

Abstract
Background
The Cornales is the basal lineage of the asterids, the largest angiosperm clade. Phylogenetic relationships within the order were previously not fully resolved. Fifteen plastid genomes representing 14 species, ten genera and seven families of Cornales were newly sequenced for comparative analyses of genome features, evolution, and phylogenomics based on different partitioning schemes and filtering strategies.
Results
All plastomes of the 14 Cornales species had the typical quadripartite structure with a genome size ranging from 156,567 bp to 158,715 bp, which included two inverted repeats (25,859–26,451 bp) separated by a large single-copy region (86,089–87,835 bp) and a small single-copy region (18,250–18,856 bp) region. These plastomes encoded the same set of 114 unique genes including 31 transfer RNA, 4 ribosomal RNA and 79 coding genes, with an identical gene order across all examined Cornales species. Two genes (rpl22 and ycf15) contained premature stop codons in seven and five species respectively. The phylogenetic relationships among all sampled species were fully resolved with maximum support. Different filtering strategies (none, light and strict) of sequence alignment did not have an effect on these relationships. The topology recovered from coding and noncoding data sets was the same as for the whole plastome, regardless of filtering strategy. Moreover, mutational hotspots and highly informative regions were identified.
Conclusions
Phylogenetic relationships among families and intergeneric relationships within family of Cornales were well resolved. Different filtering strategies and partitioning schemes do not influence the relationships. Plastid genomes have great potential to resolve deep phylogenetic relationships of plants.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4319-9) contains supplementary material, which is available to authorized users.
Keywords: Plastid genome, Phylogenomics, Cornales, Alignment, Partitioning schemes, Gene loss
Background
The Cornales is a relatively small but diverse group, representing the basal lineage of the largest angiosperm clade, the Asterids [1–4]. It comprises 42 genera and approximately 605 species in ten families, including two large families (Hydrangeaceae and Loasaceae) and eight small families. The latter contain few genera, mostly with isolated geographic ranges, i.e. Cornaceae (Cornus), Nyssaceae (Camptotheca, Nyssa), Curtisiaceae (Curtisia), Grubbiaceae (Grubbia), Hydrostachyaceae (Hydrostachys), Alangiaceae (Alangium), Davidiaceae (Davidia), and Mastixiaceae (Diplopanax, Mastixia) [3, 5–8]. Cornales contains many ecologically and economically important species, including ornamentals in Cornaceae, Davidiaceae and Hydrangeaceae; moreover Camptotheca (Nyssaceae), is the source of camptothecin. Species in the order possess different habits (evergreen, deciduous), diverse growth forms (e.g. trees, shrubs, lianas, rhizomatous and herbs) and occur in tropical, temperate and boreal ecosystems.
The circumscription and phylogenetic relationships of the order have been investigated by a number of phylogenetic analyses, mostly based on plastid DNA, beginning from the early twentieth century such as Olmstead et al. [9] and Chase et al. [10]. Increasing the amount of molecular markers has progressively improved phylogenetic resolution and branch support in Cornales [3, 5, 7, 8]. For example, based on six cpDNA regions and broader taxon sampling, Xiang et al. [3] obtained well supported but not fully resolved intra-family relationships for some families (e.g. Hydrangeaceae, Cornaceae) in this order.
Integrating genomic data into plant phylogenetic investigations is developing rapidly due to the availability of new methods of sampling genomes (e.g. genome skimming, transcriptomes, hybrid capture) facilitated by next-generation sequencing (NGS) technologies [11–14]. Complete plastid genomes have rapidly accumulated in the NCBI databases over the last few years. However, phylogenomic studies remain to be conducted for Cornales, and only a few plastid genomes within this order have been released, sporadically, on NCBI databases. The plastome is usually uniparentally inherited in seed plants [15]. It can provide an abundance of variable sites across its entire length for phylogenetic analyses [16]. Thus, plastid genomes show the potential for resolving recalcitrant phylogenetic relationships, at both high taxonomic levels such as green plants [17–19], and low taxonomic levels [20–22]. The most widely used approach for plastome phylogenomics is to analyze the concatenated coding genes as a whole [14, 20, 23, 24], but the noncoding sequences are also useful for inferring phylogenies at lower taxonomic levels when the plastid genomes are conserved [25–27]. Because different regions of plastid genomes vary in their evolutionary rates, partitioning the genome by genes or regions might be preferable for phylogenomic analysis [20]. Moreover, fast-evolving sites of plastid genomes that cannot be aligned with confidence could possibly result in misleading phylogenetic inference, and therefore removing the most variable sites or problematic regions might improve accuracy in phylogenetic inference [28–30].
Plastid genomes of angiosperms generally contain 110 to 130 distinct genes, and range in size from 120 to 160 kb. They usually show a typical quadripartite circular structure of two copies of large inverted repeat (IR) separated by the small single-copy (SSC) and the large single-copy (LSC) regions [31]. Although the plastid genome is reported as highly conserved in most angiosperms [32], it is subject to structural alterations such as extension or contraction of the IR region [33], the presence of large inversions or deletions [34, 35], pseudogenization and gene loss [36, 37]. Besides their phylogenetic utility, whole plastid genomes could also be used to investigate other aspects of molecular biology such as genome evolution on the structural and molecular level, and to develop fast evolved molecular markers for investigations of phylogeny and phytogeography [17, 27, 38–41].
In the present study, a total of 15 complete plastid genomes of 14 species representing 10 genera and seven families of Cornales were obtained. The main objectives of this study were to 1) characterize and compare the structure and gene organizations of plastid genomes in Cornales; 2) explore the potential effects of different partitioning schemes and alignment strategies on phylogenetic inferences; and 3) assess the application of the complete plastid genome sequences in Cornales, and provide genetic resources for future research.
Methods
Taxon sampling
The circumscription of families of Cornales followed Xiang et al. [3], and taxonomy within families and genera followed Flora of China [42] or the Plant List (http://www.theplantlist.org/) (accessed 1st January, 2013). A total of 15 individuals representing 14 species of 10 genera from 7 families in Cornales mainly occurring in China were sampled. Samples of three families (Grubbiaceae, Hydrostachyaceae and Loasaceae) could not be obtained for this study. The sampled species hence represented four out of the five major lineages suggested by Xiang et al. [3]. Two individuals of Cornus capitata were sequenced here to investigate the intraspecific variability within plastid genome. As outgroups, the plastid genome of Fouquieria diguetii of Ericales was newly sequenced, and the plastomes of three species within Caryophyllales (Basella alba, Talinella dauphinensis, Gisekia pharnaceoides) were obtained from another parallel work (unpublished data). These two orders are phylogenetically closest to Cornales [1–4]. Fresh leaves were collected in the field or from botanic gardens with the permission of the land owners or the botanic gardens (Table 1) and transferred to the laboratory under cool conditions (~4 °C) for total genomic DNA extraction. Voucher specimens were collected for each species, and deposited at the Herbarium of Kunming Institute of Botany (KUN), Chinese Academy of Sciences or the herbarium of the Royal Botanic Garden Edinburgh (E). Detailed information of the pecies sampled in this study is provided in Table 1.
Table 1.
