

Abstract
A key challenge for establishing a phenotypic screen for neuronal excitability is to measure membrane potential changes with high throughput and accuracy. Most approaches to probe excitability rely on methods that are invasive and low throughput, or that lack cell-specific information. These limitations have motivated the development of novel strategies for characterizing the electrical properties of cultured neurons. We have developed optogenetic technologies (Optopatch) that enable stimulation and recording of membrane voltage signals from cultured neurons with single-cell sensitivity and millisecond temporal resolution. Neuronal activity can be elicited using blue light activation of the channelrhodopsin variant, ‘CheRiff’. Action potentials and synaptic signals are measured using ‘QuasAr’, a fast and sensitive voltage-indicating protein, whose near-infrared fluorescence scales proportionately with transmembrane potential. This integrated technology of optical stimulation and recording of electrical signals enables investigation of neuronal electrical function with unprecedented scale and precision.
Keywords: Optical electrophysiology, Optopatch, CheRiff, QuasAr, disease modeling, voltage indicator, optogenetics, induced pluripotent stem cell
Unit Introduction
Disorders of the nervous system remain a major cause of disability in the US and worldwide. Progress on developing improved therapeutics has been limited, in part, by a lack of scalable in vitro assays that can inform disease mechanisms and predict therapeutic efficacy in humans. Target-based approaches to drug discovery have had limited effectiveness in finding novel treatments for diseases of the nervous system (Schoepp, 2011; IOM, 2014). Phenotypic-based screens provide an alternative strategy that is agnostic to target and can be used to probe a complex functional phenotype or interaction of a pharmacological agent with the excitability of a neuron. To this end, human pluripotent stem cell-based methods have been developed in an attempt to more closely model human neurological disorders such as ALS (Wainger et al., 2014), epilepsy (Jiao et al., 2013), and bipolar affective disorder (Mertens et al., 2015). In these disease models, different neuronal types can be produced using induced pluripotent stem (iPS) cells generated from patient somatic cells for the purpose of probing neuronal function in the context of human genetics and physiology (Han et al., 2011). This in vitro approach can become a useful complement to the array of genetically modified rodent models (e.g., (Meikle et al., 2007; Bales et al., 2014; DeMattos et al., 2001)) where specific, disease-relevant genetic alterations can be introduced in defined brain regions.
As the cellular models have continued to advance, so too have the available technologies for probing functional phenotypes and pharmacological responses. In particular, optogenetic tools now provide the capability to non-invasively stimulate neurons and record key electrophysiological parameters from many cells in parallel. Here, we focus on a platform technology termed Optopatch that rapidly and robustly characterizes single-cell electrophysiological response of multiple neuronal types using optogenetic tools. A channelrhodopsin, CheRiff, opened by blue light, stimulates action potentials in the cells while an archaerhodopsin QuasAr, excited by red light, reads out the voltage activity with millisecond temporal resolution. We describe a set of technologies and protocols employed to generate and interpret optical measurements of neuronal excitability. These methods are described in the sections listed below.
Protocol 1: Production of lentivirus encoding Optopatch components
Protocol 2: Culture and transduction of human differentiated neurons (CDI® iCell Neurons)
Protocol 3: Culture and transduction of primary rat hippocampal neurons
Protocol 4: All-optical electrophysiology of cultured neurons using Optopatch
Protocol 5: Extraction of neuronal firing properties from high-speed video recordings
Strategic Planning
The workflow for performing Optopatch measurements in both human induced pluripotent stem cell-derived neurons and rat hippocampal neurons consists of four key steps: 1) production of lentivirus encoding the Optopatch proteins, QuasAr and CheRiff; 2) culture and lentiviral transduction of neurons, 3) Optopatch imaging; and 4) extraction of neuronal firing properties from video recordings. Below we have included detailed protocols describing each step.
There are several key considerations to be made about the Optopatch constructs prior to executing the accompanying protocols. When transfecting cells with Optopatch constructs, both the channelrhodopsin voltage actuator CheRiff, and the voltage reporter QuasAr, there are critical choices regarding: i) the specific promoter used to drive their expression and; ii) the fluorescent proteins that can be fused to the Optopatch components to facilitate their localization both in terms of intracellular trafficking and imaging. The specific cell type under study will determine the optimal promoter choice while the optimal fluorescent fusion protein is determined by other fluorescent sensors or labels used in the experiment.
Neuron-specific promoters are used to avoid expression of the Optopatch components in primary glial cells, which are typically used as a supportive monolayer to drive maturation and prevent cell clumping. When driving expression with a traditional universal promoter e.g., the CMV (cytomegalovirus) sequence, the fluorescence signal in glial cells can overwhelm the signal in the neurons, therefore hindering optical measurements. The CAMK2A gene promoter provides a means to drive strong expression preferentially in excitatory, glutamatergic neurons, and has the lowest levels of expression in glial cells. When the experiment requires recordings from inhibitory neurons as well as excitatory neurons, the pan-neuronal human SYNAPSIN1 (hSYN1) promoter is used, which also drives higher expression levels in neurons, but has the drawback of increased background expression in glial cells. In order to minimize glial expression, lentiviral transduction of the cultured neurons is typically done 24–48 hours prior to the addition of primary glial cells.
In typical experiments, both the CheRiff actuator and the QuasAr2 voltage sensor are fused to a fluorescent reporter protein to aid in the identification of transduced neurons at the time of measurement as well as to provide a ready measure of transduction efficiency and cell health during culture. CheRiff and QuasAr can be successfully fused to numerous fluorescent proteins while maintaining their full function. The fluorescence of validated protein fusions spans the visible spectrum and includes EBFP2 (blue), ECFP (cyan), EGFP (green), Citrine (yellow-green), mPapaya1.0 (yellow), mOrange2 (orange), and TagRFP (red-orange). The fusion proteins, via a mechanism that is not well understood, facilitate proper trafficking of QuasAr to the cellular membrane. This trafficking augmentation also occurs with “dark” fluorescent protein variants with mutations that break the chromophore and eliminate all fluorescence. Ultimately, the selection of fusion protein depends on the planned experiment. For example, based on spectral compatibility, two distinct color channels such as EGFP paired with CheRiff and mOrange2 paired with QuasAr2 can be used in the same experiment. In instances where it may be desirable to leave a fluorescent channel available, e.g., when using post-measurement immunocytochemistry to identify particular neuronal types in the culture, a fusion of EBFP2 to CheRiff and a fusion of “dark”-mOrange2 to QuasAr might be optimal.
Protocol 1: Production of lentivirus encoding Optopatch components
Lentivirus can transduce post-mitotic cells such as cultured neurons with high efficiency as well as drive high and stable expression levels (Naldini et al., 1996; Zufferey et al., 1997; Dull et al., 1998; Zufferey et al., 1999; Sakuma et al., 2012) while inducing low cell toxicity. In our experience, lentiviral transduction efficiency approaches close to 100% of the neurons in culture. This protocol outlines the steps required for the production of lentivirus stocks through the thawing and culture of HEK293T cells, as well as the transfection of these cells with viral packaging plasmids. Unless otherwise stated, all virus production and cell culture work is carried out inside a Biological Safety Cabinet and cultures are maintained in 37°C, 5% CO2 biological incubators.
Material list
-
-
HEK293T cells (ATCC Cat# CRL3216)
-
-
Phosphate Buffered Saline (ThermoFisher Scientific Cat#10010-049)
-
-
10% FBS medium (see Reagents and Solutions section)
-
-
50mL conical tubes (Corning Cat#352050)
-
-
15mL conical tubes (Corning Cat#352196)
-
-
Neurobasal medium (ThermoFisher Scientific #10888-022)
-
-
10 cm (diameter) tissue culture dishes (Corning Cat#353003)
-
-
15 cm (diameter) tissue culture dishes (Corning Cat#352196)
-
-
Viral packaging mix containing plasmids for PsPAX2 and PMD2.G (contains VSVG gene), supplied as 250 μg in a 0.5 μg/ml solution (Cellecta Cat#CPCP-K2A)
-
-
Lentiviral plasmid with promoter and Optopatch gene
-
-
Opti-MEM reduced Serum media (ThermoFisher Scientific Cat#31985-070)
-
-
Polyethylenimine PEI MAX40000 transfection reagent (Polysciences Cat#24765-2)
-
-
0.45 μm Steriflip-HV (EMD Millipore Cat#SE1M003M00)
-
-
0.6 mL Sterile tubes (ThermoFisher Scientific Cat#3449)
-
-
Dimethyl sulfoxide (Sigma-Aldrich Cat#D2650-100ml)
-
-
1.8 mL CryoTube vials (Thermo Scientific Nunc Cat#377267)
-
-
Lenti-X concentrator (Takara Clontech Cat# 631231)
-
-
Lenti-X GoStix (Takara Clontech Cat#631243)
-
-
Lenti-X qRT-PCR Titration Kit (Takara Clontech Cat#631235)
-
-
Molecular Biology grade water (Sigma W4502)
Overall timeline:
Day 0 - Thaw 293T cell cryostock onto 1×10 cm tissue culture dish
Day 3 - Passage cells onto 2×15 cm tissue culture dishes
Day 6 - Passage cells onto 8×15 cm tissue culture dishes
Day 9 - Transfect cells with viral packaging and lentiviral plasmids
Day 12 - Harvest virus 60 hours after transfection
Preparing frozen stocks of 293T cells for virus production
To minimize batch-to-batch variability in the production of different lentiviral stocks, create a large cryobank of low passage 293T stocks that can be used to initiate multiple rounds of virus production. To prepare these frozen stocks, thaw a cryovial of 293T cells in a 37°C water bath for 3 min in preparation for culture in a 10 cm tissue culture dish.
