

Abstract
The farnesoid X receptor (FXR) agonist, a bile acid‐activated nuclear receptor, has been shown to improve the histologic features of nonalcoholic steatohepatitis (NASH); however, a satisfactory effect on hepatic fibrosis has not been achieved. We aimed to investigate the combined effect of FXR agonist and angiotensin II type 1 receptor blocker on hepatic fibrogenesis in rat models of NASH. For 8 weeks, two rat models of NASH were developed. Otsuka Long‐Evans Tokushima Fatty (OLETF) rats were administered intraperitoneal injections of 1 mL/kg pig serum (PS) twice a week, whereas Fischer‐344 rats were fed a choline‐deficient, L‐amino acid‐defined diet (CDAA). The in vitro and in vivo effects of an FXR agonist (INT747) and an angiotensin II type 1 receptor blocker (losartan) on hepatic fibrogenesis were evaluated. In PS‐administered OLETF rats, INT747 and losartan had potent inhibitory effects on hepatic fibrogenesis with suppression of hepatic stellate cell (HSC) activation and expression of transforming growth factor β1 and toll‐like receptor 4. INT747 decreased intestinal permeability by ameliorating zonula occuludens‐1 disruption, whereas losartan directly suppressed activated‐HSC (Ac‐HSC) regulation. The in vitro inhibitory effects of INT747 and losartan on messenger RNA expressions of transforming growth factor β1, toll‐like receptor 4, and myeloid differentiation factor 88 and phosphorylation of nuclear factor‐κB and mothers against decapentaplegic homolog 3 in Ac‐HSC were almost in parallel. Losartan directly inhibited the regulation of Ac‐HSC. Likewise, INT747 in combination with losartan was beneficial on hepatic fibrogenesis in rats fed with CDAA diet. The therapeutic effects of these agents were almost comparable between PS‐administered OLETF and CDAA‐treated rats. Conclusion: INT747 and losartan synergistically suppressed hepatic fibrogenesis by reversing gut barrier dysfunction and inhibiting Ac‐HSC proliferation. Combined therapy may represent a promising novel approach for NASH. (Hepatology Communications 2017;1:928–945)
Abbreviations
- α‐SMA
α‐smooth muscle actin
- Ac‐HSC
activated hepatic stellate cell
- ARB
angiotensin II type 1 receptor blocker
- AT1R
angiotensin II type1 receptor
- AT‐II
angiotensin II
- CDAA #choline‐deficient
L‐amino acid‐defined diet
- CSAA
choline‐supplemented amino acid‐defined diet
- ELISA
enzyme‐linked immunosorbent assay
- FXR
farnesoid X receptor
- HSC
hepatic stellate cell
- IFN‐γ
interferon‐γ
- IL‐6
interleukin‐6
- IR
insulin resistance
- LBP
lipopolysaccharide‐binding protein
- LETO
Long‐Evans Tokushima Otsuka
- LPS
lipopolysaccharide
- mRNA
messenger RNA
- MyD88
myeloid differentiation factor 88
- NASH
nonalcoholic steatohepatitis
- NF‐κB
nuclear factor kappa B
- NIH
National Institutes of Health
- OLETF
Otsuka Long‐Evans Tokushima Fatty
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- PPAR‐γ
peroxisome proliferator‐activated receptor
- PS
pig serum
- SHP
short heterodimer partner
- Smad
mothers against decapentaplegic
- TGF‐β1
transforming growth factor β1
- TJP
tight junction protein
- TLR4
toll‐like receptor 4
- TNF‐α
tumor necrosis factor‐α
- ZO‐1
zonula occuludens‐1
Introduction
Nonalcoholic steatohepatitis (NASH) represents a potentially progressive liver disease characterized by hepatic steatosis, inflammation, and fibrosis, possibly leading to cirrhosis. Pathogenesis of NASH progression has been recently identified as a process of multiple parallel hits,1, 2 including the production of gut‐derived endotoxin, which is profoundly related to toll‐like receptor 4 (TLR4) signaling in promoting liver fibrosis.3 Numerous pharmaceutical companies have been developing novel agents to specifically target NASH pathogenesis4; however, no evidence‐based pharmacotherapies for NASH exist.
The farnesoid X receptor (FXR) is a member of the nuclear receptor superfamily and has emerged as a key player in controlling bile acid homeostasis and multiple metabolic pathways in the liver, intestine, and peripheral tissues.5 Monotherapy with an FXR agonist significantly inhibited liver fibrogenesis in experimental models.6 FXR could critically determine fibrotic responses in mice liver, whereas FXR expression was low in human hepatic stellate cells (HSCs) and periductal myofibroblasts.7 The FXR Ligand Obeticholic Acid in NASH Treatment (FLINT) trial recently provided promising results regarding the efficacy of obeticholic acid, an FXR agonist, in ameliorating the histologic features of NASH; however, satisfactory effects on hepatic fibrosis were not achieved.8 The presence and severity of liver fibrosis on liver biopsy may be the relevant long‐term prognostic histologic features in patients with NASH and fibrosis.9, 10 The renin–angiotensin system is another process playing a crucial role in chronic liver diseases.11, 12, 13 The blockade of angiotensin II (AT‐II) signal transduction through AT‐II type 1 receptor (AT1R) inhibited hepatic fibrogenesis in rats.14, 15 This inhibitory effect of AT1R blocker (ARB) mostly coincided with the suppression of activated‐HSCs (Ac‐HSCs).16 Accordingly, combined INT747 and ARB treatment might be useful in NASH. Here, we evaluated the effect and possible underlying mechanisms of combined INT747 and ARB administration on hepatic fibrogenesis in two different NASH rat models.
Materials and Methods
ANIMALS AND REAGENTS
Two different in vivo models of liver fibrosis were used. For the first model, 6‐week‐old male Otsuka Long‐Evans Tokushima Fatty (OLETF) rats and control Long‐Evans Tokushima Otsuka (LETO) rats were used (Otsuka Pharmaceutical Co., Tokushima, Japan).17 Leptin receptor mutation was introgressed into the Nidd2/of the congenic strain in OLETF rats18 (widely used as type 2 diabetes animal models) to induce insulin resistance (IR) at 10‐15 weeks of age and noninsulin‐dependent diabetes mellitus at 25‐30 weeks of age.19 Pig serum (PS)‐induced hepatic fibrosis substantially occurs because of an intense immune response with minor hepatocyte damage.20 For the second model, 6‐week‐old male Fischer 344 (F344) rats were used (Japan SLC, Inc., Hamamatsu, Japan).
The rats were housed in stainless steel mesh cages under the following controlled conditions: temperature, 23 °C ± 3 °C; relative humidity, 50% ± 20% with 10‐15 air changes/hour; and light illumination for 12 hours/day. Animals were allowed access to tap water ad libitum. INT747 was provided by Intercept Pharmaceuticals, Inc. (New York, NY) and Sumitomo Dainippon Pharma Co. Ltd. (Tokyo, Japan). Losartan, an ARB, was purchased from Merck Ltd. (Tokyo, Japan). AT‐II and lipopolysaccharide (LPS) were purchased from WAKO Pure Chemical Industries, Ltd. (Tokyo, Japan) and Sigma‐Aldrich Corp. (Tokyo, Japan), respectively. Conventional chemical reagents were purchased from Nacalai Tesque (Kyoto, Japan). Choline‐deficient L‐amino acid‐defined (CDAA) diet and choline‐sufficient L‐amino acid‐defined (CSAA) diet were purchased from CLEA Japan, Inc. (Tokyo, Japan).