Taxa sampled in this study
| Taxa | Family | Order | Locality | Voucher | Voucher specimen | GenBank accession number |
|---|---|---|---|---|---|---|
| Nyssa wenshanensis | Nyssaceae | Conales | China, Yunnan, Kunming Botanical Garden | Cai J. & Zhang T. | 14CS9047 | MG524995 |
| Nyssa sinensis | Nyssaceae | Conales | China, Yunnan, Wenshan | Liu C., et al. | 14CS8436 | MG525000 |
| Camptotheca acuminata | Nyssaceae | Conales | China, Yunnan, Yuxi | Cai J., et al. | 13CS7273 | MG525005 |
| Davidia involucrata | Davidiaceae | Conales | China, Yunnan, Kunming Botanical Garden | Cai J. & Zhang T. | 14CS9049 | MG525002 |
| Mastixia caudatilimba | Mastixiaceae | Conales | China, Yunnan, Xishuangbannan, | Guo Y.J., et al. | 14CS9459 | MG525001 |
| Diplopanax stachyanthus | Mastixiaceae | Conales | China, Yunnan, Wenshan, | Zhang T. & Liu C. | 14CS8795 | MG524991 |
| Hydrangea heteromalla | Hydrangeaceae | Conales | China, Yunnan, Kunming | Guo Y.J. & Liu C. | 10CS1923 | MG524994 |
| Hydrangea aspera | Hydrangeaceae | Conales | China, Yunnan, Wenshan | Liu C., et al. | 14CS8432 | MG524992 |
| Deutzia crassifolia | Hydrangeaceae | Conales | China, Yunnan, Chuxiong | Guo Y.J., et al. | 14CS8216 | MG524993 |
| Alangium alpinum | Alangiaceae | Conales | China, Yunnan, Kunming Botanical Garden | Yang J.D. | 14CS9086 | MG525003 |
| Alangium chinense | Alangiaceae | Conales | China, Yunnan, Wenshan | Cai J., et al. | 14CS9130 | MG524996 |
| Cornus capitate #1 | Cornaceae | Conales | China, Yunnan, Kunming | Ya J.D., et al. | 14CS9213 | MG524990 |
| Cornus capitate #2 | Cornaceae | Conales | China, Yunnan, Kunming Botanical Garden | Liu C. & Ya J.D. | 14CS8464 | MG524998 |
| Cornus controversa | Cornaceae | Conales | China, Yunnan, Kunming Botanical Garden | Liu C. & Ya J.D. | 14CS8466 | MG525004 |
| Curtisia dentata | Curtisiaceae | Conales | UK, Royal Botanic Garden Edinburgh | Möller M. | RBGE 19240177 | MG524999 |
| Fouquieria diguetii | Fouquieriaceae | Ericales | UK, Royal Botanic Garden Edinburgh | Möller M. | RBGE 19800074 | MG524997 |
| Basella alba | Basellaceae | Caryophyllales | China, Yunnan, Kunming | Yang J.D. et al. | 14CS9526 | Unpublished |
| Talinella dauphinensis | Talinaceae | Caryophyllales | UK, Royal Botanic Garden, Kew | Yi T.S. | Yi14363 | Unpublished |
| Gisekia pharnaceoides | Gisekiaceae | Caryophyllales | China, Hainan, Lingshui, | Zhang T., et al. | 14CS8741 | Unpublished |
DNA sequencing and genome assembly
Total genomic DNA was isolated from about 100 mg fresh leaf material with a modified CTAB method [43] in which 4% CTAB was used instead of 2% CTAB and with approximately 0.1% DL-dithiothreitol (DTT) added. Subsequently, plastid DNA was selectively amplified through long-range PCR using nine or fifteen primer pairs [44, 45]. All PCR products were pooled and diluted to 0.2 ng/μL for library preparation. A short-insertion (500 bp) sequencing library was prepared following the Nextera XT Sample Preparation procedure (Illumina). The paired-end reads of 250 bp or 300 bp were generated using Illumina Miseq at the Laboratory of Molecular Biology of Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences. Four species could not be amplified through long range PCR: Camptotheca acuminata, Cornus controversa, Mastixia caudatilimba and Hydrangea heteromalla. These were sequenced instead from total DNA using Illumina Hiseq4000 after short-insert (500 bp) libraries constructed following the manufacturer’s protocol (Illumina HiSeq 4000) and 143 bp paired-end reads for Hydrangea heteromalla and 90 bp paired-end reads for the other three species, which were generated at BGI Shenzhen, China.
The raw sequence reads were assembled using following steps. First, all reads were de novo assembled into contigs with CLC Genomics Workbench 8.0.2 (CLC Bio) under a word size of 60 bp, minimum contig length of 500 bp and map reads back to contigs with default settings. Second, a closely related genome of Camellia sinensis (NC_020019.1) was used as a reference, and contigs of each individual sample were aligned to it using local BLAST, from which the contigs of plastid genome can be selected. For Cornus controversa and Hydrangea heteromalla, this process produced two and three long plastid contigs respectively, which were easily assembled into a complete genome by overlaps using Geneious v 8.1 [46]. Among the remaining 14 samples, parts of the genome were covered only by short contigs, which were hard to assemble directly. These were analyzed using the two successfully assembled species as reference sequences, and then manually concatenated by their overlaps in Geneious v 8.1.
Verification of the assembly was performed in three ways: 1) by mapping the reads to the assembled plastid genome sequences, 2) by comparing the 14 manually assembled genomes with two easily assembled ones, and 3) by obtaining the four boundary regions using newly designed primers under Sanger sequencing, which were showed in Additional file 1: Table S1.
Genome annotation and comparison
The complete genome sequences were annotated using the online program DOGMA [47] to predict protein-coding genes, transfer RNA (tRNA) genes, and ribosome RNA (rRNA) genes. Start and stop codons of protein-coding genes were determined using plastid/bacterial genetic codes, with the most closely matching reference genome as a guide. Graphical maps with annotation of genomes were drawn using OrganellarGenomeDRAWtool (OGDraw) [48].
The 15 whole plastid genomes were aligned with Mauve v 2.3.1 [49] plugin in Geneious v 8.1, including only one copy of the IR, assuming collinear genomes for the full alignment. To compare the overall similarities among different plastid genomes, pairwise alignments of the 15 genomes of Cornales were performed in the mVISTA program [50], under LAGAN mode using the annotations of Cornus controversa as reference. Plastomes of Cornales were also aligned using MAFFT [51] and manually edited in Geneious v 8.1. To observe the plastid genome divergence and determine parsimony informative sites, sliding window analysis was conducted after alignment. In order to identify some mutational hotspots, the proportion of mutational events was calculated following a modified version of the formula used by Gielly and Taberlet [52]: the proportion of mutation events = [(NS + ID)/L] * 100%, where NS is the number of nucleotide substitutions, ID is the number of indels and L is the aligned sequence length of each region. Hotspots were here defined as those regions with a value >20%. The step size was set to 200 bp, with a 600 bp window length as described by Xu et al. [27].
To test whether the abnormal gene of rpl22 is disabled or not, the ratio of nonsynonymous and synonymous (ω, dN/dS) of rpl22 for different branches was calculated in PAML v4.7 [53] using the codeml module.
Alignment and subdivision of plastid genomes
The whole plastid genomes of the 15 individuals of Cornales and the four outgroup species were aligned using the program MAFFT v 7.22 with default settings. Three primary data sets were generated for phylogenetic inference. The first data set comprised coding regions, i.e. exons of protein-coding genes, tRNAs and rRNAs; the second comprised all noncoding regions, i.e. intergenic regions and introns; the third comprised the entire plastid genome. Each gene and intergenic or intron was realigned using MAFFT v 7.22 with G-INS-i algorithm plugin in Geneious v 8.1. One of the IR regions was removed for all data sets to reduce overrepresentation of duplicated sequences.
Some regions in the whole plastome data set are highly variable and poorly aligned. So, in order to assess the effect of alignment quality on phylogeny, we compared the results from three different analysis strategies. First, the unfiltered alignment included all sequence positions of the plastomes in the alignment. Second, the lightly filtered alignment was created using the program Gblocks [54] to remove those regions that were identified as highly variable or ambiguously aligned, using the program’s default parameters; only positions where 50% or more of the sequences had a gap were retained. Third, the strictly filtered alignment was generated using the same approach as the lightly filtered alignment, but excluding all those positions that had at least one gap.