Transfer the contents of the vial to a 15 mL conical tube and rinse the cryovial with 1 mL of 10% FBS medium to collect any remaining cells, and slowly add this dropwise to the gently swirled conical tube to avoid osmotic shock to the thawing cells.
Add an additional 8 mL of 10% FBS medium, bringing the total volume to approximately 10 mL.
Centrifuge the cell mixture for 5 min at 300 g at room temperature to pellet the cells.
Aspirate the supernatant, removing all residual DMSO from the freezing solution, and re-suspend the pellet in 1 mL of 10% FBS medium by gently pipetting up and down up to 5 times with a P-1000 μL pipette.
Bring the total volume to 10 mL, and plate onto a 10 cm tissue culture dish. Culture on a level shelf, which is critical to achieve a uniform media depth and plating density in large culture dishes.
Feed 293T cells every 3 days with 10% FBS medium and passage when 80–90% confluent using the method described in the next section. From the 1×10 cm tissue culture dish, 293T cells are passaged onto 2×15 cm tissue culture dishes. Once in a 15 cm tissue culture dish, 293T cells can be passaged using a P1:4 (1 to 4) dilution factor for every passage (i.e. the cells from 1×15 cm dish can be dissociated and plated onto 4×15 cm dishes for expansion). Once the stage of 16 – 32 × 15 cm dishes is reached, cells are dissociated, counted and frozen in cryovials. On average one should obtain around 5 million 293T cells per 15 cm tissue culture dish, which are frozen in 2 cryovials. This step involves making a cell suspension of 10 million cells per mL in 10% FBS medium, adding 500 μL of this cell suspension to a cryovial, followed by drop-wise addition of 500 μL of 2× Freezing medium (20% DMSO in FBS). The cryovials are transferred immediately into a −80°C freezer for 16 hours and then transferred to a liquid nitrogen storage unit.
Passaging 293T cells for virus production
Passage 293T when they become 80–90% confluent.
Aspirate the medium from the dish and gently rinse with a sufficient volume of PBS to cover the entire surface of the dish (6 mL for 10 cm dish and 20 mL for 15 cm dish) and aspirate.
Dislodge the cells from the surface by more vigorously adding 8 mL of PBS with a 10 mL serological pipette and rinsing the plate several times until cells visibly lift from the surface.
Transfer the suspension to a 50 mL conical tube, and repeat the wash step to collect residual cells, and add the solution to the 50 mL conical tube.
Pellet the cells by centrifuging at 300 g for 5 minutes at room temperature. Aspirate the supernatant, and re-suspended in 4 mL of 10% FBS medium per starting 15 cm dish.
Divide the cell suspension into a number of 50 mL conical tubes equal to the number of 15 cm plates, 1 mL per tube, and bring the total volume in each tube up to 25 mL of 10% FBS medium.
Add each 25 mL cell suspension aliquots to its corresponding 15 cm tissue culture dish and transfer to the incubator.
Transfection of 293T cells
To ensure efficient transfection, endotoxin-free plasmid DNA is used that is typically prepared using a QIAGEN Maxiprep kit. The DNA plasmid should be in a concentration range of 1500–2000 ng/μL. Transfect 293T cells in a 15 cm tissue culture dish described in the previous section when they reach a confluency of 70–80%.
To a 50 mL conical tube, add 20 μg of plasmid containing the gene of interest, 25 μg of the mixture of packaging and envelope plasmids PSPax2 and pMD2.G, 500 μL of Opti-MEM serum free media, and 180 μL of the 1 mg/mL PEI transfection reagent.
Mix thoroughly and incubate for 10 min at room temperature
Add 25 mL of 10% FBS medium to the conical tube and mix with 5 gently inversions.
Aspirate the medium from the 15 cm dish of 293T cells to be transfected, and slowly add the 25 mL transfection mixture to the side of the dish to avoid dislodging the cells.
Culture the transfected cells in a biological incubator for 60 hours before harvesting the virus-rich supernatant.
Harvesting the virus
Lentivirus is a Biosafety Level 2 (BSL2) agent and can infect human cells. It is recommended that sleeves (in addition to the standard gloves and lab coats) be worn and that all containers and pipettes in contact with the virus be bleached prior to disposal in the biohazard waste containers. For 293T cells transfected with a fluorescent protein-expressing construct, transfection efficiency is confirmed using an epifluorescence microscope equipped with fluorescence objectives and filter cubes matched to the fused fluorescent protein (e.g. Semrock DAPI-11LP-A-000, GFP-4050B-000, LED-TRITC-A-000. Either LEDs or a mercury lamp can be used for excitation light. After 24 hours, 30–50% of 293T cells should show fluorescence, indicating PEI transfection efficiency. After 48 – 60 hours, expression efficiency should exceed 80% as released virus transfects additional cells (assuming a promoter has been used that drive expression in HEK cells). Lower transfection efficiencies are indicative of a weak batch that will yield lower viral titres.
Collect the supernatant from each plate containing the released viral particles and place into a 50 mL conical tube.
Centrifuge for 5 min at 2000 g to precipitate larger cells or cellular debris.
Filter the supernatant through a 0.45 μm Steriflip to remove residual cell debris.
Concentrating and characterizing lentivirus stocks
To minimize the volume of viral reagent needed to deliver Optopatch constructs into the neuronal cultures, as well as to eliminate any deleterious components in the 293T cell supernatant, the virus is concentrated and re-suspended in neuronal culture medium.
Mix the filtered virus suspension with the Clontech Lenti-X Concentrator reagent and centrifuge according the manufacturer’s protocol.
Re-suspend the virus in Neurobasal medium to increase viral concentration 10-fold, e.g. 25 mL of supernatant with virus particles is re-suspended in 2.5 mL of Neurobasal medium.
Aliquot the concentrated stocks in sterile 0.6 mL tubes and store at −80°C until ready to use. Repeat freeze/thaw cycles should be avoided as these lower viral titer.
The lentivirus stocks can be qualitatively titered with Clontech Lenti-X GoStix, and quantitatively with Clontech Lenti-X qRT-PCR using the manufacturer’s protocols. We typically achieve viral genome titers of 109 – 1010 copies/mL with a functional titer around 50 times lower at ~108 IFU/mL.
Protocol 2: Culture and transduction of human differentiated neurons (CDI iCell Neurons)
The goal is to culture human iPS cell-derived neurons expressing the Optopatch constructs on an optical quality substrate for subsequent optical electrophysiology recordings. This protocol guides the reader through the steps of coating glass-bottomed dishes, plating iCell Neurons, transducing cells, and adding glia to promote cell health and maturation. iCell Neurons, according to the manufacturer, represent a mixture of primarily two types of post-mitotic human iPS cell-differentiated neurons, GABAergic and glutamatergic neurons.
Material list
-
-
Glass-bottomed dishes (MatTek Cat#P35G-1.5-10-C).
-
-
Poly-D-lysine (Sigma Cat#P6407-5MG)
-
-
PBS (Life Technologies Cat#10010-049)
-
-
Laminin (Life Technologies Cat#23017-015)
-
-
iCell Neurons (Cellular Dynamics Inc. CDI Cat#NRC-100-010-001)
-
-
iCell Neuron maintenance media (CDI Cat# NRM-100-121-001)
-
-
iCell Neuron maintenance media supplement (CDI Cat #: NRM-100-031-001)
-
-
Poly-D-Lysine coated T-25 flasks (Corning Cat#356536)
-
-
Glial medium (Reagents and Solutions)
-
-
Trypsin EDTA (Sigma-Aldrich Cat#T4049-500ML)
Overview
Day 0 – Thaw and plate iCell Neurons from CDI.
Day 2 – Lentiviral delivery of Optopatch constructs
Day 3 – Add primary mouse glial cells to promote maturation and prevent clumping.
Day 21–30 – Functional imaging with Optopatch.
Coating of glass-bottomed dishes
We typically use glass-bottomed dishes as an optical quality substrate, although tissue culture treated cyclic olefin copolymer (COC) plates (Ibidi Cat#81156) often facilitate better cell adhesion and have lower autofluorescence from intense red laser illumination.
Coat 35 mm glass-bottomed dishes with a 10 mm center glass area with poly-D-lysine (PDL) by adding 150 μL of 100 μg/mL solution onto the center glass bottom area of the dish.
After overnight incubation at 4°C, aspirate the PDL solution and rinse the dishes twice with 1 mL of PBS, aspirating of the whole dish volume after each rinse.
Add 150 μL of a 20 μg/mL laminin solution to the center circle. iCell Neurons can be plated the following day after overnight incubation at 4°C, or up to three days later.
Thawing and plating of iCell Neurons onto MatTek dishes
Thaw the iCell Neurons according to the CDI iCell Neurons user’s guide:
Incubate the MatTek dishes with laminin solution overnight at 37°C.
