

Abstract
Due to the increasing prevalence of nonalcoholic steatohepatitis (NASH) and its associated health burden, there is a high need to develop therapeutic strategies for patients with this disease. Unfortunately, its long and asymptomatic natural history, the uncertainties about disease progression, the fact that most patients are undiagnosed, and the requirement for sequential liver biopsies create substantial challenges for clinical development. Adaptive design methods are increasingly used in clinical research as they provide the flexibility and efficiency for identifying potential signals of clinical benefit of the test treatment under investigation and make prompt preplanned adaptations without undermining the validity or integrity of the trial. Given the high unmet medical need and the lack of validated surrogate endpoints in NASH, the use of adaptive design methods appears reasonable. Furthermore, due to the limited number of patients willing to have multiple liver biopsies and the need for long‐term exposure to assess an impact in outcomes, a continuous seamless adaptive design may reduce the overall sample size while allowing patients to continue after each one of the phases. Here, we review strategic frameworks that include potential surrogate endpoints as well as statistical and logistical approaches that could be considered for applying adaptive designs to clinical trials in NASH with the goal of facilitating drug development for this growing medical need. (Hepatology Communications 2017;1:577–585)
Abbreviations
- EMA
European Medicines Agency
- FDA
U.S. Food and Drug Administration
- NAFLD
nonalcoholic fatty liver disease
- NAS
nonalcoholic fatty liver disease activity score
- NASH
nonalcoholic steatohepatitis
- POC
proof‐of‐concept
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in the Western world.1 In the United States, approximately 66,000 deaths are due to chronic liver disease each year. Data from the National Health and Nutrition Examination Surveys conducted between 1988 and 2008 have shown that the overall prevalence of chronic liver disease in the community rose from 12% to 15%, a change attributable to increases in rates of NAFLD.2 The principal risk factors for NAFLD include obesity, insulin resistance, and features of the metabolic syndrome.3 It is characterized by the pathologic accumulation of fat in the liver and has two major histologic phenotypes, a fatty liver or steatohepatitis. Nonalcoholic steatohepatitis (NASH) progresses to cirrhosis more frequently that nonalcoholic fatty liver4 and is rapidly rising as an etiology for end‐stage liver disease requiring liver transplantation.4, 5 In a recently published study based on longitudinal data of a Medicare multiethnic cohort, NAFLD was the most common cause of cirrhosis (29.3%).6 The histologic feature that appears to best predict mortality in NASH is the presence of significant fibrosis.7 There is currently no approved drug therapy for NASH, and development of such therapeutics is widely felt to be a public health priority.
The development pathway for drugs for NASH is complex despite recent advances in the field.3, 8, 9 The misconception that NAFLD is a benign manifestation of obesity, the heterogeneous nature of NASH with respect to the risk of progression to cirrhosis, and the lack of a validated surrogate endpoint to clinical outcomes have considerably slowed the development of drugs for NASH. In addition to specific genetic predispositions associated with more severe disease in subsets of patients, such as the patatin‐like phospholipase domain containing 3 I148M mutation,10 both disease activity and disease stage affect the risk for progression to cirrhosis. Disease activity refers to the processes (inflammation, hepatocellular injury, or death) that drive the disease toward cirrhosis, whereas disease stage (fibrosis) refers to how far the disease has already progressed to cirrhosis. Disease activity is traditionally assessed on histopathology by the presence of steatohepatitis and the NAFLD activity score (NAS),11 while the disease stage is assessed by the fibrosis stage.12 The progression from NASH without fibrosis through various stages of fibrosis is not linear; however, those with fibrosis stages 2 and 3 have a significantly higher risk of progression to cirrhosis within a 10‐year time frame and an increased mortality risk compared to lower stages (0‐1) of disease.13, 14, 15 With the ultimate goal of providing a clinically meaningful benefit, the principal objective of treatment of those with NASH at increased risk of progression to cirrhosis is to reduce disease activity in the short term and prevent progression to cirrhosis and thus adverse liver outcomes in the long term. Most liver‐related outcomes occur once cirrhosis has developed, and the progression to cirrhosis may represent a surrogate for approval.8, 9 In fact, progression to liver cirrhosis has been accepted by both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) as part of the composite primary endpoint (suggesting an impact on clinical liver outcomes) in the two ongoing NASH marketing authorization trials.16, 17 Finally, the need for an invasive methodology for histology‐based endpoints and the limited number of patients with biopsy‐confirmed NASH and/or willing to have multiple liver biopsies over the study period have been major barriers to recruitment into clinical trials and ultimately to bring a therapy option for patients with NASH.
