

Abstract
Approximately 71 million people are chronically infected with the hepatitis C virus (HCV), a potentially lethal pathogen. HCV generates oxidative stress correlating with disease severity. HCV proteins increase reactive oxygen species production by stimulating nicotinamide adenine dinucleotide phosphate oxidase (NOX) activity. Reactive oxygen species are necessary for host defense and cell signaling; however, elevated NOX activity contributes to cancer, and NOX overexpression is associated with hepatic fibrosis. Our aim was to investigate whether single nucleotide polymorphisms (SNPs) in NOX family members are associated with HCV‐related liver damage. Three hundred and thirty‐one individuals of European ancestry and 90 individuals of African ancestry, all diagnosed with HCV, were genotyped for 243 tagSNPs in NOX enzymes and their regulatory factors. Pathology scores were available for 288 Caucasians and 71 Africans, and mortality status was determined for all subjects. SNPs were tested for association with pathology scores and as predictors of mortality. In Africans, homozygosity for the A allele of rs12753665 (neutrophil cytosolic factor 2) and homozygosity for the T allele of rs760519 (neutrophil cytosolic factor 4) were associated with and predictive of higher rates of advanced fibrosis and cirrhosis compared to other genotypes after controlling for age and sex. In Caucasians, homozygosity for the T allele of rs2292464 (dual oxidase 1) was associated with and predictive of decreased periportal inflammation after controlling for age and sex. No SNPs were significant predictors of mortality. Conclusion: In this exploratory study, three NOX‐related polymorphisms in two ethnic groups were significantly associated with hepatic inflammation and fibrosis. Future studies investigating these SNPs in larger cohorts of patients with HCV are warranted. (Hepatology Communications 2017;1:973–982)
Abbreviations
- CI
confidence interval
- DUOX
dual oxidase
- FDR
false discovery rate
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HWE
Hardy‐Weinberg Equilibrium
- LD
linkage disequilibrium
- NCF
neutrophil cytosolic factor
- NOX
nicotinamide adenine dinucleotide phosphate oxidase
- OR
odds ratio
- ROS
reactive oxygen species
- SNP
single nucleotide polymorphism
Introduction
Hepatitis C is a virus that infects the liver, potentially leading to cirrhosis, hepatocellular carcinoma (HCC), and death. Approximately 71 million people worldwide and 3.5 million people in the United States are chronically infected with the hepatitis C virus (HCV).1, 2 HCV is the most common type of blood‐borne infection in the United States and a leading cause of liver disease; half of all HCC patients and one third of those needing a liver transplant are positive for HCV.3, 4, 5 Acute infection with HCV is asymptomatic, and many individuals infected with HCV are unaware of their disease status.5 The modes of HCV transmission include injection drug use, occupational exposure to contaminated blood, sexual transmission, perinatal exposure, and others. The risk of infection by injection drug use is higher than for other modes,5 and it increases substantially with time and age.6 Various demographic groups are at elevated risk for HCV infection, including male individuals, veterans, low‐income individuals, African Americans, Latinos, and those born between 1945 and 1965.5, 7
Once infected, approximately one quarter of those with HCV will spontaneously clear the virus.8 The remaining 60% to 80% of those infected with HCV will become chronically infected.9 Chronic HCV infection leads to histologic changes in the liver, including steatosis, inflammation, fibrosis, cirrhosis, and possibly HCC. HCV further generates oxidative stress and the release of proinflammatory cytokines, such as interleukin‐1β and tumor necrosis factor alpha, which contribute to local and systemic inflammation.10 Fibrosis leads to cirrhosis in about 7%‐18% of chronically infected individuals.11
Oxidative stress is defined as the production of reactive oxygen species (ROS) and reactive nitrogen species that exceeds the cell's capacity to neutralize these molecules by antioxidant mechanisms.12 Oxidative stress is well documented in patients with HCV, and ROS levels correlate with the progression of liver disease.13, 14, 15 ROS levels are highest and antioxidant levels are lowest in patients with HCV genotypes 1 and 4, the genotypes most often associated with severe disease.16 Five HCV‐derived proteins can stimulate ROS production while simultaneously activating a pathway for antioxidant production.17 The release of intracellular ROS by these proteins is attributed to multiple events, including mitochondrial dysfunction, calcium release from the endoplasmic reticulum, and the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs).18 The role of NOX in HCV is particularly intriguing as some NOX proteins are activated by calcium, and two others, NOX1 and NOX4, produce superoxide in response to HCV core protein.19, 20
The NOX family comprises seven homologous enzymes known as NOX1, NOX2, NOX3, NOX4, NOX5, dual oxidase (DUOX)1, and DUOX2. All are transmembrane proteins that use NADPH as a substrate to transfer two electrons across a membrane.21 NOXs are recognized for their roles in innate immunity. They contribute to the respiratory burst and to cell‐signaling events generating inflammatory responses. NOXs are expressed in a large number of tissue and cell types22 and have been implicated in many diseases, including cancer, gastrointestinal inflammation, atherosclerosis, thyroid dysfunction, and hearing loss.23 In the liver, hepatocytes and immune, stellate, and endothelial cells all express NOX.24 NOX expression and activity are associated with liver injury in HCV and in alcoholic hepatitis, and NOXs are up‐regulated in activated stellate cells. Accordingly, several NOX isoforms are implicated in the development of hepatic fibrosis.24
The aim of this study was to investigate whether single nucleotide polymorphisms (SNPs) in NOX members, including regulatory factors, are associated with the progression of HCV‐induced inflammation and fibrosis. A second goal was to determine whether death from any cause was associated with SNPs in the NOX family. We discovered that three SNPs in two distinct populations were significant predictors for fibrosis and inflammation in patients with HCV. This study is the first to compare the genetic makeup of the NOX family members as a functional group to the progression of liver disease.
