A heat map with hierarchical clustering was drawn with differentially expressed genes between the groups. A distance matrix among the specimens in the heat map was calculated using Spearman rank correlation. Hierarchical clustering analyses were performed using MeV_4_8_1 software (J. Craig Venter Institute, La Jolla, CA).29, 30 Heatmap gene expression values were normalized by the Z‐scaling method using the gene filter library with R. Gene ontology analysis was performed on LSKB software (World Fusion Inc., Tokyo, Japan).29 LSKB uses Fisher's exact test to estimate multiplicity between gene ontology functional classes of gene sets (http://www.geneontology.org/) and genes that are differentially expressed on microarray analysis.
STATISTICAL ANALYSIS
All normally distributed variables were expressed as mean ± SD, while variables with skewed distribution were expressed as median (quartile 1, quartile 3), unless otherwise indicated. Comparisons between groups were assessed by Welch's t test for normally distributed continuous variables, Wilcoxon rank sum test for variables with skewed distribution or scored variables, and Pearson's chi‐squared test or Fisher's exact test for categorical variables. Simple linear regression analysis was performed to assess the association between two variables. P < 0.05 was considered significant. All analyses were performed using SAS 9.3 (SAS Institute Inc., Cary, NC).
Results
CLINICAL CHARACTERISTICS OF STUDY SUBJECTS
The study subjects were 26 patients with NAFLD and intrahepatic lipid contents of >5% and 5 non‐NAFLD lean control subjects (Table 1). All patients were middle‐aged obese men and women, with an intrahepatic lipid content of 20.7% ± 13.3% and serum alanine aminotransferase twice the upper normal range (59.3 ± 27.5 IU/L). Three of the patients had NASH, while the others had simple steatosis as assessed by histopathology. The patients also had moderate hyperglycemia and hyperinsulinemia and were whole‐body insulin resistant based on the homeostatic model assessment of insulin resistance index with a large variation. Around half of the patients with NAFLD had T2DM, and half of the patients with T2DM were being treated with oral glucose‐lowering agents. Clinical characteristics of the study subjects are also shown separately for patients without T2DM and with T2DM in Supporting Table S1.
Table 1.
CHARACTERISTICS OF THE STUDY SUBJECTS
| Non‐NAFLD (n = 5) | NAFLD (n = 26) | |
|---|---|---|
| Age (years) | 35.4 ± 9.1 | 45.0 ± 11.1 |
| Sex (males/females), n (%) | 4 (80)/1 (20) | 18 (69)/8 (31) |
| Body height (cm) | 168 ± 7.7 | 168 ± 10.6 |
| Body weight (kg) | 64.5 ± 11.5 | 85 ± 17.4* |
| Body mass index (kg/m2) | 23.1 ± 3.2 | 30.1 ± 5.3* |
| Intrahepatic lipid content (%) | 1.8 ± 1.9 | 20.7 ± 13.3† |
| Alanine aminotransferase (IU/L) | 23.4 ± 8.0 | 59.3 ± 27.5† |
| NASH, n (%) | 0 (0.0) | 3 (11.5) |
| HbA1c (%) | 6.0 ± 0.9 | 6.6 ± 1.1 |
| Fasting plasma glucose (mg/dL) | 119.7 ± 28.9 | 127.0 ± 31.8 |
| Fasting plasma insulin (μU/mL) | 6.7 ± 1.4 | 18.1 ± 10.7† |
| HOMA‐IR (mg/dL) × (μU/mL) | 2.0 ± 0.6 | 5.7 ± 3.5† |
| Diabetes/no diabetes, n (%) | 2 (40.0)/3 (60.0) | 14 (53.8)/12 (46.2) |
| Glucose‐lowering medications‡ (+/–), n (%) | 1 (20.0)/4 (80.0) | 7 (26.9)/19 (73.1) |
Data are mean ± SD or number of subjects.
* P < 0.01, † P < 0.001, by Welch's t test for continuous variables and Fisher's exact test for categorical variables. All other unlabeled comparisons were not significantly different.
‡Glucose‐lowering medications were limited to α‐glucosidase inhibitor and/or rapid‐acting insulin secretagogues to minimize the effects on hepatic steatosis and insulin sensitivity.
Abbreviations: HbA1c, hemoglobin A1c; HOMA‐IR, homeostatic model assessment of insulin resistance index.
PATIENTS WITH HEPATIC INSULIN‐SENSITIVE NAFLD ARE IDENTICAL TO NON‐NAFLD SUBJECTS
We plotted hepatic insulin sensitivity with intrahepatic lipid content (Fig. 1). We divided the 26 patients with NAFLD into three groups ranked by the level of hepatic insulin sensitivity (high‐HIS group: 100%‐85.2%, n = 9; mid‐HIS group: 80.3%‐83.9%, n = 8; low‐HIS group: 55.7%‐78.5%, n = 9). The hepatic insulin‐sensitivity values of the high‐HIS group (Fig. 1, blue triangles) were identical to those of the non‐NAFLD subjects (black dots, n = 5). The respective hepatic insulin‐sensitivity values for the two groups, assessed by clamped percent suppression of endogenous glucose production, were 91.1% ± 5.2% and 91.0% ± 8.5%. In contrast, the hepatic insulin sensitivity of the low‐HIS group (66.6% ± 7.5%; Fig. 1, red triangles) was significantly lower than that of the high‐HIS group. To define the characteristics that can distinguish patients with hepatic insulin‐sensitive NAFLD from those with hepatic insulin‐resistant NAFLD, we performed a comprehensive background analysis of patients in the high‐HIS and low‐HIS groups.
Figure 1.
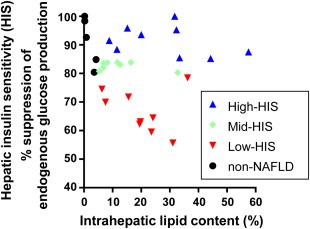
Large variation in percent suppression of endogenous glucose production and intrahepatic lipid content. Patients with NAFLD with an intrahepatic lipid content >5% (n = 26) were divided into three groups ranked by the clamped percent suppression of endogenous glucose production. Upper one third, blue triangles indicate high‐HIS; middle one third, green diamonds indicate mid‐HIS; lower one third, red triangles indicate low‐HIS. Hepatic insulin sensitivity in the upper one third (blue triangles) was identical to that of non‐NAFLD subjects (black dots).