After quickly thawing a frozen cell vial in a 37°C water bath, transfer 1 mL of the cell mixture to a 50 mL conical tube inside a biosafety cabinet.
Rinse the cryovial with 1 mL of room temperature CDI iCell Neuron complete maintenance media, and add drop-wise to the cells in the 50 mL tube to gradually dilute the DMSO from the freezing solution and minimize osmotic shock.
Add an additional 1 mL of complete maintenance media dropwise to the cells.
Finally, slowly add 7 mL of the complete maintenance media while swirling continuously to bring the total final volume to 10 mL.
Count the number of viable cells using a hemocytometer, and dilute the cells in complete maintenance medium to a concentration of 30,000 cells per 150 μL.
Immediately before adding the neuronal suspension to the center circle of the dish, aspirate the laminin solution.
Add 150 μL of the cell suspension to the Ø10 mm center circle of each MatTek dish.
On the following day, aspirate the medium and add 1.5 mL of pre-warmed iCell Neuron complete maintenance media to the sidewall of the MatTek dish, far from the plated cells.
Optopatch transduction of iCell neurons
Two days after plating, transduce the neuronal cultures with the Optopatch vectors packaged in lentivirus, typically QuasAr-Citrine and CheRiff-EBFP2.
Remove lentivirus stocks from the −80°C freezer and thaw for 10 minutes at room temperature just prior to transduction.
Mix iCell Neuron complete maintenance medium, QuasAr lentivirus stock, and CheRiff lentivirus stock at a volume ratio of 88:10:2 medium:QuasAr:CheRiff.
Just prior to addition of the virus mixture, aspirate the medium from the MatTek dishes, leaving only a small volume of medium on the center glass circle. To leave the small volume, tilt the dish to 45° and completely aspirate all liquid that flows to the dish edge, leaving a small volume captured in the center recess.
Add 120 μL of virus mixture to the cells on the central glass and incubate for 16 hours.
Remove the virus during the glial addition step described in the next section.
Addition of primary glial cells onto the transduced iCell Neurons
Following IRB approval of mouse dissection protocols, we follow the protocol detailed in (Di Giorgio et al., 2008). The cortex is dissected from P1-P2 post-natal mouse pups and glial cells are cultured in poly-D-Lysine-coated T-25 flasks for at least 10 days until cells have formed a confluent monolayer and are ready for passaging. Glial cells are cultured in “Glial medium,” described in reagents and solutions below.
In preparation for removing the glial cells from the T-25 flasks for plating onto MatTeks, pre-warm 0.25% trypsin EDTA, glial media, and iCell Neuron complete maintenance media to 37°C.
Completely aspirate the glial media from the T-25 flask and rinse the cells with 5 mL of PBS.
Aspirate the PBS and add 3 mL of 0.25% trypsin.
After 5 minutes at 37°C, gently tap the flask to dislodge the glial cells.
After most cells have lifted, mix the cell suspension 3 times with a 5 mL serological pipette to completely detach cells and bring them to a single-cell suspension.
Quench the trypsin by adding 8 mL of glial medium into the flask, and transfer the cell suspension to a 15 mL conical tube. Collect any remaining cells with a final 3 mL rinse of the flask with glial medium.
Centrifuge the cell suspension for 5 minutes at 300g in a swing-bucket centrifuge, aspirate the supernatant, and re-suspend the cells in 1 mL of iCell Neuron complete maintenance media by gently pipetting up and down 5 times to break up the cell pellet into a single cell suspension.
Count glia using a hemocytometer, and dilute to a concentration of 40,000 cells per 70 μL iCell Neuron complete maintenance media.
Retrieve the iCell Neurons from the incubator, and completely aspirate the virus.
Rinse gently with 1 mL PBS added to the side-wall of the dish and removed by aspiration.
Add 70 μL of the glial mixture dropwise onto the center circle of each MatTek dish, and incubate for 4–6 hours until the glial attach. Add 1.5 mL of iCell Neuron complete maintenance media to each MatTek dish.
Feed the culture every 3 days with complete media exchange.
Protocol 3: Culture and transduction of primary rat hippocampal neurons
Although human iPS cell-derived neurons offer significant opportunity for the generation of human disease models and have the advantage of a direct connection to a patient’s clinical history following accepted IRB protocol approval, these in vitro- differentiated cells require prolonged culture before acquisition of more mature phenotypes and specific neuronal sub-type identities. For assay development and troubleshooting of Optopatch assays, an alternative approach is to use primary rat hippocampal neurons, which in general provide good signal-to-noise-ratio (SNR) recordings and robust firing and synaptic activity. This protocol guides through the experimental steps of coating dishes, dissociation and plating of primary neurons, transduction of Optopatch constructs, and addition of glial cells.
Material list
-
-
Glass-bottomed dishes (MatTek Cat#P35G-1.5-10-C)
-
-
Poly-D-Lysine (Sigma-Aldrich Cat#P6407-5MG)
-
-
PBS (Life Technologies Cat#10010-049)
-
-
Laminin (Life Technologies Cat#23017-015)
-
-
E18 Rat Hippocampus tissue (BrainBits, Springfield, IL)
-
-
Papain enzyme with Hibernate E-CA media (Brain Bits Cat# PAP/HE)
-
-
Plating media (BrainBits Cat#NbActiv1 500)
-
-
Rat neuronal culture feeding media (See Reagents and Solutions section)
-
-
Trans retinal
Overview
Day 0 – plate E18 Rat Hippocampal neurons dissociated from tissue obtained from BrainBits.
Day 6 – Deliver lentiviral constructs encoding Optopatch proteins
Day 8 – Add primary mouse glia
Day 13–20 – Optopatch imaging
Coating dishes
Coating of dishes is performed using the same protocol as for the iCell Neurons described in coating of dishes section of Protocol 2.
Dissociation of tissue
Tissue shipped from BrainBits is stored at 4°C in HEB solution. Cell viability remains high if dissociation occurs within 2–3 days of dissection. The PDL/laminin coated dishes are equilibrated to room temperature during the dissociation of the tissue.
Prepare a 15 mL conical tube with the dissociation solution: dissolve 6 mg of papain in 3 mL Hibernate E-C for a final working concentration of 2 mg/mL papain.
Carefully transfer the HEB solution surrounding the tissue to a sterile tube for later use.
Add 2 mL of the papain solution to the tissue, transfer tissue and solution to a 15 mL conical tube, and incubated for 10 minutes in the 37°C water bath with a gentle swirl after 5 min. During the swirling, the two tissue pieces should start to adhere to each other, indicating that tissue digestion has begun.
Add the HEB medium saved from step 2 to the conical tube with the tissue, and triturate the tissue with a pipette tip for 1 min while carefully avoiding air bubbles. Let undispersed tissue pieces settle for 1 min.
Transfer the supernatant containing the dissociated cells to a sterile 15 mL tube, discarding the last ~50 μL of HEB containing solid debris. Pellet the cells by spinning at 280 g for 1 min and discard the supernatant, leaving behind ~50 μL of HEB containing the pellet.
Disperse the pellet of cells by flicking the tube bottom and re-suspend in 1 mL of NbActive1 plating media.
Plating
To plate the primary neurons, count the dissociated cells using a hemocytometer and prepare a cell suspension in the NbActive1 plating medium, at density of 20,000 cells for every 70 μL of medium. Prior to plating, aspirate the Laminin solution from the PDL/Laminin-coated MatTek dishes. Rinse each dish once with 500 μL NbAvtive1 and plate 70 μL cells onto the center circle.
Feeding
Around 16 hours after plating, confirm that neurons have attached and have started extending neuronal processes. Aspirate ~50 μL of plating media, maintaining care not to remove the cells, and add 2 mL of rat neuronal culture feeding media.
Lentiviral Transduction of Optopatch Constructs
The primary neuronal cultures are treated with lentivirus encoding the Optopatch components 7 days after plating to give the neurons enough time to firmly attach to the dishes by establishing long processes (Figure 1A). Add concentrated lentivirus stocks using 2 μL of CheRiff-encoding virus, 10 μL of QuasAr2-encoding virus, and 88 μL of feeding medium, for a final volume of 100 μL per dish. Multiply each of these volumes by the number of dishes to be transduced and create a pool of virus mixture and medium to be used for all the dishes. Incubate the neuronal cultures with the virus for at least 16 hours. After this incubation step, aspirate the medium containing the virus, and add 1.5 mL of fresh rat neuronal culture feeding medium.
Figure 1. Rat hippocampal cultures.
