

Abstract
While preclinical models such as orthotopic tumors generated in mice from patient-derived specimens are widely used to predict sensitivity or therapeutic interventions for cancer, such xenografts can be slow, require extensive infrastructure, and can make in situ assessment difficult. Such concerns are heightened in highly aggressive cancers, such as glioblastoma (GBM), that display genetic diversity and short mean survival. Biomimetic biomaterial technologies offer an approach to create ex vivo models that reflect biophysical features of the tumor microenvironment (TME). We describe a microfluidic templating approach to generate spatially graded hydrogels containing patient-derived GBM cells to explore drug efficacy and resistance mechanisms.
Introduction
Glioblastoma (GBM) is the most common and lethal form of brain cancer. Despite surgical resection and radiotherapy in combination with the alkylating agent temozolomide, which has led to increases in the median survival time (~15 months),[1] its overall low survival rate (<5% after 3 years) has remained largely unchanged for a century.[2–7] This poor prognosis is driven largely by its rapid, diffuse infiltration from the tumor margins into the surrounding brain parenchyma.[8] Molecularly targeted therapies show promise for the care of GBM patients, particularly due to the genetic diversity of the disease, yet their successful application remains a challenge.[9] The effectiveness of targeted agents such as tyrosine kinase inhibitors (TKIs) may be determined not only by the presence of the key mutant kinases, but also by other critical changes, such as loss of key tumor suppressor proteins, the selection for kinase-resistant mutants, and the deregulation of feedback loops.[10] Here, the extracellular matrix (ECM) may play an important role in tumor progression worthy of continued exploration. For instance, high levels of hyaluronic acid (HA) in tumors are a prognostic factor in several malignancies, and manipulations of HA production or interaction with cell surface receptors strongly influence tumor growth and metastasis.[11] Mechanistic studies of GBM invasion and therapeutic resistance are largely intractable in vivo due to the complex tumor microenvironment. Understanding the role cell–micro-environment interactions may play in developing combinatorial therapies to suppress resistance for GBM patients represents an exciting opportunity for continued development.
Several small-molecule inhibitors have been developed to target key molecules in GBM cell signaling, aiming at growth factors and their corresponding receptors and downstream signal transduction intermediates. The ability of engineered preclinical platforms[12] to explore the effect of these inhibitors, as well as using genetic approaches to classify signaling in these tumors, hold promise in developing new strategies for treatment. Biomimetic ex vivo tumor models can employ advanced techniques to explore the origin of failure associated with some TKIs. Moreover, it may be possible to resolve why some mono-therapies have not led to significantly improved outcomes and begin to explore the effectiveness of combinatorial approaches that address clinical resistance. For example, the high percentage of GBM tumors presenting abnormalities of the epidermal growth factor receptor (EGFR) has suggested that targeting EGFR may be a promising therapeutic strategy for decades.[13] However, their administration to GBM patients is characterized by lack of clinical benefit, and considerable efforts have been invested in underlying mechanisms that confer this resistance in glioma cells.[14,15] Here, the presence of redundant and alternative compensatory pathways, including those closely related to signals from the tumor extracellular microenvironment, may represent an important class of escape mechanism.[16]
Given the spatial, molecular, and temporal complexity of the native GBM tumor, tissue engineering approaches may provide a critical platform to systematically dissect, understand, and control features of the ECM associated with GBM expansion and therapeutic efficacy. The foundation of our effort is our recent development of a suite of methacrylamide-functionalized gelatin [gelatin methacrylamide (GelMA)] hydrogels. We have recently developed a three-dimensional hydrogel-based culture platform that is able to recapitulate key extracellular features of GBM microenvironment, notably those associated with transitions from the tumor core through the margins [Fig. 1(a)].[17] We use microfluidic forming tools and orthogonal hydrogel chemistries to generate libraries of miniaturized, optically translucent biomaterials containing overlapping patterns of cell, biophysical, and biomolecular cues inspired by the GBM margins. Using this model, we can: (1) explore the role played by transitions in the tumor microenvironment on the metabolic activity and growth of GBM tumors; (2) rapidly test the effect of therapeutic molecules; and (3) explore potential mechanisms of resistance as a means to identify combinatorial treatments. Embracing the complexity and heterogeneity of patient-derived specimens,[18–23] here we describe culture of a series of patient-derived xenografts (PDXs) that maintain patient morphologic and molecular characteristics[24–26] within GelMA hydrogels containing a linear gradient of matrix-bound HA content. This approach facilitates culture of a panel of tumor-laden hydrogels that reflect the variability of actual human brain tumors. Here we evaluate the response to TKI treatment (erlotinib) of two PDX variants that vary in the overexpression of EGFR (GBM10: EGFRwt/PTEN−, poorly invasive; GBM12: EGFR+/PTEN+, mid invasive),[24] and demonstrate the differential influence of extracellular HA in both intrinsic and acquired resistance.