DIFFERENCES IN BACKGROUND BETWEEN HIGH‐HIS AND LOW‐HIS GROUPS
We found no differences in age, sex, body mass index, and prevalence of NASH and T2DM between the high‐HIS and low‐HIS groups (Table 2). The fasting plasma insulin concentration and homeostatic model assessment of insulin resistance index were significantly lower in the high‐HIS compared to the low‐HIS group, while there was no difference in fasting plasma glucose (Table 2). These data indicate that patients with high‐HIS were whole‐body insulin sensitive. Hyperinsulinemic‐euglycemic clamp studies showed adipose tissue insulin sensitivity, assessed by the clamped percent suppression of free fatty acid, was significantly higher in the high‐HIS group (91.4% ± 3.0%) compared to the low‐HIS group (80.0% ± 10.9%; Table 2). Adipose tissue insulin sensitivity was also assessed by fasting plasma free fatty acid × fasting plasma insulin,31, 32, 33 but there was no difference in this index between the groups (Table 2). Among the putative biomarkers, only serum high molecular weight (HMW) adiponectin was significantly higher in the high‐HIS compared to the low‐HIS group (3.4 ± 1.8 μg/mL versus 1.7 ± 0.9 μg/mL, respectively; Table 2). We also assessed 356 plasma metabolites by metabolomics analysis and found only 11 metabolites were significantly different between the two groups (Table 2). Interestingly, three of the 11 metabolites were metabolites of the mitochondrial tricarboxylic acid (TCA) cycle (citric acid, cis‐aconitic acid, and succinic acid). Their concentrations were higher in the high‐HIS compared to the low‐HIS group (Table 2). Pathway analysis using MSEA indicated that the TCA cycle pathway was the only significantly different pathway between the two groups (Table 3). We also performed simple linear regression analysis using data of the 26 patients with NAFLD to assess the contribution of the above significant factors to hepatic insulin sensitivity and found that adipose tissue insulin sensitivity, HMW adiponectin, citric acid, and cis‐aconitic acid were associated with hepatic insulin sensitivity (adjusted R 2 = 0.22, P < 0.05; R 2 = 0.14, P < 0.05; R 2 = 0.38, P < 0.001; and R 2 = 0.17, P < 0.05, respectively). In contrast, other putative blood and urine biomarkers; body composition and lipid distribution, including intrahepatic lipid content (high‐HIS group versus low‐HIS group, 28.3% ± 16.1% versus 20.4% ± 9.9%); and lifestyles were comparable between the two groups (Table 4).
Table 2.
CHARACTERISTICS OF PATIENTS IN THE HIGH‐HIS AND LOW‐HIS GROUPS
| High‐HIS (n = 9) | Low‐HIS (n = 9) | P Value | |
|---|---|---|---|
| Age (years) | 44.6 ± 13.1 | 46.6 ± 12.4 | 0.75 |
| Sex (males/females), n (%) | 4 (44.4)/5 (55.6) | 7 (77.8)/2 (22.2) | 0.33 |
| Body height (cm) | 162.0 ± 7.9 | 170.5 ± 11.3 | 0.09 |
| Body weight (kg) | 81.0 ± 17.4 | 87.5 ± 22.4 | 0.51 |
| Body mass index (kg/m2) | 30.7 ± 5.5 | 29.8 ± 5.5 | 0.73 |
| NASH/non‐NASH, n (%) | 1 (11.1)/8 (88.9) | 2 (25.0)/6 (75.0)a | 0.58 |
| Diabetes/no diabetes, n (%) | 2 (22.2)/7 (77.8) | 6 (66.7)/3 (33.3) | 0.15 |
| Glucose‐lowering medications (+/–), n (%) | 0 (0)/9 (100.0) | 3 (33.3)/6 (66.7) | 0.21 |
| HbA1c (%) | 6.1 ± 1.0 | 7.0 ± 1.5 | 0.18 |
| Fasting plasma glucose (mg/dL) | 106.1 ± 6.8 | 140.6 ± 41.7 | 0.052 |
| Fasting plasma insulin (μU/mL) | 13.9 ± 3.2 | 21.4 ± 8.1 | 0.04 |
| HOMA‐IR (mg/dL) × (μU/mL) | 3.6 ± 0.9 | 7.7 ± 3.8 | 0.02 |
| Hyperinsulinemic‐euglycemic clamp data | |||
| Clamped plasma glucose (mg/dL) | 96.0 ± 2.2 | 96.7 ± 6.6 | 0.79 |
| Clamped plasma insulin (μU/mL) | 80.3 ± 9.1 | 74.2 ± 15.7 | 0.33 |
| Clamped glucose infusion rate (mg/fat‐free BW/minute) | 6.3 ± 1.8 | 4.9 ± 2.7 | 0.22 |
| Basal endogenous glucose production (mg/fat‐free BW/minute) | 2.9 ± 0.3 | 2.9 ± 0.5 | 0.87 |
| Clamped endogenous glucose production (mg/fat‐free BW/minute) | 0.3 ± 0.2 | 1.0 ± 0.2 | < 0.01 |
| Clamped percent suppression of endogenous glucose production (%) | 91.1 ± 5.2 | 66.3 ± 7.5 | < 0.01 |
| Clamped glucose disposal rate (mg/fat‐free BW/minute) | 6.5 ± 1.8 | 5.8 ± 2.6 | 0.52 |
| Clamped percent stimulation of glucose disposal rate (%) | 126.1 ± 53.4 | 113.0 ± 115.7 | 0.76 |
| Basal free fatty acid (μEq/L) | 771.1 ± 288.2 | 606.7 ± 124.3 | 0.15 |
| Clamped free fatty acid (μEq/L) | 73.0 ± 33.3 | 140.0 ± 70.9 | 0.03 |
| Clamped percent suppression of free fatty acid (%) | 91.4 ± 3.0 | 80.0 ± 10.9 | 0.01 |
| Adipose tissue‐IR (μEq/L) × (μU/mL) | 9,611.4 ± 4,452.2 | 8,608.6 ± 4,787.3 | 0.65 |
| HMW adiponectin (μg/mL) | 3.4 ± 1.8 | 1.7 ± 0.9 | 0.03 |
| Metabolomics data (relative area) | |||
| Citric acid | 1.4E‐02 ± 1.6E‐03 | 1.1E‐02 ± 1.8E‐03 | < 0.01 |
| Methionine sulfoxide | 4.7E‐04 ± 8.3E‐05 | 3.5E‐04 ± 9.9E‐05 | 0.01 |
| cis‐Aconitic acid | 8.5E‐04 ± 1.5E‐04 | 6.7E‐04 ± 1.2E‐04 | 0.02 |
| Myristoleic acid | 1.5E‐04 ± 6.8E‐05 | 8.6E‐05 ± 2.9E‐05 | 0.02 |
| Fatty acid (19:1) | 2.0E‐05 ± 5.5E‐06 | 1.5E‐05 ± 2.1E‐06 | 0.03 |
| Stachydrine | 2.3E‐03 ± 1.8E‐03 | 8.3E‐04 ± 5.0E‐04 | 0.03 |
| 2‐Aminoisobutyric acid/2‐aminobutyric acid | 6.7E‐03 ± 8.9E‐04 | 8.7E‐03 ± 2.4E‐03 | 0.04 |
| Pregnenolone sulfate | 3.1E‐05 ± 1.3E‐05 | 2.0E‐05 ± 5.4E‐06 | 0.047 |
| Threonine | 2.7E‐02 ± 2.6E‐03 | 3.0E‐02 ± 4.2E‐03 | 0.047 |
| Cholesterol sulfate | 1.6E‐04 ± 3.8E‐05 | 2.2E‐04 ± 5.9E‐05 | 0.048 |
| Succinic acid | 2.7E‐04 ± 4.1E‐05 | 2.2E‐04 ± 6.2E‐05 | 0.049 |
Data are mean ± SD. P values in bold indicate significant difference. Comparisons by Welch's t test and Fisher's exact test (for categorical data).
