

Abstract
Background
Amyotrophic lateral sclerosis (ALS) is a devastating disease whose complex pathology has been associated with a strong genetic component in the context of both familial and sporadic disease. Herein, we adopted a next-generation sequencing approach to Greek patients suffering from sporadic ALS (together with their healthy counterparts) in order to explore further the genetic basis of sporadic ALS (sALS).
Results
Whole-genome sequencing analysis of Greek sALS patients revealed a positive association between FTO and TBC1D1 gene variants and sALS. Further, linkage disequilibrium analyses were suggestive of a specific disease-associated haplotype for FTO gene variants. Genotyping for these variants was performed in Greek, Sardinian, and Turkish sALS patients. A lack of association between FTO and TBC1D1 variants and sALS in patients of Sardinian and Turkish descent may suggest a founder effect in the Greek population. FTO was found to be highly expressed in motor neurons, while in silico analyses predicted an impact on FTO and TBC1D1 mRNA splicing for the genomic variants in question.
Conclusions
To our knowledge, this is the first study to present a possible association between FTO gene variants and the genetic etiology of sALS. In addition, the next-generation sequencing-based genomics approach coupled with the two-step validation strategy described herein has the potential to be applied to other types of human complex genetic disorders in order to identify variants of clinical significance.
Electronic supplementary material
The online version of this article (10.1186/s40246-017-0126-2) contains supplementary material, which is available to authorized users.
Keywords: Sporadic amyotrophic lateral sclerosis, FTO gene, Genomic variants, Whole-genome sequencing, Founder population
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a neurodegenerative disorder that affects the upper and lower motor neurons in the motor cortex, brain stem, and spinal cord [20]. ALS was first described in the mid-1800s as a rapidly progressing motor neuron disease, with phenotypes of varying severity. Affected patients present muscle weakness, atrophy, and a general progressive paralysis. Eventually, most develop respiratory failure which leads to death on average 3 to 5 years after the onset of symptoms [45]. As yet, there is no effective treatment or pre-symptomatic diagnostic test for ALS [11].
Approximately 90–95% of ALS patients suffer from a sporadic form of ALS (sALS), which has both an environmental etiology and a likely strong genetic component [23]. The remaining 5–10% of ALS patients exhibit a clear genetic etiology for the disease, also known as familial ALS (fALS). Genomic variants in the SOD1 gene have been shown to lead to fALS, with an autosomal dominant mode of inheritance. To date, more than 170 different SOD1 genomic variants have been reported, affecting ~ 13% of fALS and ~ 1% of sALS patients [40]. Furthermore, variants in other genomic loci, such as FUS [27], TARDBP [25], and more recently C9orf72 [36], have also been implicated in fALS. In the case of C9orf72, a non-coding GGGGCC expansion appears to have a very strong association with both fALS and sALS, with an overall prevalence of 40 and 5–20% in fALS and sALS patients, respectively [39].
The etiology of sALS is quite complex. A variety of different factors, including age, overall lifestyle, occupation, and/or exposure to xenobiotics, appear to influence the risk of developing sALS, but no specific chemical or toxin has been identified as a direct cause of ALS [44]. In addition to environmental factors, sALS also has a substantial genetic component. Genome-wide association studies have so far identified over 100 genomic loci that could yet prove to be linked to ALS disease predisposition with low-to-medium penetrance, supporting the notion of pronounced heritability for ALS [reviewed in [28]]. From these genomic loci, SQSTM1 [19], UBQLN2 [14], VCP [12], ANG [29], PRPH [38], DCTN1, TAF15, and SETX [10] appear to be more frequently mutated in sALS than in fALS, with an overall prevalence of between 4 and < 1%; these findings have been confirmed in several studies/cohorts as well as in functional and animal studies. Furthermore, the co-occurrence of genomic variants in addition to the presence of de novo pathogenic variants in sALS brings the estimate of overall heritability for ALS to as high as 60% [3, 4]. The identification of the entire lexicon of underlying genetic defect(s) in both fALS and sALS patients and their families would greatly improve disease diagnosis and potentially disease management.
There is little knowledge of the genetic basis of ALS in fALS and sALS patients of Greek origin. Although the incidence of ALS in Greek patients is comparable to other European populations (~ 1–2 per 100,000), only a single study has so far been performed, highlighting a rather high prevalence of the C9orf72 GGGGCC repeat expansion in Greek ALS patients [35]. Here, we report a novel genomic locus, FTO, that appears to be associated with sALS. Genomic data comparison with sALS patients of Greek, Sardinian, and Turkish descent strongly suggest that this association may represent a founder effect that is specific to the Greek population.
Materials and methods
Case selection
Adopting a three-step strategy to determining data reliability, case selection criteria spanned all three patient cohorts studied herein (Fig. 1): (i) whole-genome sequencing analysis in sALS Greek patients (n = 6) and their healthy counterparts (n = 5), (ii) SNP genotyping (selected variants of interest) in sALS Greek patients (n = 28) and their healthy counterparts (n = 50), and (iii) SNP genotyping (selected variants of interest) in sALS patients of Greek (n = 114), Sardinian (n = 114), and Turkish (n = 148) origin and their healthy counterparts (n = 39, n = 87, n = 74, respectively). Patients were independently evaluated by at least two senior neurologists and diagnosed on the basis of revised El Escorial criteria [9]. Patients with definite, probable, probable laboratory supported, or possible ALS were included in the study. Healthy individuals were recruited after excluding the occurrence of common neurological disorders.
