

Abstract
Modifying the entry of pyruvate into mitochondria may provide a unique approach to treat metabolic disease. The pharmacology of a new class of insulin sensitizers directed against a newly identified mitochondrial target may treat many aspects of nonalcoholic steatohepatitis, including fibrosis. This commentary suggests treating nonalcoholic steatohepatitis through a newly identified mechanism consistent with pathophysiology. (Hepatology Communications 2017;1:193‐197)
Abbreviations
- MPC
mitochondrial pyruvate carrier
- mTOT
thiazolidinediones
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PPARγ
peroxisome proliferator‐activated receptor gamma
- TZD
thiazolidinedione
Classical drug discovery programs have sought to design compounds for defined cellular targets in a single disease state. A new mitochondrial drug target has been identified that provides an opportunity to treat metabolic diseases by changing the way pyruvate is metabolized, and this may have implications for treating diseases as diverse as Parkinson's disease and liver disease. Mitochondrial target of thiazolidinediones (mTOT)1, 2, 3 modulators have been selected for their ability to bind to and modulate the activity of the mTOT, which was subsequently identified as the mitochondrial pyruvate carrier (MPC). The MPC is a heterologous complex composed of two proteins, MPC1 and MPC2, in the inner mitochondrial membrane.4, 5 This complex is required for pyruvate to enter the mitochondrial matrix, where it is metabolized, from its site of synthesis in the cytosol. Recent work has suggested the utility of targeting the MPC for treatment of a variety of metabolic and inflammatory diseases, including diabetes,1, 6, 7 Parkinson's and other neurodegenerative diseases,8, 9 and now nonalcoholic fatty liver disease (NAFLD).10
Most researchers agree with the definition of NAFLD as the ectopic deposition of lipid in the liver parenchyma in the absence of excessive alcohol use. It is also clear that in some patients NAFLD progresses to nonalcoholic steatohepatitis (NASH), which increases the risk of cirrhosis, liver failure, and hepatocellular carcinoma. At the current time, NASH is diagnosed based on liver biopsies showing characteristic histologic changes, including fat deposition, inflammation, hepatocyte ballooning, and importantly in terms of prognosis, fibrotic scarring.11, 12 While there are many treatments in development that specifically address individual aspects of liver disease, epidemiologic studies have defined a strong linkage between overnutrition and incidence of NAFLD, and this linkage is driving the prevalence of this disease of the liver to pandemic proportions.13 Despite considerable efforts, there are currently no approved treatments for NAFLD or NASH.
In evaluating potential approaches that might be effective to treat NASH, it is important to consider the pathophysiology. At its core, NAFLD is caused by fat accumulation that is likely driven by nutrient excess. At the same time, the progression to NASH in mice and humans has been tightly linked to a number of factors, including insulin resistance,14, 15 abnormal mitochondrial function,16, 17, 18, 19 oxidative damage, and hepatic inflammation, that promote a state conducive to necropoptosis and replacement of dead hepatocytes with a collagen matrix secreted by hepatic stellate cells in response to these stimuli.20, 21 It is interesting to note that the entire spectrum of this pathophysiology from insulin resistance to fibrotic scarring may involve dysfunctional metabolism driven by overnutrition. Moreover, the pathology might be regulated by the delivery of pyruvate by the MPC.
We have recently shown that genetic or pharmacologic targeting of the MPC, either by selective knockout of the MPC in hepatic parenchymal cells or by using a new‐generation insulin sensitizer MSDC‐0602K, was able to reduce the NASH pathology in a mouse model fed a diet high in transfat, cholesterol, and fructose.10 Indeed, MSDC‐0602K treatment both prevented and reversed the development of NASH symptoms in this mouse model, including attenuation of fibrosis in the continued presence of the aggressive diet treatment producing the pathology. The antifibrotic effects seen in these mice involve both direct effects on parenchymal and mesenchymal (stellate) cells of the liver. The overall pharmacology was also suggested to include changes in the composition of circulating exosomes secreted by hepatocytes to influence hepatic stellate cell activation. MSDC‐0602 also potently stimulates secretion of adiponectin, which has been suggested to have beneficial effects on NASH endpoints, by adipocytes. Thus, there seem to be both direct and indirect effects of MSDC‐0602K to impact the development of fibrosis (Fig. 1).
Figure 1.