Adaptive trial designs are being increasingly examined as a way to streamline the development path and optimize the time to development without compromising the quality of the evidence needed to establish the efficacy and safety of therapeutic agents. An adaptive design provides the opportunity for prospectively planned modifications of one or more specified aspects of a study design18, 19 (i.e., making prespecified adjustments at the end of each stage) and may allow minimizing the overall number of patients required during the entire drug development process while at the same time ensuring that the minimum number of patients exposed to the drug is achieved. It may further allow the same subjects to move from one phase to another and thus reduce the need to find additional subjects for treatment trials who will be willing to accept multiple biopsies. These considerations suggest that an adaptive design‐based approach where the initial phase could demonstrate improvement in endpoints of histologic disease activity and/or improvement in disease stage followed by a second phase to demonstrate clinical outcomes may be particularly suitable for NASH. In this paper, we provide the scientific and logistical outline of such an approach.
Registration Pathways: Accelerated/Conditional Marketing Approvals
Regulatory agencies have adopted policies that can expedite drug development for serious medical conditions where few or no therapies exist.20, 21
In order to deal with the difficulties of long trials designed to demonstrate a meaningful benefit in terms of clinical outcomes, specific mechanisms have been established by the regulatory agencies. Accelerated approval (FDA) and conditional approval (European Union) pathways allow initial marketing approval based on surrogate endpoints considered reasonably likely to predict outcomes, followed by full approval based on assessment of clinical benefit in a confirmatory clinical trial.
There are three types of surrogate endpoints. Candidate surrogates are biomarkers that are under consideration for use in clinical trials but have not been accepted. Reasonably likely to predict clinical benefit are surrogates that have been accepted for use in phase 3 clinical trials but lack sufficient evidence to be used for full approval. Validated surrogates are biomarkers for which there are data to show that the biomarker does predict clinical outcomes. For example, hemoglobin A1C is a validated surrogate endpoint that has been used as the basis for full/regular approval of therapeutic agents to treat diabetes.
The accelerated and conditional approval pathways offer the potential to use surrogate endpoint(s) or intermediate clinical endpoints that are determined to be reasonably likely to predict clinical benefit. The agencies may grant accelerated/conditional approval based on these surrogate endpoints, with full approval being granted based on subsequent confirmatory studies using well‐established and well‐defined clinical outcomes.
There is no validated surrogate endpoint in NASH. Resolution of NASH (i.e., absence of ballooning with no or minimal inflammation by histology) is considered a surrogate endpoint “reasonable likely to be associated with outcomes.” Therefore, the approval process must involve a two‐step approach, i.e., an initial accelerated or conditional approval followed by a final approval after confirming clinical benefit in preventing progression to cirrhosis and liver outcomes (including cirrhosis decompensation events, overall death, and liver transplantation).
Endpoints in Clinical Trials
The NAS is a histology‐based validated scoring system (an unweighted composite of steatosis, inflammation, and cellular ballooning scores) that can be used to assess pathologic changes in clinical trials.11 The histologic endpoint of improvement in activity as assessed by a reduction in at least 2 points in NAS (with at least 1 point reduction in hepatocellular ballooning, a key feature required for the diagnosis of NASH) can be an acceptable marker of improvement if it is associated with no progression in fibrosis. However, there is no evidence to support that NAS is reasonably likely associated with outcomes; thus, resolution of NASH with no worsening of fibrosis (assessed as an increase in the fibrosis stage by histology) and/or improvement in fibrosis with no progression of steatohepatitis (i.e., no increase in activity as assessed by NAS (Table 1) is required for trials to support a marketing application.8, 9, 16, 17 Both resolution of NASH and improvement in fibrosis have been accepted16, 17 as surrogate endpoints “reasonably likely to predict clinical benefit” and have the potential to lead to an accelerated/conditional approval.