Fig. 1.
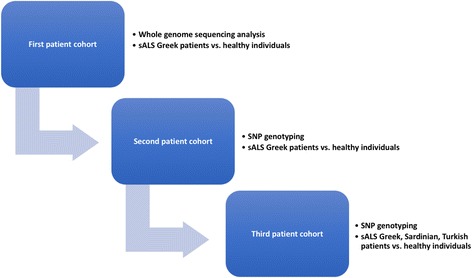
Our three-step strategy towards data reliability. Our three-step strategy involved three patient cohorts: (i) whole-genome sequencing analysis in sALS Greek patients (n = 6) and their healthy counterparts (n = 5), (ii) SNP genotyping (selected variants of interest) in sALS Greek patients (n = 28) and their healthy counterparts (n = 50), and (iii) SNP genotyping (selected variants of interest) in sALS patients of Greek (n = 114), Sardinian (n = 114), and Turkish (n = 148) origin and their healthy counterparts (n = 39, 87, 74, respectively). The first patient cohort served as the discovery dataset, while the second and third patient cohorts were the training datasets
Whole-genome sequencing and bioinformatics analyses
Genomic DNA isolation was performed either from peripheral blood using the QIAamp Blood Midi Kit (Qiagen GmbH, Hilden, Germany) or by means of the phenol-chloroform extraction method as previously described with minor modifications or saliva as previously described [1]. Whole-genome sequencing was performed using Complete Genomics’ (CA, USA) DNA nanoarray platform [18]. DNA sequencing coverage was ×110. Only high-quality call variants were included in the downstream analysis (> 93%). Genomes were aligned with the hg19 reference genome. Next-generation sequencing data (Complete Genomics Inc., CA) were analyzed using Ingenuity Variant Analysis version 3.1.2 (Ingenuity® Systems, www.ingenuity.com). This is well-established software that identifies associations between phenotypes, defined by the user in terms of the classification of the tested individuals, and variants in the sequenced genome. Upon classification by phenotype (sALS vs. healthy individuals), a number of variants were listed; the output was filtered into smaller variant lists of (i) those found only in sALS patients (n = 174 single nucleotide polymorphisms (SNPs)), (ii) those not previously annotated or annotated only once, (iii) those with matching Human Gene Mutation Database (HGMD) entries, and (iv) whole-exome findings. All variants were filtered according to the analysis required, using custom scripts and Complete Genomics Analysis Tools (CGA™ Tools).
Linkage disequilibrium and in silico analyses
Pair-wise linkage disequilibrium (LD) calculations were based on phase genotype data (SNAP v2.2) [24], utilizing the HapMap Phase II + III (release 28) [6] and the 1000 Genomes Project (http://www.1000genomes.org) dataset for Caucasians (CEU). Findings were visualized on HaploView 4.2 and by using the LDmatrix module on the LDlink web tool [30]. Τo further investigate the role of the selected FTO and TBC1D1 variants, we explored their effect on splicing motifs (including the “accept” and “donor” splice sites), the branch point, and auxiliary sequences that enhance (exonic splicing enhancers, ESE) or repress (exonic splicing silencers, ESS) splicing. To this end, in silico prediction was performed, using Human Splicing Finder (http://www.umd.be/HSF3/). This is a system that has rapidly become an international standard, as it combines 12 different algorithms [15]. Intronic variants, if “significant,” are often located within gene regions that are characterized by a reduced level of genetic variation. Conserved elements were therefore explored using Variant Effect Predictor [34].
Downstream molecular genetic analysis
The first patient cohort served as our discovery dataset, while the second and third patient cohorts constituted our training datasets (Fig. 1). For this process, selected variants were subsequently validated in sALS Greek patients (n = 28) and their healthy counterparts (n = 50) (Fig. 1, Tables 1 and 2 and Additional file 1). PCR amplification was carried out according to the KAPA2G Fast HotStart protocol (KAPABIOSYSTEMS, MA, USA); detailed information regarding SNP amplification conditions is available upon request. For FTO rs2892469 (C>T) and rs1861869 (G>C), a PCR-based conventional Sanger resequencing approach was employed. Capillary electrophoresis was performed on an ABI Prism 3130xl DNA Analyzer (Applied Biosystems). For FTO rs17217144 (C>T) and rs7186521 (A>G) as well as TBC1D1 rs6850200 (C>A), allele-specific PCR assays were developed (two alternative reverse primers hybridizing exclusively either to the wild-type or the mutant allele). Sanger sequencing was also employed to validate the allele-specific PCR method.
Table 1.
Selected FTO and TBC1D1 gene variants of prime interest
| Gene | SNPs | Alleles | MAF-all | MAF-EUR | Variant type |
|---|---|---|---|---|---|
| FTO | rs2892469 | C>T | 0.31 (T) | 0.49 (T) | Intronic |
| rs1861869 | C>G | 0.38 (G) | 0.54 (G) | ||
| rs17217144 | C>T | 0.31 (T) | 0.49 (T) | ||
| rs7186521 | A>G | 0.37 (G) | 0.49 (G) | ||
| TBC1D1 | rs6850200 | C>A | 0.41 (A) | 0.37 (A) |
Table 2.