LIVER FIBROSIS ANIMAL MODEL
Both experiments were conducted over 8 weeks. In the first model, liver fibrosis was induced by repeated intraperitoneal administration of 0.5 mL PS twice a week for 6 weeks. At 6 weeks of age, OLETF rats were randomly divided into four groups (G2‐G5); LETO rats served as negative controls (G1). Each group comprised 10 rats. To induce hepatic fibrosis, 0.5 mL PS (1 mL/kg) was intraperitoneally injected into G1 and G5 rats twice a week for 8 weeks. For assessing liver fibrosis, rats were killed at 14 weeks of age before IR developed.
In the second model, F344 rats were randomly divided into five groups (G6‐G10) of 10 rats each. G6 was designated as the negative control group and was given a CSAA diet and distilled water as a vehicle. Rats in G7‐G10 were fed a CDAA diet. Rats in G1, G2, G6, and G7 were given phosphate‐buffered saline (PBS) through daily oral gavage for 8 weeks. Rats in G3 and G8 and those in G4 and G9 were given clinically equivalent doses of INT747 (30 mg/kg/day) or losartan (30 mg/kg/day),21, 22 respectively, through daily oral gavage. Rats in G5 and G10 were given combined INT747 and losartan. Food intake was the same among the groups.
At the end of the experimental period, rats were anesthetized with diethyl ether and killed. All experiments were approved by the Animal Study Committee of Nara Medical University. The study protocol is according to the 1975 Declaration of Helsinki. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86‐23, revised 1985).
HISTOLOGIC AND IMMUNOHISTOCHEMICAL ANALYSES
In all experimental groups, 5‐μm‐thick sections of formalin‐fixed and paraffin‐embedded liver specimens were processed routinely with Sirius red staining to evaluate hepatic fibrosis. Immunohistochemical staining with α‐smooth muscle actin (α‐SMA) (DAKO, Kyoto, Japan) was performed as described.23 Stained sections of six microscopic visual fields (40 × magnification) per specimen of 10 rats were evaluated by ImageJ software (NIH) as described.23 Histologic features were semiquantitatively assessed according to the nonalcoholic fatty liver disease scoring system as described.24
QUANTITATIVE REAL‐TIME REVERSE‐TRANSCRIPTION POLYMERASE CHAIN REACTION
RNA was extracted from powdered frozen liver and intestinal tissues using RNeasy mini kit (QIAGEN, Tokyo, Japan). Total RNA (2 μg) from each sample underwent reverse transcription to its complementary DNA using the High‐Capacity RNA‐to‐cDNA kit (Applied Biosystems Inc., Foster City, CA), according to the manufacturer's instructions. Messenger RNA (mRNA) expression levels of transforming growth factor‐β1 (TGF‐β1), α1(I)‐procollagen, TLR4, LPS‐binding protein (LBP), FXR, peroxisome proliferator‐activated receptor‐γ (PPAR‐γ), small heterodimer partner (SHP), zonula occludens‐1 (ZO‐1), myeloid differentiation factor 88 (MyD88), and β‐actin were quantified by real‐time polymerase chain reaction (PCR) using SYBR Green on a sequence detection system (Step One Plus; Applied Biosystems Inc.).
PCR was performed as follows: samples were heated for 20 seconds at 95 °C and subjected to 40 cycles of 3 seconds at 95 °C for denaturing and 30 seconds at 60 °C for annealing using β‐actin as an endogenous control. The primer sequences used were as follows: 1) for TGF‐β1, forward 5′‐CGG CAG CTG TAC ATT GAC TT‐3′ and reverse 5′‐AGC GCA CGA TCA TGT TGG AC‐3′; 2) for α1(I)‐procollagen, forward 5′‐AGC TCC TGG GCC TAT CTG ATG A‐3′ and reverse 5′‐AAT GGT GCT CTG AAA CCC TGA TG‐3′; 3) for TLR4, forward 5′‐CCG CTC TGG CAT CAT CTT CA‐3′ and reverse 5′‐CCC ACT CGA GGT AGG TGT TTC TG‐3′; 4) for LBP, forward 5′‐AAC ATC CGG CTG AAC ACC AAG‐3′ and reverse 5′‐CAA GGA CAG ATT CCC AGG ACT GA‐3′; 5) for FXR, forward 5′‐TGG ACT CAT ACA GCA AAC AGA GA‐3′ and reverse 5′GTC TGA AAC CCT GGA AGT CTT TT‐3′; 6) for PPAR‐γ, forward 5′‐AAC ATC CCC AAC TTC AGC AG‐3′ and reverse 5′‐TAC TGC GCA AGA ACT CAT GG‐3′; 7) for SHP, forward 5′‐CCT GGA GCA GCC CTC GTC TCA G‐3′ and reverse 5′‐AAC ACT GTA TGC AAA CCG AGG A‐3′; 8) for ZO‐1, forward 5′‐CCC TCT GAT CAT TCC ACA CA‐3′ and reverse 5′‐TAC ATG TCA TTG CTT GGT GC‐3′; 9) for MyD88, forward, 5′‐AGG AGA TGG GTT GTT TCG AGT AC‐3′ and reverse 5′‐CTC ACG GGT CTA ACA AGG CTA‐3′; and 10) for β‐actin, forward 5′‐GGA GAT TAC TGC CCT GGC TCC TA‐3′ and reverse 5′‐GAC TCA TCG TAC TCC TGC TTG CTG‐3′.
PROTEIN EXPRESSION ANALYSIS
After protein concentration equalization, tumor necrosis factor (TNF‐α; Biosource International, Camarillo, CA), interferon‐γ (IFN‐γ; Biosource International), interleukin‐6 (IL‐6; Quantikine HS; R&D Systems, Minneapolis, MN), and ZO‐1 (LifeSpan Biosciences, Seattle, WA) were determined using enzyme‐linked immunosorbent assay (ELISA) kits according to the manufacturer's instructions.
MEASURING PORTAL ENDOTOXIN LEVELS AND INTESTINAL PERMEABILITY
Serum from the portal vein was analyzed for endotoxin using the endpoint assay method (LDL QCL 1000; BioWhittaker, Walkersville, MD). Samples were eluted for 1 hour in 30 mL pyrogen‐free PBS at room temperature. Dilutions were assayed using a 96‐well microplate reader (Thermo Fisher Scientific Inc., Minneapolis, MN).
To measure intestinal permeability, 125 mg/kg body weight of 40 kDa fluorescein isothiocyanate ‐labeled dextran (Sigma‐Aldrich, Oakville, Canada) was administered by oral gavage 4 hours before being killed. Blood was collected by portal venipuncture. Plasma fluorescence was determined with a fluorescence spectrophotometer (Shimadzu Scientific Instruments, Columbia, MD) at 490‐nm excitation wavelength and 520‐nm emission wavelength using a series of known fluorescein isothiocyanate ‐dextran concentrations diluted in rat plasma.
IMMUNOFLUORESCENCE ANALYSIS
Cryosections of intestinal tissue (10 µm) were prepared and fixed with 4% paraformaldehyde for 10 minutes at 4 °C. After blocking with 10% normal goat serum in PBS, frozen sections were incubated with rabbit polyclonal anti‐mouse ZO‐1 antibody (Invitrogen Life Technologies, Carlsbad, CA) at 1:100 at 4 °C overnight, followed by incubation with donkey anti‐rabbit secondary antibody conjugated with DyLight 488 fluorochrome (Jackson Immuno Research Laboratories, West Grove, PA) for 1 hour at room temperature. Sections were mounted on Vectashield mounting medium with 4′,6‐diamidino‐2‐phenylindole Fluoromount‐G mounting medium for fluorescent nucleic acid staining (Vector Laboratories, Burlingame, CA). Stained specimens were inspected using a digital camera‐equipped confocal scanning laser microscope (Leica TCSNT).