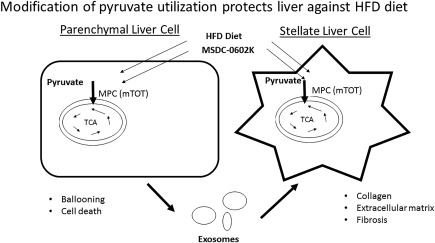
Modification of pyruvate use protects the liver against an HFC diet. A diet rich in fatty acids, fructose, and cholesterol triggers a NASH phenotype in mice. This is prevented and reversed by MSDC‐0602K treatment. There are both direct effects on parenchymal cells and stellate cells. MSDC‐0602K treatment and MPC knockout also affect factors secreted by hepatocytes in exosomes that stimulate a change to a fibrotic phenotype in stellate cells. The overall pharmacology of MSDC‐0602K involves effects in multiple cell types within the liver and in other tissues. Abbreviations: HFC, high‐fat‐cholesterol diet; HFD, high‐fat diet; TCA, tricarboxylic acid cycle.
MSDC‐0602K is a member of a new class of insulin sensitizers called mTOT modulators1 from a structural class of compounds known as thiazolidinediones (TZDs). The first‐generation insulin sensitizers, such as pioglitazone and rosiglitazone (currently marketed as a second‐line treatment for type 2 diabetes), are agonists with varying potencies for the nuclear receptor peroxisome proliferator‐activated receptor gamma (PPARγ?.22 Although there are beneficial effects attributed to PPARγ agonism, a series of PPARγ‐knockout studies have clearly shown that the side effects that limit the use of pioglitazone and rosiglitazone are driven by PPARγ.23, 24 Until recently, it has been less well appreciated that all active TZDs also attenuate mitochondrial pyruvate uptake through the MPC2 in a manner that is separable from their PPARγ agonist activity.25 It is our opinion that the focus on the PPARγ agonist activity has reduced the chances of finding TZDs or other compounds that mainly affect the mitochondrial target but avoid direct activation of PPARγ? which limits doses that can be taken to the clinic.25 We and others have hypothesized that there may be a beneficial TZD pharmacology that is separable from PPARγ agonism and that this realization could change the drug discovery approach for the treatment of metabolic diseases.23, 24 Recent data point to the modification of pyruvate metabolism as a site of this pharmacology.
Relevant to this discussion is the comparison of the two antidiabetic TZDs that are currently in use to treat type 2 diabetes and that have also been evaluated in subjects with NASH. Clinical studies of the approved doses of pioglitazone in patients with NASH have shown significant resolution of NASH pathology, including fibrosis.26, 27 On the other hand, rosiglitazone, which is a 5‐fold to 10‐fold more potent PPARγ agonist compared to pioglitazone,22 improved some NASH endpoints but failed to significantly affect fibrosis.27, 28, 29, 30, 31 The dissociation between the affinity for PPARγ and the effectiveness in treating NASH, and particularly hepatic fibrosis, could be explained by the failure of rosiglitazone to achieve adequate effects on the mitochondrial target MPC. The attenuation of the activity of the pyruvate carrier seems essential to this effect; in the studies conducted in rodents, either MSDC‐0602K or genetic MPC deletion consistently attenuated stellate cell activation and histologic evidence of fibrosis. These data and this rationale suggest that the PPARγ‐sparing MSDC‐0602K should have a preserved pharmacology for treating NASH with a diminished risk of PPARγ‐driven side effects in human patients with NASH.
Based on this rationale and efficacy in rodent models,6, 10 a phase 2 clinical trial has been initiated to evaluate the efficacy of MSDC‐0602K. This study is a 12‐month, double‐blind, placebo‐controlled evaluation of three exposures of MSDC‐0602K to determine the potential of this PPARγ‐sparing mTOT modulator to resolve NASH (EMMINENCE; NCT02784444; https://clinicaltrials.gov/ct2/show/NCT02784444). Multiple noninvasive measures are included to determine whether these provide a correlation with the histologic improvement of the liver produced by this pharmacology. Noninvasive measures include insulin sensitivity (e.g., homeostasis model assessment of insulin resistance) together with a variety of standard and novel biomarkers. This clinical study together with ongoing studies in preclinical models will provide a better understanding of how mTOT modulator pharmacology impacts NASH and should enable both the development and the practical use of such agents going forward. The preclinical approaches being used to further elucidate these mechanisms include newly available animal models with tissue‐specific knockout of the MPC.7, 8, 9, 10
There are many potential treatments in development for NASH. Current drug development is trained on several specific targets directed at specific symptoms along the natural history of the disease, including lipid synthesis, inflammation, and fibrosis.32, 33 The new data suggest that modification of the usage of pyruvate could be useful at several steps along the progression of the disease from insulin resistance6, 7 to reduction of ballooning and fibrosis.10 Such a treatment modality could be useful on its own or as a component of combination therapy.