Table 1.
ENDPOINTS AND POPULATION IN CLINICAL TRIALS IN NASH
| Phase | Primary Endpoint | Target Population |
|---|---|---|
| Early phase trials/ Proof‐of‐concept |
Endpoints should be based on mechanism of drug. Reduction in liver fat with a sustained improvement in transaminases; Improvement in biomarkers of liver inflammation, apoptosis and/or fibrosis. Consider using improvement in NAS (ballooning and inflammation) and/or fibrosis. |
Ideal to enroll patients with biopsy‐proven NASH but acceptable to enroll patients at high risk for NASH (i.e., evidence of fatty liver, two components of the metabolic syndrome, evidence of liver stiffness by imaging). |
| Dose ranging/phase 2 |
Resolution of NASH without worsening of fibrosis; alternatively, improvement in disease activity (NAS)/improvement in ballooning/ inflammation without worsening of fibrosis. |
Biopsy‐proven NASH and NAS ≥4. Include patients with NASH and liver fibrosis. Include a sufficient number of patients with NASH and fibrosis stage 2/3 to inform phase 3. |
| Trials to support a marketing application: phase 3 |
Resolution of steatohepatitis and no worsening of fibrosis. Improvement in fibrosis with no worsening of steatohepatitis. A co‐primary endpoint of the above or depending on the mode of action, either one or the other can be used. |
Patients with biopsy‐confirmed NASH with moderate/advanced fibrosis (F2/F3). |
| Trials to support a marketing application: phase 4 (postmarketing part) |
Clinical outcome trial underway by the time of submission: Composite endpoint: histopathologic progression to cirrhosis; MELD score change by >2 points or MELD increase to >15 in population enrolled with MELD ≤13; death; transplant; Cirrhosis decompensation events: |
Patients with biopsy‐confirmed NASH with moderate/advanced fibrosis (F2/F3). |
Abbreviation: MELD, model for end‐stage liver disease.
Seamless Adaptive Design: General Statistical Considerations
One of the most commonly considered adaptive designs is probably a multiple‐stage seamless adaptive design that combines several independent studies into a single one that can address study objectives of the intended individual studies. For two‐stage adaptive designs, four categories have been reported19, 22 depending upon the study objectives and endpoints at different stages. These categories include (1) a design with a common study objective and a fixed study endpoint, (2) designs with a common study objective but different study endpoints, (3) designs with different study objectives but a fixed study endpoint, and (4) designs with different study objectives and different study endpoints. In NASH programs, there is one common study objective (i.e., improvement in liver clinical outcomes) but different primary endpoints (i.e., reduction in NAS, resolution of NASH by histology, improvement in fibrosis, and reduction in progression to cirrhosis).
The use of an adaptive design has several advantages as well as some limitations23 (Table 2). From a statistical perspective, major advantages of adaptive designs include (1) an opportunity to react to negative/positive results at the time of the interim analysis, make decisions, and take corrective actions early; (2) evaluate data collected from different stages for a combined analysis with a more accurate and reliable assessment of the treatment effect in an efficient way (see therapeutix index function); and (3) shorten the clinical development process (reducing the time between finalization of one phase and the initiation of the next one, which can potentially save at least 6 months). However, it is important to consider and prevent potential operational biases that may arise during the evaluation of the interim data (e.g., identification, minimization, and control of sources of bias/variation; overall type I error control rate at a prespecified alpha level) and maintain the quality, validity, and integrity of data collected from the study with clearly outlined roles and responsibilities of a data safety monitoring committee.
Table 2.