Genotyping data of sALS patients and their healthy counterparts
| Population groups | Genotype frequency (%) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FTO rs2892469 | FTO rs1861869 | FTO rs17217144 | FTO rs7186521 | TBC1D1 rs6850200 | |||||||||||||||||
| CC | TT | CT | p value | CC | GG | CG | p value | CC | TT | CT | p value | AA | GG | AG | p value | AA | CC | CA | p value | ||
| Second patient cohort | Greek healthy individuals | 26 | 36 | 38 | 0.005 | 21 | 41 | 38 | 0.003 | 0 | 0 | 100 | PA = 1.55e−12 PB = 7.78e−13 | 0 | 0 | 100 | 1.0 | 18 | 45 | 38 | 0.017 |
| Greek sALS patients | 17 | 22 | 61 | 17 | 22 | 61 | 22 | 13 | 65 | 0 | 0 | 100 | 9 | 35 | 57 | ||||||
| Third patient cohort | Greek healthy individuals | 26 | 36 | 38 | 0.033 | 21 | 41 | 38 | PA = 0.072 PB = 0.071 |
6 | 2 | 92 | 0.002 | 0 | 0 | 100 | 5.13e−39 | 14 | 43 | 43 | 0.723 |
| Greek sALS patients | 21 | 23 | 56 | 17 | 29 | 54 | 22 | 2 | 76 | 18 | 65 | 18 | 11 | 42 | 48 | ||||||
| Sardinian healthy individuals | 38 | 25 | 38 | 0.63 | 36 | 31 | 33 | 0.61 | 24 | 36 | 40 | 0.67 | 35 | 22 | 42 | 0.64 | 20 | 41 | 39 | 0.53 | |
| Sardinian sALS patients | 31 | 28 | 41 | 29 | 34 | 37 | 27 | 30 | 43 | 29 | 25 | 46 | 15 | 39 | 46 | ||||||
| Turkish healthy individuals | 26 | 26 | 49 | 0.62 | 0 | 100 | 0 | 0.49 | 26 | 26 | 49 | 0.62 | 26 | 26 | 49 | 0.09 | 18 | 45 | 38 | 0.57 | |
| Turkish sALS patients | 24 | 21 | 55 | 0 | 99 | 1 | 26 | 20 | 54 | 21 | 26 | 53 | 13 | 44 | 43 | ||||||
Sizing PCR across the C9orf72 hexanucleotide repeats was performed using previously published primers [13] using repeat-primed PCR [32, 39]. Fragment length analysis was performed with the ABI 3500 genetic analyzer (Applied Biosystems, Foster City, Calif., USA) and analyzed using ABI GeneMapper v4.1 (Applied Biosystems).
qRT-PCR expression analysis for the FTO and TBC1D1 genes
The relative expression levels of FTO and TBC1D1 were determined using human-specific primers in a range of various human cell types; fibroblasts, embryonic stem cells (ESCs), astrocytes, neural PAX6+ progenitors, stem cell-derived purified motor neurons at three different stages of development (Hb9::GFP on days 0, 16, and 30), and stem cell-derived cortical neurons (NKX21 on days 45 and 50). Human brain and spinal cord RNA served as controls. Cell pellets were resuspended in Trizol Reagent (Life Technologies), and RNA was isolated following the manufacturer’s protocol. 0.5 to 1 μg of RNA was treated with DNase I (Invitrogen) and subsequently used for the generation of cDNA using iSCRIPT Reverse Transcription Supermix (Bio-Rad). RT-PCR was performed using SYBR green on the CFX system (Bio-Rad). All assays were performed in triplicate. The averaged cycle of threshold (Ct) value of the two housekeeping genes (ACTIN/GAPDH) was subtracted from the Ct value of the gene of interest to obtain the ΔCt. Relative gene expression was determined as 2−ΔCt (ΔΔCt) and expressed relative to undifferentiated human embryonic stem cells (hESCs).
Statistical analysis
Deviations from Hardy-Weinberg equilibrium were tested for using Pearson’s goodness-of-fit chi-square (degrees of freedom = 1), log likelihood ratio chi-square (degrees of freedom = 1), and Fisher’s Exact tests. De Finetti diagrams were also constructed [41]. Focusing on case-control (healthy individuals) phenotypes, we tested the null hypothesis of no association between rows and columns of the 2 × 3 matrix that contains the counts of the three genotypes (the two homozygotes and the heterozygote) among cases and controls. Genotype and allele frequencies were evaluated using Fisher’s exact test. We also performed the Armitage test (Monte Carlo method; it obtains results that are closer to those of an exact test, since the classical Cochran-Armitage trend test is based on approximation) [5]. All tests were performed as two-tailed, and differences were considered to be statistically significant when P < 0.05. Further calculations using contingency tables (chi-square test) were performed to explore data stratification, considering (i) the age of onset (below or above 50 years old), (ii) El Escorial criteria, and (iii) spinal or bulbar ALS onset.