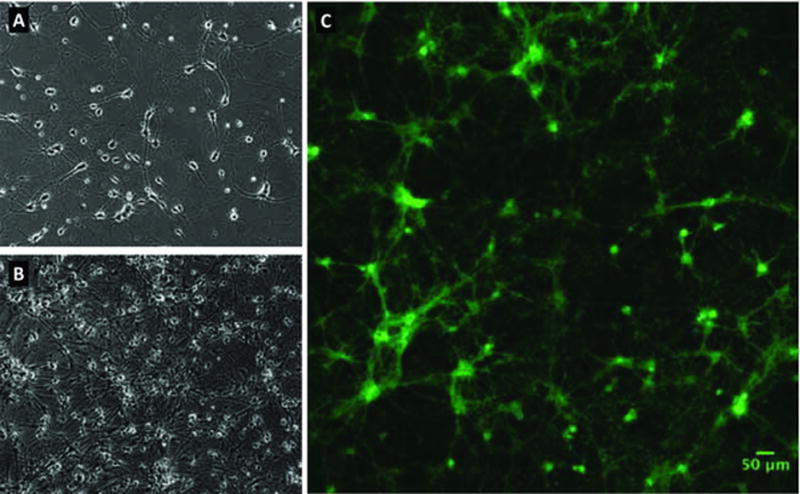
(A) A phase contrast image of rat hippocampal neurons 7 days after plating (DIV7) prior to lentiviral transduction. Cells were seeded at a density of 20k cells/cm2 on cyclic-olefin copylmer Ibidi 8 well dishes, and show early neurite growth. (B) A phase contrast image at DIV13, after transduction and addition of glial cells. (C) A fluorescence image of QuasAr3-Citrine expression in the same cells at DIV13. Gamma has been set to 0.65 to simultaneously see the bright cell bodies as well as dimmer neurites.
Glia addition
Primary glial cells are added on day 9 after plating (2 days after lentivirus transduction). Culture, dissociation and plating of primary glial cells follow the same methods described in the iCell Neuron section above. Prepare a cell suspension of dissociated glial cells using rat neuronal culture feeding medium at a concentration of 40,000 cells per 70 μL volume, which will be added to the primary neurons. Retrieve the rat neuronal cultures from the biological incubator, completely aspirate the media, and add 70 μL of the glial mixture onto the center circle of each MatTek dish. Incubate dishes for 4–6 hours or overnight until the glial cells attach, and add 1.5 mL of rat neuronal culture feeding media to each MatTek dish.
Retinal addition
As for all proteins in the rhodopsin family (Zhou et al., 2012; Fenno et al., 2011; Schneider et al., 2015), channelrhodopsin CheRiff and archaerhodopsin QuasAr bind retinal as a cofactor and require it for their function. 48 hours prior to imaging feed the rat neuronal cultures with feeding medium supplemented with 100 nM trans-retinal. Strong expression of the fluorescent reporter protein fused to the Optopatch components (Figure 1C) can be confirmed inside the tissue culture room using epifluorescence microscopy on the live cultures prior to the Optopatch imaging described in the following section.
Protocol 4: Optopatch imaging of neurons
The goal is to find differences in neuronal firing behavior (the phenotype) resulting from a disease model (genotypic differences) or from an added compound. High-speed voltage recordings are made with QuasAr2 while stimulating with CheRiff. The cells are typically stimulated with different strength steps, ramps, and combs to explore the breadth of neuronal behaviors and identify the regime where the phenotypic or pharmacological difference is most pronounced.
Material list
-
-
Neurons transfected with Optopatch
-
-
Tyrode’s imaging buffer with synaptic blockers
Custom microscope
QuasAr is a dim fluorescence sensor, 50–100× dimmer than EGFP and requires 500 – 1000 Hz frame rates for recording neuronal action potentials. These technical demands necessitate careful microscope design. The key components are:
Camera – High sensitivity and high speed are required, limiting practical selections to scientific CMOS (sCMOS) camera or electron-multiplied CCD (EMCCD) cameras. The most successful implementation has been with the Hamamatsu ORCA-Flash4.0 sCMOS camera in rolling shutter mode, which offers the best combination of high frame rate and low noise.
Microscope objective – Collecting light efficiently is critical, so the highest numerical aperture oil-immersion lenses are necessary. A 60× Olympus APON 60XOTIRF objective, which has a numerical aperture (NA) of 1.49 and enables grazing-incidence, near total internal reflection (TIR) illumination to reduce background autofluorescence is recommended.
Red excitation light – Because high intensity light is required, a high-power laser is needed for recordings. A 140 mW Coherent OBIS 637 LX laser, which has analog intensity control can be used.
Blue stimulation light – The blue light (488 nm) used to stimulate the CheRiff need not be too intense, but it must be modulated with sum-millisecond temporal resolution for full flexibility in stimulus protocols. A 50 mW Coherent OBIS 488 LX laser, which has analog intensity control can be used.
The Optopatch capable microscope can be built around a commercial inverted fluorescence microscope body such as the Olympus IX71. The objective and camera can mount on the standard ports. The red and blue laser excitation must be routed into the fluorescence excitation port, and some custom alignment is required to enable the grazing-incidence illumination. If the high-power red laser propagates up the center of the objective as is typical for epi-fluorescence, the autofluorescence from the optical components inside the objective can overwhelm QuasAr fluorescence. To minimize these issues, the beam is routed up the side of the objective, and light is coupled into the sample just shy of total internal reflection (Figure 2A). In this geometry, the red laser beam refracts at the glass-water interface and propagates nearly horizontally and parallel to the glass surface. The beam is compressed during refraction, increasing the intensity at the sample, and rapidly exits the FOV, minimizing background autofluorescence.
Figure 2. Microscope optical diagrams.
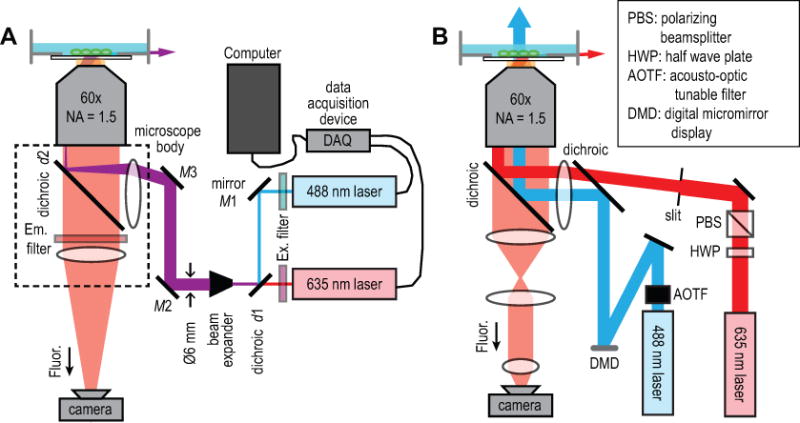
(A) The basic microscope design including required elements for Optopatch imaging. Key components include a high numerical aperture 60× oil immersion objective used to couple light into the sample at grazing incidence, a scientific CMOS camera, two lasers that can be rapidly modulated with analog control signals, and a data acquisition (DAQ) device to generate the control signals. The microscope is built using a commercial, inverted microscope body. The red and blue lasers are combined with a dichroic, and the co-propagate to the sample (indicated by a purple color). (B) A more advanced design for higher throughput and patterned illumination. To fit the highest possible number of cells onto the camera ROI that can be recorded at 1 kHz, de-magnify the image to 15×. The blue laser is routed through an acousto-optic tunable filter (AOTF), which enables rapid intensity modulation for fast stimulus paradigms (see Fig. 3) without the power fluctuations that result from the inevitable temperature instabilities that arise in the laser cavity during direct modulation of the laser drive current. The blue light is then reflected off a digital micromirror device (DMD) to enable patterned illumination for stimulating individual cells or cellular sub-compartments.
The simplest optical layout to implement Optopatch imaging is shown in Figure 2A, with the associated components in Table 1. Both lasers are Class 3b, which means they can burn the retina. Lasers should be aligned by personnel with appropriate experience and safety training using laser safety goggles, and lasers should be attenuated >100× before alignment. Particular care should be taken during alignment through the objective lens, as the laser will propagate upwards towards the eyes and will initially emerge in an unexpected direction. After alignment is fixed, the beams can be appropriately blocked so the microscope is safe to use.
Table 1.
Key parts for custom microscope assembly. Additional standard components available from Thorlabs will be needed for mounting the lasers, lenses, and mirrors, including posts (e.g. TR4), post holders (e.g. PH4), and base plates (e.g. BA1S).
| Part | Vendor | Part number |
|---|---|---|
| camera | Hamamatsu | ORCA-Flash4.0 sCMOS |
| 60× TIRF oil objective | Olympus | APON 60XOTIRF |
| inverted microscope | Olympus | IX-71 (or similar) |
| red laser | Coherent | OBIS 637 LX 140 mW |
| blue laser | Coherent | OBIS 488 LX 50 mW |
| emission dichroic, d2 | Semrock | Di03-R405/488/561/635-t1-25×36 |
| QuasAr emission filter | Semrock | FF02-736/128-25 |
| GFP emission filter | Semrock | FF02-525/40-25 |
| blue laser excitation filter | Semrock | FF01-488/10-25 |
| red laser excitation filter | Semrock | FF01-637-7-25 |
| beam-combining dichroic, d1 | Semrock | LM01-503-25 |
| mounted mirrors (M1 – M2) | Thorlabs | KM100-E02 |
| 10× beam expander | Thorlabs | GBE10-A |
| DAQ card | National Instruments | USB-6343 |
Red (637 nm) and blue (488 nm) lasers are first filtered with an excitation filter to eliminate any light away from the main laser line. The lasers are then combined collinear with one another using the dichroic mirror d1 to reflect the blue and transmit the red light. The kinematic mount for mirror M1 is adjusted until the blue laser falls directly on top of the red laser at d1. The kinematic mount of d1 is then adjusted until the blue laser lies directly on top of the red laser during propagation across the room. That is, the lasers are exactly collinear. The lasers are then routed through a 10× beam expander so the diameter of the beam is roughly 6 mm; the size will be reduced by the 60× objective magnification inside the microscope, so a 6 mm laser beam will illuminate a ~100 μm field of view. Adjust the beam expander until the lasers are collimated and the beam diameter does not change as the laser propagates across the room.