It should be noted that knockout or knockdown of the MPC results in significant compensatory changes in metabolism,7, 34 and thus compensatory metabolic pathways (increased amino acid and/or fatty acid metabolism) could reduce the efficacy of these compounds for treating NASH. However, knockout of the MPC proteins is likely to have different effects than attenuation of function by TZDs. Clearly, metabolomics studies should be performed after chronic MPC inhibition with TZDs. Another possible limitation of inhibiting MPC activity is that the MPC proteins are significantly down‐regulated in cancer cell lines, and regulation of pyruvate use is likely important in stem cells and solid tumor expansion.34, 35 In this respect, it is reassuring to know that in over 20 years of clinical use of pioglitazone, which also works through attenuation of the MPC,24 hepatocellular cancer incidence appears to be reduced.36, 37
At the present time, there are numerous questions that remain to be answered regarding this newly understood pharmacology. For example, how does the attenuation of pyruvate metabolism result in the downstream events resulting in increased insulin sensitivity, improved lipid metabolism, and reduction of hepatic fibrosis? Can the pharmacology be tied to biomarkers measurable in the clinic? Are there changes in the epigenomic regulation that can be related to the pathophysiology? What are the factors in the exosomes that transmit signals to the stellate cells, thus regulating fibrosis10?
In summary, the study of the pleotropic pharmacology of the first generation TZDs first identified empirically over 35 years ago has led to the discovery of a novel mitochondrial target and a new class of drugs called mTOT modulators. The mechanism of action for these compounds involves alterations in mitochondrial pyruvate metabolism, which leads not only to increased insulin sensitivity but also to a modification of a wide variety of cellular processes, such as differentiation, autophagy, and suppression of inflammation, all as a result of the coordinated responses involving nutrient signals.24 Although MSDC‐0602K is currently in clinical development for the treatment of NASH, one could imagine that this pharmacology would be applicable to pathologic conditions that typically occur along the natural pathology of NAFLD, as shown in Fig. 2. Most importantly, the translational preclinical and clinical studies now in progress should provide the noninvasive biomarkers needed to facilitate the rational and effective use of these compounds in clinical practice. Ongoing studies should show whether modification of pyruvate metabolism might be used to effectively treat NASH and, perhaps eventually, NAFLD in general.
Figure 2.
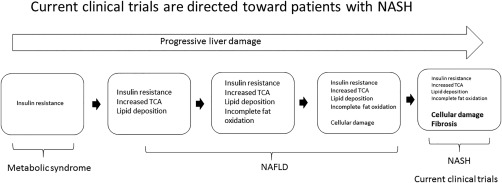
Treating the broad spectrum of NAFLD/NASH. Many current clinical trials are focused on treating specific endpoints of NASH liver injury. MSDC‐0602K could not only reverse NASH fibrosis but also prevent early stages of disease progression by affecting insulin resistance. Abbreviation: TCA, tricarboxylic acid cycle.
Authors names in bold designate shared co‐first authorship.
Potential conflict of interest: J.R.C. and W.G.M. have ownership in Metabolic Solutions Development Company (MSDC), which has interests in MSDC‐0602K; this compound is in development by Cirius Therapeutics, formerly known as Octeta Therapeutics, for the treatment of nonalcoholic steatohepatitis. J.R.C. is also an employee of Cirius Therapeutics.
REFERENCES
- 1. Colca JR, VanderLugt JT, Adams WJ, Shashlo A, McDonald WG, Liang J, et al. Clinical proof‐of‐concept study with MSDC‐0160, a prototype mTOT‐modulating insulin sensitizer. Clin Pharmacol Ther 2013;93:352‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Aad Sci USA 2013;110:5422‐5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Colca JR, McDonald WG, Cavey GS, Cole SL, Holewa DD, Brightwell‐Conrad AS, et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)‐‐relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS ONE 2013;8:e61551:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012;337:96‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012;337:93‐96. [DOI] [PubMed] [Google Scholar]
- 6. Chen Z, Vigueira PA, Chambers KT, Hall AM, Mitra MS, Qi N, et al. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator‐activated receptor gamma‐sparing thiazolidinedione. J Biol Chem 2012;287:23537‐23548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, et al. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate‐alanine cycling. Cell Metab 2015;22:682‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghosh A, Tyson T, George S, Hildebrandt EN, Steiner JA, Madaj Z, et al. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson's disease. Sci Transl Med 2016;8:368ra174. [DOI] [PubMed] [Google Scholar]
- 9. Shah RC, Matthews DC, Andrews RD, Capuano AW, Fleischman DA, VanderLugt JT, et al. An evaluation of MSDC‐0160, a prototype mTOT modulating insulin sensitizer, in patients with mild Alzheimer's disease. Curr Alzheimer Res 2014;11:564‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McCommis KS, Hodges WT, Brunt EM, Nalbantoglu I, McDonald WG, Holley C, et al. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 2016; doi:10.1002/hep.29025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686‐690. [DOI] [PubMed] [Google Scholar]