Participants and Materials
PATIENT COHORT
All participants gave written informed consent for the use of genetic information under institutional review board protocols 91‐DK‐0214 (NCT00001971) and 00‐DK‐0125 (NCT00005657). Both protocols were reviewed by the National Institute of Diabetes and Digestive and Kidney Diseases Institutional Review Board and are registered in ClinicalTrials.gov. The study cohort comprised 421 patients treated for hepatitis C at the National Institutes of Health (NIH) Clinical Center over the past two decades. The cohort included two races: patients of European descent (Caucasians, n = 331) and those of African descent (Africans, n = 90). Of these, 288 Caucasians and 71 Africans had undergone needle biopsy at least once, and all patients donated blood samples. All biopsy samples were interpreted by a single pathologist. The biopsy parameters and range of values observed in this study, which did not always reach the maxima possible, were as follows: histological activity index Ishak fibrosis score as modified by Knodell25, 26 (0–6), periportal inflammation (0–6), lobular inflammation (1–4), portal inflammation (0–4), and total inflammation (1–14). The Ishak fibrosis score was analyzed two ways: (1) in terms of the Ishak fibrosis score that was measured in the patient's first biopsy, i.e., at the onset of treatment, and (2) in terms of the highest Ishak fibrosis score achieved by a patient over the entire treatment period. All other pathology scores were measured at the onset of treatment. Demographic information was available for all patients. Information on whether patients had died at any point up until August 2014 and the date of each death was collected through a publicly available death records database (https://www.dobsearch.com/) as well as from in‐house communication and obituaries.
GENOTYPING
DNA was extracted from whole blood samples using the Qiagen Flexigene DNA Kit (Cat. No. 51206; Germantown, MD). DNA samples were checked for quality and purity and then diluted to 15 ng/µL. TagSNPs were selected from the International HapMap Project (release 27, phases I, II, and III) African ancestry in Southwest United States and Utah residents with Northern and Western European ancestry HapMap populations, using an NIH/National Institute of Environmental Health Sciences web tool (http://snpinfo.niehs.nih.gov/snpinfo/snptag.html). TagSNPs with a minor allele frequency ≥0.05 occurring in 14 genes forming the NOX complex were selected. These genes comprised NOX1, NOX2/CYBB, NOX3, NOX4, NOX5, DUOX1, DUOX2, RAC1, RAC2, CYBA, NCF2, NCF4, NOXA1, and NOXO1. No HapMap‐defined tagSNPs were available for neutrophil cytosolic factor 1 (NCF1), and tagSNPs in the 5’ and 3’ regions surrounding the target genes were not included. The DNA sequence flanking each tagSNP was acquired from the dbSNP database (National Center for Biotechnology Information) and then entered into the Sequenom (now Agena; San Diego, CA) Assay Design Suite that runs a five‐step algorithm to design primers and assemble other assay information to be used in the Sequenom MassArray system (https://www.agenacx.com/Home). Forward and reverse primers for the initial polymerase chain reaction step along with extension primers for the single base extension step were purchased from Invitrogen (now Thermo Fisher Scientific; Waltham, MA).
All patient DNA samples were genotyped using the Sequenom iPlex system. This system couples mass spectrometry with wet chemistry using automated liquid handling pipettors and other high‐throughput machinery. Multiplexed polymerase chain reactions were set up according to the Sequenom manual. Following genotyping, the data were filtered by (1) removing samples that were missing more than 10% of the targeted genotypes; (2) retaining only those tagSNPs (henceforth abbreviated SNPs) that amplified in at least 95% of the samples; and (3) dropping SNPs that did not show any variance. The final genotype data set included 243 SNPs. Because not all SNPs occur in both the Caucasian and African populations and those that do often have inherent differences in minor allele frequency, the data set was split into two, one for each race. Only those SNPs known to occur in a given race were retained in the respective race's data set. All subsequent analyses were performed separately on each data set. The Caucasian cohort contained 331 patients and 183 SNPs, while the African cohort contained 90 patients and 211 SNPs. Genotypes and mortality status were available for all members of each cohort, whereas biopsy‐based phenotypes were available for 71 and 288 African and Caucasian patients, respectively.