Phylogenetic analyses
For the unfiltered, lightly filtered and strictly filtered alignments of coding, noncoding and complete plastome data sets, jModeltest v2.1.6 [55] was used, as implemented on the Cyberinfrastructure for Phylogenetic Research (CIPRES) cluster (http://www.phylo.org/), to estimate the optimal model of molecular evolution with the Akaike Information Criterion (AIC). Maximum likelihood (ML) analyses were conducted using RAxML v8.1.11 [56] as implemented on the CIPRES cluster. These RAxML searches relied on the general time reversible model of nucleotide substitution, with the gamma model of rate heterogeneity (GTR + G) as suggested (see RAxML manual). The ML trees were inferred using the rapid bootstrap with 1000 replicates, and the best-scoring ML tree was sought. Bayesian inference (BI) analyses were conducted with MrBayes v3.2.3 [57] as implemented on the CIPRES cluster with the models estimated for the different data sets (Additional file 1: Table S2). Two runs were conducted in parallel with four Markov chains (one cold and three heated), with each running for 2,000,000 generations from a random tree and sampled every 100 generations. The convergence was checked using the average standard deviation of split frequencies (ASDFs) (<0.01). The first 25% of the trees were discarded as burn-in, and the remaining trees were used to construct majority-rule consensus trees.
To investigate the issues of data partitioning for the plastid phylogenomic analysis, an algorithmic method for estimating an optimal partitioning scheme was conducted for the complete unfiltered data set. It was partitioned into the maximum possible number of data blocks based on genomic composition. We divided the whole plastid genome into 174 subsets: each gene, intergenic region or intron was regarded as a distinct subset, while subsets of less than 200 bp, or regions that only contained invariable nucleotide sites, were combined into large data subsets according to their function (see details in Additional file 1: Table S3). Subsequently, the program PartitionFinder v1.1.1 [58] was used to identify the best partitioning schemes of these 174 subsets according to the Bayesian information criterion (BIC) using a heuristic search (search = rcluster).
For partitioned ML phylogenetic analysis, a partitioned model was used to specify the regions of alignment, for which an individual model of nucleotide substitution was estimated. Individual per-partition branch lengths were estimated using RAxML v8.1.11 software. For partitioned BI phylogeny estimation, each partition was given its own optimal model (GTR + G or GTR + G + I) (Additional file 1: Table S3). All parameters were set to be unlinked across partitions except those for branch lengths and topology; branch length rate multipliers were unlinked in MrBayes v3.2.3.
Results
Characteristics of the plastid genomes
Fifteen complete plastid genomes of Cornales, plus Fouquieria diguetii of Ericales were newly generated in this study; these genome sequences have been submitted to GenBank (Table 1). The mean coverage depth of these plastomes ranged from 383× (Alangium chinense) to 2757× (Camptotheca acuminata). Henceforth, all text describing plastid genomes refers only to Cornales unless stated otherwise. The size of the 15 Cornales plastid genomes ranged from 156,567 bp in Nyssa sinensis to 158,715 bp in Diplopanax stachyanthus, and both individuals of Cornus capitata examined had the same plastid genome size (157,200 bp) (Table 2). All of the 16 sequenced plastid genomes displayed a typical quadripartite structure (Fig. 1), comprising a pair of IRs (25,859–26,451 bp) separated by the LSC (86,089–87,835 bp) and the SSC (18,250–18,856 bp) regions (Table 2). The LSC regions exhibited the greatest standard deviation in sequence length (s.d. = 586 bp), followed by SSC regions (s.d. = 188 bp) and the IR regions (s.d. = 147 bp). The full genomes encoded 114 unique genes, which included 31 tRNA genes, four rRNA genes and 79 protein-coding genes with the same gene order. There were 16 genes duplicated in the IR regions, resulting in a total of 130 genes (Additional file 1: Table S4). Seventeen of those genes contained one intron, and two genes (ycf3 and clpP) contained two introns. The length and GC content of coding, noncoding and complete plastid genome data sets are shown in Table 2. Noncoding regions (s.d. = 789 bp) showed more variation in sequence length than coding regions (s.d. = 115 bp). Among Cornales species, the percentage of the coding regions varied from 57.2% to 58.2%. The overall GC content is similar across individuals in coding and noncoding regions of Cornales, but a little higher than that of Fouquieria diguetii of Ericales (Table 2).
Table 2.
The plastid genome features of the sequenced species
| Taxon | Full | LSC length (bp) | SSC length (bp) | IR length (bp) | Gene Number | Protein-coding | RNAs | Coding region | Noncoding region | Mean Coverage | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | GC (%) | Length (bp) | GC (%) | Length (bp) | GC | ||||||||
| Nyssa wenshanensis | 156,598 | 37.9 | 86,109 | 18,261 | 26,114 | 114 | 79 | 35 | 91,073 | 40.3 | 65,525 | 34.6 | 974 |
| Nyssa sinensis | 156,567 | 37.9 | 86,089 | 18,250 | 26,114 | 114 | 79 | 35 | 91,073 | 40.3 | 65,494 | 34.6 | 802 |
| Camptotheca acuminata | 157,811 | 37.8 | 87,333 | 18,760 | 25,859 | 114 | 79 | 35 | 91,078 | 40.3 | 66,772 | 34.4 | 2757 |
| Davidia involucrata | 158,131 | 37.8 | 87,335 | 18,856 | 25,970 | 114 | 79 | 35 | 91,037 | 40.3 | 67,094 | 34.4 | 1026 |
| Mastixia caudatilimba | 158,221 | 37.8 | 87,418 | 18,797 | 26,003 | 114 | 79 | 35 | 90,962 | 40.3 | 67,259 | 34.4 | 1889 |
| Diplopanax stachyanthus | 158,715 | 37.8 | 87,679 | 18,632 | 26,202 | 114 | 79 | 35 | 90,944 | 40.2 | 67,771 | 34.6 | 1758 |
| Hydrangea heteromalla | 157,889 | 37.8 | 86,907 | 18,738 | 26,122 | 114 | 79 | 35 | 91,138 | 40.1 | 66,751 | 34.7 | 937 |
| Hydrangea aspera | 157,637 | 37.8 | 86,815 | 18,646 | 26,088 | 114 | 79 | 35 | 91,189 | 40.2 | 66,448 | 34.5 | 766 |
| Deutzia crassifolia | 157,035 | 37.6 | 86,583 | 18,714 | 25,869 | 114 | 79 | 35 | 91,099 | 40.1 | 65,936 | 34.1 | 549 |
| Alangium alpinum | 156,673 | 37.7 | 86,181 | 18,592 | 25,950 | 114 | 79 | 35 | 90,842 | 40.2 | 65,831 | 34.2 | 2121 |
| Alangium chinense | 156,684 | 37.7 | 86,185 | 18,603 | 25,948 | 114 | 79 | 35 | 90,824 | 40.2 | 65,860 | 34.2 | 383 |
| Cornus capitate #1 | 157,200 | 38.2 | 86,564 | 18,412 | 26,112 | 114 | 79 | 35 | 90,928 | 40.5 | 66,272 | 35.0 | 1068 |
| Cornus capitate #2 | 157,200 | 38.2 | 86,564 | 18,412 | 26,112 | 114 | 79 | 35 | 90,928 | 40.5 | 66,272 | 35.0 | 2523 |
| Cornus controversa | 158,668 | 37.8 | 87,835 | 18,705 | 26,064 | 114 | 79 | 35 | 90,823 | 40.4 | 67,845 | 34.3 | 573 |
| Curtisia dentata | 158,548 | 37.7 | 87,158 | 18,490 | 26,450 | 114 | 79 | 35 | 91,018 | 40.2 | 67,530 | 34.3 | 538 |
| Fouquieria diguetii | 157,895 | 37.3 | 87,321 | 18,482 | 26,046 | 114 | 79 | 35 | 91,244 | 39.9 | 66,651 | 33.7 | 1195 |
Fig. 1.