- 12. Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander‐Tetri BA; NASH Clinical Research Network (CRN) . Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology 2011;53:810‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rinella M, Charlton M. The globalization of nonalcoholic fatty liver disease: prevalence and impact on world health. Hepatology 2016;64:19‐22. [DOI] [PubMed] [Google Scholar]
- 14. Tilg H, Moschen AR, Roden, M . NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol 2017;14:32‐42. [DOI] [PubMed] [Google Scholar]
- 15. Lomonaco R, Bril F, Portillo‐Sanchez P, Ortiz‐Lopez C, Orsak B, Biernacki D, et al. Metabolic impact of nonalcoholic steatohepatitis in obese patients with type 2 diabetes. Diabetes Care 2016;39:632‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non‐alcoholic fatty liver is lost in steatohepatitis. Cell Metab 2015;21:739‐746. [DOI] [PubMed] [Google Scholar]
- 17. Gusdon A.M., Song K.X, Qu S. Nonalcoholic fatty liver disease: pathogenesis and therapeutics from a mitochondria‐centric perspective. Oxid Med Cell Longev 2014;2014:637027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patterson RE, Kalavalapalli S, Williams CM, Nautiyal M, Mathew JT, Martinez J, et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am J Physiol Endocrinol Metab 2016;310:E484‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Satapati S, Kucejova B, Duarte JA, Fletcher JA, Reynolds L, Sunny NE, et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest 2015;125:4447‐4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friedman SL. Liver fibrosis in 2012: convergent pathways that cause hepatic fibrosis in NASH. Nat Rev Gastroenterol Hepatol 2013;10:71–72. [DOI] [PubMed] [Google Scholar]
- 21. Wallace MC, Friedman SL, Mann, DA . Emerging and disease‐specific mechanisms of hepatic stellate cell activation. Semin Liver Dis 2015;35;107‐118. [DOI] [PubMed] [Google Scholar]
- 22. Lehmann JM, Moore LB, Smith‐Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator‐activated receptor gamma (PPAR gamma). J Biol Chem 1995;270:12953‐12956. [DOI] [PubMed] [Google Scholar]
- 23. Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 2014;20:573‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Colca JR. The TZD insulin sensitizer clue provides a new route into diabetes drug discovery. Expert Opin Drug Discov 2015;10:1259‐1270. [DOI] [PubMed] [Google Scholar]
- 25. Colca JR, Tanis SP, McDonald WG, Kletzien RF. Insulin sensitizers in 2013: new insights for the development of novel therapeutic agents to treat metabolic diseases. Expert Opin Investig Drugs 2014;23:1‐7. [DOI] [PubMed] [Google Scholar]
- 26. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz‐Lopez C. et al. Long‐term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med 2016;165:305‐315. [DOI] [PubMed] [Google Scholar]
- 27. Musso G, Cassader M, Paschetta E, Gambino R. Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: a meta‐analysis. JAMA Intern Med 2017; doi: 10.1001/jamainternmed.2016.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neuschwander‐Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR‐gamma ligand rosiglitazone. Hepatology 2003;38;1008‐1017. [DOI] [PubMed] [Google Scholar]
- 29. Caldwell SH, Patrie JT, Brunt EM, Redick JA, Davis CA, Park SH, et al. The effects of 48 weeks of rosiglitazone on hepatocyte mitochondria in human nonalcoholic steatohepatitis. Hepatology 2007;46:1101‐1107. [DOI] [PubMed] [Google Scholar]
- 30. Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann‐Heurtier A, Serfaty L, et al; Lido Study Group . Rosiglitazone for nonalcoholic steatohepatitis: one‐year results of the randomized placebo‐controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008;135:100‐110. [DOI] [PubMed] [Google Scholar]
- 31. Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G, et al; LIDO Study Group . Long‐term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010;51:445‐453. [DOI] [PubMed] [Google Scholar]
- 32. Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non‐alcoholic fatty liver disease. Gut 2017;66:180‐190. [DOI] [PubMed] [Google Scholar]
- 33. Filozof C, Goldstein BJ, Williams RN, Sanyal A. Non‐alcoholic steatohepatitis: limited available treatment options but promising drugs in development and recent progress towards a regulatory approval pathway. Drugs 2015;75:1373‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, et al. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol Cell 2014;56:425‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schell JC, Olson KA, Jiang L, Hawkins AJ, Van Vranken JG, Xie J, et al. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol Cell 2014;56:400‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lai SW, Chen PC, Liao KF, Muo CH, Lin CC, Sung FC. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti‐diabetic therapy: a population‐based cohort study. Am J Gastroenterol 2012;107:46‐52. [DOI] [PubMed] [Google Scholar]
- 37. Sumie S, Kawaguchi T, Kawaguchi A, Kuromatsu R, Nakano M, Satani M, et al. Effect of pioglitazone on outcome following curative treatment for hepatocellular carcinoma in patients with hepatitis C virus infection: a prospective study. Mol Clin Oncol 2015;3:115‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]