The lasers must then be reflected off the final two mirrors and routed into the microscope. To aid near-TIR alignment, mirror M3 is located near the microscope’s back image plane, near the entrance of the fluorescence excitation port. If mirror M3 is correctly located in the image plane, adjustments will only change the angle of the laser into the sample, and not the position. To find the back image plane, place a high-contrast sample on the microscope, illuminate from above with whitelight from the microscope condenser, and bring the sample into focus on the camera. Good high-contrast samples include resolution targets (e.g. Thorlabs R1DS1P) or thin paper with black printed text face down on the microscope. The light reflected off the dichroic should be visible by eye out the back excitation port, and the image should come into sharp focus on a piece of paper located in the image plane. Place the mirror M3 as close to the center of this image plane as possible. Mirror M2 can be adjusted until the lasers hit the center of M3, and M3 can be coarsely adjusted until the laser goes straight into the microscope and comes out the objective.
The next critical part of assembly is to align the lasers to near TIR at a grazing incidence. This is most easily accomplished with a fluorescent bead sample in a glass-bottomed dish filled with water (the water is required for beam refraction and to find the correct angle). TetraSpeck beads (ThermoFisher #T7279), which will fluoresce in both the QuasAr and GFP channels, are recommended. Place the bead sample on the microscope with objective immersion oil, and bring the beads into focus on the camera using whitelight illumination from above. The pair of mirrors M2 and M3 will have to be adjusted iteratively to find correct laser alignment.
To begin laser alignment, adjust M2 to bring the illumination spot (visualized by bead fluorescence on the camera) to the center of the field of view (FOV), and adjust M3 so the laser emitted from the sample walks away from vertical towards horizontal. As this happens, the illumination will leave the center of the FOV, and M2 will have to be adjusted to bring it back. Repeat adjustments of the two mirrors until the laser propagates nearly horizontally along the bottom of the dish; if it reflects entirely at the glass/water interface the beam has gone into TIR, which is too far.
The lasers will illuminate a roughly 100 μm diameter FOV. The sCMOS camera can read out ~380 rows of pixels at 500 Hz (see ORCA-Flash manual for details), which restricts the height of the FOV to 41 μm (6.5 μm/pixel *380 pixels/60 (the magnification)). This is sufficient to image approximately one neuron at a time at typical culture densities.
A more advance optical configuration is described in Figure 2B. The image is de-magnified from 60× to 17× in order to image a larger sample area and onto the 380 pixels that can be imaged at a 500 Hz frame rate. Demagnification is implemented with a 180 mm cemented achromat (Thorlabs AC508-180-A-ML) and a 50 mm air-spaced achromat (Thorlabs ACA254-050-A). The resolution is pixilation limited at 430 nm, close to the diffraction limit. Fluorescence is separated from excitation light using a dual-band dichroic (Chroma #zt488/640rpc) and a QuasAr2-optimized emission filter (Semrock #FF02-736/128).
To account for the larger illumination area, a higher power, 500 mW red laser can be used. The 635 nm laser (CNI Lasers #MRL-III-635-500mW) power is manually controlled by rotating the half wave plate in a half wave plate/polarizing beamsplitter attenuator.
The blue light source for stimulating CheRiff is a 488 nm, 150 mW laser (Coherent OBIS LX). To enable microsecond analog control over the laser power without affecting thermal stability, the beam is routed through an acousto-optic tunable filter (AOTF; Gooch & Housego #48062-1-.55-1W). To enable patterned illumination of single cells or sub-cellular locations, the blue laser is expanded and reflected off a digital micromirror display (DMD; Texas Instruments DLP LightCrafter). The DMD is imaged onto the sample with sub-micron resolution, and each pixel can be turned “on” of “off” by loading an image. With patterned blue light and spatially high-speed voltage imaging, the instrument can stimulate and record with nearly diffraction-limited resolution anywhere within the FOV. However, for simple excitability measurements, all the DMD pixels are “on,” and full field stimulation is used.
Sample treatment prior to imaging
Samples are imaged 21–28 days post-plating (19–26 days post-transfection). Prior to imaging, the CDI complete maintenance media is removed by aspiration, cells are rinsed with 1 mL Tyrode’s imaging buffer with synaptic blockers, and then 1 mL of Tyrode’s imaging buffer is added to the dish. This buffer minimizes background autofluorescence, while maintaining stable cellular physiology at ambient CO2 levels. Cells are equilibrated to room temperature in the new buffer for 15 minutes after buffer exchange.
Compound addition
For compound addition, compounds are prepared at 2× concentration in Tyrode’s buffer such that the final compound concentration can be achieved upon addition of 1 mL to the dish and gentle pipetting up and down twice. Compound solutions are prepared fresh daily from DMSO stocks, limiting the total in-dish DMSO concentration to less than 0.2% V/V. Cells are typically incubated the compound for 10 minutes prior to imaging.
Imaging
During the measurements, cells are maintained at room temperature on the microscope. MatTek dishes are scanned manually to find FOVs containing cells with high Optopatch expression and a healthy and developed arbor. Voltage recordings in the QuasAr fluorescence channel are made while stimulating with the selected protocol using blue light. After making recordings from 10 FOVs, compounds are manually added with a pipette as described above. After incubation, a new set of 10 FOVs can be imaged. With motorized stages and control software, the process can be greatly accelerated and repeat imaging of the same cells before and after drug addition is possible. Ludl BioPrecision2 stages and custom software for this purpose can be used, but likely commercial software is available.
For primary rat hippocampal neurons, the cells are robust and the excellent SNR allows a reduced 635 nm light illumination intensity of ~100 W/cm2 during recordings. This reduced illumination power typically makes recordings of the same cells before and after drug addition the most fruitful experimental strategy. For the smaller and more delicate iCell neurons, which require a higher illumination intensity of ~ 1kW/cm2 to achieve a sufficient SNR, the first round of imaging is often perturbative and imaging new cells is preferred. In general, this must be tested for each cell type.
Stimulus Protocols
To exercise different neuronal spiking behaviors, the cells are probed with a series of user defined stimuli (Figure 3). Both blue and red lasers can be controlled with analog inputs, enabling rapid laser modulation and changes to the laser power with sub-millisecond temporal resolution. Control waveforms can be designed in LabVIEW from National Instruments, which in turn controls a multi-function data acquisition (DAQ) device (National Instruments #USB-6343). Analog control waveforms generated from the DAQ are transmitted to the lasers (Figure 2A). Blue light stimulus waveforms include spike trains at different frequencies, spontaneous recordings, steps of blue (488 nm) stimulus at increasing intensities, and intensity ramps. Pulse trains are used to probe maximum firing rate. Each pulse in the train is typically 5 ms in duration at 100 mW/cm2, and the train frequencies are at 5, 10, 20, and 40 Hz. Spontaneous recordings are implemented without any blue stimulus to probe unperturbed neuronal activity. To track firing patterns under a steady depolarization, 500 ms duration step stimuli with a logarithmic intensity ramp, typically 1, 3, 9, 27, and 81 mW/cm2 are used. In typical experiments, two intensity ramps, one fast and one slow are used. The slow ramp (0 – 10 mW/cm2 over 2 seconds) probes rheobase and the initiation of spiking. The fast ramp (0 – 100 mW/cm2 over 2 seconds) probes maximum firing rate and depolarization block at high stimuli. Between each stimulus epoch, a 5 second rest period is included to allow cell recovery, during which time both red and blue light are off. To capture the details of action potential dynamics, images are record at a 1 kHz frame rate.
Figure 3. Human iPS neurons spiking patterns under various stimulus protocols.
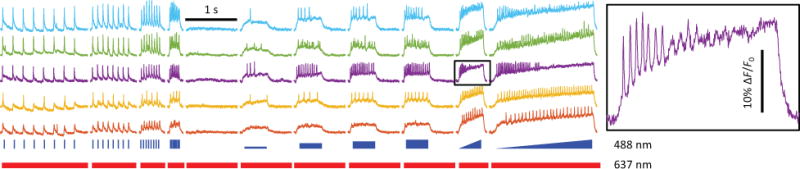
Neurons are stimulated with spike trains, left to fire spontaneously, stimulated with steps of different intensities, and stimulated with ramped intensity. The inset on the right shows a magnified view of a volley of action potentials during ramp stimulation.
Protocol 5: Extraction of neuronal firing properties from high-speed video
In high magnification (60×) videos recorded on the microscope described in Figure 2A, it will be easy to distinguish individual neurons (see Fig. 1) unless cultures are badly clumped. Measuring the average fluorescence from the cell body and proximal dendrites will yield a good approximation of the voltage time trace (see examples in Figures 3–5). We have traditionally implemented this using custom software in Matlab, but it can be done manually in ImageJ (Schneider et al., 2012). Use the polygon tool to select a (potentially contorted) a region surrounding the cell body and largest dendrites. Go to Image>Stacks>Plot Z-axis Profile to see the fluorescence time trace, which can be saved or exported for further analysis in a different program.