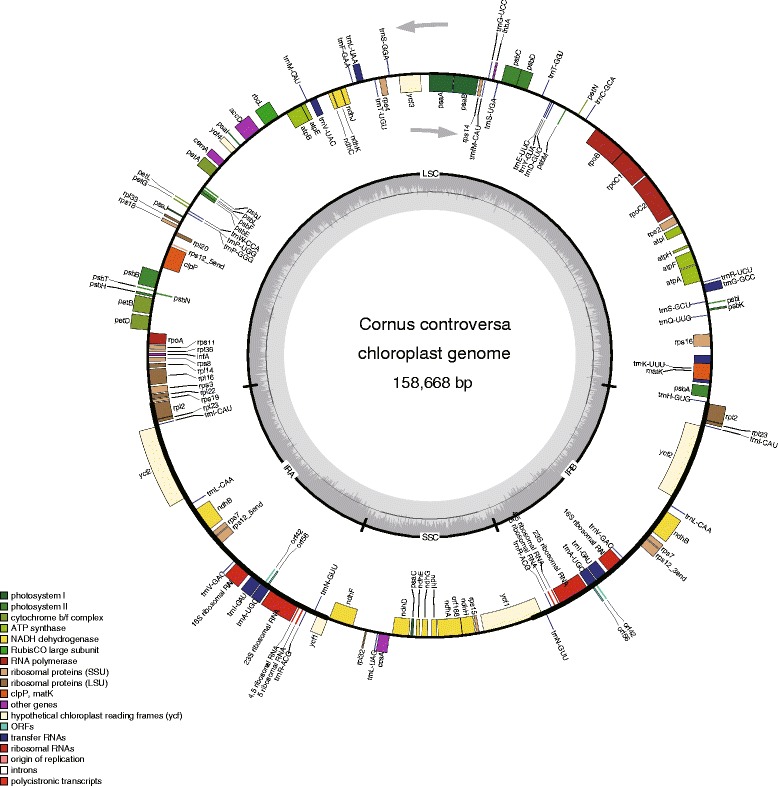
Gene map of Cornus controversa as a representative of Cornales. Genes on the inside of the outer circle are transcribed clockwise and those outsides are transcribed counterclockwise. Functional categories of genes are color-coded. The dashed area in the inner circle indicates the GC content of the plastid genome
Boundaries between the IR and SSC/LSC regions were verified by Sanger sequencing; the results were identical with the NGS sequencing. Variation in the positions of the boundaries between IR and SSC/LSC are usually considered to be the primary mechanism causing length variation among the plastid genomes of higher plants (Kim and Kim, [59]), but only slight variation was detected within Cornales (Fig. 2). The IRa/LSC junction was located within the rps19 gene in all but two species (Hydrangea davidii and Deutzia crassifolia), resulting in the presence of a part of the rps19 gene in the IRb. In Hydrangea davidii and Deutzia crassifolia, the junction was located in the rps19-rpl2 spacer. The IRb/SSC boundary positions in all species were located in the ycf1 gene, with part of this gene duplicated from 972 to 1246 bp. The ndhF gene in seven species was completely located in the SSC region, whereas in the others it extended fractionally into the IRa region (Fig. 2).
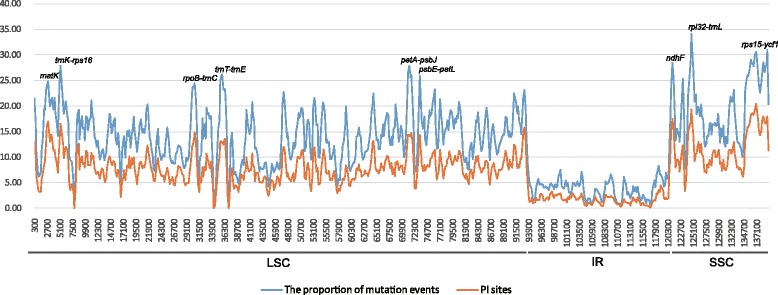
Fig. 2.

Sliding window analysis of the whole chloroplast genomes of Cornales taxa
Genome sequence divergence among Cornales
The plastid genomes within Cornales showed high sequence similarities with identities of only a few regions below 90% (Additional file 2: Fig. S1), suggesting a high conservatism of plastid genomes within Cornales. The IR regions and coding regions were more conserved than the single-copy regions and noncoding regions (Additional file 2: Fig. S1).
Slide window analysis also showed much higher proportions of both mutation events and parsimony-informative sites in single-copy regions than in the IR region. From this, nine relatively highly variable regions (mutational hotspots) were identified from the plastid genomes, which might be undergoing more rapid nucleotide substitution. These comprised 2 gene regions and 7 intergenic regions: matK, ndhF, trnK-rps16, rpoB-trnC, trnT-trnE, petA-psbJ, psbE-petL, rpl32-trnL, and rps15-ycf1 regions (Fig. 3). These regions are potential molecular markers for application in phylogeny and phytogeography investigations.
Fig. 3.
Comparison of the LSC, IR and SSC borders of 13 species of Cornales plastid genomes
Internal stop codons and putative loss of gene function
The genes rpl22 and ycf15 were interrupted by internal stop codons in seven and five Cornales species respectively. Both of them were further verified by Sanger sequencing using newly designed primers (Additional file 1: Table S1); the results were identical to the NGS-based plastid genome sequences.
For all species from Cornaceae, Alangiaceae and Curtisiaceae, a frameshift mutation generated premature termination codons within rpl22. Furthermore, Mastixia caudatilimba had one base change from G to A within rpl22, resulting in an internal termination codon (TGG to TGA) (Fig. 4a). Furthermore, this gene had a 19-bp and 5-bp insertion in Cornus capitata and C. controversa respectively, plus a 1 bp deletion in both Alangium species, and a 1 bp insertion in Curtisa dentata, all occurring upstream of the internal stop codon (Fig. 4a). rpl22 was found to be truncated in some species, with considerable length variation (384 bp to 474 bp). Despite this, the gene still exhibited nearly 80% nucleotide identities between species, with no big difference between those species with and those without internal stop codons. Furthermore, the ratio of nonsynonymous and synonymous (ω, dN/dS) of rpl22 for different branches showed similar values in both the Cornaceae-Alangiaceae-Curtisiaceae clade (ω = 0.34569) and Mastixiaceae clade (ω = 0.35594), and no significant difference with background (ω = 0.36549, P > 0.33) was found. This indicated that those genes containing stop codons have not accumulated mutations at an increased rate, and hence may not have lost their functions.
Fig. 4.

Alignment of two abnormal genes among Cornales and outgroups. a rpl22 gene; (b) ycf15 gene
The gene ycf15 varied from 102 bp to 249 bp among the 15 sequenced individuals of Cornales. For two species of Alangium, ycf15 contained a large deletion (84 bp) at the 5′ end and a 10-bp deletion near the 3′ end, potentially causing a loss of function. Additional 4-bp and 5-bp deletions within ycf15 led to internal stop codons in Davidia involucrata and Hydrangea aspera, respectively. Furthermore, in Deutzia crassifolia, a single substitution (G to A) within ycf15 likewise resulted in an internal stop codon (TAG). In the remaining nine Cornales species, ycf15 did not contain stop codons, and there was no evidence of loss of function (Fig. 4b). Because of these parallel function losses, ycf15 was not annotated in this study.
Phylogenetic analyses
The unfiltered whole plastid genome data set, with one copy of the IR region excluded, was 148,838 bp in length. Variable and parsimony informative sites of this data set were 25.5% and 15.5%, respectively. The noncoding regions were more variable than the coding regions (33.9% vs 17.5% variable sites and 20.3% vs 11.1% parsimony informative sites) (Table 3). Compared to the unfiltered alignment, a total of 22,247 sites (14.9%) in the lightly filtered data set and a total of 42,513 sites (28.6%) in the strictly filtered alignment were removed. The unfiltered and lightly filtered data sets showed similar percentages of variable and parsimony informative sites, irrespective of calculation for the different regions or the complete genome. However, the strictly filtered alignment exhibited a somewhat decreased percentage of variable and parsimony informative sites in the all data sets (Table 3).
