

Abstract
The pathogenesis of primary sclerosing cholangitis (PSC) and the mechanistic link to inflammatory bowel disease remain ill‐defined. Ectonucleoside triphosphate diphosphohydrolase‐1 (ENTPD1)/clusters of differentiation (CD) 39, the dominant purinergic ecto‐enzyme, modulates intestinal inflammation. Here, we have explored the role of CD39 in biliary injury and fibrosis. The impact of CD39 deletion on disease severity was studied in multidrug resistance protein 2 (Mdr2)–/– and 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine mouse models of sclerosing cholangitis and biliary fibrosis. Antibody‐mediated CD8+ T‐cell depletion, selective gut decontamination, experimental colitis, and administration of stable adenosine triphosphate (ATP) agonist were performed. Retinoic acid‐induced gut imprinting on T cells was studied in vitro. Over half of Mdr2–/–;CD39–/– double mutants, expected by Mendelian genetics, died in utero. Compared to Mdr2–/–;CD39+/+, surviving Mdr2–/–;CD39–/– mice demonstrated exacerbated liver injury, fibrosis, and ductular reaction. CD39 deficiency led to a selective increase in hepatic CD8+ T cells and integrin α4β7, a T‐cell gut‐tropism receptor. CD8+ cell depletion in Mdr2–/–;CD39–/– mice diminished hepatobiliary injury and fibrosis. Treatment with antibiotics attenuated, whereas dextran sulfate sodium‐induced colitis exacerbated, liver fibrosis in Mdr2–/– mice. Colonic administration of αβ‐ATP into CD39‐sufficient Mdr2–/– mice triggered hepatic CD8+ cell influx and recapitulated the severe phenotype observed in Mdr2–/–;CD39–/– mice. In vitro, addition of ATP promoted the retinoic acid‐induced imprinting of gut‐homing integrin α4β7 on naive CD8+ cells. CD39 expression was relatively low in human normal or PSC livers but abundantly present on immune cells of the colon and further up‐regulated in samples of patients with inflammatory bowel disease. Conclusion: CD39 deletion promotes biliary injury and fibrosis through gut‐imprinted CD8+ T cells. Pharmacological modulation of purinergic signaling may represent a promising approach for the treatment of PSC. (Hepatology Communications 2017;1:957–972)
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- BIDMC
Beth Israel Deaconess Medical Center
- CCL25
chemokine (C‐C motif) ligand 25
- CCR9
chemokine (C‐C motif) receptor 9
- CD
clusters of differentiation
- DSS
dextran sulfate sodium
- FVB
Friend virus B‐type
- IBD
inflammatory bowel disease
- IL
interleukin
- MAdCAM‐1
mucosal vascular addressin cell adhesion molecule 1
- Mdr2
multidrug resistance protein 2
- mRNA
messenger RNA
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- RA
retinoic acid
- WT
wild type
Introduction
Primary sclerosing cholangitis (PSC) is a progressive cholestatic liver disease characterized by inflammation and fibrosis of the intrahepatic and extrahepatic bile ducts and leads to biliary cirrhosis and end‐stage liver disease. Patients with PSC are at increased risk of developing cholangiocarcinoma, hepatocellular carcinoma, and colorectal cancer.1 Effective treatments are lacking, and liver transplantation remains the only therapeutic option for patients with end‐stage PSC.2
While the etiology of PSC remains unknown, the associations with inflammatory bowel disease (IBD) are well established, with at least 70% of patients with PSC also suffering from IBD.3 After transplantation, PSC recurrence in liver graft can be high (up to 30%) and reduced in patients who have undergone colectomy before or during liver transplantation. However, recurrent PSC is not completely prevented, putatively due to the persistence of gut‐primed long‐lived memory T cells.4 Based on these and other observations, PSC is hypothesized by some to be triggered by colitis, where gut‐primed effector T cells aberrantly home to the liver and trigger portal inflammation in PSC.5 While genome‐wide association studies suggest that IBD in patients with PSC is genetically distinct from non‐PSC‐associated IBD,6 it is likely that targeting or modulation of intestinal inflammation may result in the alleviation of liver disease in PSC.
Gut tropism of clusters of differentiation (CD)8+ T cells is determined by the expression of integrin α4β7 and chemokine (C‐C motif) receptor 9 (CCR9), which are imprinted in mesenteric lymph nodes and Peyer's patches in a retinoic acid (RA)‐dependent manner.7, 8 Aberrant hepatic expression of gut‐homing signals mucosal vascular addressin cell adhesion molecule 1 (MAdCAM‐1) and chemokine (C‐C motif) ligand 25 (CCL25) have been reported in patients with PSC, and these allow recruitment of intestinal T cells to the liver.9 However, the extent by which the (auto)immune system contributes to PSC pathogenesis remains obscure and controversial. Interestingly, PSC is associated with several autoimmunity susceptibility loci and is strongly linked to human leukocyte antigen10 but does not respond to steroids or immunosuppressive therapies.11 On the other hand, recent data link the immunomodulatory effects of vitamin D supplementation to promising clinical responses in patients with PSC.12
The purinergic system is a critical modulator of immune responses given the differential properties of proinflammatory adenosine triphosphate (ATP) and its immunosuppressive derivatives, such as adenosine. Extracellular ATP is released during inflammation and is rapidly hydrolyzed to adenosine diphosphate, adenosine monophosphate (AMP), and adenosine by an ecto‐enzymatic cascade involving CD39 and CD73. The ectonucleotidase CD39/ENTPD1 is expressed on immune cells: T cells, B cells, monocytes/macrophages, natural killer cells, and dendritic cells. More recent data indicate that CD39 expression on TH17 cells dictates immunomodulatory effects and immune plasticity in inflammatory disease.13, 14
In PSC, impaired immunoregulatory T‐cell function15 as well as increased TH17 responses16 support a mechanism mediated by T cells. Whether CD39 is involved in the regulation of underlying immune‐mediated responses leading to tissue injury in PSC is not known. Here, we studied the potential contribution of purinergic signaling in driving liver injury and fibrosis in mouse models of PSC, using genetic and pharmacologic approaches.
Materials and Methods
ANIMALS
All animals were housed in a specific‐pathogen‐free facility at Beth Israel Deaconess Medical Center (BIDMC; Boston, MA) with a 12‐hour light–dark cycle and were permitted ad libitum consumption of water and a standard chow diet unless otherwise stated. All animal procedures were approved by the Institutional Animal Care and Use Committee at BIDMC (protocols 004‐2012 and 010‐2015).
GENERATION OF CONGENIC C57BL/6.MDR2–/– MOUSE
Friend virus B‐type (FVB).multidrug resistance protein (Mdr2)–/– mice (FVB.129P2‐Abcb4tm1Bor/J; Jackson Laboratory) were backcrossed at BIDMC onto a C57Bl/6 background using an accelerated microsatellite marker‐assisted protocol (“speed congenics”). Briefly, backcrossing Mdr2–/– (FVB.129P2‐Abcb4tm1Bor/J) on the C57Bl/6J background was carried out by mating mice heterozygous for the Mdr2 deletion with wild‐type (WT) C57Bl/6J mice obtained from Jackson Laboratory (Bar Harbor, ME). A minimum of 19 male Mdr2 mutation carriers were produced in each generation, and their genome screened using microsatellite marker differences (simple sequence length polymorphisms) at Jackson Laboratory (https://www.jax.org/jax-mice-and-services/breeding-and-rederivation-services/speed-congenic) for the percentage of C57Bl/6J background. Individual carriers with the highest percentage of the C57Bl/6J genome were selected for the next round of breeding until the fully congenic state was reached at generation N5 (99.9% of C57Bl/6J as assessed by a complete genome scan). N5 heterozygotes were intercrossed to obtain fully congenic C57Bl/6J.Mdr2–/– mice (herein referred to as Mdr2–/–) as founders of our breeding colony.