Green fluorescence at 490‐nm excitation wavelength and 510‐nm emission wavelength corresponded to localized ZO‐1 on the intestinal epithelial cell tight junctions. Five images were randomly selected for each sample; the mean fluorescence intensity of five equally sized image regions was measured for each image using ImageJ software (https://imagej.nih.gov/ij/). The mean fluorescence intensity of negatively stained sections was subtracted from that of positively stained sections.
IN VITRO PRIMARY HSC ASSAYS
HSCs were isolated from OLETF rats by sequential digestion of livers with pronase and collagenase as described.25 Freshly isolated HSCs were plated on uncoated plastic dishes at a density of 5 × 105 cells/mL. After 5 days of culture, HSCs became myofibroblast‐like, with reduced lipid vesicles and increased α‐SMA expression. After HSC activation by 7 days of culture, all cells became uniformly distributed and α‐SMA‐positive.
The effect of INT747 and losartan on Ac‐HSC proliferation was evaluated using a colorimetric assay (Roche Applied Science, Laval, Canada), based on cleaving a tetrazolium salt (WST‐1) by mitochondrial dehydrogenases to form formazan in viable cells. Briefly, 5,000 Ac‐HSCs/well were seeded on 96‐well plates containing 100 μL fetal bovine serum‐free media and incubated in the presence or absence of AT‐II (10−6 M), LPS (10−5 M), INT747 (2 × 10−6 M), and/or losartan (10−5 M) at 37 °C for 48 hours followed by addition of WST‐1 (10 μL). After 1 hour incubation, the absorbance of each well was measured at 450 nm using a microplate reader at a reference wavelength of 630 nm.
WESTERN BLOT ANALYSIS
We further examined the in vitro effects of INT747 and losartan on nuclear factor kappa B (NF‐κB) and mothers against decapentaplegic (Smad) signaling pathways in Ac‐HSC, using western blot analysis. A 50‐μg protein was resolved on 10% acrylamide sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and transferred to polyvinylidene fluoride membranes by semidry transfer. Membranes were blocked using 5% skim milk in Tris‐buffered saline with Tween 20 (TBST) for 1 hour at room temperature, followed by overnight incubation with 1/1,000 dilution of specific antibodies against phosphorylated (p) ‐NF‐κB (3031), NF‐κB (8242) (Bioss Inc., Woburn, MA), p‐Smad2 (3101), p‐Smad3 (9520), Smad2/3 (3102), and Smad3 (9523) (Cell Signaling Technology Inc., Beverly, MA) at 4 °C; the membranes were then washed in TBST before incubating with horseradish peroxidase‐conjugated secondary antibodies (diluted in 3% [weight per volume] bovine serum albumin in TBST; 1:1,000) for 1 hour at room temperature. After additional washing in TBST, signals were detected using enhanced chemiluminescence, and densitometric scans were quantified using Image J software (NIH).
STATISTICAL ANALYSES
One‐way analysis of variance followed by Bonferroni or Tukey's post‐hoc tests (in vitro study) were performed. Statistical analyses were performed using GraphPad Prism version 6.04 (GraphPad Software, Inc., La Jolla, CA). All tests were two‐tailed; P < 0.05 was considered significant.
Results
INHIBITORY EFFECT OF INT747 AND LOSARTAN ON HEPATIC FIBROGENESIS
We examined the effects of clinically comparable INT747 and losartan doses on hepatic fibrogenesis. Significant inhibitory effects on hepatic fibrogenesis were observed in G3 and G4 compared with those in G2 (Fig. 1A,B). G5 had marked synergistic inhibitory effects on hepatic fibrogenesis compared to either agent alone; combination treatment with both agents almost completely inhibited hepatic fibrogenesis. No hepatic fibrosis occurred in G1.
Figure 1.
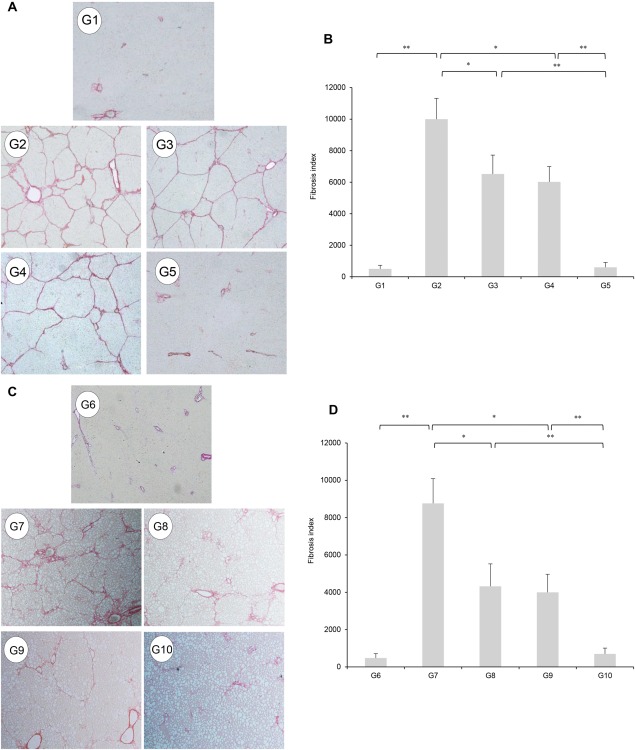
(A) Photomicrographs of Sirius red‐stained liver sections (original magnification × 40). Extensive liver fibrosis is observed in PS‐injected OLETF rats (Group 2). Treatment with INT747 (G3) or ARB (losartan; G4) demonstrated significant inhibitory effects, but combined INT747 and losartan (G5) had more potent inhibitory effects. There was no fibrosis in PS‐injected LETO rats (G1). (C) CDAA treatment (G7) demonstrated the development of extensive fibrosis accompanied by fat accumulation significantly inhibited by individual treatment with INT747 (G8) or losartan (G9). Combination treatment with both agents (G10) exerted a stronger inhibitory effect than single‐agent treatment (G8 and G9). No fibrosis development is observed in the CSAA‐fed control rats (G6). (B,D) Semiquantitative analysis confirmed the histologic findings. Values represent mean ± SD (bars, n = 10). Asterisks indicate significant differences between experimental groups (*P < 0.05, **P < 0.01).
We examined whether these inhibitory effects were also present in CDAA‐treated rats. The therapeutic effects of these agents on hepatic fibrogenesis in PS‐administered OLETF rats were nearly comparable to the inhibitory effects in CDAA‐treated rats (Fig.1C,D). A significantly reduced number of α‐SMA‐positive Ac‐HSCs were observed after administering INT747 and losartan (Fig. 2A,C). Computer‐assisted semiquantitative analysis showed that the number of α‐SMA‐positive cells in INT747‐ and losartan‐treated groups significantly decreased compared with that in the control group (Fig. 2B,D). Moreover, INT747 and losartan together exerted stronger inhibitory effects on both hepatic TGF‐β1 and α1(I)‐procollagen mRNA expressions compared with either single agent (Fig. 3A,G and Fig. 3B,H, respectively). In both experimental models, the drug combination strongly and synergistically inhibited TGF‐β1 and α1(I)‐procollagen expressions, closely coinciding with reduced fibrotic area.
Figure 2.