HARDY‐WEINBERG EQUILIBRIUM
The SNPs occurring in each data set were tested for Hardy‐Weinberg Equilibrium (HWE) using Pearson's chi square test in SVS version 8.4.1 (Golden Helix, Bozeman, MT, through the NIH Library Bioinformatics Support Program). SNPs located on the X chromosome were tested for HWE only in the female individuals of the appropriate race. In all cases, a Bonferroni correction was applied. The thresholds for significance were as follows: for non‐X chromosome African SNPs: 0.00025; for X chromosome African SNPs: 0.004; for non‐X chromosome Caucasian SNPs: 0.0003; and for X chromosome Caucasian SNPs: 0.004. Only rs5917471, occurring on the X chromosome in the African population, was significant for HWE (P = 0.0020); it was therefore dropped from further analysis, reducing the African data set to 210 SNPs.
DATA REDUCTION AND RECODING
All SNPs were bi‐allelic. The pathology scores and mortality status represented the phenotypes of interest, while the SNPs served as independent variables. With 210 and 183 SNPs in the African and Caucasian data sets, respectively, along with small sample sizes, we recognized that the dimensions of the data sets might lead to low statistical power. Therefore, the linkage disequilibrium (LD) Pruning tool in SVS (Golden Helix) was used to remove SNPs with redundant information by virtue of LD with other SNPs. For the African cohort, a genetic marker map was applied and the LD Pruning tool was executed using the expectation–maximization method and a window size of 210, an increment set to 2, and the LD threshold set to 0.7. For the Caucasian cohort, a genetic maker map was applied and the same settings as before were used to run the LD Pruning tool, except that the window size was set to 183. Using this tool, 21 SNPs were dropped from the African data set, leaving 189 SNPs for analysis, while 64 SNPs were dropped from the Caucasian data set, with 119 SNPs remaining (Supporting Table S1). All subsequent tests of association and the survival analysis were conducted using the LD pruned version of the African and Caucasian data sets.
All phenotypes were recoded into binary factors except for total inflammation. The recoding strategy separated patients with advanced disease from the rest of the cohort for each phenotype, as shown in Table 1. We also questioned whether there were trends in SNP composition among Ishak scores when the samples were divided to include a middle range of scores. Consequently, we conducted a second round of recoding on the first‐observed and highest achieved Ishak fibrosis scores to divide them into three categories (Table 1).
Table 1.
RECODING STRATEGY FOR ALL PHENOTYPES EXCEPT TOTAL INFLAMMATION
| Phenotype | Original Range of Values | Recoded Values |
|---|---|---|
| First observed Ishak score (binary) | 0‐4 | 0 |
| 5‐6 | 1 | |
| First observed Ishak score (3 levels) | 0‐2 | 0 |
| 3‐4 | 1 | |
| 5‐6 | 2 | |
| Highest observed Ishak score (binary) | 0‐4 | 0 |
| 5‐6 | 1 | |
| Highest observed Ishak score (3 levels) | 0‐2 | 0 |
| 3‐4 | 1 | |
| 5‐6 | 2 | |
| Periportal inflammation | 0‐3 | 0 |
| 4‐6 | 1 | |
| Portal inflammation | 0‐1 | 0 |
| 3‐4 | 1 | |
| Lobular inflammation | 1‐3 | 0 |
| Lobular inflammation | 4 | 1 |
STATISTICAL ANALYSIS
The association between each SNP and each recoded phenotype was assessed by Fisher's exact test. After all SNPs were compared with a given phenotype, the false discovery rate (FDR) multiple test correction was applied to each P value. The observed values for total inflammation spanned 14 levels, so this variable was treated as continuous, and SNPs were tested for association using the Kruskal‐Wallis test, followed by the FDR multiple test correction. The predictive value of each SNP that was significantly associated with a phenotype was assessed by logistic regression. Age and sex were included as covariates because these factors potentially affect the progression of HCV‐related liver disease.27, 28
To assess the risk of death associated with each SNP, all genotypes were converted to numeric values using the additive model and Cox proportional hazards regression was executed for each SNP. Two approaches, using different covariates, were used. In the first approach, periportal inflammation, the first‐observed and the highest achieved Ishak fibrosis scores (recoded as binary factors), were included as covariates. In the second approach, the same phenotypes were used as covariates but with the first‐observed and highest achieved Ishak fibrosis scores recoded to three levels. For all models, the overall survival (or time to death) was measured from the date of birth (because the diagnosis of HCV may come years after infection) to the date of death or to the last date of the year when the latest death occurred. The FDR multiple test correction was applied to all results.
For tests of association, FDR‐corrected P values < 0.05 were considered significant. For logistic regression, P values < 0.05 were considered significant. The Fisher exact test, Kruskal‐Wallis Test, FDR multiple test correction, and logistic regression were performed in R (version 3.1.2). Cox proportional hazard models were conducted using SAS (version 9.4).
Results
Clinical and pathologic attributes of the study cohorts are provided in Table 2. Both at the time of the first biopsy and at the time the highest Ishak score was reached, African patients were, on average, older than Caucasian patients and had significantly more fibrosis. The two cohorts were not significantly different in terms of inflammation.
Table 2.