Results
Whole-genome sequencing analysis of Greek sALS patients reveals a positive association with FTO and TBC1D1 variants
Our next-generation sequencing-based genomics approach yielded a total of 174 SNPs that were found only in the sALS patients studied; these were variously intergenic (n = 144), intronic (n = 23), and/or were present in upstream (n = 2), downstream (n = 1), and 3′UTR (n = 2) regions or non-coding RNAs (n = 2). Next, genes were clustered by metabolic or disease network and the most prominent ones are shown in Table 3. Our data analysis revealed three genomic variants of prime interest: FTO rs2892469 (C>T) and rs1861869 (G>C) and TBC1D1 rs6850200 (C>A). Findings were further explored in Greek sALS patients and their healthy counterparts (second patient cohort). FTO intronic variants, rs2892469 (C>T) and rs1861869 (G>C), reached statistical significance (p = 0.005 and p = 0.003, respectively). For TBC1D1 rs6850200 (C>A), a strong association also became evident (p value of 0.017). Data are summarized in Tables 1 and 2 and Additional file 1.
Table 3.
Gene clustering per metabolic or disease network using Ingenuity Pathway Analysis (IPA) to assist decision-making
| Disease or metabolic network | Genes | p value |
|---|---|---|
| Neural cell proliferation | ISL1, FTO, ADCYAP1, ΟΤΡ, SPP1 | 6.02 × 10–5 |
| Motor neuron proliferation | ISL1, FTO | 9.53 × 10–5 |
| Motor neuron dysfunction | ISL1, WNT4 | 1.28 × 10–3 |
| Development of neuromuscular junction | PDZRN3 | 1.65 × 10–3 |
| Branching of sensory neurons | PRKG1 | 1.65 × 10–3 |
| ALS | FUS | 1.65 × 10–3 |
| Development of P and M cells of visual nervous system | ΟΤΡ | 1.65 × 10–3 |
| Growth of cerebellar cortex, regulation of upper cervical ganglia | ADCYAP1 | 1.65 × 10–3 |
| Cranial nerve development | ISL1, ERBB4 | 1.9 × 10–3 |
| Brain development | ADCYAP1, ERBB4, FTO, ΟΤΡ, PRKG1 | 2.56 × 10–3 |
| Development of specific nerves of the hand, area of dendritic trees | ISL1 | 3.3 × 10–3 |
| Movement of the cranial neural crest, participation in the formation of the central nervous system, astrocyte attachment | ERBB4 | 3.3 × 10–3 |
| Neural cell number | ADCYAP1, ERBB4, WNT4, SPP1 | 3.75 × 10–3 |
| Nervous system morphology | ERBB4, FTO, WNT4, SPP1, YES1, ISL1 | 6.33 × 10–3 |
| Oligodendrocyte development, excitatory synapse formation | ERBB4 | 6.6 × 10–3 |
| Central nervous system development, dendritic cell development | ERBB4, ADCYAP1 | 7.4 × 10–3 |
| Motor neuron sprouting | FTO | 9.88 × 10–3 |
| v3 spinal cord motor neuron sprouting | FTO | 9.88 × 10–3 |
| Midbrain size | FTO | 9.88 × 10–3 |
| Neuronal withdrawal | WNT4 | 1.32 × 10–2 |
| Brain morphology | ERBB4, FTO, SPP1, YES1 | 1.35 × 10–2 |
| Neuronal branching | ERBB4, ADCYAP1, PRKG1 | 1.58 × 10–2 |
| DNA repair | APTX, FTO, FUS | 1.65 × 10–2 |
In addition, a C9orf72 hexanucleotide expansion was identified in 1 of 28 patients with sALS (3.5%; Additional file 1: Table S4). This female patient was diagnosed with unequivocal ALS but was devoid of a positive family history for ALS.
Linkage disequilibrium analyses suggest a haplotype of FTO gene variants
Our findings support a disease-associated haplotype constituting FTO rs2892469 (C>T), rs1861869 (G>C), rs17217144 (C>T), and rs7186521 (A>G) (Fig. 2). SNAP analysis (HapMap datasets) was in agreement with the HaploView v4.2 analysis (our Greek cohort). The same result was produced for the LDmatrix module on the LDlink web tool. rs2892469 and rs1861869 are in perfect linkage disequilibrium (LD) (D′ = 1, LOD = 23.66, r 2 = 0.884), while rs17217144 and rs7186521 exhibit a strong LD (D′ = 0.882, LOD = 10.1, r 2 = 0.738). Block1 is based on confidence intervals, i.e., it was created due to 95% of informative comparisons being in “strong LD.” Such findings serve as a paradigm as to how information technology tools coupled to experimental data may empower discovery.
Fig. 2.