Figure 1.
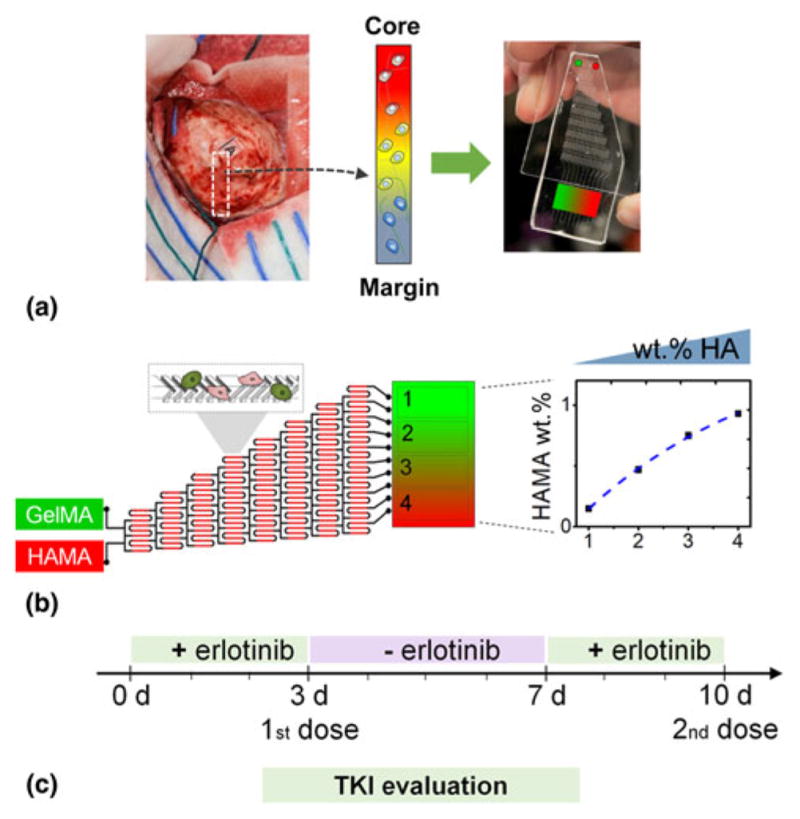
Biomaterial platforms allow us to recreate tumor tissues into hydrogel-based biochips to examine patient-specific responses to a range of therapeutic strategies. (a) Glioblastoma microenvironment transitions in hyaluronic acid content can be recreated into a (b) gelatin-HA gradient hydrogel by using a microfluidic device. (c) Response to erlotinib tyrosine kinase inhibitor is analyzed in the gradient platforms.
Experimental
Hydrogel fabrication and cell encapsulation
Gelatin (type A, 300 bloom from porcine skin, Sigma Aldrich, St. Louis, Missouri) and HA (60 kDa, Lifecore Biomedical, Chaska, Minnesota) were functionalized with methacrylate groups as previously described[27] to obtain GelMA and HA methacrylate (HAMA). The degree of functionalization was quantified by using proton nuclear magnetic resonance (1H NMR) and established at 60%, defining the ratio between the methacrylate peaks area at 5.5 ppm and the area of aromatic peaks at 7.3 ppm. 1H NMR analysis was also performed to determine the degree of methacrylation of HA, calculated from the relative integrations of the methacrylate to carbohydrate protons. We determined a modification of about 30 methacrylate groups per hundred disaccharides.