Analyzed number of subjects is 8. A total of 356 metabolites were detected by metabolomics analysis, and only 11 metabolites were significantly different between the two groups and shown in this table.
Clamped percent suppression of endogenous glucose production (EGP) = (basal EGP – clamped EGP) / (basal EGP) × 100. Clamped percent stimulation of glucose disposal rate (Rd) = (clamped Rd – basal Rd) / (basal Rd) × 100. Percent suppression of free fatty acids (FFA) = (basal FFA – clamped FFA) / (basal FFA) × 100. Adipose tissue‐IR = (fasting plasma free fatty acid) × (fasting plasma insulin).
Abbreviations: BW, body weight; HbA1c, hemoglobin A1c; HOMA‐IR, homeostatic model assessment of insulin resistance index; IR, insulin resistance.
Table 3.
RESULTS OF METABOLITE SET ENRICHMENT ANALYSIS
| Metabolic Pathway | Total Metabolites | Detected Metabolites | Selected Metabolites | P Value (Fisher's Exact Test)a | Q Value (BH Method) |
|---|---|---|---|---|---|
| Tricarboxylic acid cycle | 13 | 6 | 3 | 0.0024 | 0.0636 |
| Methionine metabolism | 25 | 3 | 1 | 0.1743 | 1.0000 |
| Glutamic acid and glutamine metabolism | 11 | 5 | 1 | 0.2759 | 1.0000 |
| Shikimic acid metabolism | 27 | 5 | 1 | 0.2759 | 1.0000 |
| Tyrosine metabolism | 76 | 5 | 1 | 0.2759 | 1.0000 |
| Glycine, serine and threonine metabolism | 42 | 17 | 1 | 0.6917 | 1.0000 |
| Glycolysis/gluconeogenesis | 11 | 3 | 0 | 1.0000 | 1.0000 |
| Pentose phosphate pathway | 10 | 2 | 0 | 1.0000 | 1.0000 |
| Alanine, aspartic acid, and asparagine metabolism | 11 | 6 | 0 | 1.0000 | 1.0000 |
| Lysine metabolism | 55 | 6 | 0 | 1.0000 | 1.0000 |
| Valine, leucine, and isoleucine metabolism | 22 | 8 | 0 | 1.0000 | 1.0000 |
| Cysteine metabolism | 30 | 11 | 0 | 1.0000 | 1.0000 |
| Histidine metabolism | 39 | 8 | 0 | 1.0000 | 1.0000 |
| Urea cycle | 23 | 7 | 0 | 1.0000 | 1.0000 |
| Proline metabolism | 22 | 5 | 0 | 1.0000 | 1.0000 |
| Polyamine metabolism | 20 | 3 | 0 | 1.0000 | 1.0000 |
| Tryptophan metabolism | 80 | 4 | 0 | 1.0000 | 1.0000 |
| Beta‐alanine metabolism | 31 | 7 | 0 | 1.0000 | 1.0000 |
| Taurine and hypotaurine metabolism | 20 | 6 | 0 | 1.0000 | 1.0000 |
| Creatine metabolism | 11 | 8 | 0 | 1.0000 | 1.0000 |
| Purine metabolism | 87 | 12 | 0 | 1.0000 | 1.0000 |
| Pyrimidine metabolism | 56 | 8 | 0 | 1.0000 | 1.0000 |
| Ribonucleotide metabolism | 18 | 6 | 0 | 1.0000 | 1.0000 |
| Deoxyribonucleotide metabolism | 19 | 0 | 0 | 1.0000 | 1.0000 |
| Conjugated bile acid metabolism | 5 | 1 | 0 | 1.0000 | 1.0000 |
| Nicotinic acid metabolism | 43 | 6 | 0 | 1.0000 | 1.0000 |
All significantly different metabolites found between the high‐HIS and low‐HIS groups by Welch's t test were later analyzed by the metabolite set enrichment analysis, which identified the tricarboxylic acid cycle pathway as the only significant pathway. P values in bold indicate a significant difference.
Abbreviation: BH method, Benjamini and Hochberg method.
Table 4.