Figure 5. Analysis workflow.
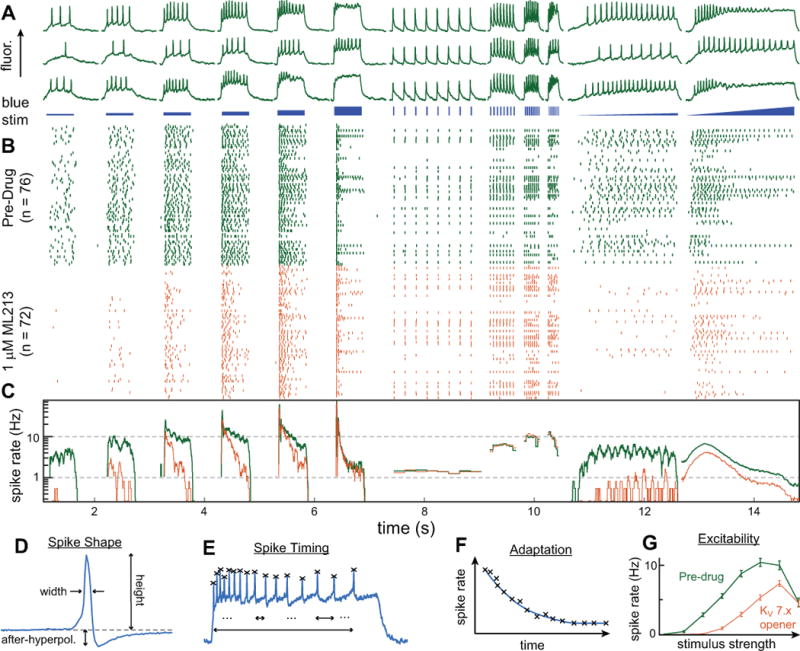
An overview of the cascade of analyses used to extract data from the segmented images. Here the data from 76 cells are pooled. (A) Time traces from three exemplary neurons in response to the blue stimulus pattern shown just underneath. The stimulus includes 500 ms blue steps of different intensities, pulse trains to probe maximal firing rates, and ramps where the stimulus strength is smoothly increased. (B) A raster plot showing the aggregated firing pattern of all the cells. Each row in the raster is one cell, and each tick in the row represents one action potential. Green represents the spikes before compound addition and orange represents spikes from the same cells after the addition of ML213, a KV 7.x potassium channel agonist that hyperpolarizes the cell and reduces firing. (C) The spike rate, calculated by averaging over all the spikes in (B), reveals clear drug-induced reduction in firing during the steps and ramps. In addition to examining overall firing rate, we extract many parameters related to spike shape and timing from each cell. (D). A magnified view of a single spike showing typical extracted spike shape parameters including width, height, and after-hyperpolarization. (E) Extracted spike timing properties, including adaptation (F), which is the gradual slowing of spike rate after the onset of stimulus displayed by many neurons. (G) Aggregated data showing a highly significant reduction in spike rate for each stimulus step in the staircase.
To implement a more scalable process that can operate at higher throughput with minimal manual intervention, a more advanced analysis is required. For example, neurons can be closely spaced and have overlapping neurites (Figure 4), making manual selection difficult. When more neurons are present, as is the case for the lower magnification microscope design in Figure 2B, manual selection becomes tedious. The goal of an automated analysis is to extract the voltage recording from every cell in the high speed video, identify all action potentials, and characterize spike timing and action potential shape properties for each type of stimulus (Figure 5). This reduced parameter set is used to detect statistically significant differences between different cell genotypes or changes induced by compound addition.
Figure 4. Image segmentation in CDI iCell neurons.
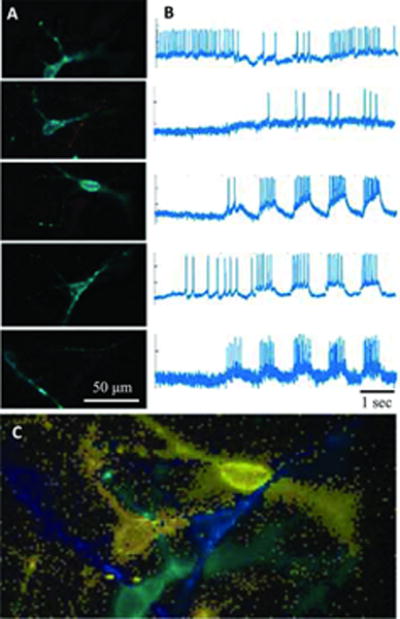
At high magnification, neurons can be identified manually, but at lower magnification or in high-density cultures, automated analysis is required. Principal component analysis (PCA) identifies significant sources of variation in the movie, representative of neuronal firing, but time traces and pixel weight maps are typically mixtures of multiple neurons. Independent component analysis (ICA) “unmixes” the signal, identifying individual time traces even in crowded images where cells are partially overlapped. This figure shows results from one such field of view, where 5 overlapped neurons are present. (A) Identified neuronal sources and (B) their action potential trains resulting from PCA/ICA analysis. Red indicates negative pixel weights. (C) The composite image with all sources combined.
Segmenting movies to identify individual cells
Movie segmentation can be performed via temporal Principle Component Analysis (PCA) (Peason, 1901; Hotelling, 1933, 1936) followed by spatial-temporal Independent Component Analysis (ICA) (Hérault and Ans, 1984; Ans et al., 1985; Hérault et al., 1985; Herault et al., 1986; Bell and Sejnowski, 1997). PCA decomposes multiple observed responses (e.g. the observed intensities in different pixels) into a series of co-varying signals or “components” (e.g. each component is a putative neural source, which causes many pixels to vary synchronously in response to that source’s activity). PCA provides a measure of how important each signal is, so the signals can be ranked. The PCA components (initially there is one component for each pixel in the movie) just represent noise, and in practice most are discarded to greatly compress the dataset. One downside of PCA is that it tends to mix neural signals together due to the fact that each PCA component must be orthogonal. These mixed signals clearly cannot be voltage traces from individual neurons: different PCA components will often contain action potentials of different heights, have substantial physical overlap with multiple cells, and exhibit non-physical negative fluorescent intensities. ICA is used to “unmix” the PCA components into new independent components that capture the true voltage activity physical structure of individual neurons. While PCA is an exact mathematical transformation, ICA has adjustable parameters to pull out components that are most physical. One effective strategy is to select pixel weight maps that are connected (see Figure 4A) and pike waveforms where all the action potentials are positive and maximally sparse in their response (typically as measured by signal skewness or kurtosis). Figure 4B shows the extracted voltage traces and Fig. 4C shows the overlay of the cell weights in false color.
To implement PCA/ICA, the movies are first reshaped into a #frames × #pixels matrix. Medium-pass temporal filtering is applied using a difference of Gaussians: the “fast” Gaussian was chosen with σ = 1/6 expected spike width, and the “slow” Gaussian was chosen to be 10 times wider. Performing this medium-pass filtering accentuated the detection of spiking sources over blobs that may have had a large but slow change in fluorescence over the course of the movie.
Temporal PCA was performed by finding the 16 largest eigenvalues of the product of the filtered movie times its transpose. For each #frames × 1 temporal eigenvector, a corresponding 1 × #pixels spatial coefficients vector was obtained by multiplying the transposed movie by the temporal eigenvector. The results of the PCA were “unmixed” by spatial-temporal ICA. Each temporal eigenvector and spatial coefficients vector was z-scored (mean-subtracted, standard-deviation divided) and packed into a 16 × (#frames + #pixels) matrix. Each row of the matrix was constructed by multiplying the z-scored eigenvector by a coefficient 1 – μ, and concatenating it with the z-scored coefficients vector multiplied by μ. For this process μ = 0.025 was used, which heavily emphasized the temporal information. Finally, the FastICA package was used to perform ICA with symmetric convergence, keeping eight independent components. These eight components were unpacked by multiplying the resulting ICA unmixing matrix into the PCA spatial coefficients to obtain final coefficients vectors (per-pixel weights). Final source traces were obtained by multiplying these weight vectors into the unfiltered original movie matrix. Figure 4 shows an example of this process on one field of view containing 5 CDI iCell Neurons.
Spike detection
Putative spikes were identified by medium-pass filtering spike traces and finding intervals where the trace was positive. The distribution of maximum heights during these intervals was quantified with a kernel density estimator. Most such heights are background fluctuations, and their distribution is roughly gaussian and close to zero. The width of this gaussian was estimated and set a probability threshold of 0.001 using the inverse of the erfc function (i.e. the height threshold is found where the probability that a random gaussian fluctuation is larger than the threshold is less than 0.001). Final spikes are those that surpass the threshold. To fully bracket the spike waveform (e.g. to find spike shape parameters), intervals are expanded until the beginning and end of each interval were at a local minimum of the mean-filtered trace (the filter width was 3 frames).