We produced GelMA scaffolds (7 wt.%) that contained gradients from 0 or 1 wt.% HAMA in phosphate-buffered saline (PBS; Invitrogen) by UV photopolymerization in the presence of 0.1 wt.% Irg2959 as photoinitiator. GBM PDX cells (provided by Mayo Clinic, Rochester, Minnesota) were encapsulated within these hydrogels at an initial concentration of 4 million cells/mL.
Fabrication of the microfluidic device
We employed a microfluidic approach to generate GelMA hydrogels containing linear gradient of matrix-immobilized HAMA. This mixer was based on a device containing micro-fluidic channels (100 μm deep × 200 μm wide) with eight sequential layers of mixing containing herringbone structures to aid mixing via chaotic advection.[28] We employed a silicon (Si) master for the microfluidic device mixer was based on this previously described design.[29] Microfluidic devices were created using the Sylgard 184 Silicone elastomer kit (Dow Corning, Midland, Michigan, USA) from the Si master.[28] Briefly, 10 parts polydimethylsiloxane (PDMS) were mixed with one part curing agent, mixed vigorously and degassed to remove all bubbles. The mixture was poured over the Si master and allowed to cure at 65 °C for at least 1 h. The PDMS piece was gently peeled off the Si wafer and access ports were punched into the device inlets using a 20-gauge blunt syringe needle. PDMS was poured on to a blank Si wafer, cured, and a rectangular blocked was carved out, functioning as a cell culture media reservoir for the final device. The PDMS pieces were cleaned with tape (Scotch™ 3 M), treated with oxygen plasma (Harrick Plasma, Ithaca, New York), and immersed in a 1% solution of (3-aminopropyl)-trimethoxysilane (Sigma-Aldrich, St. Loius, Missouri). A transparent polycarbonate membrane (5 μm pores; EMD Millipore, Billerica, Massachusetts) was treated with oxygen plasma and immersed in a 1% solution of (3-glycidoxypropyl)-methyldiethoxysilane (Sigma-Aldrich, St. Loius, Missouri). All components were washed with deionized water and dried on a lint-free cloth. A three-layer sandwich was created from the PDMS mixer layer, the polycarbonate membrane, and the PDMS media well and placed in the 65 °C oven overnight before use.
Generation of the gradient hydrogel
GelMA hydrogels containing gradients of HA were created by mixing two distinct pre-polymer solutions through the micro-fluidic device using a computer-controlled syringe pump (Harvard Apparatus, Micro-Liter OEM syringe pump, Holliston, Massachusetts, USA). GelMA becomes more viscous at lower temperatures causing poor flow within the narrow microfluidic channels [Fig. 1(b)]. Therefore, all gradient experiments were performed within a temperature-controlled environment maintained at 37 °C. Two 1 ml syringes were loaded on the pumps; the pre-polymer solutions (containing PDX cells) were withdrawn into the syringes (100 μL/min), and later infused into the microfluidic devices at 30 μL/min. Upon filling the hydrogel chamber with the polymer/PDX cell solution, the syringe pumps were stopped, the device disconnected from the syringes, and placed under UV light at 10 mW/cm2 (25 cm between the light source and the sample) for 10 min (Mercury lamp 365 nm, UVP Blak-Ray). Upon polymerization, media was added to the reservoir chamber and the devices were placed in the incubator (37 °C and 5% CO2) for cell culture.
Cell culture
PDX GBM specimens (GBM10: EGFRwt/PTEN−, poorly invasive; GBM12: EGFR+/PTEN+, mid invasive)[24] were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/mL and 100 μg/mL) at 37 °C in a 5% CO2 environment and used immediately upon receipt from Mayo Clinic. GBM-seeded hydrogels (4 million cells/mL) were incubated on an orbital shaker in low adhesion well plates containing standard culture media for 48 h. Subsequently, the culture media was supplemented with a 2, 10, or 100 μM dose of erlotinib [0.2 mM dimethyl sulfoxide (DMSO)], using DMSO as a control. After 3 days of continuous erlotinib exposure, GBM specimens were analyzed for metabolic activity, viability, and gene expression. Alternatively, GBM-seeded hydrogels were placed back into fresh (erlotinib-free) media for an additional 4 days, where specimens were isolated to examine recovery after the removal of erlotinib. The remainder of the GBM-seeded specimens was re-exposed to a second dose of erlotinib (10 μM) for an additional 3 days to examine acquired resistance [Fig. 1(c)].