CLINICAL AND PATHOLOGIC ATTRIBUTES OF THE AFRICAN AND CAUCASIAN COHORTS
| Parameter | African Cohort | Caucasian Cohort | P Value |
|---|---|---|---|
| Males (number, % of cohort)a | 56 (62) | 196 (59) | 0.606 |
| Deceased (number, % yes)a | 16 (18) | 43 (13) | 0.246 |
| Age at first biopsy (mean, range)b | 51 (34–74) | 47 (20–76) | <0.001 |
| Age at biopsy with highest Ishak fibrosis score (mean, range)b | 53 (36–74) | 49 (20–76) | 0.002 |
| Ishak fibrosis score, first biopsy (median)c | 3 | 2 | 0.016 |
| Ishak fibrosis score, highest achieved over treatment period (median)c | 3 | 2 | 0.040 |
| Periportal inflammation, first biopsy (median)c | 3 | 3 | 0.799 |
| Lobular inflammation, first biopsy (median)c | 4 | 4 | 0.269 |
| Portal inflammation, first biopsy (median)c | 1 | 1 | 0.520 |
| Total inflammation, first biopsy (median)c | 8 | 8 | 0.871 |
Pearson's chi square test;
Two‐sample Student t test, two‐tailed, pooled variances;
Mann‐Whitney U test.
In the African cohort, rs12753665, located in an intron of NCF2, was significantly associated with the highest Ishak score achieved over the course of treatment (FDR P = 0.018). All patients who were homozygous for the A allele of rs12753665 (n = 7) reached advanced fibrosis or cirrhosis (Ishak scores 5‐6) during their treatment period versus 22% to 25% of patients who were heterozygous or homozygous for the G allele (Fig. 1). This SNP was a significant predictor (P = 0.005) for the highest Ishak score after controlling for age and sex. Odds ratios (ORs) and confidence intervals (CIs) for the risk of advanced fibrosis or cirrhosis could not be generated due to a lack of convergence of regression models. Interestingly, rs12753665 also was associated with the Ishak fibrosis score at the onset of treatment, but the relationship was not significant (FDR P = 0.100).
Figure 1.
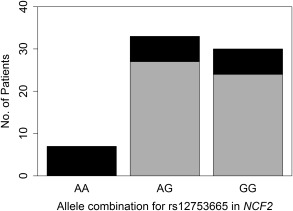
Number of patients with HCV in the African cohort, by rs12753665 genotype, who reached advanced fibrosis/cirrhosis versus none, mild, or moderate fibrosis over the course of treatment. Gray represents none, mild, or moderate fibrosis (Ishak fibrosis score 0‐4); black represents advanced fibrosis or cirrhosis (Ishak fibrosis score 5‐6).
Also in the African cohort, an intronic SNP occurring in the NCF4 gene, rs760519, was significantly associated with the Ishak fibrosis score at the onset of treatment (FDR P = 0.017). Over two thirds (69%) of African patients who were homozygous for the T allele had Ishak scores between 3 and 6 (moderate fibrosis through cirrhosis) at the onset of treatment compared to 16% of those who were heterozygous (C/T; Fig. 2). None of the African patients were homozygous for the C allele of rs760519. The rs760519 genotype was a significant predictor (P = 0.0003) for the Ishak fibrosis score at the onset of treatment after controlling for age and sex. Patients who were homozygous for the T allele of rs760519 had 36 times higher risk of moderate fibrosis (versus none or mild) than patients who were heterozygous (C/T) for rs760519 (OR, 35.8; 95% CI, 3.98‐321). Furthermore, patients who were homozygous for the T allele had 5 times higher risk of advanced fibrosis (versus none or mild) than patients who were heterozygous (C/T) for rs760519 (OR, 4.99; 95% CI, 0.897‐27.8). Thus, homozygosity for the T allele appeared to predispose African patients to greater levels of fibrosis.
Figure 2.
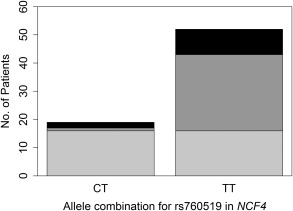
Number of patients with HCV in the African cohort, by rs760519 genotype, who had none to mild, moderate, or advanced fibrosis/cirrhosis at the onset of treatment. Light gray represents none to mild fibrosis (Ishak fibrosis score 0‐2); dark gray represents moderate fibrosis (Ishak fibrosis score 3‐4); black represents advanced fibrosis or cirrhosis (Ishak fibrosis score 5‐6).
In the Caucasian cohort, rs2292464, a SNP located in an intron of DUOX1, was significantly associated with periportal inflammation (FDR P = 0.041). Only 4% of patients who were homozygous for the T allele presented with advanced periportal inflammation at the onset of treatment, whereas 26% of patients who were heterozygous (C/T) and 29% of patients who were homozygous for the C allele had advanced periportal inflammation when treatment was initiated (Fig. 3). The rs2292464 genotype was a significant predictor (P = 0.002) for periportal inflammation after controlling for age and sex. Patients who were homozygous for the C allele of rs2292464 had 11 times higher risk of having advanced periportal inflammation compared to patients who were homozygous for the T allele (OR, 10.6; 95% CI, 2.38‐47.3). Similarly, Caucasian patients who were heterozygous (C/T) for rs2292464 had 9 times higher risk of having advanced periportal inflammation compared to patients who were homozygous for the T allele (OR, 8.54; 95% CI, 1.98‐36.9). Thus, homozygosity for the T allele appeared to protect Caucasian patients from developing advanced periportal inflammation prior to treatment.