Source rejection and acceptance
The PCA ICA procedure always finds as many sources as there are ICA components. Usually the majority of them are due to noise rather than detected neurons. The quality of sources are measured using several different metrics, the most important being: signal to noise ratio (height of 3rd-highest spike divided by standard deviation of high-pass filtered trace), skew (3rd moment) of trace, and diameter and area of soma (detected as the largest ellipsoidal blob detected from the source image). The thresholds are adjusted depending on circumstances of the project, but typical minimum values were SNR >= 3, Skew >= 0.25, Diameter >= 10 μm, and Area >= 75 μm2. If any of the metrics fell below threshold, the source was rejected. Figure 5(a) shows some example traces extracted using these criteria.
Calculation of properties
Source properties are calculated based on the timing and shape of the detected spikes. For each temporal waveform, each spike is identified and plotted as a raster (Figure 5B). Each cell is a row, and each tick is an action potential. By averaging over all cells, one obtains the average spike rate (Figure 5C), showing how the cells respond to stimulus. Often, this is one of the most sensitive parameters for detecting phenotypes or pharmacological effects. Figure 5B and C show the difference in cellular behavior before (green) and after (orange) the addition of a potassium channel agonist that hyperpolarizes the cell and reduces the firing rate.
After each spike is identified, further characterization of action potential shape and timing parameters is done as depicted in Figure 5D-F. A multitude of parameters is measured, which are listed here as examples. Spike onset is defined as the point of maximum 2nd derivative before the spike peak (an alternative approach would be to fit two straight lines with a “hinge” and use the time point of the hinge). Action potential width was measured at 80% below the action potential peak towards the onset. Spike after-hyperpolarization potential was assessed as the maximal extent of the trace below onset after a spike, as a fraction of spike height. First spike time was the time to first spike after a stimulus was applied, and last spike time was likewise measured relative to stimulus onset. Frequency was total number of spikes divided by the duration of the stimulus. The maximum and minimum derivative were used to calculate the upstroke and downstroke, respectively. The first instantaneous frequency was computed as the inverse of the time between the first two spikes following the stimulus. The spike frequency adaptation was the difference in spike rate between the first and second half of the stimulus. Phasic firing was defined as a single spike event throughout the entirety of a stimulus.
Identifying significant changes in properties
Properties of interest were selected manually. One can test whether each property changed significantly in response to drug concentration, adjusting for multiple comparisons within each drug. The degree of difference was measured using the Kolmogorov–Smirnov (KS) statistic. Subsequently, the data was scrambled (across drug concentrations) and the KS statistic assessed for the scrambled data. This scrambling procedure was carried out Nscramble = 1000 times, to develop the distribution of KS statistics for random data (i.e. the distribution implied by the null hypothesis). To adjust for multiple comparisons, an iterative procedure was followed. The comparison Cmax with the greatest KS statistic in the real data was compared to the maximum difference from each scrambled trial (regardless of which scrambled property was being compared). If ngreater was the number of scrambled trials with a greater difference ratio, then the p-value for this change in mean values was p = (1 + ngreater)/(Nscramble + 2). If this p-value was less than 0.05, the difference was deemed significant. Then Cmax was removed from the list of pairwise comparisons (including the scrambled data), and the testing continued iteratively. If p-value was greater than 0.05, the difference was not significant and the procedure terminated.
Reagents and Solutions
10% FBS medium:
To make 500 mL of medium, combine 440 mL of high glucose DMEM (Life Technologies Cat#11995-073), 5 mL of GlutaMax (Life Technologies Cat#35050-061), 5 mL of non-essential amino-acids NEAA (Life Technologies Cat#11140-050), and 50 mL of heat-inactivated Fetal Bovine Serum (HyClone Cat# SH30071.02HI). Mix and vacuum filter inside the biosafety cabinet. Do not add penicillin/streptomycin as that may lower virus yield. Typical shelf life is 14 days at 4°C.
Glia medium: MEM basal medium (Life Technologies Cat#11095-080), 10% Donor Equine Serum (heat inactivated at 55°C for 30 minutes, Hyclone Cat#SH30074.03) and 3% v/v of a 20% stock D-glucose solution in PBS solution (Sigma-Aldrich Cat#G-8270). Typical shelf life is 14 days at 4°C.
Rat neuronal culture feeding media: To make 500 mL of media, mix 475 mL Neurobasal A (ThermoFisher Cat#10888-022), 5 mL GlutaMax (ThermoFisher Scientific Cat#35050-061), 5 mL Pen-Strep (Corning Cat#30-001-CI), 10 mL 50× B-27 Supplement (ThermoFisher Scientific Cat#17504044), and 5 mL 100× N2 Supplement (ThermoFisher Scientific Cat#17502048). Typical shelf life is 14 days at 4°C. Immediately prior to use on the primary rat neuronal cultures, add 1 μg/mL of a laminin solution (ThermoFisher Scientific Cat#23017-015) and the neurotrophic factors BDNF (R&D Systems Cat#248-BD-025) and GDNF (R&D Systems Cat# 212-GD-010), each at a final concentration of 10 ng/mL.
Tyrode’s imaging buffer: 125 mM NaCl, 2 mM KCl, 3 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 30 mM Glucose. In addition to the buffer, for excitability measurements, add three synaptic blockers to uncouple the cells and get an un-biased measure of singe-cell excitability: 25 μM D-AP5 (blocks NMDA channel), 10 μM NBQX (blocks AMPA channel), 20 μM Gabazine (blocks GABA channel). The sterile-filtered Tyrode’s buffer can be stored at 4°C for years. Synaptic blockers are added to the buffer on the day of imaging, and can be stored in aqueous solution at 100× at −20°C for at least 1 month.
Preparation of PEI MAX40000 transfection reagent
Polyethylenimine (PEI) stock solution is prepared at 1 mg/mL in molecular biology-grade water by adding 10 mg of PEI into 9 mL water. Bring the PEI solution to a pH of 7.10 using 1N sodium hydroxide (NaOH) base, since the solution is initially acidic (pH, 2.0 – 3.0). Top off the solution, bringing the final volume to 10 mL to reach the target concentration of 1 mg/mL. Divide the solution into 1 mL aliquots and store at −80°C.
Commentary
Background Information
All-optical electrophysiology first became practical after the initial publication by Hochbaum et al., (2014) which described key improvements introduced in both the voltage sensor and the channelrhodopsin activator, that enabled faithful recordings of neurons and a negligible amount of optical crosstalk between the red and blue channels. While the gold standard for electrophysiological measurement in terms of data quality and SNR has been manual patch clamp, the process is difficult and slow; a skilled operator can record from roughly 2 cells/hour. Optopatch enables voltage recording from several cells per minute with minimal training, and there is a clear path forward to much higher throughput using wide-field imaging.
Several other approaches have been developed to enable higher throughput measurements of electrophysiology. Automated patch clamp has a much higher throughput for immortalized cell lines like HEK293, but is essentially incompatible with neurons because the flow sheers away neurites and synaptic connections. Multielectrode arrays can make extracellular recordings of spontaneous activity in neural networks in a parallelized, multi-well format, but cannot be used to stimulate and record from defined cells, cannot be paired with other fluorescent imaging technologies, and are insensitive to sub-threshold events. Ca2+ imaging enables very high-throughput optical measurements with single-cell resolution. However, it has insufficient temporal resolution to detect action potentials and is difficult to pair with channelrhodopsin because of optical crosstalk. Voltage-sensitive dyes are an alternative sensor to QuasAr. New red dyes (Yan et al., 2012; Huang et al., 2015) are compatible with channelrhodopsin, and provide high SNR recordings when loaded into single cells with a patch pipette. However, when incubated with the dye, all cells including glia and neurites from distant neurons are stained, masking and complicating recordings. In addition, high dye loading can change membrane capacitance, and radical mechanisms can cause phototoxicity. Nonetheless, this will remain a complimentary approach to QuasAr as dyes and analysis mechanisms continue to improve.
Critical Parameters
There are several critical parameters to consider in establishing robust implementation of the above-described methods. These are discussed briefly here to alert the reader.
Choice of Optopatch construct
As discussed at the beginning of the chapter, the choice of Optopatch construct is a critical decision that depends on the experiment of interest. Considerations include the fluorescent fusion protein for both QuasAr2 and CheRiff, as well as the choice of promoter depending on the neuronal type under investigation. Promoters do not always demonstrate perfect specificity for neuronal sub-types and should be used with caution or cross-validated with immunocytochemistry.
Transfection method
Attaining high expression levels of the Optopatch constructs while maintaining cell health is critical for performance. Lipofection and cationic polymer chemical transfection methods have a low efficiency and adversely affect cell health. Calcium phosphate transfection and nucleofection yield low transfection efficiency, but the cells that do express are bright and healthy. Sparse, high expression is desirable in many situations so these methods should be considered. Lentiviral expression gives both high efficiency and high expression levels, and is typically our method of choice in spite of the additional effort required for lentivirus production.
Culture with glia
Culturing both human iPS-cell derived neurons and primary rat hippocampal neurons with glia significantly improves performance. Glia aid cell maturation and growth, prevent clumping, and extend total time before in vitro cultures begin to degrade.