Table 3.
Sequence alignment information and support values for key nodes under different alignment strategies
| Data set | Blocks | Number of sites | Variable sites | Parsimony informative sites | Support value (LB/PP) | |
|---|---|---|---|---|---|---|
| (Cornaceae-Alangiaceae)-Curtisiaceae | (Mastixiaceae-Davidiaceae-Nyssaceae) - Hydrangeaceae | |||||
| Coding | Unfiltered | 75,334 | 13,147(17.5%) | 8336(11.1%) | 81/1.0 | 99/1.0 |
| Light filtered | 74,352 | 13,040(17.5%) | 8305(11.2%) | 82/1.0 | 100/1.0 | |
| Strict filtered | 72,369 | 12,267(17.0%) | 7809(10.8%) | 78/1.0 | 100/1.0 | |
| Noncoding | Unfiltered | 72,056 | 24,406(33.9%) | 14,643(20.3%) | 99/1.0 | 100/1.0 |
| Light filtered | 51,343 | 18,974(37.0%) | 12,232(23.8%) | 99/1.0 | 100/1.0 | |
| Strict filtered | 32,852 | 10,167(30.9%) | 6545(19.9%) | 100/1.0 | 100/1.0 | |
| Complete | Unfiltered | 148,838 | 37,928(25.5%) | 23,136(15.5%) | 100/1.0 | 100/1.0 |
| Light filtered | 126,591 | 32,266(25.5%) | 20,678(16.3%) | 99/1.0 | 100/1.0 | |
| Strict filtered | 106,325 | 22,745(21.4%) | 14,551(13.7%) | 100/1.0 | 100/1.0 | |
Using both ML and BI methods without data partitioning, the phylogenetic inference of Cornales from the whole unfiltered data set provided complete resolution of relationships among all species sampled, with maximum support (100%/1.0) for all nodes (Fig. 5). Nyssaceae was monophyletic and sister in turn to Davidiaceae, then a monophyletic Mastixiaceae, then a monophyletic Hydrangeaceae. Cornales comprised this clade plus another, in which Cornaceae and Alangiaceae (both monophyletic) were together sister to Curtisiaceae (Fig. 5). The phylogenetic topology of Cornales based on unfiltered coding and noncoding regions were consistent with that from the complete plastome data set. Only the sister relationship of Curtisiaceae and Cornaceae-Alangiaceae received support values below 99% or 1.0 from unfiltered data sets, with 81%/1.0 support from coding regions (Table 3, Fig. 5, Additional file 2: Fig. S2).
Fig. 5.
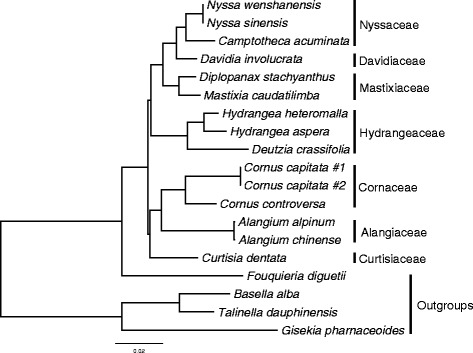
Phylogenetic relationships of Cornales based on unfiltered whole plastid genome alignment. Nodes without values represent maximal support in both ML and BI methods
Likewise, using lightly filtered and strictly filtered data sets, both the topology and support values were almost identical (Additional file 2: Fig. S3, S4). However, when only coding data sets are used, the bootstrap support value for the sister relationship of Curtisiaceae and Cornaceae-Alangiaceae drops to 78% with strictly filtered alignment (Additional file 2: Fig. S4A).
When partitioning was applied using the program PartitionFinder, the whole unfiltered data set was divided into 13 partitions (Table S3). Topology and support values obtained from this analysis were consistent with unpartitioned analysis, except for a decrease in BS support value from 100% to 95% for the sister relationship of Curtisiaceae to Cornaceae-Alangiaceae in ML analysis (Additional file 2: Fig. S2C).
Discussion
Structure of plastome and comparative analyses
In the present study, the complete plastid genomes of 14 species of Cornales were obtained for the first time. They showed the typical quadripartite structure of most angiosperms, including a pair of IR regions, separated by an LSC and an SSC region. The Cornales plastid genome was highly conserved in structure compared to most angiosperms [32], with all sampled species encoding the same set of 114 unique genes in same gene order (Table 2). The GC content was around the average for plant plastomes (GC = 37%) [60], but was slightly higher than that of the outgroup taxa. The length variation of the Cornales plastid genomes observed here was low (156–159 kpb), and differences were mainly due to variation in noncoding regions (65–68 kbp). Length variation of plastid genomes was previously shown to result from expansion and contraction of the inverted repeat regions [61]. Here, we also found that the IR/SSC boundary located differently among the 14 species, but the location of boundary and length of IR regions only showed moderate variation (Fig. 2). Furthermore, there was no obvious phylogenetic implication of extension/contraction of IRs among the Cornales plastomes.
Premature stop codons in two genes, but no apparent loss of function in rpl22
The gene content is highly conserved among plastid genomes of land plants, although gene loss has been reported in several angiosperm lineages [17]. Two genes, rpl22 and ycf15, contained premature termination codons in several species in the present study. rpl22 showed premature termination codons in Alangium, Cornus, Curtisia, and Mastixia, making it about 20% shorter in these species compared to others. This gene appears to be absent from plastids in some taxa such as legumes [62] and was reported to have been transferred to the nucleus in both Pisum and Fagaceae [63, 64]. In Cornales, rpl22 in those species with internal stop codons have not undergone a detectable increase in mutation rate compared to those without them, whereas such an increase would be expected if the gene was disabled in the former but functional in the latter. Moreover, the former group still contain nearly 80% of the normal gene sequence. This implies that rpl22 in the plastid either functions as a gene in all examined species, or in none of them. If the former, truncation does not remove its function. If the latter, then it might be a pseudogene in all Cornales, as it is in Citrus sinensis [65]; if so it might have a non-coding function in the plastid. Possibly, a functional copy of rpl22 might exist in the nucleus, as in Pisum and Fagaceae [63, 64], removing any selective disadvantage to loss of function in the chloroplast. Therefore, Cornales might be in the early stages of a process of losing rpl22 from the plastid. Hence more data is needed, regarding function of rpl22 in the chloroplast and whether a full copy exists in the nucleus.
The nucleotide sequence of ycf15 has been shown to vary among angiosperm plastid genomes, with conserved motifs at 5′ to 3′ ends in some taxa (like tobacco) and an intervening region of about 250 bp in some other taxa (like Eucalyptus globulus) that renders it as a pseudogene [66]. A comparative study of ycf15 transcripts in taxa with or without the insertion suggested that this gene may not be a protein-coding gene even when it is intact [67]. Although transcripts of ycf15 were detected in some taxa like Camellia, it may have been removed from the pre-mRNA after transcription in order to activate the function of other genes, thus ycf15 is possibly an intergenic sequence without function [68]. The non-coding ycf15 hypothesis to some extent is supported by data from Cornales, within which four independent mutation events within ycf15 either inserted stop codons (Davidia, Hydrangea and Deutzia) or deleted parts of the gene (Alangium). The evolutionary patterns of ycf15 showed that they evolved in a discontinuous fashion across angiosperms [68] [69]. It shows an intact and conserved structure in nine Cornales species, but cannot be translated normally in species of Alangium, Davidia and Deutzia. These three genera belong to distinct clades, implying separate and independent alterations in each case; hence ycf15 might not provide phylogenetic implication (Fig. 5).