PUTATIVE BIOMARKERS, BODY COMPOSITION AND LIPID DISTRIBUTION, AND LIFESTYLES OF THE TWO GROUPS
| High‐HIS (n = 9) | Low‐HIS (n = 9) | P Value | |
|---|---|---|---|
| Putative blood and urine biomarkers | |||
| Glucagon (pg/mL) | 174 ± 26 | 185 ± 54 | 0.60 |
| Cortisol (U/mL) | 15.6 ± 6.7 | 11.7 ± 4.6 | 0.16 |
| Alanine aminotransferase (IU/L) | 74.3 ± 34.0 | 56.6 ± 18.4 | 0.19 |
| High‐sensitive C‐reactive protein (mg/dL) | 0.34 (0.2,0.4) | 0.17 (0.1,0.6) | 0.93 |
| Ferritin (ng/mL) | 229 ± 164 | 361 ± 222 | 0.19 |
| Procollagen III peptide (U/mL) | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.66 |
| Type IV collagen 7s (ng/mL) | 4.7 ± 0.9 | 5.0 ± 1.0 | 0.48 |
| Hyaluronic acid (ng/mL) | 17.0 (4.5,49.0) | 46.0 (20.0,61.0) | 0.18 |
| Cytokeratin‐18 fragment (U/L) | 583 (245,803) | 458 (393,660) | 1.00 |
| Fetuin‐A (μg/mL) | 260 ± 48 | 280 ± 28 | 0.29 |
| Fibroblast growth factor 21 (pg/mL) | 243 (195,348) | 240 (227,410) | 0.86 |
| Urine 8‐OHdG (ng/mL) | 12.7 ± 8.3 | 17.1 ± 8.6 | 0.29 |
| Body composition and lipid distribution | |||
| Whole body skeletal muscle mass index (kg/m2) | 18.6 ± 3.0 | 20.3 ± 2.7 | 0.21 |
| Whole body fat ratio (%) | 35.5 ± 7.5 | 29.5 ± 7.8 | 0.11 |
| Abdominal visceral fat area (cm2) | 182.1 ± 65.7 | 180.4 ± 56.9 | 0.95 |
| Abdominal subcutaneous fat area (cm2) | 276.0 ± 120.5 | 268.5 ± 117.8 | 0.90 |
| Subcutaneous fat/visceral fat ratio | 1.5 ± 0.6 | 1.6 ± 0.7 | 0.96 |
| Abdomen circumference (cm) | 98.4 ± 13.0 | 103.9 ± 13.4 | 0.41 |
| Intrahepatic lipid content (%) | 28.3 ± 16.1 | 20.4 ± 9.9 | 0.23 |
| Extramyocellular lipid content (S‐fat/Cr) | 29.3 ± 25.1 | 16.6 ± 13.8 | 0.21 |
| Intramyocellular lipid content (S‐fat/Cr) | 6.3 ± 4.2 | 4.2 ± 2.6 | 0.23 |
| Diet | |||
| Total dietary intake (kcal/day) | 1749 ± 634 | 2030 ± 702 | 0.40 |
| Alcohol (g/day) | 0 (0,0) | 0 (0,4.8) | 0.44 |
| Protein intake ratio (%) | 15.5 ± 2.1 | 16.0 ± 2.3 | 0.67 |
| Fat intake ratio (%) | 26.5 ± 5.3 | 28.6 ± 6.1 | 0.48 |
| Carbohydrate intake ratio (%) | 56.0 ± 6.5 | 52.8 ± 8.5 | 0.40 |
| Exercise | |||
| Physical activity (MET[Link] hours/day) | 2.7 ± 1.0 | 4.3 ± 1.9 | 0.06 |
| Total daily energy expenditure (MET[Link] hours/day) | 25.3 ± 3.3 | 27.1 ± 4.2 | 0.34 |
| Sleep | |||
| Morning–Evening Questionnaire score | 54.3 ± 8.0 | 52.3 ± 12.4 | 0.69 |
| Pittsburg Sleep Quality Index | 6.6 ± 4.4 | 6.1 ± 4.7 | 0.85 |
| Sleep time (minutes) | 392.4 ± 56.4 | 440.9 ± 79.1 | 0.18 |
| Well‐being score | 22.7 ± 5.8 | 23.0 ± 4.3 | 0.90 |
| Beck Depression Inventory II | 13.4 ± 12.9 | 10.3 ± 8.9 | 0.56 |
Data are mean ± SD or medians (quartile 1, quartile 3). Comparisons by the Welch's t test or Wilcoxon's rank sum test.
Intake ratio was expressed as calories.
Abbreviations: 8‐OHdG, 8‐hydroxy‐2′‐deoxyguanosine; MET, metabolic equivalent of task; S‐fat, soluble fat (methylene signal intensity).
Histopathologic conditions are also known to be associated with insulin sensitivity,34, 35 but total NAFLD activity score did not differ between the high‐HIS and low‐HIS groups (Fig. 2A). Steatosis and hepatocellular ballooning scores, which are subscores of the NAFLD activity score, were also comparable between the two groups (Fig. 2B,C). The lobular inflammation score, which is another subscore of the NAFLD activity score, trended to be lower in the high‐HIS compared to the low‐HIS group (P = 0.06; Fig. 2D). Fibrosis stage was comparable between the two groups (Fig. 2E).
Figure 2.
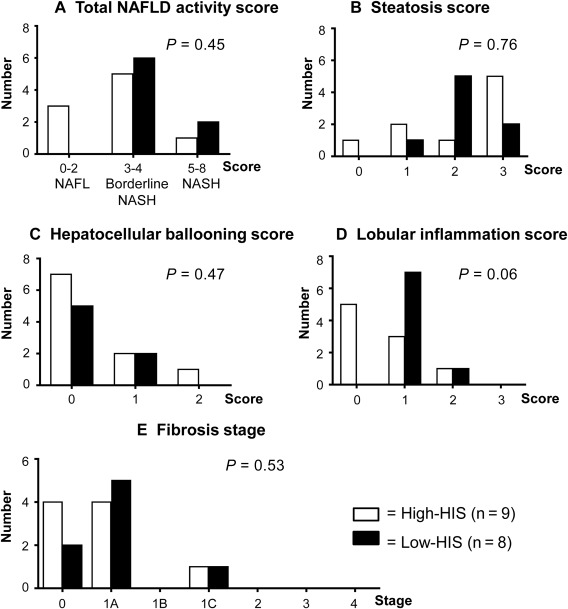
Histopathologic comparison between high‐HIS and low‐HIS groups.
The NAFLD activity score was assessed by histologic features of steatosis (0‐3), lobular inflammation (0‐2), and hepatocellular ballooning (0‐2). (A) Total NAFLD activity score. (B) Steatosis score. (C) Hepatocellular ballooning score. (D) Lobular inflammation score. The total NAFLD activity score (A) was calculated by summation of the scores of B, C, and D. (E) Fibrosis stage was also assessed in addition to the NAFLD activity score. Data represent the number of patients for each score. Wilcoxon rank sum test was used for comparing the two groups.