RELATIVE MERITS AND LIMITATION OF TWO‐STAGE ADAPTIVE DESIGN IN NASH
| Characteristic | Two Independent Trials | Two‐Stage Adaptive Design | ||
|---|---|---|---|---|
| Power | 90% 90% (81%) | 90% | ||
| Sample size |
|
|
||
| Operational bias | Less | Moderate to severe | ||
| Data analysis | By study analysis | Combined analysis | ||
| Efficiency | 6 to 12 months lead time between studies | Reduced lead time between trials | ||
| Flexibility/long‐term follow‐up | New study design based on previous data. New patients are enrolled | Adaptations based on IA, e.g., stop one or more study arms/randomize more patients. Continue follow‐up | ||
| Regulatory aspects | Standard practice | Requires buy‐in by global authorities prior to initiation | ||
| Statistical perspective | Valid statistical methods are well established | Evolving statistical methods | ||
| Operational complexity | Low | High |
Depends on adaptations; N is total number of subjects for both studies; N1 is the sample size for trial 1; N2 is the sample size for trial 2.
THERAPEUTIC INDEX FUNCTION FOR ENDPOINT SELECTION
In order to link all endpoints for the analysis of adaptive designs with distinct study endpoints at each stage (as needed in NASH), a therapeutic index (or utility) function that is either scalar or vector is proposed, depending on whether the study includes single or co‐primary endpoints.24, 25, 26 A mathematical model allows an overall analysis linking all NASH endpoints at different stages (e.g., changes in noninvasive biomarkers, reduction in at least 2 points in NAS, resolution of NASH without worsening of fibrosis, improvement in fibrosis, etc.). Furthermore, a prespecified weight for each of the study endpoints can be allocated (the mathematical proposed model is included in the Supporting Material). Although the endpoints are different at each stage, we work under the assumption that there is a well‐established relationship between them.
ADAPTIVE DESIGN IN EARLY PHASES
Biopsy‐driven endpoints are, in general, not feasible in short‐term proof‐of‐concept (POC) trials. In these studies, designed mainly to assess safety and tolerability of new drugs and to look for early futility signals to direct decisions regarding further development, different biomarkers can be used. An improvement of hepatic steatosis as determined by magnetic resonance technology is one such biomarker and might be suitable because improvement in steatohepatitis is generally associated with a reduction in liver fat.15 This reduction in liver fat should be associated with improvement in liver aminotransferases and/or other noninvasive biomarkers of insulin sensitivity, inflammation, apoptosis, and fibrosis that could help in decision making. The description of these biomarkers is beyond the scope of this article but can be found elsewhere.27 However, it is important to note that the use of noninvasive biomarkers is still considered experimental, and there are no noninvasive biomarkers that are considered to meet the requirements for a surrogate reasonably likely to predict clinical benefit.
An early‐phase POC dose‐finding trial can be a stand‐alone study or part of an adaptive POC/phase 2b study (Fig. 1). A stand‐alone POC study can enroll subjects with a high risk of NASH (e.g., patients with fatty liver or metabolic risk factors, such as type 2 diabetes and/or the metabolic syndrome). Results obtained with this “high risk of NASH” approach provide a degree of confidence for further development, but the limitation of the patient population not being biopsy confirmed for NASH would remain.
Figure 1.
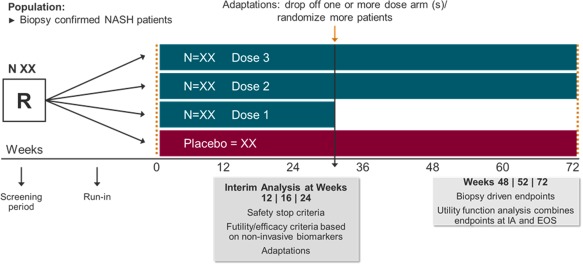
Proof‐of‐concept/dose‐ranging adaptive design trial. A single proof‐of‐concept dose‐finding study seamless adaptive trial design can enroll patients with biopsy‐confirmed NASH and allow adaptations, rolling over those patients on the most promising doses. The evaluation of changes in liver fat and other noninvasive biomarkers of liver function, inflammation, and fibrosis may help in decision making during the IA after a prespecified period. At this point, efficacy and futility analysis allows adaptations (i.e., drop‐off study arm/s, randomize more patients, stop the study if a safety concern arises). The continuous follow‐up allows the evaluation of changes in liver histology in the selected dose/s. Abbreviations: EOS, end of study; IA, interim analysis; N, number of subjects per study arm; R, randomized patients.