Influence of data set subdivision, alignment filtering, and data partitioning on phylogeny
In addition to the complete plastid genome, two data subsets were generated, one comprising all coding genes, and the other only noncoding regions. We conducted three filtering strategies (none, light and strict) on each of these three data sets. Phylogenetic inference from BI and ML analyses based on all data sets provided the same topology. All data sets supported the sister relationship between Curtisiaceae and Cornaceae-Alangiaceae, support for this clade from the coding region and its filtered data subsets was relatively low, e.g. 81%/1.0 for unfiltered (Additional file 2: Fig. S2A). Although the noncoding regions are usually excluded for phylogenomic analyses at high taxonomic levels [14, 24], the phylogenetic resolution within Cornales obtained from noncoding regions in all three strategies was high (Additional file 2: Fig. S2, S3, S4; Table 3). This might because plastid genomes within the order have a conserved collinear structure, and the noncoding regions can provide more phylogenetic signals. The treatment of problematic or ambiguous regions in alignments can affect the final phylogenetic relationships, and for alignments that are long enough, removal of problematic regions leads to better phylogenetic resolution [69, 70]. Conversely, in this study, alignment filtering has no influence in any of the coding, noncoding and complete alignment data sets (Table 3), which may also be due to the conservation of plastid genomes within Cornales.
When a genome-scale approach is adopted in phylogenetic analyses, partitioning is one of the most popular methods used to model the heterogeneity of molecular evolution among regions in an alignment for phylogenetic inference [71]. In the present study, however, data partitioning by PartitionFinder had no effect on the topology of the resulting phylogenetic trees compared to unpartitioned plastid genome data set. The phylogenetic relationships of Cornales were robustly resolved based on both partitioned and unpartitioned datasets. It was indicated that the longer the data set was, the less likely that the results will be affected by partitioning scheme [71]. This is perhaps because the whole plastid genome contains sufficient amount of phylogenetic signals (while noise is randomly dispersed) and may converge on the correct phylogenetic tree, irrespective of partitioning. It was also observed, in a previous phylogenetic study of Cornales with six plastid fragments, that the partitioned data sets presented the same topology as the unpartitioned ones with only some differences among the branch support values [3]. Irrespective of different regions or data subsets, partitioned or unpartitioned data sets used, our results suggested that the plastid genome as a whole contains sufficient phylogenetic signals in different regions within Cornales to fully resolve the phylogenetic relationships. The conservatism in genome structure and gene content along with abundance of phylogenetic signal of Cornales plastid genomes, both coding and noncoding regions, make it a valuable phylogenetic tool, at and below family level.
Phylogenetic implication among Cornales with plastid genome
Preliminary phylogenetic frameworks for Cornales have previously been provided based on a few molecular markers, but relationships among families of the order tended to be poorly resolved [5, 7, 72]. A later study [3], based on six plastid loci (rbcL, matK, ndhF, atpB, trnL-F and trnH-K) and a broader taxon sampling, recovered five major clades: Cornaceae-Alangiaceae, Curtisiaceae-Grubbiaceae, Mastixiaceae-Nyssaceae-Davidiaceae, Hydrostachyaceae, and Hydrangeaceae-Loasaceae; relationships between these clades were well supported. However, cpDNA-based relationships were contradicted by 26S rDNA data in the study, and relationships within some families (e.g. Hydrangeaceae, Cornaceae) were not fully recovered [3]. Therefore, further work is needed on relationships within these families. Our own work recovered identical relationships with even higher support, demonstrating that greater genome coverage can compensate for reduced taxon sampling, at least in some cases. The sequence and structure of the whole plastid genome has been recognized for its great potential to resolve relationships for phylogenetically recalcitrant plant groups [14, 21, 40, 73, 74]. Given that resources will seldom permit full genome sequencing across large numbers of taxa, the best strategy for wide taxonomic sampling is to identify marker regions that contain a high proportion of phylogenetically useful information. To this end, our study identified two genes (matK, ndhF) and seven regions trnK-rps16, rpoB-trnC, trnT-trnE, petA-psbJ, psbE-petL, rpl32-trnL, and rps15-ycf1 that are mutational hotspots, and are hence recommended as phylogenetic markers within Cornales, and perhaps beyond it.
Conclusions
Phylogenomic data have rapidly accumulated and been broadly used for resolving phylogenetic relationships in the last few years. In the present study, fifteen full plastid genomes of 14 Cornales species were sequenced to investigate the phylogenetic relationships and plastome evolution of Cornales. Comparative analysis of the plastid genomes revealed that plastomes of the order have a conserved collinear structure with identical gene content and order. Two genes (rpl22 and ycf15) contained premature stop codons in seven and five species respectively. Plastid genomes showed strong potential for resolving phylogenetic relationships within Cornales, both for the interfamily and intrafamily relationships, with very strong support. Different partitioning schemes and filtering strategies (none, light and strict) of sequence data sets have no effect on phylogenetic relationships. The topology recovered from coding and noncoding data sets was likewise identical to that for the whole plastome. However, the coding data set provided lower support values than the latter two data sets. Mutational hotspots and highly informative regions of Cornales were identified. All data presented here are fundamental to phylogenomic analyses of Cornales, and will be a useful genomic resource for future studies of evolutionary biology.
Additional files
The primers newly designed in this study for four junctions and two genes (rpl22, ycf15). Table S2. Molecular models selected for all the data sets of the three alignment strategies. Table S3. Model selected for each data partition identified by software PartitionFinder for unfiltered complete plastid genomes. Table S4. Gene category and gene contained in plastid genomes of Cornales. (DOC 147 kb)
A percent identity plot showing the overall sequence similarity of the fourteen Cornales plastid genomes. Fig. S2. Phylogenetic relationships of Cornales based on three different data sets with light filtered alignment. Fig. S3. Phylogenetic relationships of Cornales based on three different data sets with strict filtered alignment. (PDF 1121 kb)
Acknowledgements
We are grateful to Jun-Bo Yang, Xiang-Qin Yu, Jian-Jun Jin, Gang Yao and Zhi-Rong Zhang for their help with data analysis and laboratory work. We also thank Dr. Michael Möller from Royal Botanic Garden Edinburgh for the constructive comments and English editing for the early version of the MS. We thank Jie Cai, Ting-Shuang Yi, Cheng Liu, Yong-Jie Guo and Ji-Dong Ya from the CAS Kunming Institute of Botany, and the Royal Botanic Garden Edinburgh and Royal Botanic Gardens, Kew for providing samples.
Funding
This study was supported by the National Key Basic Research Program of China (2014CB954100), the Large-scale Scientific Facilities of the Chinese Academy of Sciences (Grant No: 2017-LSFGBOWS-01), Major program of CAS Kunming Institute of Botany (2014KIB02), and the Applied Fundamental Research Foundation of Yunnan Province (2014GA003).
Availability of data and materials
Sequence data that support the findings of this study have been deposited in GenBank (accession numbers were provided in Table 1). The phylogenetic matrix and trees are available in the TreeBASE repository (http://purl.org/phylo/treebase/phylows/study/TB2:S21874).
Abbreviations
- AIC
Akaike information criterion
- BI
Bayesian inference
- BIC
Bayesian information criterion
- GC
Guaninecytosine
- IR
Inverted repeat region
- LB
Likelihood bootstrap
- LSC
Large single copy region
- ML
Maximum likelihood
- PP
Posterior probability
- SSC
Small single region
Authors’ contributions
LG and DL designed the research, TZ, CF, LG and DL collected samples, CF, JY and HL collected the data, CF and PM analyzed the data, CF, LG, PM, RM and DL wrote and revised the manuscript. CF performed the study, participated in the data analysis and wrote the manuscript. HL participated in the data analysis. RM revised the manuscript. TZ participated in samples collection. PM participated in the data analysis and manuscript revising. JY participated in the DNA sequencing. DL designed the research and revised the manuscript. LG designed the research, collected study materials and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All the samples were collected with the permission of the land owners or the botanic gardens.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4319-9) contains supplementary material, which is available to authorized users.