We also assessed hepatic mRNA expression levels using liver biopsies by quantitative RT‐PCR and by microarray analysis. mRNA expression levels of two genes involved in hepatic gluconeogenesis, a major factor that promotes hepatic glucose production in T2DM,36 were significantly lower in the high‐HIS compared to the low‐HIS group (pyruvate carboxylase [PC], –27.1%; fructose‐1,6‐bisphosphatase, –27.0%; each P < 0.05) (Fig. 3A). Because fasting plasma insulin concentration was significantly lower in the high‐HIS group, these results suggest that mRNA expression of hepatic gluconeogenic genes was more effectively down‐regulated by lower fasting plasma insulin in patients with high‐HIS than in those with low‐HIS. This is consistent with the higher clamped percent suppression of endogenous glucose production in high‐HIS compared to low‐HIS groups. Consistent with no difference in intrahepatic lipid content between the two groups, hepatic mRNA expression levels of lipogenesis and lipid oxidation were comparable between the two groups (Fig. 3B). Finally, microarray analysis indicated that the mRNA expression levels of 43 genes were significantly different (log2 ratio >0.585) between the two groups (Supporting Table S1; Supporting Fig. S1). Gene ontology analysis with these 43 genes indicated that genes involved in the steroid metabolic process were up‐regulated while genes involved in the oxidation–reduction process and inflammatory response were down‐regulated in the high‐HIS group compared to those in the low‐HIS group (Supporting Table S2).
Figure 3.
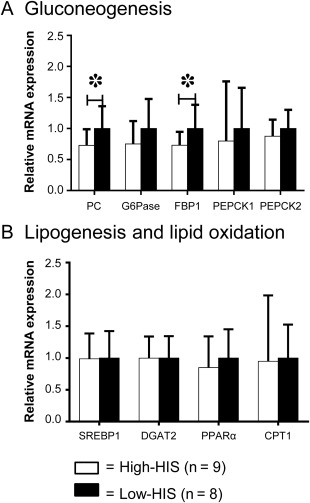
Comparison of hepatic mRNA expression between high‐HIS and low‐HIS groups. (A) Gluconeogenesis. (B) Lipogenesis and lipid oxidation. The average values of the low‐HIS group were set as 1. Data are mean ± SD. *P < 0.05, by unpaired t test.
Abbreviations: CPT1, carnitine palmitoyltransferase I; DGAT2, diacylglycerol O‐acyltransferase 2; FBP1, fructose‐1,6‐bisphosphatase; G6Pase, glucose‐6‐phosphatase; PEPCK, phosphoenolpyruvate carboxykinase; PPARα, peroxisome proliferator‐activated receptor α; SREBP1, sterol regulatory element binding transcription factor 1.
Discussion
This is the first study that comprehensively explored the characteristics of patients with NAFLD with hepatic insulin sensitivity in moderately obese Asian individuals. In the present study, our patients with NAFLD exhibited a wide range of hepatic insulin sensitivity and hepatic lipid content, but these parameters did not correlate with each other. Thus, we were able to identify a subgroup of patients with NAFLD with hepatic insulin sensitivity and define the specific characteristics that distinguish such patients from patients with NAFLD with hepatic insulin resistance. Finally, we demonstrated that high levels of adipose tissue insulin sensitivity, serum HMW adiponectin, and plasma TCA cycle metabolites are unique characteristics of patients with hepatic insulin‐sensitive NAFLD.
Importantly, hepatic insulin‐sensitive NAFLD was the focus in this study, but this study is not just a mirror study of hepatic insulin‐resistant NAFLD. Most studies have demonstrated the impact of intrahepatic lipid content on NAFLD‐associated hepatic insulin resistance,3, 4 but our study had a cohort of patients with NAFLD with hepatic insulin sensitivity who were hepatic insulin sensitive regardless of the intrahepatic lipid content. Consistent with our findings, Bril et al.37 reported that hepatic insulin sensitivity decreased with only a small lipid accumulation in the liver (∼1.5%) but then the mean value of hepatic insulin sensitivity remained constant regardless of the amount of intrahepatic lipid content; no association was observed between intrahepatic lipid content and hepatic insulin sensitivity in 352 Caucasians, including T2DM and/or NASH patients, using 1H‐MRS and hyperinsulinemic‐euglycemic clamp studies. In this context, we further explored the factors that associate with hepatic insulin sensitivity despite having intrahepatic lipid accumulation. Accordingly, our study may provide a new therapeutic approach for hepatic insulin resistance in patients with NAFLD without any reduction in intrahepatic lipid content.
We identified some characteristics linked to hepatic insulin sensitivity in NAFLD. First, adipose tissue insulin sensitivity was associated with hepatic insulin sensitivity. Although the causality is unclear in this cross‐sectional observational study, previous reports suggested a contribution of adipose tissue insulin sensitivity to hepatic insulin sensitivity.38, 39 Insufficient suppression of lipolysis by insulin in adipose tissue, i.e., adipose tissue insulin resistance, leads to increased fatty acids and glycerol delivery to the liver. Increased fatty acid delivery to the liver increases hepatic acetyl‐coenzyme A content, leading to activation of PC, which in turn increases hepatic gluconeogenesis.38, 39 In this study, patients in the high‐HIS group showed down‐regulation of PC mRNA accompanied by adipose tissue insulin sensitivity. In contrast to adipose tissue insulin sensitivity, muscle insulin sensitivity did not differ between patients of the high‐HIS and low‐HIS groups (Table 2). Because muscle insulin resistance is known to be associated with intrahepatic lipid content,14, 40 the lack of difference in muscle insulin sensitivity may reflect the lack of difference in intrahepatic lipid content between the high‐HIS and low‐HIS patients.
Second, hyper‐HMW adiponectinemia was observed in patients in the high‐HIS group compared to the low‐HIS group, and this might be another mechanism linking adipose tissue insulin sensitivity to hepatic insulin sensitivity. A high level of adipose tissue insulin sensitivity is associated with both suppressed fatty acid release and increased HMW adiponectin secretion from adipose tissue, which could cooperatively lead to higher hepatic insulin sensitivity.41, 42, 43 In contrast to the positive association between HMW adiponectin and hepatic insulin sensitivity, the intrahepatic lipid content did not differ between the high‐HIS and low‐HIS groups. Although adiponectin is known to reduce hepatic steatosis44 by suppression of lipogenesis45 and stimulation of lipid oxidation,46 hepatic mRNA expression levels of lipogenesis or lipid oxidation were comparable between the two groups (Fig. 3B). This may explain, at least in part, the lack of difference in intrahepatic lipid content between the two groups. It is likely that other unknown factors could operate in favor of hepatic steatosis against HMW adiponectin in patients with hepatic insulin‐sensitive NAFLD.