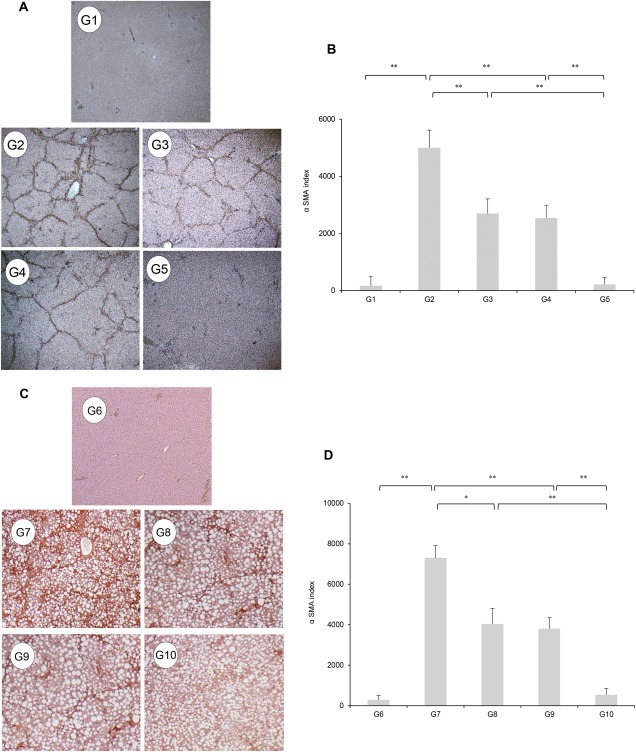
(A,C) Immunohistochemical analysis of α‐SMA expression in the liver. (B,D) Cells stained by α‐SMA immunohistochemistry were measured using image analysis software. Treatment with INT747 (G3) or losartan (G4) resulted in a marked inhibitory effect on α‐SMA expression in the liver compared with PS‐injected OLETF rats (G2). Combination treatment with both agents (G5) exerted a stronger inhibitory effect. No α‐SMA‐positive cells are observed in liver sections from the LETO rats (G1). Compared with G7, monotherapy with INT747 (G8) or losartan (G9) markedly inhibited hepatic α‐SMA expression. Combined INT747 and losartan (G10) exerted a stronger inhibitory effect. No α‐SMA‐positive cells are observed in the hepatic sections from G6 rats. Values represent mean ± SD (bars, n = 10). Asterisks indicate significant differences between indicated experimental groups (*P < 0.05, **P < 0.01).
Figure 3.
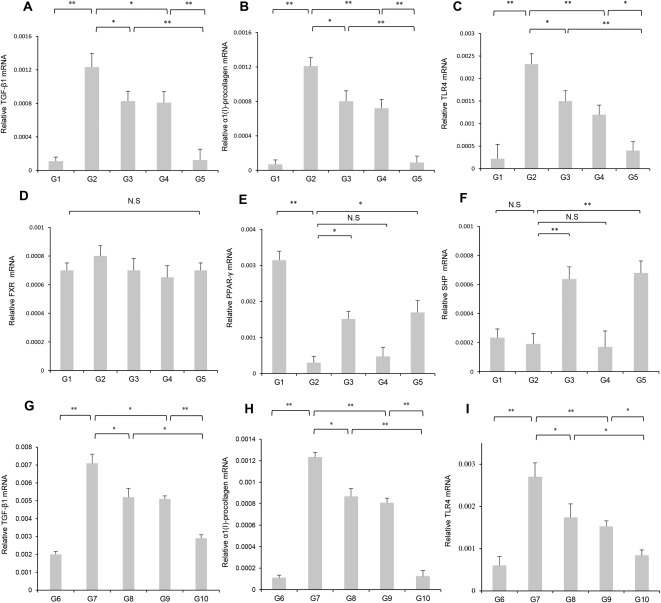
Effects of INT747 and losartan on the hepatic mRNA expression of TGF‐β1, α1(I)‐procollagen, TLR4, FXR, PPAR‐γ, and SHP in the liver from OLETF and LETO rats after PS administration. Compared with single‐agent treatment groups (G3 and G4), combined INT747 and losartan (G5) significantly inhibited hepatic mRNA expression of (A) TGF‐β1, (B) α1(I)‐procollagen, and (C) TLR4. These inhibitory effects closely matched the changes in fibrosis development and α‐SMA. (D) Hepatic FXR expression remained significantly unchanged between the experimental groups. Hepatic expressions of (E) PPAR‐γ and (F) SHP were significantly enhanced in G3 and G5 but were not significantly altered in G4. mRNA expressions of (G) TGF‐β1, (H) α1(I)‐procollagen, and (I) TLR4 were markedly increased in CDAA‐fed rats (G7) compared with those in CSAA‐fed rats (G6). Treatment with INT747 (G8) and losartan (G9) significantly suppressed the hepatic expression of three genes compared with the control group (G7). The suppressive effect of combination treatment (G10) was more potent than that of either agent alone. Values represent mean ± SD (bars, n = 10). Asterisks indicate significant differences between indicated experimental groups (*P < 0.05, **P < 0.01). Abbreviation: N.S, not significant.
INHIBITORY EFFECTS OF INT747 AND LOSARTAN ON TLR4 AND FXR‐SHP SIGNALING
Because the LPS‐TLR4 signaling cascade plays a pivotal role in hepatic fibrogenesis, we evaluated the effects of INT747 and losartan on hepatic and intestinal TLR4 mRNA expression in both models. Hepatic expression of these three genes was markedly augmented in G2 and G7 compared with that in G1 and G6 (Fig. 3C,I). In PS‐administered OLETF rats, G3 and G4 significantly attenuated increases in hepatic TLR4 expression. G8 and G9 significantly attenuated hepatic TLR4 expression in response to the CDAA diet. Combined INT747 and losartan treatment (G5 and G10) resulted in greater inhibition of hepatic TLR4 expression than with either agent alone. The magnitude of the inhibitory effects of INT747 and losartan on hepatic TLR4 expression was comparable with the observed effects on hepatic fibrogenesis between PS‐administered OLETF and CDAA‐fed rats. Intestinal TLR4 was significantly enhanced in G2 compared with that in G1. Intestinal TLR4 expression was significantly inhibited in both G3 and G5 compared with that in G2 and was not significantly reduced in G4 (Fig. 4B). Alternatively, no significant difference in intestinal TLR4 expression was observed between the experimental groups (Fig. 4E). Hepatic FXR expression remained significantly unchanged between the groups (Fig. 3D). Hepatic PPAR‐γ expression was markedly reduced in G2 compared with that in G1. Hepatic SHP expression remained unchanged between G1 and G2. Hepatic PPAR‐γ and SHP expressions were significantly enhanced in G3 and G5 but were not significantly altered in G4 (Fig. 3E,F).
Figure 4.
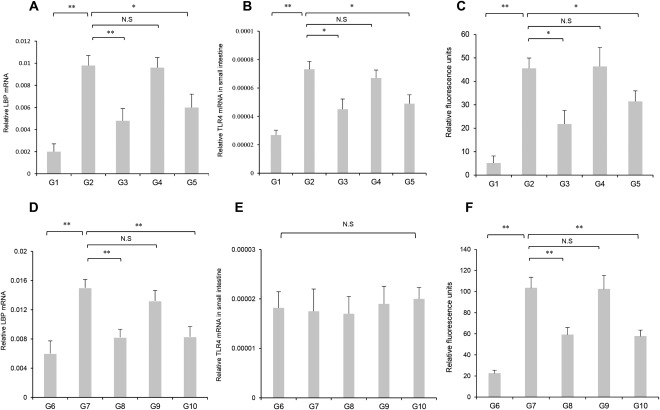
Effect of INT747 and losartan on mRNA expression of hepatic LBP and intestinal TLR4 and intestinal permeability. (A) Hepatic LBP, (B) intestinal TLR4 mRNA expression, and (C) intestinal permeability were drastically augmented in OLETF rats (G2) compared with those in LETO rats (G1). After PS administration in OLETF rats, all three parameters were significantly attenuated in the INT747 (G3) and INT747 combined with losartan (G5) groups but were not significantly altered in the losartan‐treated group (G4). (D) Hepatic LBP expression and (F) intestinal permeability were also augmented in CDAA‐fed rats (G7) compared with those in CSAA‐fed rats (G6). Hepatic LBP expression and intestinal permeability were significantly attenuated in INT747 (G8) and INT747 combined with losartan (G10) groups but were not significantly altered in the losartan‐treated group (G9). (E) No significant differences in intestinal TLR4 expression are observed between the experimental groups (G6‐G10). Values represent mean ± SD (bars, n = 10). Asterisks indicate significant differences between indicated experimental groups (*P < 0.05, **P < 0.01). Abbreviation: N.S, not significant.