With a biopsy‐proven NASH population enrolled into an adaptive POC/phase 2b study, an interim analysis allows for an early look at the data and can result in preplanned adaptations to the study (e.g., discontinuation of study arms, randomization of more patients, stopping the study). This early or interim look at the data also allows for a futility analysis of the entire study or specific doses of drug, benefiting both patients and the sponsor. Furthermore, for studies that continue with fewer selected doses of drug, patients in the discontinued arms can be switched to the doses selected. While these “switched” patients are not intended to be included in the efficacy analysis, they do provide additional safety information, and the patients receive continued evaluations throughout the study. For all patients, the continuous follow‐up enables evaluations of liver histology at later time points and the potential use of a therapeutic index function in the analysis of the final data. This continuity of follow‐up in the POC/phase 2b study avoids the need to recruit a new set of patients with biopsy‐confirmed NASH in a separate study.
ADAPTIVE DESIGN IN THE PIVOTAL TRIAL
The required two‐step approach (i.e., an initial accelerated or conditional approval followed by a final approval after confirming improvement in outcomes) creates an opportunity to use an adaptive design with potential preplanned adaptations after the interim analysis and the use of a utility function in the analysis.
For phase 3/4 NASH trials in which the intent is for the drug to be approved for marketing based on a surrogate endpoint, the sample size needs to be planned by considering the interim alpha (control of the false‐positive rate) at the end of phase 3 as well as the final alpha at phase 4. Although the primary endpoints for both phase 3 and phase 4 are supposed to be strongly correlated, we recommend splitting the overall study alpha (e.g., 0.01 and 0.04) into two phases to ensure type I error control of the entire study as well as assessments of other potential key secondary endpoints. Because accelerated approval in NASH trials is considered on the basis of a (nonvalidated) surrogate endpoint and therefore some doubt remains about its validity, we recommend that a much smaller alpha is allocated to the first test relative to the second one because the phase 3 portion of the study will lead to the accelerated or conditional approval.
In this phase 3/4 two‐stage adaptive design (Fig. 2), the proposed utility function results in a composite endpoint (which accounts for both improvement in the disease activity and resolution of NASH with no worsening of fibrosis or an actual improvement in liver fibrosis) for a responder analysis. A co‐primary endpoint of improvement in fibrosis can be incorporated, and thus the proposed utility function becomes a vector of endpoints. At the second stage, the study endpoint of interest could be composed of all‐cause mortality, progression to liver cirrhosis, and cirrhosis decompensation events. In this case, the proposed utility function could result in a composite endpoint to account for these criteria for a responder analysis. The derived endpoints based on the proposed utility function at different stages are expected to be correlated.
Figure 2.
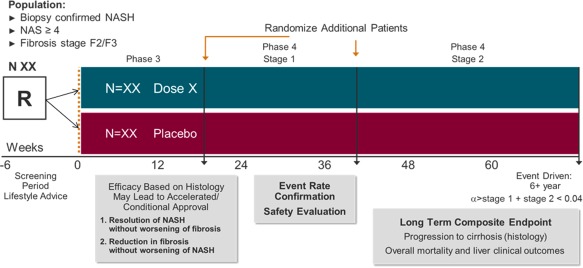
Phase 3/4 adaptive design. A single seamless adaptive trial design allows for continuous exposure and long‐term follow‐up. A therapeutic index (or utility) function can be adopted to link all NASH endpoints at different stages. Furthermore, different prespecified weights can be allocated in the function. Endpoints at the interim analysis are: i) resolution of NASH by histology without worsening of fibrosis and/or ii) improvement in fibrosis without worsening of NASH. If positive, a long‐term follow‐up to confirm efficacy in reduction in clinical outcome is mandatory. It is important to ensure type 1 error control. At present, because marketing authorization is based on a surrogate endpoint that is “reasonably likely to predict benefit on morbidity or mortality,” based on epidemiologic data but not “surrogates that are validated by definitive studies,” a smaller alpha is allocated to the first test compared to the second one (e.g., 0.01 and 0.04, respectively). Abbreviations: N, number of subjects per study arm; R, randomized patients.