Light illumination intensities
The intensity of red light for QuasAr2 excitation and the intensity of blue light for CheRiff stimulation are both critical for Optopatch performance. To measure intensity (W/cm2), the optical illumination power (W) incident on the sample must be measured with a power meter (e.g. Thorlabs #PM100A) and the illumination area (cm2) in the sample must be measured with the aid of a length calibration target (e.g. Thorlabs #R1L1S4P). Red excitation light should typically be in the 100 – 1000 W/cm2 range. If the light is too intense, the cells will overheat and die; if the light is too dim, the SNR will be too poor to make action potential recordings. Blue stimulus light should typically be in the 1 – 100 mW/cm2 range, although the optimal range depends on cell type. If the light is too intense, the cells will go into depolarization block and stop firing; if the light is too weak, cells will not be stimulated sufficiently to record action potentials. In typical experiments, the blue light intensity is stepped across the suggested range in a logarithmic scale to ensure appropriate stimulus intensity.
Troubleshooting
See the critical parameters above as likely causes for troubleshooting. The choice of red laser source (≥ 500 mW), microscope objective (60× oil immersion), and camera (EMCCD or sCMOS) will be critical for achieving sufficient SNR. We recommend first getting the system working in a cell type that has larger cells and gives higher SNR, such as rodent hippocampal cells or dorsal root ganglion (DRG) cells. Typically, it is possible to anticipate Optopatch performance without functional recording by looking at fluorescence images of the fused proteins, Citrine or EBFP2 (see Figure 1C). If cells are dim or clumped, the experiment is unlikely to work well. If the constructs are not visible in the neurites or localized to the cell membrane, again the experiment is unlikely to work and culture and transfection protocols should be re-visited. Finally, if there is observable electrical activity in the cells but it is hard to interpret, check the synaptic blockers in the imaging medium. Network activity makes measurements of intrinsic neuronal activity much more complex and difficult to analyze.
Anticipated Results
Neuronal recordings should yield traces of action potentials that look like those in Figures 3–5. Drug effects on the neuronal spike trains should be easily detectable, as shown in Figure 5.
Time Considerations
CDI iCell neurons have a large functional imaging window when cultured with glia; functional activity can be recorded from day 21 – day 40, and significant degradation is not seen at 40 days. The rat hippocampal cells can be imaged between DIV 12 and 25. Once neurons are removed from the incubator and placed on the microscope, they maintain functional activity for at least 1 hour at room temperature with minimal degradation in performance. Good microscope automation control software can significantly increase the number of cells that can be recorded in this time window.
Key References with Annotations.
Hochbaum, D. R., Zhao, Y., Farhi, S. L., Klapoetke, N., Werley, C. A., Kapoor, V., Zou, P., Kralj, J. M., Maclaurin, D., Smedemark-Margulies, N., et al. 2014. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nature Methods 11:825–833. Introduces the Optopatch system for the first time, with control experiments in the supplement.
Acknowledgments
We would like to acknowledge Prof. Adam Cohen, who helped us implement Optopatch measurements, and Prof. Kevin Eggan, who helps us develop stem-cell models. This work was supported in part by an SBIR grant from NINDS (5R43NS087714-02).
Footnotes
Conflicts of Interest: Authors on this paper work for Q-State Biosciences, a startup biotechnology company using the Optopatch technology and stem-cell disease models in drug discovery.
Literature cited
- Ans B, Hérault J, Jutten C. Architectures neuromim{é}tiques adaptatives: D{é}tection de primitives. Proceedings of “Cognitiva. 1985;85:593–597. [Google Scholar]
- Bales KL, Solomon M, Jacob S, Crawley JN, Silverman JL, Larke RH, Sahagun E, Puhger KR, Pride MC, Mendoza SP. Long-term exposure to intranasal oxytocin in a mouse autism model. Translational psychiatry. 2014;4:e480. doi: 10.1038/tp.2014.117. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4259989&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AJ, Sejnowski TJ. The “independent components” of natural scenes are edge filters. Vision research. 1997;37:3327–3338. doi: 10.1016/s0042-6989(97)00121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell stem cell. 2008;3:637–48. doi: 10.1016/j.stem.2008.09.017. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19041780 [Accessed August 25, 2014] [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8850–5. doi: 10.1073/pnas.151261398. Available at: /pmc/articles/PMC37524/?report=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey GT, Chaudhary KW, Atwater N, Nguyen C, Brown BS, McNeish JD, Cohen AE, Kralj JM. Cardiotoxicity screening with simultaneous optogenetic pacing, voltage imaging and calcium imaging. Journal of Pharmacological and Toxicological Methods. 2016 doi: 10.1016/j.vascn.2016.05.003. Available at: http://dx.doi.org/10.1016/j.vascn.2016.05.003. [DOI] [PubMed]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. Journal of virology. 1998;72:8463–71. doi: 10.1128/jvi.72.11.8463-8471.1998. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9765382%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC110254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annual review of neuroscience. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SSW, Williams LA, Eggan KC. Constructing and Deconstructing Stem Cell Models of Neurological Disease. Neuron. 2011;70:626–644. doi: 10.1016/j.neuron.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Hérault J, Ans B. R{é}seau de neurones {à} synapses modifiables: D{é}codage de messages sensoriels composites par apprentissage non supervis{é} et permanent. (S{é}rie 3, Sciences de la vie).Comptes rendus des s{é}ances de l’Acad{é}mie des sciences. 1984;299:525–528. [PubMed] [Google Scholar]
- Hérault J, Jutten C, Ans B. D{é}tection de grandeurs primitives dans un message composite par une architecture de calcul neuromim{é}tique en apprentissage non supervis{é} 10 Colloque sur le traitement du signal et des images, FRA. 1985 1985. [Google Scholar]
- Herault J, Jutten C, Denker JS. Space or time adaptive signal processing by neural network models. AIP conference proceedings. 1986:206–211. [Google Scholar]
- Hotelling H. Analysis of a complex of statistical variables into principal components. Journal of educational psychology. 1933;24:417. [Google Scholar]
- Hotelling H. Relations between two sets of variates. Biometrika. 1936;28:321–377. [Google Scholar]
- Huang YL, Walker AS, Miller EW. A Photostable Silicon Rhodamine Platform for Optical Voltage Sensing. Journal of the American Chemical Society. 2015;137:10767–10776. doi: 10.1021/jacs.5b06644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey SM, Pankevich DE, Davis M, Altevogt BM, editors. Institute of Medicine of the National Academies. Improving and accelerating therapeutic development for nervous system disorders: workshop summary. 2014. [PubMed] [Google Scholar]
- Jiao J, Yang Y, Shi Y, Chen J, Gao R, Fan Y, Yao H, Liao W, Sun XF, Gao S. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Human Molecular Genetics. 2013;22:4241–4252. doi: 10.1093/hmg/ddt275. [DOI] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A Mouse Model of Tuberous Sclerosis: Neuronal Loss of Tsc1 Causes Dysplastic and Ectopic Neurons, Reduced Myelination, Seizure Activity, and Limited Survival. Journal of Neuroscience. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. Available at: http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, Zheng Y, Diffenderfer KE, Zhang J, Soltani S, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015;527:95–9. doi: 10.1038/nature15526. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26524527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science (New York, NY) 1996;272:263–7. doi: 10.1126/science.272.5259.263. Available at: http://science.sciencemag.org/content/272/5259/263%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/8602510. [DOI] [PubMed] [Google Scholar]
- Peason K. On lines and planes of closest fit to systems of point in space. Philosophical Magazine. 1901;2:559–572. [Google Scholar]
- Sakuma T, Barry MA, Ikeda Y. Lentiviral vectors: basic to translational. Biochem J. 2012;443:603–618. doi: 10.1042/BJ20120146. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22507128. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. Available at: http://dx.doi.org/10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Grimm C, Hegemann P. Biophysics of Channelrhodopsin. Annual review of biophysics. 2015;44:167–186. doi: 10.1146/annurev-biophys-060414-034014. Available at: http://www.annualreviews.org/doi/full/10.1146/annurev-biophys-060414-034014. [DOI] [PubMed] [Google Scholar]
- Schoepp DD. Where will new neuroscience therapies come from? Nature Reviews Drug Discovery. 2011;10:715–716. doi: 10.1038/nrd3559. Available at: http://dx.doi.org/10.1038/nrd3559. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SSW, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell reports. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4023477&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, Acker CD, Zhou WL, Lee P, Bollensdorff C, Negrean A, Lotti J, Sacconi L, Antic SD, Kohl P, et al. Palette of fluorinated voltage-sensitive hemicyanine dyes. Proceedings of the National Academy of Sciences. 2012;109:20443–20448. doi: 10.1073/pnas.1214850109. Available at: http://www.pnas.org/cgi/doi/10.1073/pnas.1214850109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XE, Melcher K, Xu HE. Structure and activation of rhodopsin. Acta pharmacologica Sinica. 2012;33:291–9. doi: 10.1038/aps.2011.171. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3677203&tool=pmcentrez&rendertype=abstract%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/22266727%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3677203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Donello JE, Trono D, Hope TJ. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J virology. 1999;73:2886–92. doi: 10.1128/jvi.73.4.2886-2892.1999. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10074136%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC104046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. Available at: http://dx.doi.org/10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]