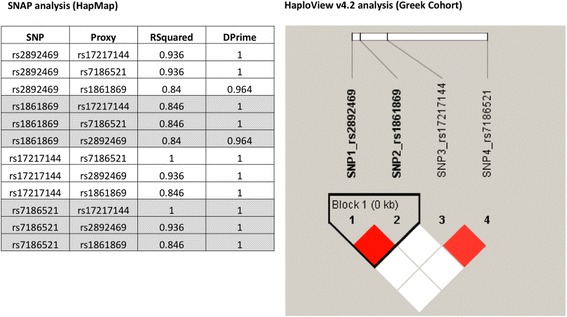
Linkage disequilibrium and haplotype block analysis for FTO rs2892469, rs1861869, rs17217144, and rs7186521. Data suggest a haplotype for the FTO genomic variants studied herein. SNAP analysis refers to HapMap datasets, while the HaploView v4.2 analysis corresponds to our Greek cohort. The HaploView 4.2 color scheme denotes the following: white (D′ < 1, LOD < 2) and red (D′ = 1, LOD ≥ 2). rs2892469 and rs1861869 are in perfect LD (D′ = 1, LOD = 23.66, r 2 = 0.884); rs17217144 and rs7186521 exhibit a strong LD (D′ = 0.882, LOD = 10.1, r 2 = 0.738). Block1 is based on confidence intervals, i.e., it was created through 95% of informative comparisons being in “strong LD”
In silico analyses suggest that FTO and TBC1D1 variants affect splicing
TBC1D1 rs6850200 (C>A) and FTO rs2892469 (C>T) were predicted to create a splicing enhancer site which would potentially alter splicing. In the case of FTO rs1861869 (G>C), a putative exonic splicing silencer site is disrupted, while FTO rs7186521 (A>G) appears to have created a new donor splice site. No impact on splicing was predicted for FTO rs17217144 (C>T). Using Variant Effect Predictor, PhyloP, and PhastCons data showed that the selected FTO variants are evolutionarily conserved.
Replication analyses
Replication analyses were carried out in sALS patients of Greek origin (to ensure data reliability) and Sardinian and Turkish descent (to account for population differences (third patient cohort)). We found that four FTO intronic variants, rs2892469 (C>T) and rs1861869 (G>C), rs17217144 (C>T), and rs7186521 (A>G), reached statistical significance (p = 0.033, p = 0.07, p = 0.002, and p < 0.001, respectively), when Greek sALS patients were compared to their healthy counterparts. For TBC1D1 rs6850200 (C>A), the aforementioned strong association (p value of 0.017) no longer held upon replication in Greek sALS patients (p value of 0.723). Data stratification based upon (i) the age of onset (below or above 50 years old), (ii) El Escorial criteria, and (iii) spinal or bulbar ALS onset did not alter our findings. It should be noted that all FTO and TBC1D1 variants failed to reach statistical significance, even when stratified for SOD1, when Sardinian and Turkish sALS patients were considered. Data are summarized in Tables 1 and 2. The lack of association of these variants with sALS in patients of Sardinian and Turkish descent may suggest a founder effect in the Greek population.
FTO is highly expressed in motor neurons
To begin defining genotype-to-phenotype associations for FTO, TBC1D1, and ALS, we determined the relative mRNA levels for both genes across several human cell types. We specifically analyzed fibroblasts, undifferentiated embryonic stem cells (ESCs), astrocytes, neural PAX6+ progenitors, and stem cell-derived spinal cord motor neurons (MNs) and cortical neurons (CNs). We used the HUES3-Hb9::GFP cell line, which contains a GFP reporter under the control of the MN-specific Hb9 enhancer [17] to differentiate and FACS-purify MNs as described previously [26], on three different stages of development (days 0, 16, and 30). We also used the NKX2.1::GFP ESC line to differentiate and FACS-purify cortical neurons as described previously [33] on three different stages of development (days 45 and 50). Our qRT-PCR data show that FTO is relatively neural specific and notably is most highly expressed in motor neurons (Fig. 3). TBC1D1 seems to be rather ubiquitously expressed.
Fig. 3.
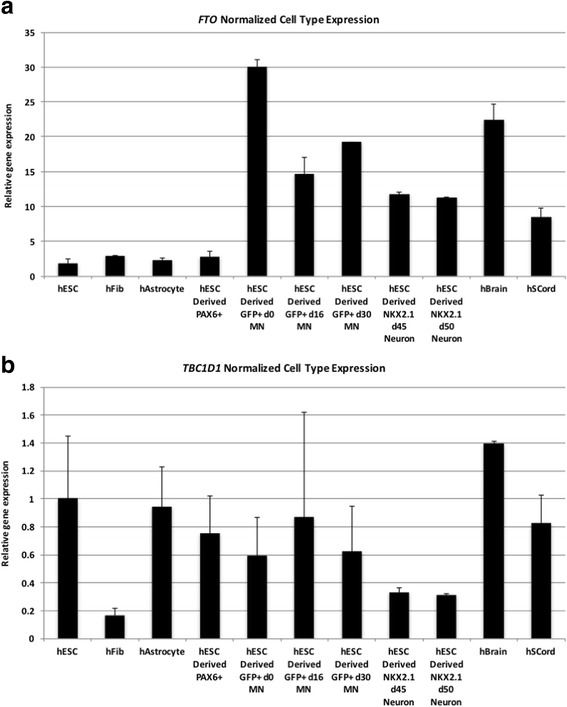
a, b FTO and TBC1D1 expression levels in a range of various human cell types. FTO and TBC1D1 mRNA levels were determined using human-specific primers in various cell types; fibroblasts, embryonic stem cells (ESCs), astrocytes, neural PAX6+ progenitors, stem cell-derived purified motor neurons at three different stages of development (Hb9:GFP on days 0, 16, and 30), and stem cell-derived cortical neurons (NKX21 on days 45 and 50). Human brain and spinal cord RNA served as controls. Normalized RT-qPCR data are expressed as averaged sample replicates. Error bars represent standard deviations
Discussion
sALS is polygenic with a complex interplay with environmental and other factors. Previous studies have proposed genetic associations of a variety of genes with the etiology of sALS. In this article, we adopted a three-step approach to delineate the association of genomic variants with sALS in patients of Greek origin.