The total number of events in phase 4 can be estimated assuming an average progression rate of 5%‐7% based on the natural history cohorts in patients with NASH with moderate or advanced fibrosis.7, 13, 14, 28, 29 However, the final statistical inference will be evaluated when a certain number of events are reached. At this stage, an interim analysis would allow the following decisions to be considered: (1) terminate the trial due to futility; (2) continue the trial to the final analysis without any changes; (3) continue the trial to the final analysis and increase the target number of events in the primary analysis. Additionally, an analysis of the interim safety data is recommended and should be performed by an independent unblinded data safety monitoring committee.
A phase 2/3/4 seamless adaptive design (Fig. 3) could also be considered. In these trials, one could adopt the commonly used seamless design but control the overall alpha after the alpha has been adjusted for phase 3 and phase 4. For this type of phase 2/3/4 adaptive design, sample size is often selected to power the study for detecting a meaningful treatment effect at the end of phase 3. Therefore, there is usually not sufficient power for critical decision making (e.g., detecting clinically meaningful difference for dose selection or dropping the inferior treatment groups) at earlier stages. In this case, a precision analysis is recommended to ensure that the selected dose has achieved statistical significance (i.e., the observed difference is not by chance alone) based on the prospectively defined significance level required at the interim analysis. For example, the following criteria are often considered to facilitate decisions: 1) the dose with highest confidence level for achieving statistical significance will be selected; 2) the doses with confidence levels for achieving statistical significance less than 75% will be dropped.
Figure 3.
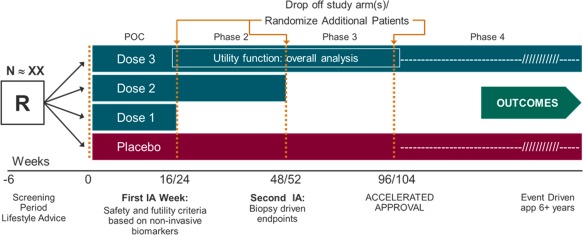
Phase 2/3/4 adaptive design. A single seamless 2/3/4 adaptive trial design allows adaptations, continuous exposure, and long‐term follow‐up. Endpoints Ad interim analysis are reduction of at least 2 points in NAS, resolution of NASH by histology without worsening of fibrosis, and/or improvement in fibrosis without worsening of NASH. One (the most promising dose) or two doses may continue to the next phase. A postmarketing phase 4 with demonstration of improvement in clinical outcomes will lead to final marketing authorization. Because only one trial would lead to approval, a very small overall alpha (i.e., <0.001) is recommended to ensure proper control of a type I error. Abbreviations: IA, interim analysis; N, number of subjects per study arm; R, randomized patients.
It should also be noted that precision assessment in terms of the confidence interval approach is operationally equivalent to hypotheses testing for comparing means. Of note, if the interim analysis conducted at phase 2 is mainly to assess the dose response and to further study the highest dose in subsequent phases, a trend test, such as the Cochran–Armitage test, may be considered. However, to protect the trial integrity due to the unblinding of the interim data, a very small alpha (e.g., 0.0001) needs to be allocated.
Adaptive Design, the Regulatory Framework
Both the FDA and the EMA have released documents describing the use of adaptive designs in clinical studies. In 2007, the EMA published a reflection paper on methodological issues in confirmatory clinical trials planned with an adaptive design,30 and in 2010 the FDA released draft guidance on adaptive designs for drugs and biologics.20 In addition, the Japanese Pharmaceuticals and Medical Devices agency in Japan has published papers describing its experiences with companies discussing adaptive designs.31 Fortunately, the concept for the use of adaptive designs appears similar among these regulatory agencies, with an emphasis that the designs must be supported by sound rationale.