Third, the levels of mitochondrial TCA cycle metabolites in plasma were significantly higher in patients in the high‐HIS compared to the low‐HIS group. Adiponectin reportedly stimulates hepatic mitochondrial biogenesis in mice,43 and interestingly, there is a hepatic mitochondrial functional adaptation in patients with NAFLD but the adaptation was lost in patients with hepatic insulin‐resistant NASH.47 Taken together, hyper‐HMW adiponectinemia could contribute to increased hepatic mitochondrial function through mitochondrial biogenesis, which results in increased hepatic insulin sensitivity. In this study, the higher concentration of plasma TCA cycle metabolites in the high‐HIS group may reflect higher hepatic mitochondrial function stimulated by hyper‐HMW adiponectinemia. In this regard, inhibition of the citrate transporter, which regulates the plasma citrate flux into the liver,48, 49 was also reported to increase plasma citrate,48 increase hepatic mitochondrial biogenesis,48 and prevent diet‐induced hepatic steatosis and hepatic insulin resistance.48, 50 Thus, it would be another potential therapeutic option to increase plasma TCA cycle metabolites, specifically citric acid, in patients with NAFLD.
Comprehensive gene expression analysis was performed using liver biopsies to explore genes associated with hepatic insulin‐sensitive NAFLD. Significantly higher (almost double) levels of hepatic cytochrome P450, family 2, subfamily C, polypeptide 19 (CYP2C19) were found in patients in the high‐HIS group compared to the low‐HIS group (Supporting Table S1). The enzymatic activity and expression level of CYP2C19 protein were reported to decrease with progression of NAFLD,51 and the expression level of its gene is reportedly influenced by the glycemic state.52 Further studies are needed to determine the therapeutic potential of CYP2C19 in NAFLD‐associated hepatic insulin resistance. Gene ontology analysis showed down‐regulation of mRNA expression of genes involved in the oxidation–reduction process and inflammatory response in the high‐HIS group (Supporting Table S2), which may associate with the lower lobular inflammation score as assessed by liver histopathologic examination (P = 0.06; Fig. 2D).
While our study clearly demonstrated the characteristics of patients with hepatic insulin‐sensitive NAFLD, the study had certain limitations that need to be discussed. First, we could not assess the accumulated lipid species in the liver due to the limited size of the liver needle biopsy. Comprehensive assessment in morbidly obese Caucasians with NAFLD who underwent bariatric surgery demonstrated a strong association between hepatic diacylglycerol content/protein kinase C epsilon activation and hepatic insulin resistance.5, 53 Thus, reduction of the hepatic diacylglycerol content would be another potential therapeutic option for NAFLD‐associated hepatic insulin resistance in humans, although this hypothesis remains to be tested in moderately obese patients with NAFLD. Second, this study included some patients with T2DM and NASH. Although the prevalence of T2DM and NASH was not different between high‐HIS and low‐HIS groups and the distribution of hepatic insulin sensitivity and intrahepatic lipid content did not change significantly when T2DM and NASH patients were excluded (Supporting Fig. S2), it is difficult to determine whether diabetes rather than innate differences between patients with NAFLD with high‐ and low‐HIS was responsible for the observed differences between high‐HIS and low‐HIS groups. We originally planned to include both NAFLD with diabetes and NAFLD without diabetes in the present study, but there are already numerous published studies that have compared non‐NAFLD and NAFLD,33, 47, 54, 55, 56, 57 simple steatosis and steatohepatitis,47, 54, 56 and non‐diabetes and diabetes.14, 57 Thus, importantly, even in the cohort of individuals with T2DM and NASH, the above characteristics became significant factors that distinguished hepatic insulin‐sensitive NAFLD from insulin‐resistant NAFLD. Additionally, we separately analyzed NAFLD with diabetes and NAFLD without diabetes and have placed these data in the supporting files (Supporting Figs. S3‐S6; Supporting Tables S4‐S7), but because it is a secondary subanalysis with only a few participants after the separation of diabetes and no diabetes, we cannot interpret or draw a conclusion from the findings. Third, our study included a small number of Japanese patients, and consequently the two groups of high‐HIS and low‐HIS were not well matched for sex. Thus, our findings need to be tested in a large number of patients of different ethnicities. Fourth, due to the cross‐sectional study design, this study is unable to provide any mechanisms for why some people with NAFLD have high‐HIS. There is no evaluation of potential genetic mutations (e.g., familial hypobetalipoproteinemia) that cause NAFLD but are not associated with insulin resistance. Finally, the mean insulin concentration during the clamp tended to be higher in the high‐HIS than in the low‐HIS group, suggesting it was much lower in some low‐HIS subjects compared to high‐HIS subjects given the high values of SD. It is possible that this has contributed to the differences in the percent suppression of glucose production between the groups. To check this possibility, we reanalyzed Fig. 1 by expressing percent suppression of glucose production divided by clamped plasma insulin concentration to potentially correct for differences in insulin concentration. Interestingly, we found that the difference between low‐HIS and high‐HIS was small and there was a negative correlation tendency between intrahepatic lipid content and percent suppression of endogenous glucose production divided by clamped plasma insulin (Supporting Fig. S7). Because the insulin infusion rate was the same between groups, the insulin clearance rate or some other factor might be causing the difference in the clamped plasma insulin concentration between the groups. Further investigation is needed for understanding these observations.
In conclusion, this is the first study that explored the characteristics of patients with hepatic insulin‐sensitive NAFLD. Hepatic insulin sensitivity in these patients was identical to that of non‐NAFLD subjects. Our data demonstrated that patients with hepatic insulin‐sensitive NAFLD were characterized by high levels of adipose tissue insulin sensitivity, serum HMW adiponectin, and plasma TCA cycle metabolites. These factors might cooperatively play a crucial role in the pathogenesis of hepatic insulin sensitivity despite the presence of NAFLD. In this study, we focused on hepatic insulin action because hepatic insulin resistance plays a central role in the pathogenesis of T2DM and HCC.2, 3 Future longitudinal or interventional studies should elucidate whether preserving hepatic insulin sensitivity through the above characteristics can prevent the development of T2DM or HCC in patients with NAFLD.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1077/suppinfo.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Supporting Information Figure 7.
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.
Supporting Information Table 5.
Supporting Information Table 6.
Supporting Information Table 7.
Supporting Information
Acknowledgment
We thank the volunteers for participating in this study. We also thank Hirokazu Yamada and Tomokazu Shibamiya for their excellent technical assistance with the statistical analysis, Teppei Matsui, Yasushi Matsukiyo, and Koujirou Kobayashi for their assistance with the liver biopsy, and Ken Kanazawa, Mai Hijikata, Hiroyuki Igarashi, Naofumi Miwa, Kaori Awano, Azusa Fujimura, Sachiko Mizuno, Saori Kakehi, Takehiko Oka, Motoharu Ohtsu, Ichikawa Kouji, and Hisanobu Harada for their excellent technical assistance.