CD39–/– mice on the C57Bl/6 background were described,17 and Mdr2–/–;CD39–/– double‐mutant mice (C57Bl/6 background) were generated by crossing C57Bl/6J.Mdr2–/– with C57Bl/6.CD39–/– mice to obtain F1 double‐heterozygous mutants and intercrossing F1 mice to obtain F2 progeny (n = 148). F2 progeny were genotyped at weaning, and mice of both sexes were phenotyped at age 8 weeks as described.18 For further studies and long‐term outcomes, three Mdr2–/–;CD39–/– mice (one male, two females) in the F2 generation were bred to establish a colony of double knockouts.
In vivo CD8+ T‐cell depletion was performed using a single intraperitoneal dose of anti‐CD8 antibody at 120 μg/mouse, as described,19 in age‐matched Mdr2–/–; CD39–/– mice challenged with a low‐dose cholic acid (0.1% diet) feeding for 1 week. Efficacy of depletion was confirmed by immunofluorescence in CD8‐depleted livers and fluorescence‐activated cell sorting analysis of splenocytes 3 days after anti‐CD8 antibody injection.
ATP agonist administration of stable ATP analogue αβ‐ATP (1 mg/mouse/day; Tocris Bioscience) was dispensed rectally into 5‐week‐old Mdr2–/– mice for 6 days to mimic the effects of gut flora‐derived ATP as described.20
Naive CD8+ T‐cell isolation and in vitro gut imprinting was performed as described.7 Briefly, naive CD8+ cells were purified from spleens and peripheral lymph nodes from WT mice by red blood cell lysis followed by negative immunomagnetic selection (Miltenyi Biotec, Auburn, CA) using MACS LS columns (Miltenyi Biotec). T cells were cultured with CD3/CD28 beads in a 1:1 ratio either with dimethyl sulfoxide, with 50 nM RA (Sigma, St. Louis, MO) or with 50 nM RA plus 250 μM ATP (Sigma). Surface expression of α4β7 and CCR9 were assessed after 3‐5 days of culture.
HUMAN LIVER AND COLON SAMPLES
Human colon samples were obtained from patients with IBD (ulcerative colitis, n = 7) undergoing colon resection. Human explant liver samples were obtained from patients with end‐stage PSC (n = 5) or primary biliary cholangitis (PBC; n = 4) undergoing orthotopic liver transplantation. Wedge biopsies of normal human colon (n = 7) or livers (n = 2) served as normal controls. All patients gave written informed consent, and the study protocol was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat‐sen University (Guangzhou, China). Samples were de‐identified before analysis for this study.
STATISTICAL ANALYSIS
Data were expressed as mean ± SEM and analyzed using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA). Statistical comparisons between two groups were performed using the Student t test. P < 0.05 was considered significant. Additional methods can be found in Supporting Material.
Results
CD39 DELETION AGGRAVATES LIVER INJURY AND PERIBILIARY FIBROSIS IN MOUSE MODELS
To determine the role of purinergic signaling in biliary injury and fibrosis, we crossed mice with global CD39 deletion (CD39–/–) with Mdr2–/– mice that spontaneously develop PSC‐like biliary disease (see Materials and Methods for details). The double‐null mutants (Mdr2–/–;CD39–/–) on a C57Bl/6 background experienced high rates of fetal wastage and were not born at the expected Mendelian ratio; only 43% of the expected double mutants were born (4 out of 9 expected from a total of 148 F2 pups). In surviving Mdr2–/–;CD39–/– mice, liver injury and biliary fibrosis were evaluated at the age of 8 weeks when fibrotic lesions are well developed in the Mdr2–/– model21 compared to the Mdr2–/–;CD39+/+ and Mdr2–/–;CD39+/– littermates. Sirius Red staining demonstrated substantially more advanced portal fibrosis with frequent bridging in Mdr2–/–;CD39–/– mice compared to Mdr2–/–;CD39+/+ mice (Fig. 1A). This was accompanied by a more pronounced ductular reaction in Mdr2–/–;CD39–/– mice, with a 2‐fold increase in pan‐cytokeratin‐positive ductal cell counts compared to their CD39‐sufficient littermates (P = 0.0034) (Fig. 1A,C). Hepatic collagen content increases (above the normal baseline levels of 230.9 ± 28.91 μg/liver in WT controls) were significantly greater by 48% in CD39‐deficient Mdr2–/– mice (652.8 ± 50.87 μg/liver versus 515 ± 21.48 μg/liver; P < 0.05, analysis of variance) (Fig. 1B). Mdr2–/–;CD39–/– mice had 2‐fold elevated serum alanine aminotransferase (ALT) levels compared to the control CD39‐sufficient littermates (P < 0.01), indicating increased hepatocyte injury in the double‐null mice (Fig. 1D). While Mdr2–/– female mice demonstrated slightly more severe liver disease, no sex difference was observed in Mdr2–/– mice with CD39 deficiency (not shown) compared to male littermates (Fig. 1A‐G).
Figure 1.
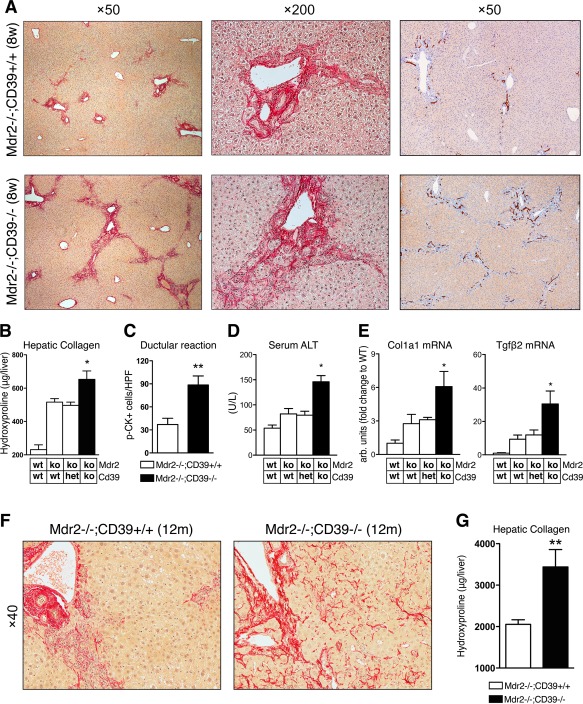
CD39 gene disruption aggravates liver injury and fibrosis in Mdr2–/– mice. Mdr2–/–;CD39–/– double‐mutant mice were generated as described in Materials and Methods, and pups of both sexes phenotyped at 8 weeks of age in comparison to their Mdr2–/–;CD39+/+ and Mdr2–/–;CD39+/– littermates (males are shown). (A) Representative pictures of connective tissue (Sirius Red, left and middle) and p‐CK (right) staining demonstrate portal fibrosis and pronounced ductular reaction in Mdr2–/– mice, respectively, which is exacerbated in Mdr2–/–;CD39–/– mice. Magnification ×50 (left and right panel) and ×200 (middle panel, portal area). (B) Liver hydroxyproline content, (C) p‐CK‐positive ductal cell counts (quantified from 10 random portal HPF area/liver at ×200, n = 3/bar; *P = 0.0034, t test), and (D) serum ALT levels are significantly increased in Mdr2–/–;CD39–/– compared to Mdr2–/– mice. (E) CD39 deletion leads to an up‐regulation of Col1a1 and Tgfβ2 mRNA expression. qRT‐PCR data are shown as fold increase compared to age‐matched WT controls. Data (B‐E,G) are mean ± SEM (n = 3‐6 mice/bar). *P < 0.05 compared to Mdr2–/–;CD39+/+ controls (ANOVA followed by Dunnett's posttest). (F) Mdr2–/–;CD39–/– mice at 12 months old demonstrate severe periductular scarring that extends into liver parenchyma (sinusoidal fibrosis), while fibrotic lesions in Mdr2–/– mice are conferred to the periportal region (Sirius Red, ×200, portal area). (G) Total hepatic hydroxyproline content in Mdr2–/–;CD39–/– mice (n = 8) compared to Mdr2–/– mice (n = 12) at the age of 12 months (**P = 0.0013, t test). Abbreviations: ANOVA, analysis of variance; het, heterozygous; HPF, high‐power field; ko, knockout; p‐CK, pan‐cytokeratine; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; Tgfβ2, transforming growth factor β2; wt, wild type.