INHIBITORY EFFECTS OF INT747 AND LOSARTAN ON LBP, INTESTINAL PERMEABILITY, PORTAL ENDOTOXIN LEVEL, AND HEPATIC INFLAMMATORY CYTOKINES
The LBP‐mediated transfer of LPS to TLR4 is closely associated with portal endotoxin levels through mechanisms involving exaggerated bacterial translocation by increased intestinal permeability.3 To elucidate the protective role of INT747 against gut‐derived endotoxins, the effects of INT747 and losartan on hepatic LBP mRNA expression, intestinal permeability, and portal endotoxin level were evaluated. Similar to serum alanine aminotransferase level, levels of these three variables were higher in G2 and G7 than in G1 and G6, respectively, but were significantly attenuated by INT747 (G3 and G8) and combination treatment (G5 and G10) (Fig. 4A,C,D,F; Tables 1 and 2). Conversely, losartan treatment did not significantly influence these variables.
Table 1.
CHARACTERISTICS OF THE EXPERIMENTAL GROUPS IN LETO AND OLETF RATS ADMINISTERED WIp/TH PIG SERUM
| LETO (G1) | OLETO (G2) | INT747 (G3) | LO (G4) | INT747+LO (G5) | |
|---|---|---|---|---|---|
| Number of rats | 10 | 10 | 10 | 10 | 10 |
| Body weight, g | 362 ± 13.0 | 478 ± 29.5† | 431 ± 11.5†§ | 468 ± 34.1† | 442 ± 20.7†§ |
| Liver/body weight ratio, g/100g BW | 2.9 ± 0.1 | 3.7 ± 0.4† | 3.2 ± 0.4*‡ | 3.8 ± 0.4† | 3.3 ± 0.2†‡ |
| ALT, IU/L | 38.6 ± 5.2 | 70.2 ± 13.6† | 50.8 ± 19.7†‡ | 68.3 ± 26.4† | 48.0 ± 20.3‡ |
| ALB, g/dL | 4.1 ± 0.3 | 3.9 ± 0.4 | 4.1 ± 0.5 | 4.0 ± 0.4 | 3.9 ± 0.4 |
| T‐Bil, mg/dL | 0.13 ± 0.1 | 0.12 ± 0.05 | 0.09 ± 0.07 | 0.11 ± 0.05 | 0.12 ± 0.03 |
| Glucose, mg/dL | 167.2 ± 17.1 | 199.4 ± 11.3† | 184.5 ± 19.9 | 193.5 ± 20.8† | 187.2 ± 27.2 |
| Insulin, μU/mL | 5.2 ± 1.1 | 7.0 ± 0.9† | 6.2 ± 1.8 | 7.2 ± 1.3† | 6.3 ± 2.0 |
| QUICKI | 0.20 ± 0.08 | 0.15 ± 0.09 | 0.17 ± 0.04 | 0.14 ± 0.04 | 0.16 ± 0.05 |
| TG, mg/dL | 143 ± 18.3 | 134.5 ± 19.1 | 131.2 ± 28.3 | 135.2 ± 17.3 | 152.2 ± 22.1 |
| NEFA, μg/L | 302 ± 30.5 | 298 ± 28.5 | 305 ± 32.5 | 279 ± 25.1 | 311 ± 34.1 |
| Portal endotoxin | 0.05 ± 0.01 | 0.76 ± 0.12† | 0.54 ± 0.27†‡ | 0.71 ± 0.20† | 0.43 ± 0.25†§ |
Data are mean ± SD. G1‐G5, Groups 1‐5. Significant compared to G1 (*P < 0.05, † P < 0.01) and G2 (‡ P < 0.05, § P < 0.01).
Abbreviations: ALT, alanine aminotransferase; ALB, albumin; BW, body weight; LO, Losartan; NEFA, nonesterified fatty acids; QUICKI, quantitative insulin sensitivity check index; T‐Bil, total bilirubin; TG, triglyceride.
Table 2.
CHARACTERISTICS OF THE RESPECTIVE GROUPS IN RATS FED THE CSAA‐ OR CDAA‐DIET
| CSAA (G6) | CDAA (G7) | INT747 (G8) | LO (G9) | INT747+LO (G10) | |
|---|---|---|---|---|---|
| Number of rats | 10 | 10 | 10 | 10 | 10 |
| Body weight, g | 317.8 ± 4.2 | 261.8 ± 14.5* | 238.0 ± 17.3*† | 231.2 ± 15.3*† | 207.6 ± 25.1*† |
| Liver/body weight ratio, g/100g BW | 3.3 ± 0.0 | 4.5 ± 0.5* | 4.8 ± 0.2* | 4.7 ± 0.4* | 4.8 ± 0.2* |
| ALT, IU/l | 29.0 ± 7.9 | 391.4 ± 33.6* | 232.8 ± 22.6*† | 356.2 ± 29.5* | 192.3 ± 44.6*† |
| ALB, g/dL | 4.7 ± 0.3 | 4.4 ± 0.5 | 4.5 ± 0.2 | 4.7 ± 0.4 | 4.9 ± 0.2 |
| T‐Bil, mg/dL | 0.14 ± 0.03 | 0.12 ± 0.06 | 0.11 ± 0.03 | 0.11 ± 0.05 | 0.11 ± 0.03 |
| Glucose, mg/dL | 127.2 ± 17.1 | 123.6 ± 24.4 | 145 ± 27.2 | 131.2 ± 25.6 | 125.0 ± 19.5 |
| Insulin, μU/mL | 0.9 ± 0.3 | 0.7 ± 0.5 | 1.0 ± 0.5 | 1.2 ± 0.7 | 1.1 ± 0.4 |
| QUICKI | 1.1 ± 0.3 | 1.2 ± 0.4 | 1.0 ± 0.4 | 0.8 ± 0.5 | 0.9 ± 0.3 |
| TG, mg/dL | 124.0 ± 11.3 | 132.5 ± 18.1 | 136.2 ± 18.6 | 135.2 ± 17.1 | 142.2 ± 19.1 |
| NEFA, μg/L | 309 ± 32.6 | 303 ± 27.5 | 284 ± 37.6 | 296 ± 22.1 | 286 ± 36.4 |
| Portal endotoxin | 0.19 ± 0.1 | 1.15 ± 0.22* | 0.72 ± 0.27*† | 1.01 ± 0.20* | 0.63 ± 0.25*† |
Data are represented as mean ± SD. G6‐G10, Groups 6‐10. Significant compared to G6 (*P < 0.01) and G7 († P < 0.01).
Abbreviations: ALT, alanine aminotransferase; ALB, albumin; BW, body weight; LO, Losartan; NEFA, nonesterified fatty acids; QUICKI, quantitative insulin sensitivity check index; T‐Bil, total bilirubin; TG, triglyceride.
We elucidated the inhibitory effect of INT747 and losartan on proinflammatory cytokine expression using ELISA. TNF‐α and IL‐6 levels in the liver and IFN‐γ levels in the liver and intestine were significantly increased in PS‐administered OLETF rats (G2) compared with PS‐administered LETO rats (G1) (Fig. 5A‐D). IFN‐γ levels in the intestine were drastically augmented in CDAA‐fed rats (G6) compared with CSAA‐fed rats (G7) (Fig. 5E). The levels of these proinflammatory cytokines were significantly suppressed in INT747‐treated (G3 and G8) and combination treatment (G5 and G10) groups but were not significantly altered after losartan treatment (G4 and G9). These data suggest that the inhibitory effect of INT747 on hepatic fibrogenesis was mediated by cytoprotective effects during endotoxin‐triggered hepatic inflammatory response.