Potential conflict of interest: N. Kumashiro received lecture fees from Novo Nordisk Inc.; Y. Tamura received lecture fees from Takeda Pharmaceutical Co., Ltd., Merck & Co., Inc., Eli Lilly Japan K.K., Kissei Pharmaceutical Co., Ltd., and AstraZeneca; H. Watada received research funds from Merck & Co., Inc., Astellas Pharma Inc., AstraZeneca, Ono Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Sanofi K.K., Daiichi‐Sankyo Co., Ltd., Dainippon Sumitomo Pharma Co., Ltd., Takeda Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Terumo Corporation, Boehringer Ingelheim Pharmaceuticals, Inc., Novo Nordisk Inc., Pfizer Japan Inc., Benefit One Healthcare K.K., Mochida Pharmaceutical Co., Ltd., Nittoubouseki K.K., Novartis Pharma K.K., Eli Lilly Japan K.K., and Taisho Pharmaceutical Co., Ltd. and received lecture fees from Astellas Pharma Inc., AstraZeneca, Kowa Company, Ltd., Sanofi K.K., Takeda Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Novartis Pharma K.K., Novo Nordisk Inc., Boehringer Ingelheim Pharmaceuticals, Inc., Merck & Co., Inc., and Dainippon Sumitomo Pharma Co., Ltd.; Y. Sumino received research funds from Daiichi‐Sankyo Co.; T. Hirose received research funds from AstraZeneca, Boehringer Ingelheim Pharmaceuticals, Inc., Astellas Pharma Inc., Ono Pharmaceutical Co., Ltd., Novo Nordisk Inc., Sanofi‐Aventis Deutschland GmbH, Daiichi‐Sankyo Co., Ltd., Eli Lilly Japan K.K., Takeda Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Dainippon Sumitomo Pharma Co., Ltd., Kissei Pharmaceutical Co., Ltd., and Johnson & Johnson and received lecture fees from Sanofi‐Aventis Deutschland GmbH, Eli Lilly Japan K.K., Novo Nordisk Inc., Takeda Pharmaceutical Co., Ltd., Daiichi‐Sankyo Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Merck & Co., Inc., Dainippon Sumitomo Pharma Co., Ltd., Novartis Pharma K.K., Kissei Pharmaceutical Co., Ltd., Boehringer Ingelheim Pharmaceuticals, Inc., Ono Pharmaceutical Co., Ltd., and AstraZeneca. The funding agencies and corporations had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supported by the Japan Society for the Promotion of Science KAKENHI Grant Numbers 26702032 (N. K.) and JP17H02180 (N. K.).
REFERENCES
- 1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 2. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 2010;51:1820‐1832. [DOI] [PubMed] [Google Scholar]
- 3. Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014;510:84‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 2010;51:679‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2011;108:16381‐16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014;59:713‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010;140:900‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007;132:2169‐2180. [DOI] [PubMed] [Google Scholar]
- 9. Kumashiro N, Tamura Y, Uchida T, Ogihara T, Fujitani Y, Hirose T, et al. Impact of oxidative stress and peroxisome proliferator‐activated receptor gamma coactivator‐1alpha in hepatic insulin resistance. Diabetes 2008;57:2083‐2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kantartzis K, Peter A, Machicao F, Machann J, Wagner S, Konigsrainer I, et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin‐like phospholipase 3 gene. Diabetes 2009;58:2616‐2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stefan N, Haring HU. The metabolically benign and malignant fatty liver. Diabetes 2011;60:2011‐2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lehmann R, Franken H, Dammeier S, Rosenbaum L, Kantartzis K, Peter A, et al. Circulating lysophosphatidylcholines are markers of a metabolically benign nonalcoholic fatty liver. Diabetes Care 2013;36:2331‐2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeno K, Tamura Y, Kawaguchi M, Kakehi S, Watanabe T, Funayama T, et al. Relation between insulin sensitivity and metabolic abnormalities in Japanese men with BMI of 23‐25 kg/m2 . J Clin Endocrinol Metab 2016;101:3676‐3684. [DOI] [PubMed] [Google Scholar]
- 14. Kato K, Takamura T, Takeshita Y, Ryu Y, Misu H, Ota T, et al. Ectopic fat accumulation and distant organ‐specific insulin resistance in Japanese people with nonalcoholic fatty liver disease. PLoS One 2014;9:e92170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, et al. Insulin resistance in non‐diabetic patients with non‐alcoholic fatty liver disease: sites and mechanisms. Diabetologia 2005;48:634‐642. [DOI] [PubMed] [Google Scholar]
- 16. Kotronen A, Seppala‐Lindroos A, Bergholm R, Yki‐Jarvinen H. Tissue specificity of insulin resistance in humans: fat in the liver rather than muscle is associated with features of the metabolic syndrome. Diabetologia 2008;51:130‐138. [DOI] [PubMed] [Google Scholar]
- 17. Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, et al. Hepatic diacylglycerol‐associated protein kinase Cepsilon translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep 2017;19:1997‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vozarova B, Stefan N, Lindsay RS, Saremi A, Pratley RE, Bogardus C, et al. High alanine aminotransferase is associated with decreased hepatic insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 2002;51:1889‐1895. [DOI] [PubMed] [Google Scholar]
- 19. Kelley DE, McKolanis TM, Hegazi RA, Kuller LH, Kalhan SC. Fatty liver in type 2 diabetes mellitus: relation to regional adiposity, fatty acids, and insulin resistance. Am J Physiol Endocrinol Metab 2003;285:E906‐E916. [DOI] [PubMed] [Google Scholar]
- 20. Korenblat KM, Fabbrini E, Mohammed BS, Klein S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008;134:1369‐1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maggs DG, Buchanan TA, Burant CF, Cline G, Gumbiner B, Hsueh WA, et al. Metabolic effects of troglitazone monotherapy in type 2 diabetes mellitus. A randomized, double‐blind, placebo‐controlled trial. Ann Intern Med 1998;128:176‐185. [DOI] [PubMed] [Google Scholar]
- 22. Sato F, Tamura Y, Watada H, Kumashiro N, Igarashi Y, Uchino H, et al. Effects of diet‐induced moderate weight reduction on intrahepatic and intramyocellular triglycerides and glucose metabolism in obese subjects. J Clin Endocrinol Metab 2007;92:3326‐3329. [DOI] [PubMed] [Google Scholar]
- 23. Ohashi Y, Hirayama A, Ishikawa T, Nakamura S, Shimizu K, Ueno Y, et al. Depiction of metabolome changes in histidine‐starved Escherichia coli by CE‐TOFMS. Mol Biosyst 2008;4:135‐147. [DOI] [PubMed] [Google Scholar]
- 24. Ooga T, Sato H, Nagashima A, Sasaki K, Tomita M, Soga T, et al. Metabolomic anatomy of an animal model revealing homeostatic imbalances in dyslipidaemia. Mol Biosyst 2011;7:1217‐1223. [DOI] [PubMed] [Google Scholar]