Figure 3.
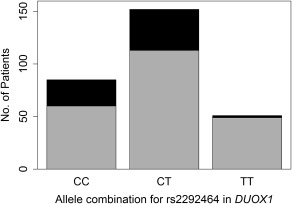
Number of patients with HCV in the Caucasian cohort, by rs2292464 genotype, who had none to moderate versus advanced periportal inflammation. Gray represents none to moderate periportal inflammation (score 0‐3); black represents advanced periportal inflammation (score 4‐6).
Interestingly, rs2292464 was associated with the highest Ishak fibrosis score achieved over the course of treatment, but the relationship was not significant (P = 0.100). No SNPs in either cohort were associated with portal, lobular, or total inflammation.
By Cox regression, no SNPs were significant risk factors for death. However, in the Caucasian cohort, a SNP in an intron of the NOX4 gene, rs10830277, was mildly associated with the risk of death (P = 0.104), using periportal inflammation and the first‐observed and highest achieved Ishak fibrosis scores, recoded into three levels, as covariates.
Discussion
In this exploratory study, we assessed whether polymorphisms in the genes encoding the seven members of the NOX family, and their regulatory factors, were associated with the progression of liver disease in patients with HCV. We also tested whether these polymorphisms were risk factors for death, given a patient's state of hepatic inflammation and fibrosis. We conducted our analyses race specifically and discovered that three SNPs were significantly associated with the progression of liver disease.
In the African cohort, rs12753665 and rs760519 were significantly associated with Ishak score. rs12753665 is located in an intron of NCF2, while rs760519 falls in an intron of NCF4. All African patients who were homozygous for the A allele of rs12753665 had progressed to advanced fibrosis or cirrhosis over their course of treatment compared to only about one quarter of those who were heterozygous (A/G) or homozygous for the G allele, respectively (Fig. 1). This SNP remained a significant predictor for fibrosis after controlling for age and sex and was mildly but not significantly associated with the Ishak fibrosis score at the onset of treatment. Similarly, only 16% of patients who were heterozygous (C/T) for rs760519 presented with moderate to advanced fibrosis or cirrhosis at the onset of treatment compared to 69% of those who were homozygous for the T allele (Fig. 2). After controlling for age and sex, the rs760519 genotype was a significant predictor for the Ishak fibrosis score at the onset of treatment. Patients who were homozygous for the T allele of rs760519 had 36 times and 5 times the risk of having moderate or advanced fibrosis, respectively, relative to patients who were heterozygous (C/T) for rs760519.
To our knowledge, no significant associations between rs12753665 or rs760519 and any chronic disease have been reported nor have the protein products of NCF2 and NCF4 been studied for their roles in human liver disease. However, the importance of these genes in innate immunity and inflammation is evident from studies of nonhepatic diseases. Missense and nonsense variants in NCF2 and NCF4 along with mutations in other NOX family members cause autosomal recessive chronic granulomatous disease.29, 30, 31, 32, 33, 34 Missense variants in NCF2 have been linked to lupus35 and very early onset inflammatory bowel disease,36 and NCF2 gene expression is significantly elevated in patients with chronic rhinosinusitis who have Staphylococcus aureus infection and nasal polyps.37 Several variants in NCF4 were identified as susceptibility factors for autoimmune diseases. The variant rs8137602, not tested in our study, was associated with ulcerative colitis in Caucasians.38 Likewise, an intronic variant, rs4821544, has been implicated in Crohn's disease in Caucasians.39, 40 This variant was tested in our Caucasian cohort but was not significantly associated with any measures of inflammation or fibrosis. It was not tested in the African cohort because it was dropped during the LD pruning step. Similarly, another intronic SNP, rs729749, was linked to rheumatoid arthritis in autoantibody‐negative Swedish men.41 In our study, it was not significantly associated with inflammation or fibrosis in the African cohort and it was not tested in the Caucasian cohort as it was dropped during the LD pruning step.