Location-specific analysis of cell response
After culture, the membrane separating the hydrogel from the media reservoir was gently removed using a scalpel to expose the hydrogel. The entire gradient hydrogel was gently lifted from the exposed device, placed on a glass slide, and dissected into four equal parts across the width of the gradient (45 μL) for localized analyses. To obtain identical samples, a template was used to establish the dimensions (4 × 8 mm2) of every section (1–4) of the hydrogel gradient to be analyzed. The gradient was then sectioned by using a surgical scalpel.
Analysis of cell viability
The metabolic activity of encapsulated GBM cells was assessed for discrete samples isolated from four regions across the spatially graded hydrogel via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Vybrant®, ThermoFisher Scientific) as previously described.[30] Briefly, isolated regions of hydrogel were placed into medium containing 12 mM MTT salt, which is reduced by viable cells to a colored, water-insoluble formazan. The precipitated salt was subsequently dissolved in DMSO followed by measure of absorbance at 540 nm. Cell viability was observed using a laser scanning confocal microscope (Zeiss LSM710), using Z-stacks through the hydrogel to a depth of ~ 400 μm. Prior to imaging, gels were incubated in PBS containing 4 mM calcein AM (λem ~ 515 nm, Invitrogen) to stain viable cells and 1.5 mM propidium iodide (λem ~ 617 nm, Invitrogen) to stain dead cells.
Gene expression analysis
RNA was extracted from cell-laden sub-regions isolated from across gradient hydrogels via an RNeasy Plant Mini kit (Qiagen, Valencia, California, USA).[31] Isolated RNA was subsequently reverse transcribed to cDNA in a Bio-Rad S1000 thermal cycler using the QuantiTect Reverse Transcription kit (Qiagen). RT–PCR reactions were performed in triplicate (QuantiTect SYBR Green PCR, Qiagen) with 10 ng of cDNA per reaction using an Applied Biosystems 7900HT Fast Real-Time PCR System (Carlsbad, California, USA). Primers were synthesized by Integrated DNA Technologies (Coralville, Iowa, USA). Expression levels of CD44, HAS3, VEGF, and MMP-2 were examined relative to the housekeeping gene GAPDH (Supplementary Table S1), with primer sequences derived from literature and gene specificity confirmed via BLAST searches. Results were generated using the Δ−ΔCt (ΔΔCt) method,[32] with all results presented as a relative fold induction, calculated as 2−ΔΔCT, normalized to expression levels of cells in section 1 of gradient platform as the control.
Statistical analysis
All analyses were performed using a one-way analysis of variance followed by Tukey’s honest significant difference post hoc test. Significance level was set at P < 0.05. At least n = 3 samples were examined for cell viability and metabolic assays, and at least n = 3 samples were examined for PCR analysis. Error was reported in figures as the standard deviation unless otherwise noted.
Results and discussion
Hydrogel gradients replicate transitions in tumor microenvironment
We fabricated gelatin hydrogels containing a linear gradient of matrix-immobilized HA concentration to mimic some of the spatially graded biophysical features[33] of the GBM tumor microenvironment. We previously showed that gradients of matrix-immobilized HA contribute to the malignant phenotype of GBM-associated cell lines.[17] As a result, here we examined metabolic activity and gene expression profiles of patient-derived GBM10 and GBM12 PDX specimens using a library of genes related to poor GBM prognoses (CD44, MMP-2, VEGF) and endogenous HA production (HAS-3) [Fig. 2(a)]. From the perspective of this investigation, these two GBM specimens contain molecular differences relevant for our study of resistance to TKIs: overexpression of EGFR (EGFR+) and PTEN (phosphatase and tensin homolog) deletion (PTEN−). PTEN is a tumor suppressor gene, playing important roles in the regulation of cell proliferation, adhesion and invasion, and its deletion, together with EGFR amplification, is strongly correlated with poor prognosis in glioma patients.[34] Epidermal growth factor receptor overexpression, present in 40–60% of all GBM tumors, plays an important oncogenic role in cancer. Regular EGFR protein levels are increased in tumor cells and the receptor then becomes constitutively active. It has been reported that EGFR co-localizes and co-immunoprecipitates with CD44 in head and neck cancer[35] and that HA–CD44 interactions constitutively promote activation of several receptor tyrosine kinases (RTKs), including EGFR, as well as formation of large RTK-containing signaling complexes in malignant carcinoma cells.[11] The combination of these two mutations underlies previously observed differences in response to erlotinib treatment in vivo in heterotopic and orthotropic tumors in mice. Notably, GBM12 (EGFR+, PTENwt) remains sensitive to erlotinib treatment while GBM10 (EGFR−/PTEN−) as GBM14 shows no survival benefit from erlotinib treatment.[24]
Figure 2.