Conclusions
The increasing prevalence, associated health burden, and lack of approved therapy for patients with NASH represent a substantial unmet medical need. At the same time, statistical methods in seamless adaptive clinical designs are evolving and becoming better understood. This development paradigm has the potential to bring additional therapeutic options to patients earlier without compromising the quality of the evidence needed to establish the efficacy and safety of therapeutic agents. Maintaining patients in long‐term clinical trials by progressing them through the various stages of the development program offers prolonged drug exposure with the flexibility of enabling additional patients to be enrolled at each stage of the study to support robust statistical analyses. Additionally, for a condition like NASH where there is a limited number of biopsy‐proven study subjects, adaptive methods can reduce the need to find full cohorts of new patients with histologically confirmed NASH. The use of a scalar or vector utility function may also allow all endpoints to be linked for the overall analysis, providing a reliable method to assess the treatment effect. Nevertheless, adaptive designs have some limitations. Due to potential operational biases, an adaptive design program needs to be accepted by global authorities prior to initiation. Additionally, the application of an adaptive strategy for registration does not avoid the need for multiple liver biopsies, and long‐term follow‐up of phase 2/3/4 seamless trials may limit the number of patients who are willing to participate. The need for type 1 error control with a very small alpha requires a large number of patients to be enrolled. Finally, it is important to note that trial designs are based on many factors and an adaptive design strategy may not be appropriate for all NASH development programs.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1079/suppinfo.
Supporting Information
Acknowledgment
The authors gratefully acknowledge the thoughtful critique and comments on the manuscript by Dr. Victor Crentsil (FDA).
Potential conflict of interest: Dr. Filozof is employed by and owns stock in Covance. Dr. Goldstein is employed by and owns stock in Covance. Dr. Sanyal is president of Sanyal Biotechnologies. He advises and received grants from Intercept, Merck, Bristol‐Myers Squibb, Immuron, Novo Nordisk, Cirius, and Gilead. He advises Galectin, Boehringer Ingelheim, ChemomAb, Afimmune, Protalix, and Nitto Denko. He received grants from Tobira, Allergan, AstraZeneca, and Shire. He owns stock in Genfit, Arkana, Tiziana, Indalo, Durect, Exhalenz, and HemoShear.
The discussions and recommendations made herein are the opinions of the authors and do not represent the official position or opinions of the FDA.
REFERENCES
- 1. Sanyal AJ. NASH: A global health problem. Hepatol Res 2011;41:670‐674. [DOI] [PubMed] [Google Scholar]
- 2. Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol 2011;9:524‐530.e1. [DOI] [PubMed] [Google Scholar]
- 3. Filozof C, Goldstein BJ, Williams RN, Sanyal A. Non‐alcoholic steatohepatitis: limited available treatment options but promising drugs in development and recent progress towards a regulatory approval pathway. Drugs 2015;75:1373‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Misra VL, Khashab M, Chalasani N. Nonalcoholic fatty liver disease and cardiovascular risk. Curr Gastroenterol Rep 2009;11:50‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global Epidemiology of non‐alcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 6. Setiawan VW, Stram DO, Porcel J, Lu SC, Le Marchand L, Noureddin M. Prevalence of chronic liver disease and cirrhosis by underlying cause in understudied ethnic groups: the multiethnic cohort. Hepatology 2016;64:1969‐1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanyal AJ, Friedman SL, McCullough AJ, Dimick‐Santos L; American Association for the Study of Liver Diseases; United States Food and Drug Administration . Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases‐U.S. Food and Drug Administration joint workshop. Hepatology 2015;61:1392‐1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sanyal A, Brunt E, Kleiner D, Kowdley KV, Chalasani N, Lavine JE, et al. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011;54:344‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Valenti L, Al‐Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, et al. Homozygosity for the patatin‐like phospholipase‐3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1209‐1217. [DOI] [PubMed] [Google Scholar]
- 11. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 12. Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander‐Tetri BA; NASH Clinical Research Network (CRN) . Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology 2011;53:810‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol 2015;13:643‐54.e1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Argo CK, Northup PG, Al‐Osaimi AM, Caldwell SH. Systematic review of risk factors for fibrosis progression in non‐alcoholic steatohepatitis. J Hepatol 2009;51:371‐379. [DOI] [PubMed] [Google Scholar]
- 15. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta‐analysis. Hepatology 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. ClinicalTrials.gov. Phase 3 study to evaluate the efficacy and safety of elafibranor versus placebo in patients with nonalcoholic Steatohepatitis (NASH) (RESOLVE‐IT). https://clinicaltrials.gov/ct2/show/NCT02704403?term=genfit&rank=3. Accessed October 2016.