Using a next-generation sequencing-based genomics approach, we identified 174 variants that were only present in the initial sALS patients studied. We opted to focus on the FTO rs2892469 (C>T) and rs1861869 (G>C) and TBC1D1 rs6850200 (C>A) variants, based on pathway analysis. Our replication analyses confirmed that the FTO but not the TBC1D1 genomic variants maintained their statistical significance as far as their association with sALS was concerned. Data stratification considering (i) the age of onset (below or above 50 years old), (ii) El Escorial criteria, and (iii) spinal or bulbar ALS onset did not affect our findings (not shown).
We subsequently attempted to explore whether the FTO gene variants identified as being associated with sALS in Greek patients might also be associated with sALS in patients of Turkish and Sardinian descent. However, genetic analysis of all FTO and TBC1D1 variants failed to reach statistical significance, even when stratified for SOD1, when Sardinian and Turkish sALS patients were considered. This may be due to a founder effect in the Greek population.
In order to explore the functional significance of our findings, we measured the FTO and TBC1D1 gene expression levels in several human cell types, namely fibroblasts, embryonic stem cells (ESCs), astrocytes, neural PAX6+ progenitors, stem cell-derived purified motor neurons at three different stages of development (Hb9::GFP on days 0, 16, and 30), and stem cell-derived cortical neurons (NKX21 on days 45 and 50). Our qRT-PCR data demonstrated that FTO gene expression is relatively neuron-specific and notably is highly expressed in motor neurons (Fig. 3). By contrast, TBC1D1 would appear to be rather ubiquitously expressed.
Interestingly, long distance interactions have been reported for the FTO intronic region: (i) obesity-associated non-coding sequences within FTO interact with the promoter regions of IRX3 and FTO in human, mouse, and zebrafish, and (ii) obesity-associated FTO gene variants influence IRX3 expression in the human brain. The latter gene was associated with body mass, as IRX3-deficient mice displayed a decrease in their body weight by 25–30% [42]. Nutritional surveillance of ALS patients has been well established to be of fundamental importance, both in bulbar and spinal onset patients, and nutritional status is a prognostic factor for survival in ALS patients [16]. Jawaid et al. [22] reported that a decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS patients. Moreover, an LD block of 49 kb in the first intron of FTO contains numerous highly conserved non-coding elements (HCNEs) and lies within an extended region of conserved synteny involving the IRX3/5/6 cluster, the closest gene of which is the transcription factor-encoding IRX3 [37]. IRX3 is a member of the Iroquois homeobox gene family and plays a role in the early stages of neural development [7].
We also identified an example of C9orf72 hexanucleotide expansion in 1 of 28 patients with sALS (3.5%; Additional file 1: Table S4). This female patient, harboring this expansion, was diagnosed with unequivocal ALS but was lacked a positive family history of ALS. This massive hexanucleotide repeat expansion mutation in C9orf72, which has been linked to fALS, sALS, and frontotemporal dementia, has a single founder who is thought to have originated approximately 6300 years ago in Europe [31, 43]. Our findings differ somewhat from those reported by Mok and coworkers [35], the only study to date in fALS and sALS patients of Greek descent, in which 11 of 136 sALS patients (8.2%) were carriers of the expansion. Such a discrepancy may reflect inter-individual differences not as far as statistical power is concerned, but rather related to the patients’ genetic background, as the aforementioned study examined only individuals from Athens, whereas we recruited individuals from across the entire Greek mainland (north, central, and south Greece). Differential exposure to environmental factors are known to contribute to an increased risk of ALS; thus, exposure to lead, mercury, selenium, zinc, and copper is known to increase the risk of ALS [46], while high-risk populations include those who collect their domestic water from surface water [8], possibly reflecting an association between the neurotoxin b-methylamino-l-alanine (BMAA) produced by cyanobacteria and the risk of ALS. Al-Chalabi and Hardiman [4] have proposed a disease model, according to which ALS is the net result of environmental risks and time acting on a pre-existing genetic load, followed by an automatic, self-perpetuating decline until death. Akimoto et al. [2] reported marked differences in genotyping results for GGGGCC-repeat expansions in C9orf72. Herein, amplicon length analysis and repeat-primed PCR were combined as a recommended minimum requirement in a research setting [2].
Small sample sizes and data variability, plus our ancestrally diverse patient cohorts, should be taken into account when interpreting our findings. Yet, our datasets may assist future efforts of replicating these findings in independent ALS patient cohorts.