These findings were accompanied by up‐regulation of profibrogenic messenger RNAs (mRNAs), including Col1α1 and transforming growth factor β2 (P < 0.05; Fig. 1E). Interestingly, deletion of a single functional CD39 copy in Mdr2–/–;CD39+/– mice did not affect collagen deposition, serum ALT, or gene expression.
The CD39 deletion‐related increase in disease severity persisted over the lifetime of Mdr2–/–;CD39–/– mice. No mortality was observed in adult mice of either genotype up to 1 year of age. At 12 months of age, Mdr2–/–;CD39–/– mice exhibited more severe fibrosis/cirrhosis than Mdr2–/– mice histologically and biochemically, with significantly greater increases (by 67.7%) in collagen levels (P = 0.0013, t test) (Fig. 1F,G). Notably, a much more pronounced panlobular perisinusoidal fibrosis was observed histologically throughout the liver parenchyma in aged Mdr2–/–;CD39–/– mice, consistent with a cirrhosis‐like stage in this model.18, 22
Importantly, CD39 deletion produced a similar profibrotic phenotype in a second mechanistically different model of biliary injury and putative sclerosing cholangitis, as induced by 3 weeks of 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine feeding. Here, CD39 deficiency resulted in more severe fibrosis histologically, with an almost 2‐fold increase in hepatic collagen accumulation (P = 0.0009; Supporting Fig. S1).
LOSS OF CD39 LEADS TO CD8+ T‐CELL INFILTRATION IN THE LIVER
Next, we characterized the impact of CD39 deficiency on immune cells in livers of Mdr2–/– and Mdr2–/–;CD39–/– mice. Flow cytometry analysis of intrahepatic lymphocytes showed a selective increase in CD8+ T cells from 20.7% ± 1.62% in Mdr2–/– mice to 36.4% ± 3.2% in CD39 deficient Mdr2–/– mice (P = 0.0143) (Fig. 2A). Quantitative reverse‐transcription polymerase chain reaction analysis also confirmed an increase in hepatic CD8 mRNA expression (P = 0.037), while CD3 and CD4 mRNA levels remained unchanged (Fig. 2B). Furthermore, loss of CD39 resulted in elevated hepatic expression of the gut‐tropism T‐cell marker integrin β7 (P = 0.045), while the other gut‐homing marker CCR9 demonstrated a trend (P = 0.127, not significant) toward increased expression (Fig. 2C). Immunostaining of Mdr2–/– livers revealed ectopic expression of the corresponding adhesion molecules for gut‐primed lymphocytes MAdCAM1 (ligand for integrin α4β7) and CCL25 (receptor for CCR9), with marked hepatic immunopositivity for MAdCAM1 (and to a lesser extent CCL25) irrespective of CD39 genotype (Fig. 2D). Of note, F4/80 staining revealed pronounced macrophage infiltration within periportal fibrotic lesions in Mdr2–/– mice but no apparent differences were observed due to CD39 ablation (Supporting Fig. S2).
Figure 2.
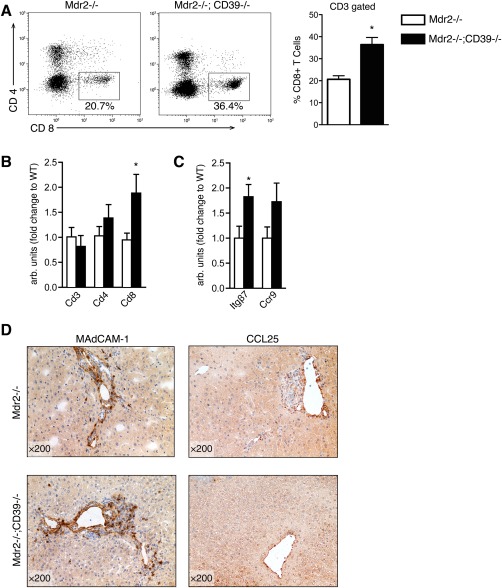
Loss of CD39 is associated with CD8+ T‐cell infiltration in the liver. (A) FACS analysis of liver‐infiltrating lymphocytes isolated from 8‐week‐old male mice. Representative dot plots of CD3‐gated hepatic lymphocytes (average values shown) and mean ± SEM for CD8+ T‐cell quantification (gated on CD3) in Mdr2–/– (n = 3) versus Mdr2–/–;CD39–/– (n = 5). (B,C) qRT‐PCR analysis of T‐cell subsets and T‐cell‐related tropism markers shown as fold relative to Mdr2–/–. (D) Immunohistochemical staining of liver for MAdCAM‐1 and CCL25. Values are presented as mean ± SEM. *P < 0.05 for Mdr2–/– versus Mdr2–/–;CD39–/– (t test). Abbreviations: FACS, fluorescence‐activated cell sorting; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction.
CD8+ T‐CELL DEPLETION AMELIORATED LIVER INJURY AND FIBROGENESIS IN Mdr2–/– MICE WITH CD39 DEFICIENCY
In order to determine whether increased CD8+ T cells are bystanders or functionally modulate severity of sclerosing cholangitis in CD39–/– mice, we performed CD8‐selective cell depletion using anti‐CD8 antibody in Mdr2–/–;CD39–/– mice. In the liver, CD8+ cells were virtually undetectable by immunofluorescence in antibody‐treated mice (Fig. 3A). Flow cytometry analysis of splenocytes 3 days after a single anti‐CD8 antibody injection confirmed highly selective and efficient (>98%) CD8+ T‐cell depletion, with no changes in CD4+ T cells (Fig. 3B). Mice with CD8+ cell depletion showed a trend to a decreased mean serum ALT level (1152 ± 84.93 U/L versus 1410 ± 146.7 U/L; P = 0.167, not significant) and a significant reduction in alkaline phosphatase (ALP; 764 ± 65.70 U/L versus 1076 ± 109.4 U/L; P = 0.04) (Fig. 3C). This was accompanied by the following significant changes in fibrosis‐related hepatic gene expression: 2‐fold increase in collagenases matrix metalloproteinase (Mmp)8 and Mmp13 and a corresponding decrease in endogenous MMP inhibitor tissue inhibitor of metalloproteinase‐1 (Timp1) (Fig. 3D). The observed “profibrolytic” gene expression signature has previously been associated with a favorable fibrosis outcome in long‐term experiments performed in Mdr2–/– mice.22 Collectively, these data suggest that liver‐infiltrating CD8+ T cells directly contribute to increased liver injury and fibrogenesis in Mdr2–/–;CD39–/– mice.