In the Caucasian cohort, rs2292464, a SNP found in an intron of DUOX1, was significantly associated with periportal inflammation. At the onset of treatment, only 4% of patients who were homozygous for the T allele had advanced periportal inflammation compared to 26%‐29% of patients who were heterozygous (C/T) or homozygous for the C allele (Fig. 3). After controlling for age and sex, the rs2292464 genotype remained a significant predictor for periportal inflammation. Patients who were homozygous for the C allele or heterozygous (C/T) had, respectively, 11 times and 9 times greater risk of having advanced periportal inflammation compared to those who were homozygous for the T allele. Interestingly, rs2292464 also was mildly but not significantly associated with the highest Ishak fibrosis score achieved over the course of treatment. The association of rs2292464 with both periportal inflammation and fibrosis is in accordance with studies demonstrating that periportal inflammation and necrosis increase with progressive stages of fibrosis.42
There are no studies to our knowledge testing rs2292464 as a risk factor for any human or other mammalian disease. However, DUOX1 has been implicated in HCC, and several studies show that DUOX1 may act as a tumor suppressor gene. For example, Ling et al.43 found that DUOX1 messenger RNA expression was significantly decreased in primary HCC tissues, which was due to hypermethylation of the DUOX1 promoter. Overexpression of DUOX1 inhibited the growth of cultured cancer cells by arresting the cell cycle and increasing intracellular ROS. A follow‐up study confirmed that in patients with HCC, DUOX1 expression was decreased in liver tumors relative to adjacent healthy tissues.44 Furthermore, elevated levels of DUOX1 transcripts were associated with longer survival, making DUOX1 prognostic for HCC‐related mortality.44, 45 However, another group studying HCC following hepatectomy found that DUOX1 messenger RNA expression was higher in neoplastic versus adjacent non‐neoplastic tissues and was undetectable in normal liver tissue.46 Elevated DUOX1 expression rather than decreased expression was prognostic of a poor outcome in patients with HCC. Aside from the liver, DUOX1 is highly expressed in the thyroid and lung and to a lesser extent in other adult tissues, including prostate, heart, kidney, pancreas, and placenta.47 Its physiological role is unclear, and there is disagreement regarding whether it can compensate for loss of function in DUOX2.48, 49
Although none of the variants in this study were significant predictors of mortality, a SNP in the intron of NOX4, rs10830277, was mildly associated with the risk of death. To our knowledge, there are no reports evaluating this SNP for its role in liver disease. There are contradictory studies regarding NOX4 as a prognostic factor for HCC. Ha et al.50 found that patients with low NOX4 protein expression had significantly lower recurrence‐free and overall survival compared to those with high protein expression. However, Lu et al.46 measured NOX4 transcripts in HCC tumor tissues and found that they were not associated with recurrence‐free or overall survival. Further studies are needed to elucidate the effect of NOX4 expression on survival in patients with liver disease.
A limitation to our study is the small number of study participants given the number of SNPs tested; this reduced statistical power. We attempted to maximize power by objectively reducing the number of candidate SNPs, collapsing patients into fewer phenotypic groups based on recoding strategies, and using stringent statistical methods. Nonetheless, we recognize the need to validate this study in a larger cohort of African and Caucasian patients with HCV. We also acknowledge that there could be a confounding effect of treatment on the association between SNP composition and Ishak score; however, this confounding could only exist for the highest Ishak score achieved because that was the only phenotype (potentially) measured after treatment had begun. All other phenotypes, including the first Ishak score, were measured prior to treatment.
We anticipate that polymorphisms in the NOX gene family are one of many factors that contribute to the progression of liver disease. Our study suggests that NOX polymorphisms may be deleterious in some cases but protective in others. Further studies are necessary to confirm the roles of our target SNPs and to determine whether additional NOX polymorphisms are related to disease progression. Nonetheless, it is likely that NOX family proteins and the polymorphisms affecting their structure or function are universally involved in liver disease, regardless of etiology. NOX proteins are strongly implicated in the development of hepatic fibrosis.51, 52 They mediate, in part, the activation of hepatic stellate cells, which in turn release collagen to the extracellular matrix, forming the scar tissue that is the hallmark of fibrosis. NOX‐derived ROS also promote hepatic stellate cell proliferation and hepatocyte apoptosis. NOX inhibitors are therefore currently being tested as therapeutics for hepatic fibrosis.51, 52
This study was the first to explore the relationship between the genetic makeup of NOX genes as a functional group and measures of liver damage. Our results suggest that, in the context of hepatitis C, genetic variants in NOX family members may act as risk factors for disease. We identified two SNPs that are significantly associated with hepatic fibrosis and one that is associated with periportal inflammation. None of these SNPs have been previously associated with any chronic human disease. Future studies investigating these SNPs in larger cohorts of patients with HCV and in patients with other chronic diseases are warranted. Although treatments for HCV are becoming more widespread and effective, knowledge of the genetic risk for disease progression could prove extremely helpful in stratifying patients into different treatment groups, especially at earlier stages of disease.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1103/full.
Supplemental Table 1. SNPs analyzed for associations with liver fibrosis and inflammation in the African (ASW) and Caucasian (CEU) cohorts. This list includes only those SNPs that were retained after the LD pruning step.
Potential conflict of interest: Nothing to report.
Supported by intramural National Institute of Diabetes and Digestive and Kidney Diseases, National Cancer Institute, and National Human Genome Research Institute funding and was conducted independently without input from sponsors.
REFERENCES
- 1. Edlin BR, Eckhardt BJ, Shu MA, Holmberg SD, Swan T. Toward a more accurate estimate of the prevalence of hepatitis C in the United States. Hepatology 2015;62:1353‐1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization . Hepatitis C. http://www.who.int/mediacentre/factsheets/fs164/en/. Accessed June 24, 2017.