Conclusions
There is no single predominant genetic association underlying sALS. Instead, sALS may be considered to be the net result of the complex interplay of environmental factors and numerous low-risk susceptibility loci. We propose that under this polygenic model, a large number of alleles, each conferring a small genotypic risk, combine additively or multiplicatively to confer a range of susceptibilities in the general population [21, 47].
To our knowledge, this has been the first study to present a possible association between FTO variants and the genetic basis of sALS. These findings need to be further validated in a larger sALS patient cohort of Greek origin to overcome the small sample size bias, while a gene ontology analysis to look at links between FTO and the other genes known to be associated with ALS could support the validity of these findings.
Acknowledgements
The authors are grateful to the Greek ALS Patients Association for their continuous support. We thank Ms. Aikaterini Dimitropoulou, Vasiliki Delatolla, Maria-Valsamo Founta, Chrysavgi Gritzapi, Venetia Karathanassi, Anna Karra, Dimitra Taroussi, and Paraskevi Tassopoulou for their expert technical assistance.
Funding
Our work was partly funded by grants from the European Commission [RD-Connect (FP7-305444), U-PGx (H2020-668353)], the Greek General Secretariat of Research and Technology [eMoDiA (ΠΔΕ11_0415)], and the Golden Helix Foundation to GPP. VK and IN are funded by the Serbian Ministry of Education, Science and Technology development Grant No. ON175090 and Grant No. ON17509, respectively. BRA is funded by UAEU grants (31M135 and 31M187).
Availability of data and materials
Data supporting the results reported in this study are available in the Supplementary material.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- ESCs
Embryonic stem cells
- fALS
Familial amyotrophic lateral sclerosis
- HCNEs
Highly conserved non-coding elements
- sALS
Sporadic amyotrophic lateral sclerosis
- SNPs
Single nucleotide polymorphisms
Additional file
Supplementary Tables S1–3. (PDF 784 kb)
Authors’ contributions
GPP conceived the study. KM, EMP, GX, TK and GPP designed the study. GX, ED, SR, GD, SO, NB, and GPP provided the samples. KM, EMP, GX, AB, KC, VG, KVZ, AJ, KK, EP, AP, FA, AL, VD, MB, EET, and TK performed the experiments. AD and EK generated the samples, performed and analyzed the qRT-PCR experiments. AP, FA, AL, VD, CM, BAP, and TK analyzed the data. BRA and GPP provided funding for this work. GX, IN, VSK, NB, SO, EK, DNC, SG, GMH, RD, BRA, TK, and GPP wrote the manuscript. All authors read and approved the manuscript.
Ethics approval and consent to participate
Patient recruitment and sample collection were performed with informed consent. The study was approved by the University of Patras Bioethics Committee.
Consent for publication
“Not applicable” in this section.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40246-017-0126-2) contains supplementary material, which is available to authorized users.
References
- 1.Aidar M, Line SRP. A simple and cost-effective protocol for DNA isolation from buccal epithelial cells. Braz Dent J. 2007;18:148–152. doi: 10.1590/S0103-64402007000200012. [DOI] [PubMed] [Google Scholar]
- 2.Akimoto, C, Volk, AE, van Blitterswijk, M, Van den Broeck, M, Leblond, CS, Lumbroso, S, Camu, W, Neitzel, B, Onodera, O and van Rheenen, W. (2014). A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J. Med. Genet. jmedgenet-2014-102360. [DOI] [PMC free article] [PubMed]
- 3.Al-Chalabi, A, Fang, F, Hanby, MF, Leigh, PN, Shaw, CE, Ye, W and Rijsdijk, F. (2010). An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry jnnp 2010.207464. [DOI] [PMC free article] [PubMed]
- 4.Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 5.Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet. 2006;7:781. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- 6.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2004;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 7.Bellefroid EJ, Kobbe A, Gruss P, Pieler T, Gurdon JB, Papalopulu N. Xiro3 encodes a Xenopus homolog of the Drosophila Iroquois genes and functions in neural specification. EMBO J. 1998;17:191–203. doi: 10.1093/emboj/17.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradley WG, Borenstein AR, Nelson LM, Codd GA, Rosen BH, Stommel EW, Cox PA. Is exposure to cyanobacteria an environmental risk factor for amyotrophic lateral sclerosis and other neurodegenerative diseases? Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2013;14:325–333. doi: 10.3109/21678421.2012.750364. [DOI] [PubMed] [Google Scholar]
- 9.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J. Neurol. Sci. 1994;124:96–107. doi: 10.1016/0022-510X(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 10.Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, Mitra RD, Ravits J, Harms MB, Baloh RH. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol. 2015;77:100–113. doi: 10.1002/ana.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu Y-F, Wang Q, Krueger BJ. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeJesus-Hernandez M, Desaro P, Johnston A, Ross OA, Wszolek ZK, Ertekin-Taner N, Graff-Radford N, Rademakers R, Boylan K. Novel p. Ile151Val mutation in VCP in a patient of African American descent with sporadic ALS. Neurology. 2011;77:1102–1103. doi: 10.1212/WNL.0b013e31822e563c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng H-X, Chen W, Hong S-T, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H. Mutations in UBQLN2 cause dominant X-linked juvenile and adult onset ALS and ALS/dementia. Nature. 2011;477:211. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desport J, Preux P, Truong T, Vallat J, Sautereau D, Couratier P. Nutritional status is a prognostic factor for survival in ALS patients. Neurology. 1999;53:1059. doi: 10.1212/WNL.53.5.1059. [DOI] [PubMed] [Google Scholar]