- 17. ClinicalTrials.gov. Randomized global phase 3 study to evaluate the impact on NASH with fibrosis of obeticholic acid treatment (REGENERATE). https://clinicaltrials.gov/ct2/show/NCT0254 8351?term=intercept+NASH&rank=1. Accessed October 2016.
- 18.Guidance for Industry. Adaptive design clinical trials for drugs and biologics. Food and Drug Administration . February 2010. Available at: http://www.fda.gov/downloads/Drugs/.../Guidances/ucm201790.pdf.
- 19. Chow SC, Lin M. Analysis of two‐stage adaptive seamless trial design. Pharm Anal Acta 2015;6:341. [Google Scholar]
- 20. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research (CBER) . Guidance for industry expedited programs for serious conditions – drugs and biologics. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/U CM358301.pdf. Published May 30, 2014.
- 21.European Medicines Agency launches adaptive licensing pilot project [press release]. European Medicines Agency/430892/2013; March 19, 2014. Available at: www.ema.europa.eu.
- 22. Chow SC, Tu YH. On two‐stage seamless adaptive design in clinical trials. J Formos Med Assoc 2008;107(12 Suppl.):52‐60. [DOI] [PubMed] [Google Scholar]
- 23. Chow SC, Corey R. Benefits, challenges and obstacles of adaptive clinical trial designs. Orphanet J Rare Dis 2011;6:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu Q, Chow SC, Tse SK, Chi Y, Yang LY. Sample size estimation based on event data for a two‐stage survival adaptive trial with different durations. J Biopharm Stat 2009;19:311‐323. [DOI] [PubMed] [Google Scholar]
- 25. Lu Q, Tse SK, Chow SC. Analysis of time‐to‐event data under a two‐stage survival adaptive design in clinical trials. J Biopharm Stat 2010;20:705‐719. [DOI] [PubMed] [Google Scholar]
- 26. Lu Q, Tse SK, Chow SC, Lin M. Analysis of time‐to‐event data with nonuniform patient entry and loss to follow‐up under a two‐stage seamless adaptive design with weibull distribution. J Biopharm Stat 2012;22:773‐784. [DOI] [PubMed] [Google Scholar]
- 27. Buzzetti E, Lombardi R, De Luca L, Tsochatzis EA. Noninvasive assessment of fibrosis in patients with nonalcoholic fatty liver disease. Int J Endocrinol 2015;2015:343828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long‐term follow‐up of patients with NAFLD and elevated liver enzymes. Hepatology 2006;44:865‐873. [DOI] [PubMed] [Google Scholar]
- 29. Pagadala MR, McCullough AJ. The relevance of liver histology to predicting clinically meaningful outcomes in nonalcoholic steatohepatitis. Clin Liv Dis 2012;16:487‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. European Medicines Agency . Reflection paper on methodological issues in confirmatory clinical trials planned with an adaptive design. Committee for Medicinal Products for Human Use (CHMP). CHMP/EWP/2459/02. Available at: http://www.emea.europa.eu. Published October 18, 2007.
- 31. Ando Y, Hirakawa A, Uyama Y. Adaptive clinical trials for new drug applications in Japan. Eur Neuropsychopharmacol 2011;21:175‐179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1079/suppinfo.
Supporting Information