Contributor Information
De-Zhu Li, Phone: +86-0871-65223503, Email: dzl@mail.kib.ac.cn.
Lian-Ming Gao, Phone: +86-0871-65225286, Email: gaolm@mail.kib.ac.cn.
References
- 1.Soltis DE, Soltis PS, Chase MW, Mort ME, Albach DC, Zanis M, et al. Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot J Linn Soc. 2000;133:381–461. doi: 10.1111/j.1095-8339.2000.tb01588.x. [DOI] [Google Scholar]
- 2.The Angiosperm Phylogeny Group An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc. 2016;181(1):1–20. doi: 10.1111/boj.12385. [DOI] [Google Scholar]
- 3.Xiang QY, Thomas DT, Xiang QP. Resolving and dating the phylogeny of Cornales--effects of taxon sampling, data partitions, and fossil calibrations. Mol Phylogenet Evol. 2011;59(1):123–138. doi: 10.1016/j.ympev.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 4.Zeng L, Zhang N, Zhang Q, Endress PK, Huang J, Ma H. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 2017;214(3):1338–1354. doi: 10.1111/nph.14503. [DOI] [PubMed] [Google Scholar]
- 5.Fan CZ, Xiang QY. Phylogenetic analyses of Cornales based on 26S rRNA and combined 26S rDNA-matK-rbcL sequence data. Am J Bot. 2003;90(9):1357–1372. doi: 10.3732/ajb.90.9.1357. [DOI] [PubMed] [Google Scholar]
- 6.Xiang QY. Cornales (dogwood). In: eLS. Chichester: John Wiley & Sons, Ltd; 2005. http://www.els.net
- 7.Xiang QY, Moody ML, Soltis DE, Fan CZ, Soltis PS. Relationships within Cornales and circumscription of Cornaceae-matK and rbcL sequence data and effects of outgroups and long branches. Mol Phylogen Evol. 2002;24(1):35–57. doi: 10.1016/S1055-7903(02)00267-1. [DOI] [PubMed] [Google Scholar]
- 8.Xiang QY, Manchester SR, Thomas DT, Zhang W, Fan CZ. Phylogeny, biogeography, and molecular dating of cornelian cherries (Cornus, Cornaceae): tracking tertiary plant migration. Evolution. 2005;59(8):1685–1700. doi: 10.1111/j.0014-3820.2005.tb01818.x. [DOI] [PubMed] [Google Scholar]
- 9.Olmstead RG, Kim KJ, Jansen RK, Wagstaff SJ. The phylogeny of the Asteridae Sensu late based on chloroplast ndhF gene sequences. Mol Phylogen Evol. 2000;16(1):96–112. doi: 10.1006/mpev.1999.0769. [DOI] [PubMed] [Google Scholar]
- 10.Chase MW, Soltis DE, Olmstead RG, Morgan D, Les DH, Mishler BD, et al. Phylogenetics of seed plants - an analysis of nucleotide-sequences from the plastid gene rbcL. Ann Mo Bot Gard. 1993;80(3):528–580. doi: 10.2307/2399846. [DOI] [Google Scholar]
- 11.Straub SCK, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am J Bot. 2012;99(2):349–364. doi: 10.3732/ajb.1100335. [DOI] [PubMed] [Google Scholar]
- 12.Zeng LP, Zhang Q, Sun RR, Kong HZ, Zhang N, Ma H. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat Commun. 2014;5:12. doi: 10.1038/ncomms5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Twyford AD, Ness RW. Strategies for complete plastid genome sequencing. Mol Ecol Resour. 2017;17(5):858–68. [DOI] [PMC free article] [PubMed]
- 14.Barrett CF, Baker WJ, Comer JR, Conran JG, Lahmeyer SC, Leebens-Mack JH, et al. Plastid genomes reveal support for deep phylogenetic relationships and extensive rate variation among palms and other commelinid monocots. New Phytol. 2016;209(2):855–870. doi: 10.1111/nph.13617. [DOI] [PubMed] [Google Scholar]
- 15.Birky CW. Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc Natl Acad Sci U S A. 1995;92(25):11331–11338. doi: 10.1073/pnas.92.25.11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tonti-Filippini J, Nevill PG, Dixon K, Small I. What can we do with 1000 plastid genomes? Plant J. 2017;90(4):808–818. doi: 10.1111/tpj.13491. [DOI] [PubMed] [Google Scholar]
- 17.Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J, et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci U S A. 2007;104(49):19369–19374. doi: 10.1073/pnas.0709121104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore MJ, Bell CD, Soltis PS, Soltis DE. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc Natl Acad Sci U S A. 2007;104(49):19363–19368. doi: 10.1073/pnas.0708072104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruhfel BR, Gitzendanner MA, Soltis PS, Soltis DE, Burleigh JG. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol Biol. 2014;14:26. doi: 10.1186/1471-2148-14-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xi ZX, Ruhfel BR, Schaefer H, Amorim AM, Sugumaran M, Wurdack KJ, et al. Phylogenomics and a posteriori data partitioning resolve the cretaceous angiosperm radiation Malpighiales. Proc Natl Acad Sci U S A. 2012;109(43):17519–17524. doi: 10.1073/pnas.1205818109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma PF, Zhang YX, Zeng CX, Guo ZH, Li DZ. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (poaceae) Syst Biol. 2014;63(6):933–950. doi: 10.1093/sysbio/syu054. [DOI] [PubMed] [Google Scholar]
- 22.Zhang SD, Jin JJ, Chen SY, Chase MW, Soltis DE, Li HT, et al. Diversification of Rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 2017;214(3):1355–1367. doi: 10.1111/nph.14461. [DOI] [PubMed] [Google Scholar]
- 23.Stull GW. Duno de Stefano R, Soltis DE, Soltis PS. Resolving basal lamiid phylogeny and the circumscription of Icacinaceae with a plastome-scale data set. Am J Bot. 2015;102(11):1794–1813. doi: 10.3732/ajb.1500298. [DOI] [PubMed] [Google Scholar]
- 24.Zhang N, Wen J, Zimmer EA. Another look at the phylogenetic position of the grape order Vitales: chloroplast phylogenomics with an expanded sampling of key lineages. Mol Phylogen Evol. 2016;101:216–223. doi: 10.1016/j.ympev.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 25.Cai J, Ma PF, Li HT, Li DZ. Complete plastid genome sequencing of four Tilia species (malvaceae): a comparative analysis and phylogenetic implications. PLoS One. 2015;10(11):e0142705. doi: 10.1371/journal.pone.0142705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reginato M, Neubig KM, Majure LC, Michelangeli FA. The first complete plastid genomes of Melastomataceae are highly structurally conserved. PeerJ. 2016;4:16. doi: 10.7717/peerj.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu C, Dong W, Li W, Lu Y, Xie X, Jin X, et al. Comparative analysis of six lagerstroemia complete chloroplast genomes. Front Plant Sci. 2017;8:15 [DOI] [PMC free article] [PubMed]
- 28.Zhong BJ, Deusch O, Goremykin VV, Penny D, Biggs PJ, Atherton RA, et al. Systematic error in seed plant phylogenomics. Genome Biol Evol. 2011;3:1340–1348. doi: 10.1093/gbe/evr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goremykin VV, Nikiforova SV, Biggs PJ, Zhong BJ, Delange P, Martin W, et al. The evolutionary root of flowering plants. Syst Biol. 2013;62(1):50–61. doi: 10.1093/sysbio/sys070. [DOI] [PubMed] [Google Scholar]
- 30.Goremykin VV, Nikiforova SV, Cavalieri D, Pindo M, Lockhart P. The root of flowering plants and total evidence. Syst Biol. 2015;64(5):879–891. doi: 10.1093/sysbio/syv028. [DOI] [PubMed] [Google Scholar]
- 31.Sugiura M. The chloroplast genome. Plant Mol Biol. 1992;19:149–168. doi: 10.1007/BF00015612. [DOI] [PubMed] [Google Scholar]
- 32.Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3):273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu A, Guo W, Gupta S, Fan W, Mower JP. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 2015;209(4):1747–1756. doi: 10.1111/nph.13743. [DOI] [PubMed] [Google Scholar]
- 34.Schwarz EN, Ruhlman TA, Sabir JSM, Hajrah NH, Alharbi NS, Al-Malki AL, et al. Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J Syst Evol. 2015;53(5):458–468. doi: 10.1111/jse.12179. [DOI] [Google Scholar]