Figure 3.
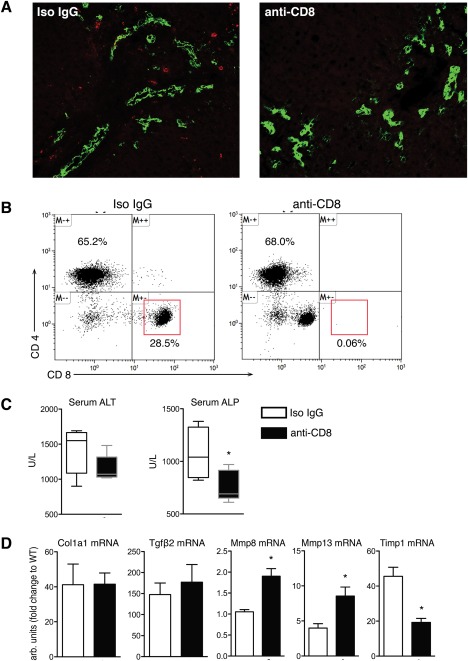
CD8+ T‐cell depletion ameliorates liver injury and has beneficial effects on fibrosis‐related gene expression. (A) Immunofluorescent staining of CD8+ (red) and bile ducts visualized by pan‐CK (green) in liver sections from male Mdr2–/–;CD39–/– mice 3 days after administration of anti‐CD8 monoclonal antibody or the respective isotype control (original magnification, ×200). (B) Flow cytometry analysis of CD8 expression in splenocytes isolated from Mdr2–/–;CD39–/– mice treated for 3 days with anti‐CD8 or isotype control. Cells were gated on CD3+ subsets. (C) CD8+ T‐cell depletion resulted in a significant decrease in serum ALT and ALP. (D) CD8+ T‐cell depletion led to a profibrolytic shift in gene expression with significantly reduced TIMP‐1 expression and increased MMP‐8 and MMP‐13 expression. Data are mean ± SEM (n = 3‐5 mice/bar). *P < 0.05 compared to isotype‐treated control mice (t test). Abbreviations: CK, cytokeratine; IgG, immunoglobulin G; Tgfβ2, transforming growth factor β2; TIMP‐1, tissue inhibitor of metalloproteinase 1.
INTESTINAL BACTERIA PROMOTE SCLEROSING CHOLANGITIS IN Mdr2–/– MICE
Because of the strong association of PSC with colitis and the major role of gut flora‐derived purines in immune cell homeostasis,20 we assessed the role of gut flora in the regulation of liver disease severity in Mdr2–/– mice. First, colitis was induced by 3% and 5% dextran sulfate sodium (DSS)‐containing drinking water for 7 days, and the effect on liver fibrosis was evaluated 7 days after DSS discontinuation. Administration of 3% and 5% DSS resulted in clinically moderate (mild diarrhea) and severe (weight loss, diarrhea, blood in stool) colitis, respectively, which was confirmed histologically (Fig. 4A).
Figure 4.
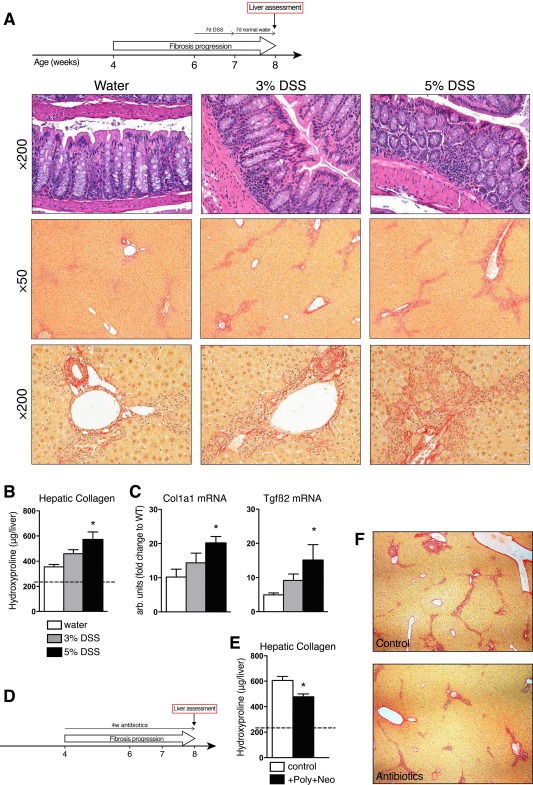
Intestinal bacteria promote sclerosing cholangitis in Mdr2–/– mice. (A) Experimental design of experimental colitis model in 6‐week‐old male FVB.Mdr2–/– mice. After 7 days of DSS administration, colitis was assessed, the DSS‐supplemented water was replaced by normal drinking water, and livers assessed 7 days later. Hematoxylin and eosin staining of colons (×200) and Sirius Red staining of liver specimens from Mdr2–/– mice receiving drinking water (left), 3% DSS (middle), or 5% DSS (right) for 7 days. Magnification ×50 (middle panel) and ×200 (lower panel, portal area). (B) Hydroxyproline content in livers from control and DSS‐treated Mdr2–/– mice. (C) Hepatic expression of Col1a1 and Tgfβ2. (D) Schematic outline for the antibiotic treatment in male Mdr2–/– mice. (E) Hepatic hydroxyproline content and (F) representative connective tissue stain (Sirius Red, ×50) in livers from 8‐week‐old Mdr2–/– mice after 4 weeks of antibiotic treatment with polymyxin B (100 mg/kg) + neomycin (220 mg/kg). Data are shown as mean ± SEM (n = 4‐5 mice/bar). *P < 0.05 compared to age‐matched Mdr2–/– controls (analysis of variance followed by Dunnett's posttest). Abbreviations: DSS, dextran sulfate sodium; FVB, Friend virus B‐type; Tgfβ2, transforming growth factor β2.
DSS‐induced colitis exacerbated sclerosing cholangitis with increased ductular reaction and bridging fibrosis (Fig. 4A). Consistent with histology, hepatic collagen levels significantly increased in a dose‐dependent manner in response to experimental colitis, with up to 60% hydroxyproline increase (Fig. 4B) and 2‐fold to 3‐fold increases in profibrogenic transcripts Col1a1 and transforming growth factor β2 (Fig. 4C).