- 25. Coloff JL, Murphy JP, Braun CR, Harris IS, Shelton LM, Kami K, et al. Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab 2016;23:867‐880. [DOI] [PubMed] [Google Scholar]
- 26. Sakurai Y, Tamura Y, Takeno K, Kumashiro N, Sato F, Kakehi S, et al. Determinants of intramyocellular lipid accumulation after dietary fat loading in non‐obese men. J Diabetes Investig 2011;2:310‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 28. Pfaffl MW. A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oba J, Nakahara T, Hashimoto‐Hachiya A, Liu M, Abe T, Hagihara A, et al. CD10‐equipped melanoma cells acquire highly potent tumorigenic activity: A plausible explanation of their significance for a poor prognosis. PLoS One 2016;11:e0149285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dharmawardhana P, Brunner AM, Strauss SH. Genome‐wide transcriptome analysis of the transition from primary to secondary stem development in Populus trichocarpa. BMC Genomics 2010;11:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sondergaard E, Espinosa De Ycaza AE, Morgan‐Bathke M, Jensen MD. How to measure adipose tissue insulin sensitivity. J Clin Endocrinol Metab 2017;102:1193‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sondergaard E, Jensen MD. Quantification of adipose tissue insulin sensitivity. J Investig Med 2016;64:989‐991. [DOI] [PubMed] [Google Scholar]
- 33. Lomonaco R, Ortiz‐Lopez C, Orsak B, Webb A, Hardies J, Darland C, et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012;55:1389‐1397. [DOI] [PubMed] [Google Scholar]
- 34. Ballestri S, Nascimbeni F, Romagnoli D, Lonardo A. The independent predictors of non‐alcoholic steatohepatitis and its individual histological features. Insulin resistance, serum uric acid, metabolic syndrome, alanine aminotransferase and serum total cholesterol are a clue to pathogenesis and candidate targets for treatment. Hepatol Res 2016;46:1074‐1087. [DOI] [PubMed] [Google Scholar]
- 35. Grigorescu M, Crisan D, Radu C, Grigorescu MD, Sparchez Z, Serban A. A novel pathophysiological‐based panel of biomarkers for the diagnosis of nonalcoholic steatohepatitis. J Physiol Pharmacol 2012;63:347‐353. [PubMed] [Google Scholar]
- 36. Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest 1992;90:1323‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bril F, Barb D, Portillo‐Sanchez P, Biernacki D, Lomonaco R, Suman A, et al. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology 2017;65:1132‐1144. [DOI] [PubMed] [Google Scholar]
- 38. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 2016;126:12‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015;160:745‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Flannery C, Dufour S, Rabol R, Shulman GI, Petersen KF. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 2012;61:2711‐2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Musso G, Cassader M, De Michieli F, Rosina F, Orlandi F, Gambino R. Nonalcoholic steatohepatitis versus steatosis: adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology 2012;56:933‐942. [DOI] [PubMed] [Google Scholar]
- 42. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 2006;116:1784‐1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Handa P, Maliken BD, Nelson JE, Morgan‐Stevenson V, Messner DJ, Dhillon BK, et al. Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology 2014;60:133‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med 2007;13:332‐339. [DOI] [PubMed] [Google Scholar]
- 45. Awazawa M, Ueki K, Inabe K, Yamauchi T, Kaneko K, Okazaki Y, et al. Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem Biophys Res Commun 2009;382:51‐56. [DOI] [PubMed] [Google Scholar]
- 46. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty‐acid oxidation by activating AMP‐activated protein kinase. Nat Med 2002;8:1288‐1295. [DOI] [PubMed] [Google Scholar]
- 47. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non‐alcoholic fatty liver is lost in steatohepatitis. Cell Metab 2015;21:739‐746. [DOI] [PubMed] [Google Scholar]
- 48. Birkenfeld AL, Lee HY, Guebre‐Egziabher F, Alves TC, Jurczak MJ, Jornayvaz FR, et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab 2011;14:184‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li Z, Erion DM, Maurer TS. Model‐based assessment of plasma citrate flux into the liver: implications for NaCT as a therapeutic target. CPT Pharmacometrics Syst Pharmacol 2016;5:132‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pesta DH, Perry RJ, Guebre‐Egziabher F, Zhang D, Jurczak M, Fischer‐Rosinsky A, et al. Prevention of diet‐induced hepatic steatosis and hepatic insulin resistance by second generation antisense oligonucleotides targeted to the longevity gene mIndy (Slc13a5). Aging (Albany NY) 2015;7:1086‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fisher CD, Lickteig AJ, Augustine LM, Ranger‐Moore J, Jackson JP, Ferguson SS, et al. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos 2009;37:2087‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Davidson MD, Ballinger KR, Khetani SR. Long‐term exposure to abnormal glucose levels alters drug metabolism pathways and insulin sensitivity in primary human hepatocytes. Sci Rep 2016;6:28178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012;142:1444‐1446.e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lomonaco R, Bril F, Portillo‐Sanchez P, Ortiz‐Lopez C, Orsak B, Biernacki D, et al. Metabolic impact of nonalcoholic steatohepatitis in obese patients with type 2 diabetes. Diabetes Care 2016;39:632‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Portillo‐Sanchez P, Bril F, Maximos M, Lomonaco R, Biernacki D, Orsak B, et al. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J Clin Endocrinol Metab 2015;100:2231‐2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rosso C, Mezzabotta L, Gaggini M, Salomone F, Gambino R, Marengo A, et al. Peripheral insulin resistance predicts liver damage in nondiabetic subjects with nonalcoholic fatty liver disease. Hepatology 2016;63:107‐116. [DOI] [PubMed] [Google Scholar]
- 57. Ortiz‐Lopez C, Lomonaco R, Orsak B, Finch J, Chang Z, Kochunov VG, et al. Prevalence of prediabetes and diabetes and metabolic profile of patients with nonalcoholic fatty liver disease (NAFLD). Diabetes Care 2012;35:873‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1077/suppinfo.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Supporting Information Figure 7.
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.
Supporting Information Table 5.
Supporting Information Table 6.
Supporting Information Table 7.
Supporting Information