- 3. Kim WR, Terrault NA, Pedersen RA, Therneau TM, Edwards E, Hindman AA, et al. Trends in waiting list registration for liver transplantation for viral hepatitis in the United States. Gastroenterology 2009;137:1680‐1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. El‐Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012;142:1264‐1273.e1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ward JW. The epidemiology of chronic hepatitis C and one‐time hepatitis C virus testing of persons born during 1945 to 1965 in the United States. Clin Liver Dis 2013;17:1‐11. [DOI] [PubMed] [Google Scholar]
- 6. Hagan H, Des Jarlais DC, Stern R, Lelutiu‐Weinberger C, Scheinmann R, Strauss S, et al. HCV synthesis project: preliminary analyses of HCV prevalence in relation to age and duration of injection. Int J Drug Policy 2007;18:341‐351. [DOI] [PubMed] [Google Scholar]
- 7. Attar BM, Van Thiel DH. Hepatitis C virus: a time for decisions. Who should be treated and when? World J Gastrointest Pharmacol Ther 2016;7:33‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Micallef JM, Kaldor JM, Dore GJ. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J Viral Hepat 2006;13:34‐41. [DOI] [PubMed] [Google Scholar]
- 9. Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009;119:1745‐1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zampino R, Marrone A, Restivo L, Guerrera B, Sellitto A, Rinaldi L, et al. Chronic HCV infection and inflammation: clinical impact on hepatic and extra‐hepatic manifestations. World J Hepatol 2013;5:528‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thein HH, Yi Q, Dore GJ, Krahn MD. Estimation of stage‐specific fibrosis progression rates in chronic hepatitis C virus infection: a meta‐analysis and meta‐regression. Hepatology 2008;48:418‐431. [DOI] [PubMed] [Google Scholar]
- 12. Persson T, Popescu BO, Cedazo‐Minguez A. Oxidative stress in Alzheimer's disease: why did antioxidant therapy fail? Oxid Med Cell Longev 2014;2014:427318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valgimigli M, Valgimigli L, Trere D, Gaiani S, Pedulli GF, Gramantieri L, et al. Oxidative stress EPR measurement in human liver by radical‐probe technique. Correlation with etiology, histology and cell proliferation. Free Radic Res 2002;36:939‐948. [DOI] [PubMed] [Google Scholar]
- 14. Vidali M, Tripodi MF, Ivaldi A, Zampino R, Occhino G, Restivo L, et al. Interplay between oxidative stress and hepatic steatosis in the progression of chronic hepatitis C. J Hepatol 2008;48:399‐406. [DOI] [PubMed] [Google Scholar]
- 15. Fujita N, Sugimoto R, Ma N, Tanaka H, Iwasa M, Kobayashi Y, et al. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J Viral Hepat 2008;15:498‐507. [DOI] [PubMed] [Google Scholar]
- 16. Khadem Ansari MH, Omrani MD, Kheradmand F. Oxidative stress response in patients infected by diverse hepatitis C virus genotypes. Hepat Mon 2015;15:e22069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ivanov AV, Smirnova OA, Ivanova ON, Masalova OV, Kochetkov SN, Isaguliants MG. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS‐dependent and independent mechanisms in HUH7 cells. PLoS One 2011;6:e24957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ivanov AV, Bartosch B, Smirnova OA, Isaguliants MG, Kochetkov SN. HCV and oxidative stress in the liver. Viruses 2013;5:439‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ivanov AV, Smirnova OA, Petrushanko IY, Ivanova ON, Karpenko IL, Alekseeva E, et al. HCV core protein uses multiple mechanisms to induce oxidative stress in human hepatoma Huh7 cells. Viruses 2015;7:2745‐2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Mochel NS, Seronello S, Wang SH, Ito C, Zheng JX, Liang TJ, et al. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010;52:47‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245‐313. [DOI] [PubMed] [Google Scholar]
- 22. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity‐associated pathologies. Cell Mol Immunol 2015;12:5‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maraldi T. Natural compounds as modulators of NADPH oxidases. Oxid Med Cell Longev 2013;2013:271602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal 2014;20:2854‐2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22:696‐699. [DOI] [PubMed] [Google Scholar]
- 26. Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology 1981;1:431‐435. [DOI] [PubMed] [Google Scholar]
- 27. Nasta P. “Immune activation, aging and gender” and progression of liver disease. Acta Biomed 2011;82:115‐123. [PubMed] [Google Scholar]
- 28. Lee MH, Yang HI, Yuan Y, L'Italien G, Chen CJ. Epidemiology and natural history of hepatitis C virus infection. World J Gastroenterol 2014;20:9270‐9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ben‐Farhat K, Ben‐Mustapha I, Ben‐Ali M, Rouault K, Hamami S, Mekki N, et al. A founder effect of c.257 + 2T > C mutation in NCF2 gene underlies severe chronic granulomatous disease in eleven patients. J Clin Immunol 2016;36:547‐554. [DOI] [PubMed] [Google Scholar]