Gut microflora is considered critical for the development of DSS‐induced colitis by triggering inflammatory immune responses.23 We further interrogated the contribution of commensal bacteria to liver fibrosis in Mdr2–/– mice using selective gut decontamination with an antibiotic cocktail to suppress Gram‐negative bacteria (neomycin + polymixin B) during active disease progression (4‐8 weeks of age) (Fig. 4D). Antibiotic‐treated mice demonstrated a significant reduction of hepatic collagen deposition histologically and biochemically (Fig. 4E,F). These results suggest that intestinal microbiota (and intestinal inflammation) significantly contribute to liver disease pathogenesis in our system, recapitulating the gut–liver axis in the pathogenesis of PSC.
COLONIC ATP ADMINISTRATION RECAPITULATES THE PHENOTYPE OF EXACERBATED SCLEROSING CHOLANGITIS DUE TO LOSS OF CD39
CD39 controls the rate‐limiting step (proinflammatory) extracellular ATP conversion to AMP resulting in the generation of (anti‐inflammatory) adenosine. Thus, the effects we observed following the loss of CD39 in Mdr2–/– mice can be due to either increased extracellular ATP signaling or the result of lower adenosine generation. We hypothesized that exacerbation of the sclerosing cholangitis‐type lesions in double‐null mice is the consequence of intestinal immune cell activation through luminal ATP derived from either gut bacteria or epithelial cells. To test this, Mdr2–/– mice were rectally administered the stable ATP analogue αβ‐ATP (1 mg/mouse) for 6 days to mimic the effects of increased luminal ATP levels on CD39 deficiency.
In αβ‐ATP‐treated Mdr2–/– mice, immunostaining revealed accumulation of CD8+ T cells in portal areas in close proximity to pan‐cytokeratine‐positive biliary epithelium, with a >2‐fold increase in hepatic CD8+ T‐cell counts (10.32 ± 0.38 versus 3.85 ± 0.30 cells/high‐power field) and elevated CD8 mRNA when compared to vehicle controls (Fig. 5A,B). Furthermore, colonic ATP administration resulted in significantly elevated serum ALT (P = 0.001) and ALP (P = 0.018) levels (Fig. 5C). A trend toward increased profibrogenic genes (Fig. 5D) and hepatic collagen deposition in animals treated with αβ‐ATP was also observed (564.0 ± 44.38 μg versus 488.3 ± 16.48 μg of hydroxyproline/liver; P = 0.131; data not shown), but these changes did not reach statistical significance, presumably due to the relatively short (6 days) duration of this experiment. Thus, colonic ATP administration in Mdr2–/– mice mostly recapitulates the central features of exacerbated sclerosing cholangitis phenotype due to CD39 deficiency in Mdr2–/–;CD39–/– mice.
Figure 5.
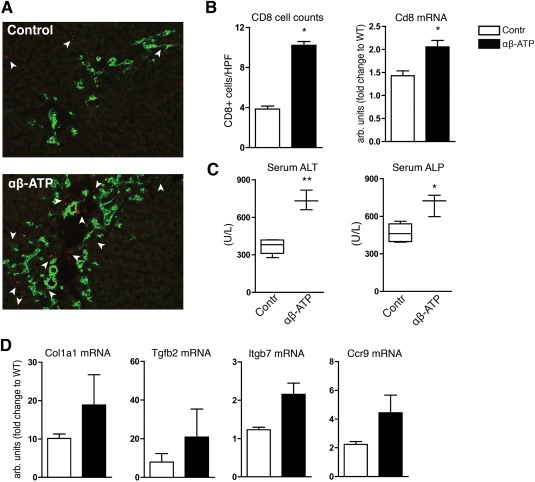
Colonic ATP administration to Mdr2–/– mice recapitulates the liver phenotype of Mdr2–/–;CD39–/– mice. Stable ATP agonist (αβ‐ATP, 1 mg/kg/day) or vehicle (RPMI medium) was rectally administered into 5‐week‐old male Mdr2–/– mice daily for 6 days. (A) Double immunofluorescence for CD8+ (red) and biliary marker p‐CK (green) showed remarkable CD8+ T‐cell (arrowheads) infiltration in portal areas in Mdr2–/– mice challenged with αβ‐ATP (original magnification, ×200). (B) CD8+ T cells were quantified by counting 10 randomly selected high‐power fields and by measuring hepatic CD8 mRNA expression. (C) Elevation of serum ALT and ALP in mice treated with αβ‐ATP. (D) mRNA expression of Col1a1, Tgfb2, as well as the gut‐homing markers Itgb7, Ccr9 (n.s.). Data are mean ± SEM (n = 3 mice/bar). *P < 0.05 compared to vehicle‐treated Mdr2–/– (t test). Abbreviations: HPF, high‐power field; Itgb7, integrin b7; n.s., not significant; p‐CK, pan‐cytokeratine; RPMI, Roswell Park Memorial Institute; Tgfb2, transforming growth factor β2.
EXTRACELLULAR ATP PROMOTES RA‐DEPENDENT IMPRINTING OF GUT‐HOMING RECEPTOR α4β7 ON NAIVE CD8+ T CELLS
Aberrant homing of gut‐specific lymphocytes to the liver has been reported in patients with PSC24, 25 and is consistent with increased CD8+ T‐cell and gut‐tropism T‐cell markers in the livers of Mdr2–/–; CD39–/– mice. Only gut‐associated dendritic cells in mesenteric lymph nodes or Peyer's patches express the retinal dehydrogenase required to produce RA, which is necessary for the imprinting of gut specificity on lymphocytes.8 High expression of CD39 by immunofluorescence was found in the intestine of Mdr2–/– mice where it was abundantly present in nonepithelial cells of the villi and Peyer's patches. Indeed, a significant subset of CD39 cells within Peyer's patches co‐expressed dendritic cell marker CD11c (Fig. 6A).
Figure 6.
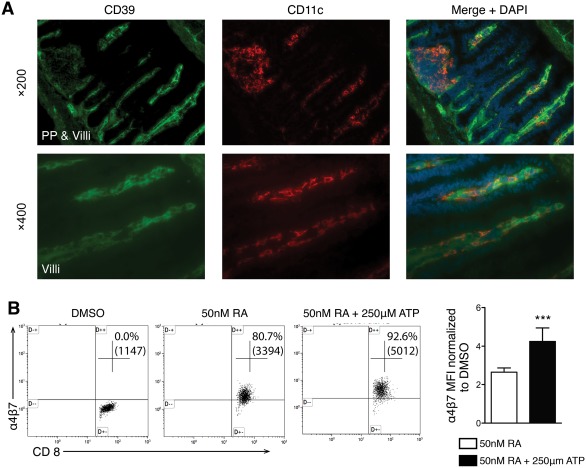
Extracellular ATP promotes RA‐dependent imprinting of gut‐homing receptor α4β7 on naive CD8+ T cells. (A) Frozen tissue sections of intestine from male Mdr2–/– mice were double stained for CD39 (green) and CD11c (red) using DAPI for nuclei visualization. Photographs in the upper panel (magnification ×200) show CD39‐positive cells in the mucosa, villi, and Peyer's patch. CD11c‐positive cells were detected in villi and the Peyer's patch. CD11c cells expressing CD39 were found in the Peyer's patch and in the villi. The lower panel at a higher magnification (×400) clearly demonstrates that a subset of DC is CD11c/CD39 double positive. (B) Flow cytometry analysis of α4β7 expression in naive CD8+ T cells cultured for 4 days with CD3/CD28 beads in the presence of DMSO, 50 nM RA, or 50 nM RA + 250 μM ATP. Representative dot plots of CD45+ gated cells are shown with numbers indicating the percentage of cells positive for CD8 and α4β7; in parentheses is the mean fluorescence intensity for α4β7 in CD8+ cells. Results are shown from three different experiments (n = 9). Bar graph: α4β7 expression (MFI) in CD45+/CD8+ cells stimulated with 50 nM RA with or without ATP was normalized to DMSO‐stimulated cells (mean ± SD). ***P < 0.001. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; DC, dendritic cell; DMSO, dimethyl sulfoxide; MFI, mean fluorescence intensity; PP, Peyer's patch.