- 17.Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 19.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 20.Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- 21.Houlston RS, Peto J. The search for low-penetrance cancer susceptibility alleles. Oncogene. 2004;23:6471. doi: 10.1038/sj.onc.1207951. [DOI] [PubMed] [Google Scholar]
- 22.Jawaid A, Murthy SB, Wilson AM, Qureshi SU, Amro MJ, Wheaton M, Simpson E, Harati Y, Strutt AM, York MK. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph Lateral Scler. 2010;11:542–548. doi: 10.3109/17482968.2010.482592. [DOI] [PubMed] [Google Scholar]
- 23.Ji, A-L, Zhang, X, Chen, W-W and Huang, W-J. (2017). Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J Med Genet jmedgenet-2016-104271. [DOI] [PubMed]
- 24.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'donnell CJ, De Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Velde CV, Bouchard J-P, Lacomblez L, Pochigaeva K, Salachas F. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 26.Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 2014;14:781–795. doi: 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwiatkowski TJ, Bosco D, Leclerc A, Tamrazian E, Vanderburg C, Russ C, Davis A, Gilchrist J, Kasarskis E, Munsat T. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 28.Lattante S, Ciura S, Rouleau GA, Kabashi E. Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD) Trends Genet. 2015;31:263–273. doi: 10.1016/j.tig.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 29.Lattante S, Conte A, Zollino M, Luigetti M, Del Grande A, Marangi G, Romano A, Marcaccio A, Meleo E, Bisogni G. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology. 2012;79:66–72. doi: 10.1212/WNL.0b013e31825dceca. [DOI] [PubMed] [Google Scholar]
- 30.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. The Lancet Neurology. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandic-Stojmenovic G, Stefanova E, Dobricic V, Novakovic I, Stojkovic T, Jesic A, Kostic V. Screening for C9orf72 expansion mutation in Serbian patients with early-onset dementia. Dement Geriatr Cogn Disord. 2015;40:358–365. doi: 10.1159/000438748. [DOI] [PubMed] [Google Scholar]
- 33.Maroof AM, Keros S, Tyson JA, Ying S-W, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mok, KY, Koutsis, G, Schottlaender, LV, Polke, J, Panas, M and Houlden, H. (2012). High frequency of the expanded C9ORF72 hexanucleotide repeat in familial and sporadic Greek ALS patients. Neurobiol Aging 33, e1851-1851. [DOI] [PMC free article] [PubMed]
- 36.Prudencio, M, Gonzales, PK, Cook, CN, Gendron, TF, Daughrity, LM, Song, Y, Ebbert, MT, van Blitterswijk, M, Zhang, Y-J and Jansen-West, K. (2017). Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. [DOI] [PMC free article] [PubMed]
- 37.Ragvin A, Moro E, Fredman D, Navratilova P, Drivenes Ø, Engström PG, Alonso ME, de la Calle Mustienes E, Skarmeta JLG, Tavares MJ. Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc Natl Acad Sci. 2010;107:775–780. doi: 10.1073/pnas.0911591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ratti A, Corrado L, Castellotti B, Del Bo R, Fogh I, Cereda C, Tiloca C, D'Ascenzo C, Bagarotti A, Pensato V. C9ORF72 repeat expansion in a large Italian ALS cohort: evidence of a founder effect. Neurobiol Aging. 2012;33:e2527–e2528. doi: 10.1016/j.neurobiolaging.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, Van Swieten JC, Myllykangas L. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosen DR, Siddiquef T, Patterson D, Figlewicz DA, Sapp P, Hentatif A, O’Regan JP, Dengf H-X, McKenna-Yasek D, Cayabyabi A. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic. Nature. 1993;362:4. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 41.Sasieni PD. From genotypes to genes: doubling the sample size. Biometrics. 1997;1:1253–1261. doi: 10.2307/2533494. [DOI] [PubMed] [Google Scholar]
- 42.Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, Miller J, Lee Y, Troakes C, Scott KM. The C9ORF72 expansion mutation is a common cause of ALS+/− FTD in Europe and has a single founder. Eur J Hum Genet. 2013;21:102. doi: 10.1038/ejhg.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su F-C, Goutman SA, Chernyak S, Mukherjee B, Callaghan BC, Batterman S, Feldman EL. Association of environmental toxins with amyotrophic lateral sclerosis. JAMA neurology. 2016;73:803–811. doi: 10.1001/jamaneurol.2016.0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10:661–670. doi: 10.1038/nrneurol.2014.184. [DOI] [PubMed] [Google Scholar]
- 46.Trojsi F, Monsurrò MR, Tedeschi G. Exposure to environmental toxicants and pathogenesis of amyotrophic lateral sclerosis: state of the art and research perspectives. Int J Mol Sci. 2013;14:15286–15311. doi: 10.3390/ijms140815286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN. Deleterious-and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet. 2012;91:1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data supporting the results reported in this study are available in the Supplementary material.