- 30. Chou J, Hsu JT, Bainter W, Al‐Attiyah R, Al‐Herz W, Geha RS. A novel mutation in NCF2 associated with autoimmune disease and a solitary late‐onset infection. Clin Immunol 2015;161:128‐130. [DOI] [PubMed] [Google Scholar]
- 31. Koker MY, Sanal O, van Leeuwen K, de Boer M, Metin A, Patiroglu T, et al. Four different NCF2 mutations in six families from Turkey and an overview of NCF2 gene mutations. Eur J Clin Invest 2009;39:942‐951. [DOI] [PubMed] [Google Scholar]
- 32. Noack D, Rae J, Cross AR, Munoz J, Salmen S, Mendoza JA, et al. Autosomal recessive chronic granulomatous disease caused by novel mutations in NCF‐2, the gene encoding the p67‐phox component of phagocyte NADPH oxidase. Hum Genet 1999;105:460‐467. [DOI] [PubMed] [Google Scholar]
- 33. Leiding JW, Holland SM. Chronic granulomatous disease In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., eds. GeneReviews(R). Seattle, WA, University of Washington: 1993:1–2. [PubMed] [Google Scholar]
- 34. Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood 2009;114:3309‐3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, et al. Lupus‐associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A 2012;109:E59‐E67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muise AM, Xu W, Guo CH, Walters TD, Wolters VM, Fattouh R, et al. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut 2012;61:1028‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jardeleza C, Jones D, Baker L, Miljkovic D, Boase S, Tan NC, et al. Gene expression differences in nitric oxide and reactive oxygen species regulation point to an altered innate immune response in chronic rhinosinusitis. Int Forum Allergy Rhinol 2013;3:193‐198. [DOI] [PubMed] [Google Scholar]
- 38. Tyler AD, Milgrom R, Stempak JM, Xu W, Brumell JH, Muise AM, et al. The NOD2insC polymorphism is associated with worse outcome following ileal pouch‐anal anastomosis for ulcerative colitis. Gut 2013;62:1433‐1439. [DOI] [PubMed] [Google Scholar]
- 39. Wu PB, Dai JF, Wang Q, Zhang G, Tan SY, Li M, et al. Association between NCF4 rs4821544T/C polymorphism and inflammatory bowel disease risk in Caucasian: a meta‐analysis. Inflamm Res 2015;64:825‐831. [DOI] [PubMed] [Google Scholar]
- 40. Roberts RL, Hollis‐Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn's disease in a population‐based cohort. Genes Immun 2008;9:561‐565. [DOI] [PubMed] [Google Scholar]
- 41. Olsson LM, Lindqvist AK, Kallberg H, Padyukov L, Burkhardt H, Alfredsson L, et al. A case‐control study of rheumatoid arthritis identifies an associated single nucleotide polymorphism in the NCF4 gene, supporting a role for the NADPH‐oxidase complex in autoimmunity. Arthritis Res Ther 2007;9:R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ghany MG, Kleiner DE, Alter H, Doo E, Khokar F, Promrat K, et al. Progression of fibrosis in chronic hepatitis C. Gastroenterology 2003;124:97‐104. [DOI] [PubMed] [Google Scholar]
- 43. Ling Q, Shi W, Huang C, Zheng J, Cheng Q, Yu K, et al. Epigenetic silencing of dual oxidase 1 by promoter hypermethylation in human hepatocellular carcinoma. Am J Cancer Res 2014;4:508‐517. [PMC free article] [PubMed] [Google Scholar]
- 44. Chen S, Ling Q, Yu K, Huang C, Li N, Zheng J, et al. Dual oxidase 1: A predictive tool for the prognosis of hepatocellular carcinoma patients. Oncol Rep 2016;35:3198‐3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen SS, Yu KK, Ling QX, Huang C, Li N, Zheng JM, et al. The combination of three molecular markers can be a valuable predictive tool for the prognosis of hepatocellular carcinoma patients. Sci Rep 2016;6:24582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu CL, Qiu JL, Huang PZ, Zou RH, Hong J, Li BK, et al. NADPH oxidase DUOX1 and DUOX2 but not NOX4 are independent predictors in hepatocellular carcinoma after hepatectomy. Tumour Biol 2011;32:1173‐1182. [DOI] [PubMed] [Google Scholar]
- 47. Edens WA, Sharling L, Cheng G, Shapira R, Kinkade JM, Lee T, et al. Tyrosine cross‐linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J Cell Biol 2001;154:879‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carvalho DP, Dupuy C. Role of the NADPH oxidases DUOX and NOX4 in thyroid oxidative stress. Eur Thyroid J 2013;2:160‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sirokmany G, Donko A, Geiszt M. Nox/Duox family of NADPH oxidases: lessons from knockout mouse models. Trends Pharmacol Sci 2016;37:318‐327. [DOI] [PubMed] [Google Scholar]
- 50. Ha SY, Paik YH, Yang JW, Lee MJ, Bae H, Park CK. NADPH oxidase 1 and NADPH oxidase 4 have opposite prognostic effects for patients with hepatocellular carcinoma after hepatectomy. Gut Liver 2016;10:826‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crosas‐Molist E, Fabregat I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol 2015;6:106‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang S, Kisseleva T, Brenner DA. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front Physiol 2016;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1103/full.
Supplemental Table 1. SNPs analyzed for associations with liver fibrosis and inflammation in the African (ASW) and Caucasian (CEU) cohorts. This list includes only those SNPs that were retained after the LD pruning step.