Figure 5.

Inhibitory effect of INT747 and losartan on hepatic proinflammatory cytokine expression. Using ELISA, (A) TNF‐α, (B) IFN‐γ levels in the liver, (C) IL‐6 levels in the liver, and (D) IFN‐γ levels in the intestine were significantly increased in PS‐administered OLETF rats (G2) compared with PS‐administered LETO rats (G1). (E) IFN‐γ levels in the intestine were drastically augmented in CDAA‐fed rats (G6) compared with CSAA‐fed rats (G7). The levels of these proinflammatory cytokines were significantly suppressed in INT747‐treated (G3 and G8) and combination treatment (G5 and G10) groups but were not significantly altered after losartan treatment (G4 and G9), respectively. Values represent mean ± SD (bars, n = 10). Asterisks indicate significant differences between indicated experimental groups (*P < 0.05, **P < 0.01). Abbreviation: N.S, not significant.
SEMIQUANTITATIVE DETERMINATION OF INTESTINAL ZO‐1 EXPRESSION
Intestinal epithelial permeability is regulated by intercellular tight junction protein (TJP) complexes comprising several components. We evaluated the effect of INT747 and losartan on ZO‐1 expression to identify mechanisms underlying decreased portal endotoxin levels, hepatic LBP expression, and intestinal permeability in response to INT747 treatment. TJPs were reportedly localized on the apical plasma membrane of epithelial cells and maintain epithelial barrier integrity (Fig. 6A,C).26 Semiquantitative immunofluorescence microscopy revealed a marked down‐regulation of ZO‐1 expression in G2 and G7 compared with that in G1 and G6, respectively (Fig. 6B,D). These findings were confirmed using ELISA (Fig. 6E,F) and PCR assays (Fig. 6G,H). INT747 (G3 and G8) and combination treatment (G5 and G10) attenuated ZO‐1 expression down‐regulation, whereas losartan had no significant effect on ZO‐1 expression (G4 and G9).
Figure 6.

Effect of INT747 and losartan on intestinal ZO‐1 expression. Confocal immunofluorescence microscopy was used to evaluate the effect of INT747 and losartan on ZO‐1 expression in intestinal tissues in (A) OLETF and LETO rats and (C) CDAA‐ and CSAA‐diet‐fed rats. Semiquantitative immunofluorescence microscopy, ELISA, and PCR assays revealed (C,E,G) decreased ZO‐1 expression in OLETF rats (G2) compared with that in LETO rats (G1) and (D,F,H) decreased ZO‐1 expression in CDAA‐fed rats (G7) compared with that in LETO rats (G6). INT747 alone (G3 and G8) and combined INT747 and losartan (G5 and G10) significantly enhanced ZO‐1 expression that was reduced in G2 and G7, respectively. No significant reduction in liver fibrosis was observed in losartan‐treated rats (G4 and G9). Values represent mean ± SD (bars, n = 10). Asterisks indicate significant differences between indicated experimental groups (*P < 0.05, **P < 0.01). Abbreviation: N.S, not significant.
INHIBITORY EFFECT OF INT747 AND LOSARTAN ON ACTIVATED HSCs IN VITRO
Microscopy examination of the direct effect of INT747 and losartan on AT‐II‐ and LPS‐induced proliferation of Ac‐HSC in vitro did not show any significant morphologic changes among the groups during the culture period. AT‐II and LPS treatment resulted in a 1.5‐fold increase in Ac‐HSC proliferation (Fig. 7A). The administration of losartan alone or combined INT747 and losartan significantly inhibited the effects of AT‐II and LPS. However, there was no significant effect of INT747 on Ac‐HSC proliferation. AT‐II and LPS treatment significantly increased mRNA expression of TGF‐β1, TLR4, and Myd88 and phosphorylation of NF‐κB, Smad2, and Smad3. Contrary to the effects on Ac‐HSC proliferation, AT‐II‐ and LPS‐induced mRNA expression of TGF‐β1, TLR4, and Myd88 and phosphorylation of NF‐κB and Smad3 were individually suppressed by INT747 or losartan (Fig. 7B‐E,G). Combined therapy exerted more potent suppression than monotherapy with either agent. The administration of losartan alone or combined therapy significantly reduced AT‐II‐ and LPS‐induced Smad2 phosphorylation, whereas INT747 alone had no significant effect on Smad2 phosphorylation (Fig. 7F). These results indicated that an attenuation of hepatic fibrogenesis is, at least in significant part, mediated by interfering with HSC proliferation by losartan and with the intracellular signaling pathways in HSC by both agents.
Figure 7.
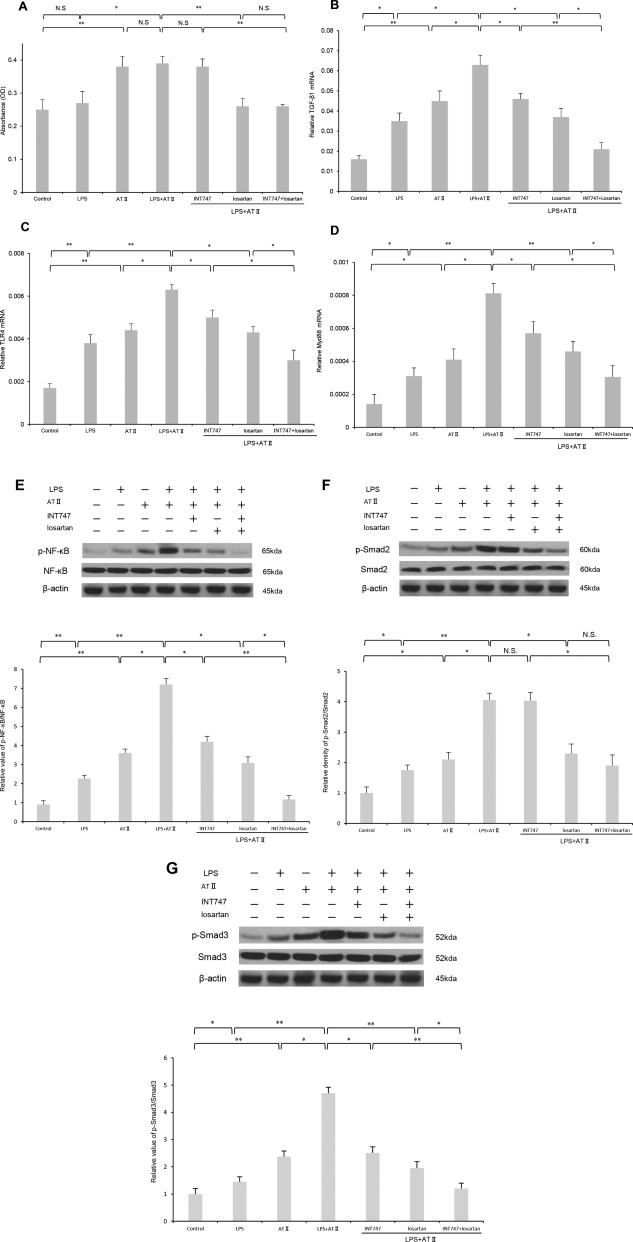
In vitro effects of INT747 and losartan on the AT‐II‐ and LPS‐induced proliferation, mRNA expressions of TGF‐β1, TLR4, and Myd88, and NF‐κB and Smad signaling pathways in Ac‐HSCs. (A) AT‐II and LPS treatment resulted in a significant increase in Ac‐HSC proliferation. The administration of losartan alone or combined INT747 and losartan significantly inhibited the effects of AT‐II and LPS. Treatment with INT747 did not significantly attenuate Ac‐HSC proliferation. AT‐II and LPS treatment significantly increased mRNA expressions of (B) TGF‐β1, (C) TLR4, and (D) Myd88 and phosphorylation of (E) NF‐κB, (F) Smad2, and (G) Smad3. AT‐II‐ and LPS‐induced mRNA expression of TGF‐β1, TLR4, and Myd88 and phosphorylation of NF‐κB and Smad3 were individually suppressed by INT747 or losartan. Combined therapy exerted more potent suppression than monotherapy with either agent. The administration of losartan alone or combined therapy significantly reduced AT‐II‐ and LPS‐induced Smad2 phosphorylation, whereas INT747 alone had no significant effect on Smad2 phosphorylation. Values represent mean ± SD (bars, n = 8). Asterisks indicate significant differences between indicated experimental groups (*P < 0.05, **P < 0.01). Abbreviation: N.S, not significant.