We hypothesized that CD39 on dendritic cells may regulate imprinting of gut tropism on CD8 T cells. To test this, naive CD8+ T cells were “gut imprinted” with CD3/CD28 beads in the presence of 50 nM RA in vitro and analyzed by flow cytometry. By day 4, CCR9 was expressed on all CD8+ T cells (99.6%, data not shown) whereas only 71.11% of the CD8+ T cells were α4β7 positive (Fig. 6B). If cells were activated in the presence of RA + ATP, the expression of α4β7 was further enhanced to 91.8% (Fig. 6B) while addition of adenosine had the opposite effect (data not shown). RA‐dependent gut‐tropism imprinting in CD8 T cells isolated from CD39–/– mice was not impaired compared to WT mice (data not shown).
CD39 IS EXPRESSED BY IMMUNE CELLS IN HUMAN COLON AND IS FURTHER UP‐REGULATED IN IBD
To survey CD39 expression in human colon, immunostaining was performed in diagnostic biopsies from healthy patients and patients with IBD. We observed abundant CD39 expression by immune and endothelial cells in control colon biopsies. In intestinal tissue from patients with IBD, CD39 expression was increased in terms of extent and intensity (Fig. 7A). Immunohistochemical staining of CD39 in colon biopsies showed a significantly enhanced immunoreactivity in IBD colon compared to control tissue samples (2.9 versus 1.4; P = 0.0036) (Fig. 7B). We investigated the expression of CD39 in healthy livers and contrasted this in PSC and PBC livers. CD39 expression was detected on sinusoidal endothelial and macrophage‐like septal cells and did not differ significantly between healthy and diseased (PSC, PBC) livers when assessed semiquantitatively (Fig. 7C,D). In general, CD39 immunoreactivity in liver tissues was notably lower than in colon samples.
Figure 7.
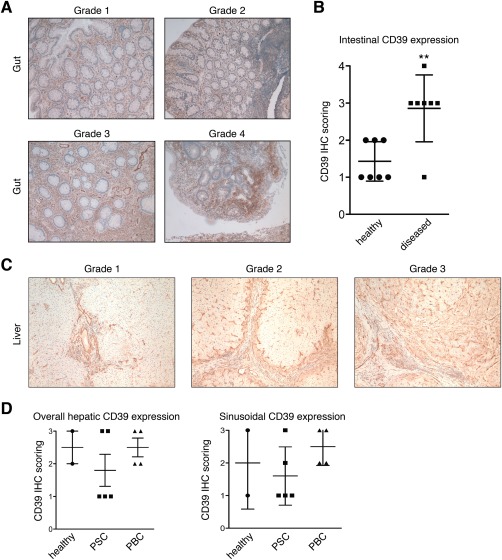
Survey of CD39 expression in healthy and diseased human liver and colon. Formalin‐fixed intestinal tissue sections from patients with IBD and healthy controls were immunohistochemically stained for CD39. (A) Representative images showing CD39 IHC scores 1‐4 based on intensity and area of CD39 expression in human colon tissue. (B) Semiquantitative assessment of CD39 immunoreactivity in healthy and diseased intestinal tissue samples (mean ± SD). **P < 0.05. (C) CD39 immunohistochemistry in formalin‐fixed liver sections from PSC, PBC, and healthy livers. Representative pictures show examples for CD39 expression scored as grades 1, 2, and 3. (D) Dot plot showing semiquantitatively assessed CD39 expression as overall expression in the liver (left) and as sinusoidal CD39 expression (right). Each dot represents an individual specimen score. Abbreviation: IHC, immunohistochemistry.
Discussion
By genetically deleting CD39, we provide evidence that purinergic signaling regulates biliary injury and fibrosis in a murine PSC‐like model of sclerosing cholangitis induced by Mdr2 deficiency. Worsening of liver histopathology in CD39‐deficient Mdr2–/– mice appears to be driven by extracellular ATP‐dependent hepatic influx of CD8 T cells. In human samples, CD39 expression is relatively low in normal and PSC livers but is abundantly present on intestinal immune cells in healthy colon and is further up‐regulated in IBD. In vitro, extracellular ATP promoted RA‐dependent gut‐tropism imprinting on naive CD8+ T cells, a lymphocyte population known to contribute to liver injury in PSC.
The strong clinical association of PSC with IBD and the shared genetic susceptibility loci provide the rationale that PSC and IBD have common underlying pathophysiologic mechanisms. In both cases, the precise etiology remains unknown. In recent years, the gut microbiome has been increasingly recognized as a critical factor in numerous immune‐mediated diseases, including IBD and PSC.26 It is now generally accepted that microbial dysbiosis is one of the key players in the inflammatory process in IBD.27, 28
Using experimental colitis and selective gut decontamination in the PSC‐like Mdr2–/– mouse model, we demonstrated that intestinal microbes contribute, at least in part, to driving the liver injury and fibrosis in our model. This is in line with results of a recent, randomized, double‐blind study demonstrating significant improvements in serum ALP levels in patients with PSC after 12 weeks treatment with vancomycin.29 However, another recent study reported paradoxical worsening of cholangitis in germ‐free Mdr2–/– mice on a Friend virus B‐type background, arguing for a protective role of the gut microbiota.30 This is in direct contradiction to our data derived from specific‐pathogen‐free Mdr2–/– mice, which clearly show improvement in their liver disease with antibiotic‐mediated gut decontamination (and worsening on induction of experimental colitis). The discrepancy is difficult to explain, but the profound differences in immune system development in mice kept under germ‐free conditions may be a major confounder.31 It is clear that further studies are required to decipher harmful versus protective effects of microbial flora in the pathogenesis of PSC.
The role of purinergic signaling in chronic liver disease remains unclear in that both ATP and adenosine may promote liver fibrosis. For example, adenosine A2A receptor‐deficient mice are protected from CCl4– or thioacetamide‐induced fibrosis. Similar effects were observed in WT animals treated with adenosine receptor antagonists.32 Moreover, it has been shown that adenosine generated by CD73 plays a critical role in the development of hepatotoxin‐induced fibrosis.33 While these two studies imply that adenosine is profibrogenic, Dranoff and colleagues34 recently examined the effects of ATP signaling and showed that blocking of purinergic receptors with a synthetic P2 receptor inhibitor attenuates CCl4‐induced liver fibrosis. Given that CD39 impacts P2‐mediated nucleotide signaling through ATP degradation and generates AMP, a precursor for adenosine generation, it was uncertain which pathway would be dominant in regulating sclerosing cholangitis in our system.