- 35.Hsu CY, Wu CS, Chaw SM. Birth of four chimeric plastid gene clusters in Japanese umbrella pine. Genome Biol Evol. 2016;8(6):1776–1784. doi: 10.1093/gbe/evw109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cusimano N, Wicke S. Massive intracellular gene transfer during plastid genome reduction in nongreen Orobanchaceae. New Phytol. 2016;210(2):680–693. doi: 10.1111/nph.13784. [DOI] [PubMed] [Google Scholar]
- 37.Graham SW, Lam VKY, Merckx VSFT. Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol. 2017;214(1):48–55. doi: 10.1111/nph.14398. [DOI] [PubMed] [Google Scholar]
- 38.Yi TS, Jin GH, Wen J. Chloroplast capture and intra- and inter-continental biogeographic diversification in the Asian – new world disjunct plant genus Osmorhiza (Apiaceae). Mol Phylogen Evol 2015; 85(0):10–21. [DOI] [PubMed]
- 39.Huang H, Shi C, Liu Y, Mao SY, Gao LZ. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 2014;14:151. doi: 10.1186/1471-2148-14-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knox EB. The dynamic history of plastid genomes in the Campanulaceae Sensu Lato is unique among angiosperms. Proc Natl Acad Sci U S A. 2014;111(30):11097–11102. doi: 10.1073/pnas.1403363111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Logacheva MD, Schelkunov MI, Nuraliev MS, Samigullin TH, Penin AA. The plastid genome of mycoheterotrophic monocot Petrosavia stellaris exhibits both gene losses and multiple rearrangements. Genome Biol Evol. 2014;6(1):238–246. doi: 10.1093/gbe/evu001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Z, Raven PH. Flora of China. Beijing: Science Press; Missouri Botanical Garden; 2005. [Google Scholar]
- 43.Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bul. 1987;19:11–15. [Google Scholar]
- 44.Yang JB, Li DZ, Li HT. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol Ecol Resour. 2014;14(5):1024–1031. doi: 10.1111/1755-0998.12251. [DOI] [PubMed] [Google Scholar]
- 45.Zhang T, Zeng CX, Yang JB, Li HT, Li DZ. Fifteen novel universal primer pairs for sequencing whole chloroplast genomes and a primer pair for nuclear ribosomal DNAs. J Syst Evol. 2016;54(3):219–227. doi: 10.1111/jse.12197. [DOI] [Google Scholar]
- 46.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- 48.Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52(5–6):267–274. doi: 10.1007/s00294-007-0161-y. [DOI] [PubMed] [Google Scholar]
- 49.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(Web Server issue):W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gielly L, Taberlet P. The use of chloroplast DNA to resolve plant phylogenies: noncoding versus rbcL sequences. Mol Biol Evol. 1994;11(5):769–777. doi: 10.1093/oxfordjournals.molbev.a040157. [DOI] [PubMed] [Google Scholar]
- 53.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 54.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 55.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 57.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 58.Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29(6):1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- 59.Kim JS, Kim JH. Comparative genome analysis and phylogenetic relationship of order Liliales insight from the complete plastid genome sequences of two Lilies (Lilium longiflorum and Alstroemeria aurea) PLoS One. 2013;8(6):11. doi: 10.1371/annotation/c5f432f6-1e92-42c0-8727-7ae972215d6f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ravi V, Khurana J, Tyagi A, Khurana P. An update on chloroplast genomes. Plant Syst Evol. 2008;271(1–2):101–122. doi: 10.1007/s00606-007-0608-0. [DOI] [Google Scholar]
- 61.Kim KJ, Lee HL. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004;11(4):247–261. doi: 10.1093/dnares/11.4.247. [DOI] [PubMed] [Google Scholar]
- 62.Doyle JJ, Doyle JL, Palmer JD. Multiple independent losses of two genes and one intron from legume chloroplast genomes. Syst Bot. 1995;20(3):272–294. doi: 10.2307/2419496. [DOI] [Google Scholar]
- 63.Bausher MG, Singh ND, Lee SB, Jansen RK, Daniell H. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 2006;6:11. doi: 10.1186/1471-2229-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gantt JS, Baldauf SL, Calie PJ, Weeden NF, Palmer JD. Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J. 1991;10(10):3073–3078. doi: 10.1002/j.1460-2075.1991.tb07859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jansen RK, Saski C, Lee SB, Hansen AK, Daniell H. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol Biol Evol. 2011;28(1):835–847. doi: 10.1093/molbev/msq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steane DA. Complete nucleotide sequence of the chloroplast genome from the Tasmanian blue gum, Eucalyptus globulus (Myrtaceae) DNA Res. 2005;12(3):215–220. doi: 10.1093/dnares/dsi006. [DOI] [PubMed] [Google Scholar]
- 67.Raubeson LA, Peery R, Chumley TW, Dziubek C, Fourcade HM, Boore JL, et al. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics. 2007;8 [DOI] [PMC free article] [PubMed]
- 68.Shi C, Liu Y, Huang H, Xia EH, Zhang HB, Gao LZ. Contradiction between plastid gene transcription and function due to complex posttranscriptional splicing: an exemplary study of ycf15 function and evolution in angiosperms. PLoS One. 2013;8(3):e59620. [DOI] [PMC free article] [PubMed]
- 69.Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 70.Som A. Causes, consequences and solutions of phylogenetic incongruence. Brief Bioinform. 2015;16(3):536–548. doi: 10.1093/bib/bbu015. [DOI] [PubMed] [Google Scholar]
- 71.Kainer D, Lanfear R. The effects of partitioning on phylogenetic inference. Mol Biol Evol. 2015;32(6):1611–1627. doi: 10.1093/molbev/msv026. [DOI] [PubMed] [Google Scholar]
- 72.Xiang QY, Soltis DE, Soltis PS. Phylogenetic relationships of cornaceae and close relatives inferred from matK and rbcL sequences. Am J Bot. 1998;85(2):285–297. doi: 10.2307/2446317. [DOI] [PubMed] [Google Scholar]
- 73.Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci U S A. 2010;107(10):4623–4628. doi: 10.1073/pnas.0907801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang DI, Hefer CA. Kolosova N, Douglas CJ, Cronk QC. Whole plastome sequencing reveals deep plastid divergence and cytonuclear discordance between closely related balsam poplars, Populus balsamifera and P. trichocarpa (Salicaceae) New Phytol. 2014;204(3):693–703. doi: 10.1111/nph.12956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The primers newly designed in this study for four junctions and two genes (rpl22, ycf15). Table S2. Molecular models selected for all the data sets of the three alignment strategies. Table S3. Model selected for each data partition identified by software PartitionFinder for unfiltered complete plastid genomes. Table S4. Gene category and gene contained in plastid genomes of Cornales. (DOC 147 kb)
A percent identity plot showing the overall sequence similarity of the fourteen Cornales plastid genomes. Fig. S2. Phylogenetic relationships of Cornales based on three different data sets with light filtered alignment. Fig. S3. Phylogenetic relationships of Cornales based on three different data sets with strict filtered alignment. (PDF 1121 kb)
Data Availability Statement
Sequence data that support the findings of this study have been deposited in GenBank (accession numbers were provided in Table 1). The phylogenetic matrix and trees are available in the TreeBASE repository (http://purl.org/phylo/treebase/phylows/study/TB2:S21874).