EFFECT OF INT747 ON ZO‐1 AND FXR EXPRESSIONS IN INTESTINAL EPITHELIAL CELLS
We evaluated the in vitro effect of INT747 on FXR and ZO‐1 mRNA expressions in LPS‐induced intestinal epithelial cells (IEC‐6). INT747 prevented the down‐regulation of ZO‐1 mRNA expression in LPS‐induced IEC‐6 (Fig. 8A), whereas INT747 did not significantly alter FXR expression (Fig. 8B), suggesting that TJP was not directly regulated by INT747 in IEC‐6.
Figure 8.

Effects of INT747 on ZO‐1 and FXR expressions in intestinal epithelial cells. (A) INT747 prevented the down‐regulation of ZO‐1 mRNA expression in LPS‐IEC‐6, whereas (B) INT747 did not significantly alter FXR expression. Values represent mean ± SD (bars, n = 10). Asterisks indicate significant differences between indicated experimental groups (*P < 0.05, **P < 0.01). Abbreviation: N.S, not significant.
Discussion
FXR functions as an endogenous sensor for bile acids to orchestrate inflammatory processes at hepatic and extrahepatic sites. The liver is the main site of inflammatory responses to gut lumen‐derived bacterial products and foreign antigens crossing the intestinal epithelial barrier, playing a crucial role in protecting the intestine from bacterial overgrowth‐induced damage. We demonstrated that treatment with clinically equivalent doses of INT747 (30 mg/kg/day) or losartan (30 mg/kg/day) successfully ameliorated hepatic fibrosis and suppressed Ac‐HSCs in rats. In these NASH experimental models, combined INT747 and losartan treatment exerted a marked synergistic inhibitory effect and almost completely attenuated hepatic fibrogenesis. IR was not associated with liver fibrosis progression in two NASH rat models, while IR is the pathophysiologic hallmark of NASH. We evaluated hepatic and intestinal TLR4 expressions in NASH to elucidate the mechanisms underlying hepatic fibrogenesis amelioration by INT747. TLR4‐mediated signaling in intestinal epithelial cells regulated hepatic fibrogenesis in PS‐administered OLETF rats. LPS augmented intestinal epithelial tight junction permeability in vitro and in vivo by TLR4‐dependent increase in colocalization of the membrane‐associated protein CD14.27 Furthermore, LPS regulated tight junction permeability through TLR4‐dependent activation of the focal adhesion kinase‐MyD88 signaling pathway in mice.28 INT747 also normalized TLR4‐dependent NF‐κB activation and inflammation‐related cytokine expressions in the intestine in carbon tetrachloride‐induced liver cirrhosis.29 However, consistent with our observations,3, 30 hepatic fibrogenesis was independently associated with intestinal TLR4 expression in CDAA‐treated rats. The reason for the difference in the role for TLR4 signaling in liver fibrogenesis between two models is unclear but may be explained with an assumption that TLR4 expression in intestinal epithelial cells varies between different species and strains. The effects of intestinal TLR4‐mediated signaling on hepatic fibrogenesis warrant further investigation. Evaluation of tight junctions, which regulate epithelial paracellular pathways in response to endotoxin, demonstrated that INT747 decreased intestinal permeability. Proinflammatory cytokines, including TNF‐α and IFN‐γ, induced TJP disruption through alterations of the apical junctional complex.31 Verbeke et al.32 reported that natural killer cells promote epithelial TJP internalization by secreting IFN‐γ in a bile duct ligation‐induced model of hepatic fibrosis, implying that the LPS‐TLR4 signaling cascade in the liver may be, at least in part, a prerequisite to hepatic fibrogenesis. The inhibitory effect of INT747 on liver fibrosis was mediated by preventing intestinal permeability through restoration of TJP expression and inhibition of the systemic inflammatory response caused by LPS translocation.
HSC activation remains a central event during hepatic fibrosis and involves a complex network of autocrine/paracrine fibrogenic signals that promote transdifferentiation of quiescent HSCs to myofibroblastic phenotypes; these are characterized by abundant expression of intracellular filaments, including α‐SMA, and deposition of collagen type 1.
FXR and PPAR‐γ are ligand‐mediated transcription factors exerting counter‐regulatory effects on HSCs.33 FXR activation restricted HSC transdifferentiation from a quiescent phenotype into a myofibroblast‐like phenotype.6 Consistent with our results, one report showed that INT747 did not affect the in vitro proliferation of HSCs but reduced hepatic α‐SMA and TGF‐β expressions through the hepatic FXR‐SHP axis.34 However, FXR expression remained unchanged among the experimental groups, consistent with our observations.34 Other in vitro and in vivo studies revealed that PPAR‐γ activation inhibited collagen synthesis and HSC activation.35 PPAR‐γ expression was attenuated at the early stages of HSC activation, and PPAR‐γ depletion was accompanied by HSC myofibroblastic transdifferentiation.33 These findings indicate that FXR and PPAR‐γ cascades could exert counter‐regulatory effects on HSC activation and modify HSC function at different points.
Losartan markedly inhibited liver fibrogenesis and reduced Ac‐HSC numbers and TGF‐β1 levels. The suppressive effect of losartan on hepatic fibrogenesis was due to directly inhibiting Ac‐HSC proliferation. We recently demonstrated that AT‐II was critically involved in TLR4 expression up‐regulation in Ac‐HSC through AT1R stimulation.36 AT‐II augmented the LPS‐TLR4 signaling cascade by TLR4 up‐regulation but could not stimulate the signaling pathways downstream of TLR4 without LPS because TLR4 was not the downstream receptor of AT‐II. AT‐II significantly promoted LPS‐induced activation and losartan significantly suppressed LPS‐AT‐II‐mediated TLR4/Myd88/NF‐κB and Smad signaling pathways, leading to significantly reduced TGF‐β1 expressions in Ac‐HSCs. These findings reinforce the notion that AT‐II and LPS‐TLR4 interaction in Ac‐HSCs plays a pivotal role in developing liver fibrosis by regulating TGF‐β1 production in Ac‐HSCs.
In our two liver fibrosis experimental models, simultaneously administering INT747 and losartan exerted a more potent and synergistic inhibitory effect on hepatic fibrogenesis than either agent alone by reversing endotoxin‐induced gut barrier dysfunction and suppressing Ac‐HSC proliferation, respectively. This synergistic effect was achieved by clinically equivalent doses of INT747 and losartan. A combination of both therapeutic agents is likely required to yield a substantial therapeutic benefit of slowing NASH progression in future clinical applications.
Potential conflict of interest: Nothing to report.
Supported by Grants‐in‐Aid for Scientific Research (15K08077 to H.Y., M.K., R.N.).