Compared to Mdr2–/– mice, Mdr2–/–;CD39–/– mice exhibited a worsened PSC phenotype with more severe liver injury and fibrosis and an increase in liver‐infiltrating CD8+ T cells (Figs. 1 and 2). Administration of the stable ATP analogue αβ‐ATP into CD39‐sufficient Mdr2–/– mice recapitulated the phenotype observed with CD39 loss. These results suggest that excess extracellular ATP rather than diminished adenosine is responsible for hepatic influx of CD8+ T cells and worsened disease phenotype in Mdr2–/– due to the loss of CD39. Selective CD8+ cell depletion in Mdr2–/–;CD39–/– further indicates that sequelae of CD39 deficiency are primarily CD8+ T cell mediated. Interestingly, exogenous ATP directly promoted RA‐induced gut‐tropism imprinting on naive CD8+ T cells in vitro, providing a possible mechanistic explanation for aberrant liver homing of gut‐primed CD8+ T cells observed in human PSC.5, 25
Although the exact pathogenesis of PSC remains elusive, there is a growing body of evidence that dysregulated immune responses represent a critical mechanism in PSC. Genome‐wide association studies have identified numerous immune‐related susceptibility loci for PSC in the human leukocyte antigen, macrophage stimulating 1 gene, interleukin‐2 (IL‐2), and its receptor IL‐2RA.35 Several immune‐mediated diseases share some of the genetic risk factors, demonstrating an immunologic component to be involved in PSC development. Immunohistochemical studies demonstrated a mixed inflammatory cell infiltrate predominantly consisting of T cells, natural killer cells, and macrophages.36, 37, 38 Several studies suggested that T cells are the major infiltrating cells in PSC; however, conflicting findings have been reported on the CD4+ or CD8+ T cells being the prevalent subset.12, 37, 39 In our study, we showed that the hepatic lymphocytic infiltrate is due to increased recruitment of CD8+ T cells that drive worsened liver disease in CD39‐deficient Mdr2–/– mice. Remarkably, immunosuppressive therapy is not effective in PSC despite it having several features of an immune‐mediated disease.40 On the other hand, immunomodulation approaches (such as vitamin D supplementation) have shown beneficial results and may hold promise for PSC.12
Our Mdr2–/–;CD39–/– mice showed an increased number of CD8+ T cells in the liver compared to Mdr2–/– together with an up‐regulation of the gut‐homing markers integrin α4β7 in the liver. MAdCAM‐1 and CCL25, the respective binding partners of α4β7 and CCR9 that mediate lymphocyte gut homing, were also both expressed in livers from Mdr2–/– and Mdr2–/–;CD39–/– mice, generally consistent with aberrant homing of T cells to the liver in human PSC.9, 41 While none of these mice presented signs of overt colitis, unlike the majority of patients with PSC, it is conceivable that ATP‐dependent T‐cell activation20 in the intestine, which is a major site of T‐cell differentiation,42 may promote distant T‐cell‐mediated injury as observed in PSC or in Mdr2–/– mice.
ATP limits lymphocyte motility during antigen recognition and thereby potentiates lymphocyte activation.43 Bacteria‐derived ATP has been identified as a critical driver of intestinal TH17 cell generation by activating CD11c+ lamina propria cells to produce IL‐6 and IL‐23 and thereby promoting TH17 differentiation.20 In our study, we showed a worsened liver phenotype accompanied by increased CD8+ T‐cell influx after rectal ATP administration. Moreover, we demonstrated for the first time that extracellular ATP directly impacts imprinting of gut specificity by RA. This observation supports the concept that ATP signaling is involved in T‐cell activation and gut‐liver trafficking, thus promoting the progression of sclerosing cholangitis. This concept is further supported by two sets of experiments that show 1) either CD39 deletion or rectal ATP administration led to an increase in CD8+ T cells and worsened biliary injury and fibrosis in Mdr2–/– mice and 2) conversely, CD8+ depletion attenuate the sclerosing cholangitis in Mdr2–/–;CD39–/– mice. Both approaches support our hypothesis that purinergic mechanisms control CD8+ T‐cell responses and modulate fibrosis progression in PSC. Based on our results, we propose the novel concept that extracellular ATP in the lumen, under control of CD39, promotes activation and gut‐liver trafficking of CD8+ effector T cells, thereby driving progression of sclerosing cholangitis (summarized in Fig. 8). Furthermore, we developed and characterized robust experimental tools that will allow further dissecting and interrogating this purinergic pathway pharmacologically.
Figure 8.
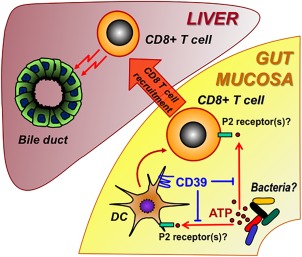
Scheme of putative purinergic pathway regulating the pathophysiologic gut–liver axis in PSC. Luminal ATP (released from intestinal bacteria or injured mucosal cells) activates gut DC/CD8+ T cells as regulated by the cellular ectonucleotidase CD39. CD8+ T cells, once activated in the gut, traffic to the liver. In the liver, CD8+ T cells (by effector functions) target biliary epithelia, promoting cholangitis and periductular fibrosis. Abbreviation: DC, dendritic cell.
In summary, our findings demonstrate a previously unrecognized role of purinergic signaling in regulating biliary injury and fibrosis. We showed that gut flora promote progression of cholangitis and that manipulation of purinergic pathways either through CD39 deletion or by boosting exogenous ATP levels in the colon leads to an influx of CD8+ T cells to the liver and aggravates liver injury and fibrosis in animal models. Pharmacological modulation of the purinergic system may represent a promising new approach for the treatment of PSC.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1084/full.
Supporting Information
Potential conflict of interest: Nothing to report.
Supported by an institutional grant from the Department of Medicine (Beth Israel Deaconess Medical Center to Y.P.) and the Leona M. and Harry B. Helmsley Charitable Trust (281574.5069091.0010 to S.C.R.). Z.W.P. was a recipient of career development awards from the First Affiliated Hospital of Sun Yat‐sen University (Guangzhou, China), National Natural Science Foundation of China (81301842), and Pearl River S&T Nova Program (2014J2200087). S.R. was a recipient of a fellowship from the Swiss National Science Foundation (P300PB_161098).
Contributor Information
Simon C. Robson, Email: srobson@bidmc.harvard.edu
Yury V. Popov, Email: ypopov@bidmc.harvard.edu.
REFERENCES
- 1. Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology 2011;54:1842‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campsen J, Zimmerman MA, Trotter JF, Wachs M, Bak T, Steinberg T, et al. Clinically recurrent primary sclerosing cholangitis following liver transplantation: a time course. Liver Transpl 2008;14:181‐185. [DOI] [PubMed] [Google Scholar]
- 3. Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol 2012;56:1181‐1188. [DOI] [PubMed] [Google Scholar]
- 4. Alabraba E, Nightingale P, Gunson B, Hubscher S, Olliff S, Mirza D, et al. A re‐evaluation of the risk factors for the recurrence of primary sclerosing cholangitis in liver allografts. Liver Transpl 2009;15:330‐340. [DOI] [PubMed] [Google Scholar]
- 5. Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet 2002;359:150‐157. [DOI] [PubMed] [Google Scholar]
- 6. Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al.; UK‐PSC Consortium; International IBD Genetics Consortium ; International PSC Study Group . Genome‐wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet 2017;49:269‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, et al. Selective imprinting of gut‐homing T cells by Peyer's patch dendritic cells. Nature 2003;424:88‐93. [DOI] [PubMed] [Google Scholar]
- 8. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut‐homing specificity on T cells. Immunity 2004;21:527‐538. [DOI] [PubMed] [Google Scholar]
- 9. Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra‐intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol 2006;6:244‐251. [DOI] [PubMed] [Google Scholar]
- 10. Karlsen TH, Franke A, Melum E, Kaser A, Hov JR, Balschun T, et al. Genome‐wide association analysis in primary sclerosing cholangitis. Gastroenterology 2010;138:1102‐1111. [DOI] [PubMed] [Google Scholar]
- 11. Trivedi PJ, Hirschfield GM. Treatment of autoimmune liver disease: current and future therapeutic options. Ther Adv Chronic Dis 2013;4:119‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liaskou E, Jeffery LE, Trivedi PJ, Reynolds GM, Suresh S, Bruns T, et al. Loss of CD28 expression by liver‐infiltrating T cells contributes to pathogenesis of primary sclerosing cholangitis. Gastroenterology 2014;147:221‐232.e227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007;204:1257‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fernandez D, Flores‐Santibanez F, Neira J, Osorio‐Barrios F, Tejon G, Nunez S, et al. Purinergic signaling as a regulator of Th17 cell plasticity. PLoS One 2016;11:e0157889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sebode M, Peiseler M, Franke B, Schwinge D, Schoknecht T, Wortmann F, et al. Reduced FOXP3(+) regulatory T cells in patients with primary sclerosing cholangitis are associated with IL2RA gene polymorphisms. J Hepatol 2014;60:1010‐1016. [DOI] [PubMed] [Google Scholar]
- 16. Katt J, Schwinge D, Schoknecht T, Quaas A, Sobottka I, Burandt E, et al. Increased T helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology 2013;58:1084‐1093. [DOI] [PubMed] [Google Scholar]
- 17. Enjyoji K, Sevigny J, Lin Y, Frenette PS, Christie PD, Esch JS 2nd. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med 1999;5:1010‐1017. [DOI] [PubMed] [Google Scholar]
- 18. Peng ZW, Ikenaga N, Liu SB, Sverdlov DY, Vaid KA, Dixit R, et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016;63:217‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009;15:914‐920. [DOI] [PubMed] [Google Scholar]
- 20. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature 2008;455:808‐812. [DOI] [PubMed] [Google Scholar]
- 21. Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)–/– mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro‐ and antifibrogenic genes. J Hepatol 2005;43:1045‐1054. [DOI] [PubMed] [Google Scholar]
- 22. Yoshida S, Ikenaga N, Liu SB, Peng ZW, Chung J, Sverdlov DY, et al. Extrahepatic platelet‐derived growth factor‐beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology 2014;147:1378‐1392. [DOI] [PubMed] [Google Scholar]
- 23. Rath HC, Schultz M, Freitag R, Dieleman LA, Li F, Linde HJ, et al. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect Immun 2001;69:2277‐2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grant AJ, Lalor PF, Hubscher SG, Briskin M, Adams DH. MAdCAM‐1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM‐1 in chronic inflammatory liver disease). Hepatology 2001;33:1065‐1072. [DOI] [PubMed] [Google Scholar]
- 25. Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hubscher SG, et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut‐homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med 2004;200:1511‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eksteen B. Advances and controversies in the pathogenesis and management of primary sclerosing cholangitis. Br Med Bull 2014;110:89‐98. [DOI] [PubMed] [Google Scholar]
- 27. Bellaguarda E, Chang EB. IBD and the gut microbiota‐‐from bench to personalized medicine. Curr Gastroenterol Rep 2015;17:15. [DOI] [PubMed] [Google Scholar]
- 28. Hold GL, Smith M, Grange C, Watt ER, El‐Omar EM, Mukhopadhya I. Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years? World J Gastroenterol 2014;20:1192‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tabibian JH, Weeding E, Jorgensen RA, Petz JL, Keach JC, Talwalkar JA, et al. Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis ‐ a pilot study. Aliment Pharmacol Ther 2013;37:604‐612. [DOI] [PubMed] [Google Scholar]
- 30. Tabibian JH, O'Hara SP, Trussoni CE, Tietz PS, Splinter PL, Mounajjed T, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016;63:185‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009;9:313‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan ES, Montesinos MC, Fernandez P, Desai A, Delano DL, Yee H, et al. Adenosine A(2A) receptors play a role in the pathogenesis of hepatic cirrhosis. Br J Pharmacol 2006;148:1144‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng Z, Fernandez P, Wilder T, Yee H, Chiriboga L, Chan ES, et al. Ecto‐5′‐nucleotidase (CD73) ‐mediated extracellular adenosine production plays a critical role in hepatic fibrosis. FASEB J 2008;22:2263‐2272. [DOI] [PubMed] [Google Scholar]
- 34. Dranoff JA, Kruglov EA, Abreu‐Lanfranco O, Nguyen T, Arora G, Jain D. Prevention of liver fibrosis by the purinoceptor antagonist pyridoxal‐phosphate‐6‐azophenyl‐2′,4′‐disulfonate (PPADS). In Vivo 2007;21:957‐965. [PubMed] [Google Scholar]
- 35. Mells GF, Kaser A, Karlsen TH. Novel insights into autoimmune liver diseases provided by genome‐wide association studies. J Autoimmun 2013;46:41‐54. [DOI] [PubMed] [Google Scholar]
- 36. Chandok N, Hirschfield GM. Management of primary sclerosing cholangitis: conventions and controversies. Can J Gastroenterol 2012;26:261‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hashimoto E, Lindor KD, Homburger HA, Dickson ER, Czaja AJ, Wiesner RH, et al. Immunohistochemical characterization of hepatic lymphocytes in primary biliary cirrhosis in comparison with primary sclerosing cholangitis and autoimmune chronic active hepatitis. Mayo Clin Proc 1993;68:1049‐1055. [DOI] [PubMed] [Google Scholar]
- 38. Ponsioen CY, Kuiper H, Ten Kate FJ, van Milligen de Wit M, van Deventer SJ, Tytgat GN. Immunohistochemical analysis of inflammation in primary sclerosing cholangitis. Eur J Gastroenterol Hepatol 1999;11:769‐774. [DOI] [PubMed] [Google Scholar]
- 39. Whiteside TL, Lasky S, Si L, Van Thiel DH. Immunologic analysis of mononuclear cells in liver tissues and blood of patients with primary sclerosing cholangitis. Hepatology 1985;5:468‐474. [DOI] [PubMed] [Google Scholar]
- 40. Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet 2013;382:1587‐1599. [DOI] [PubMed] [Google Scholar]
- 41. Seidel D, Eickmeier I, Kuhl AA, Hamann A, Loddenkemper C, Schott E. CD8 T cells primed in the gut‐associated lymphoid tissue induce immune‐mediated cholangitis in mice. Hepatology 2014;59:601‐611. [DOI] [PubMed] [Google Scholar]
- 42. Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, et al. Control of TH17 cells occurs in the small intestine. Nature 2011;475:514‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang CM, Ploia C, Anselmi F, Sarukhan A, Viola A. Adenosine triphosphate acts as a paracrine signaling molecule to reduce the motility of T cells. EMBO J 2014;33:1354‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1084/full.
Supporting Information