REFERENCES
- 1. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52:1836‐1846. [DOI] [PubMed] [Google Scholar]
- 2. Zhu L, Baker RD, Baker SS. Editorial: from multiple hits to multiple therapeutic targets of non‐alcoholic fatty liver disease. Curr Drug Targets 2015;16:1272‐1273. [DOI] [PubMed] [Google Scholar]
- 3. Douhara A, Moriya K, Yoshiji H, Noguchi R, Namisaki T, Kitade M, et al. Reduction of endotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non‐alcoholic steatohepatitis model. Mol Med Rep 2015;11:1693‐1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ratziu V, Goodman Z, Sanyal A. Current efforts and trends in the treatment of NASH. J Hepatol 2015;62(Suppl.):S65‐S75. [DOI] [PubMed] [Google Scholar]
- 5. Blumberg B, Evans RM. Orphan nuclear receptors‐‐new ligands and new possibilities. Genes Dev 1998;12:3149‐3155. [DOI] [PubMed] [Google Scholar]
- 6. Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L, et al. Cross‐talk between farnesoid‐X‐receptor (FXR) and peroxisome proliferator‐activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther 2005;315:58‐68. [DOI] [PubMed] [Google Scholar]
- 7. Fickert P, Fuchsbichler A, Moustafa T, Wagner M, Zollner G, Halilbasic E, et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol 2009;175:2392‐2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al.; NASH Clinical Research Network . Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, et al. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver‐related mortality. Hepatology 2011;53:1874‐1882. [DOI] [PubMed] [Google Scholar]
- 10. Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology 2015;61:1547‐1554. [DOI] [PubMed] [Google Scholar]
- 11. Yoshiji H, Yoshii J, Ikenaka Y, Noguchi R, Tsujinoue H, Nakatani T, et al. Inhibition of renin‐angiotensin system attenuates liver enzyme‐altered preneoplastic lesions and fibrosis development in rats. J Hepatol 2002;37:22‐30. [DOI] [PubMed] [Google Scholar]
- 12. Yoshiji H, Kuriyama S, Noguchi R, Fukui H. Angiotensin‐I converting enzyme inhibitors as potential anti‐angiogenic agents for cancer therapy. Curr Cancer Drug Targets 2004;4:555‐567. [DOI] [PubMed] [Google Scholar]
- 13. Yoshiji H, Kuriyama S, Fukui H. Angiotensin‐I‐converting enzyme inhibitors may be an alternative anti‐angiogenic strategy in the treatment of liver fibrosis and hepatocellular carcinoma. Possible role of vascular endothelial growth factor. Tumour Biol 2002;23:348‐356. [DOI] [PubMed] [Google Scholar]
- 14. Kitade M, Yoshiji H, Noguchi R, Ikenaka Y, Kaji K, Shirai Y, et al. Crosstalk between angiogenesis, cytokeratin‐18, and insulin resistance in the progression of non‐alcoholic steatohepatitis. World J Gastroenterol 2009;15:5193‐5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, et al. Angiotensin‐II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 2001;34:745‐750. [DOI] [PubMed] [Google Scholar]
- 16. Yoshiji H, Noguchi R, Ikenaka Y, Kaji K, Aihara Y, Shirai Y, et al. Cocktail therapy with a combination of interferon, ribavirin and angiotensin‐II type 1 receptor blocker attenuates murine liver fibrosis development. Int J Mol Med 2011;28:81‐88. [DOI] [PubMed] [Google Scholar]
- 17. Sato T, Asahi Y, Toide K, Nakayama N. Insulin resistance in skeletal muscle of the male Otsuka Long‐Evans Tokushima Fatty rat, a new model of NIDDM. Diabetologia 1995;38:1033‐1041. [DOI] [PubMed] [Google Scholar]
- 18. Kanemoto N, Hishigaki H, Miyakita A, Oga K, Okuno S, Tsuji A, et al. Genetic dissection of “OLETF”, a rat model for non‐insulin‐dependent diabetes mellitus. Mamm Genome 1998;9:419‐425. [DOI] [PubMed] [Google Scholar]
- 19. Nakao‐Hayashi J, Ito H, Kanayasu T, Morita I, Murota S. Stimulatory effects of insulin and insulin‐like growth factor I on migration and tube formation by vascular endothelial cells. Atherosclerosis 1992;92:141‐149. [DOI] [PubMed] [Google Scholar]
- 20. Bhunchet E, Eishi Y, Wake K. Contribution of immune response to the hepatic fibrosis induced by porcine serum. Hepatology 1996;23:811‐817. [DOI] [PubMed] [Google Scholar]
- 21. Michel MC, Foster C, Brunner HR, Liu L. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol Rev 2013;65:809‐848. [DOI] [PubMed] [Google Scholar]
- 22. Remuzzi A, Perico N, Amuchastegui CS, Malanchini B, Mazerska M, Battaglia C, et al. Short‐ and long‐term effect of angiotensin II receptor blockade in rats with experimental diabetes. J Am Soc Nephrol 1993;4:40‐49. [DOI] [PubMed] [Google Scholar]
- 23. Noguchi R, Yoshiji H, Ikenaka Y, Kaji K, Aihara Y, Shirai Y, et al. Dual blockade of angiotensin‐II and aldosterone suppresses the progression of a non‐diabetic rat model of steatohepatitis. Hepatol Res 2013;43:765‐774. [DOI] [PubMed] [Google Scholar]
- 24. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 25. Aihara Y, Yoshiji H, Noguchi R, Kaji K, Namisaki T, Shirai Y, et al. Direct renin inhibitor, aliskiren, attenuates the progression of non‐alcoholic steatohepatitis in the rat model. Hepatol Res 2013;43:1241‐1250. [DOI] [PubMed] [Google Scholar]
- 26. Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr 2011;141:769‐776. [DOI] [PubMed] [Google Scholar]
- 27. Guo S, Al‐Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR‐4 and CD14. Am J Pathol 2013;182:375‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo S, Nighot M, Al‐Sadi R, Alhmoud T, Nighot P, Ma TY. Lipopolysaccharide regulation of intestinal tight junction permeability is mediated by TLR4 signal transduction pathway activation of FAK and MyD88. J Immunol 2015;195:4999‐5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ubeda M, Lario M, Munoz L, Borrero MJ, Rodriguez‐Serrano M, Sanchez‐Diaz AM, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol 2016;64:1049‐1057. [DOI] [PubMed] [Google Scholar]
- 30. Namisaki T, Noguchi R, Moriya K, Kitade M, Aihara Y, Douhara A, et al. Beneficial effects of combined ursodeoxycholic acid and angiotensin‐II type 1 receptor blocker on hepatic fibrogenesis in a rat model of nonalcoholic steatohepatitis. J Gastroenterol 2016;51:162‐172. [DOI] [PubMed] [Google Scholar]
- 31. Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis‐independent mechanisms. J Immunol 2003;171:6164‐6172. [DOI] [PubMed] [Google Scholar]
- 32. Verbeke L, Farre R, Verbinnen B, Covens K, Vanuytsel T, Verhaegen J, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol 2015;185:409‐419. [DOI] [PubMed] [Google Scholar]
- 33. Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C, et al. Peroxisome proliferator‐activated receptor gamma transcriptional regulation is involved in platelet‐derived growth factor‐induced proliferation of human hepatic stellate cells. Hepatology 2000;31:101‐108. [DOI] [PubMed] [Google Scholar]
- 34. Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004;127:1497‐1512. [DOI] [PubMed] [Google Scholar]
- 35. Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati‐Baroni G, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology 2002;122:1924‐1940. [DOI] [PubMed] [Google Scholar]
- 36. Shirai Y, Yoshiji H, Noguchi R, Kaji K, Aihara Y, Douhara A, et al. Cross talk between toll‐like receptor‐4 signaling and angiotensin‐II in liver fibrosis development in the rat model of non‐alcoholic steatohepatitis. J Gastroenterol Hepatol 2013;28:723‐730. [DOI] [PubMed] [Google Scholar]