(a) Gene expression analysis of GBM10 and GBM12 cells encapsulated in GelMA/HAMA hydrogel gradients at 48 h. Fold change is normalized to section 1. GAPDH is used as housekeeping gene. (b) Metabolic activity (MTT) of GBM cells over time with increasing concentrations of hyaluronic acid. Data are normalized to MTT value after seeding (day 0). *P < 0.05 compared with section 1.
Analysis of gene expression profiles across a linear gradient of matrix-immobilized HA within a GelMA hydrogel reveals markedly different gene expression profiles for distinct PDX GBM specimens. GBM10 cells (EGFRwt PTEN−) show a significant overexpression of HAS3 with increasing matrix-bound HA [Fig. 2(a)]. Interestingly, HAS-3 expression decreases with matrix-bound HA for GBM12 cells (EGFR+ PTENwt), while we instead observe a significant increase in CD44 expression with HA concentration. HA biosynthesis has previously been identified as a critical pathway for tumor survival, with intracellular HA signals influenced by the presence of HA in the ECM.[36] Specifically, HAS-3 produces predominantly low molecular weight (<300 kDa) HA that have been related to inhibition of CD44–HA interactions and subsequent EGFR inhibition.[37,38] Hence, we believe it likely for these two PDX GBM specimens to exhibit disparate responses to TKIs when cultured in GelMA hydrogels containing spatially graded matrix-immobilized HA. Recent reports have suggested that GBM invasion is dependent on matrix HA content,[39] and survival in mice and humans depends biphasically on extracellular transmembrane CD44 levels with intermedia HA levels associated with the highest invasion potential.[40] Hence, our observed shifts in CD44 expression in GBM12 PDX cells across the hydrogels containing HA gradients suggest that this xenograft should exhibit increased invasive potential and malignancy in vivo, a conclusion borne out in vivo.[25]
While the metabolic activity of both PDX specimens increased with time over the 10-day culture [Fig. 2(b)], only GBM12 PDX cells showed an influence of HA, with reduced metabolic activity with the highest region of matrix-immobilized HA at day 10. This finding is consistent with previous efforts in our laboratory where GBM cell lines showed decreased proliferation and metabolic activity but increased invasion in HA-containing hydrogels.[39]
Changes in intrinsic resistance to erlotinib in HA-decorated hydrogels
The epidermal growth factor receptor is a transmembrane RTK whose activation triggers mechanisms that are involved in cell proliferation and survival. Strategies that disrupt EGFR signal transduction, such as the orally active, reversible small-molecule TKIs erlotinib (Tarceva; Genentech, Inc., San Francisco, California) and gefitinib (Iressa; AstraZeneca Pharmaceuticals, Cambridge, United Kingdom), have been evaluated as antitumor therapies.[41] The efficacy of these inhibitors has not met expectations for GBM therapy, even for EGFR-overexpressing tumors. An increasing number of in vitro and in vivo studies are aimed at overcoming this resistance by combinatorial approaches using anti-EGFR treatment together with one or more additional drugs. In this regard, our ex vivo platform suggests an approach to rapidly profile ECM-associated mechanisms mediating resistance to anti-EGFR exposure as a means to identify combinatorial strategies that may represent better therapeutic approaches for future study in vivo.
We first profiled the initial response of GBM10 and GBM12 PDX specimens to erlotinib within GelMA hydrogels containing margin-inspired gradients of matrix-immobilized HA (Fig. 3). Here, GBM cells embedded in the hydrogel were exposed to discrete soluble concentrations of erlotinib (2, 10, 100 μM) for 3 days, with cell response measured via changes in metabolic activity, as erlotinib is known to inhibit cell-cycle progression and proliferation and initiate apoptosis.[42] Erlotinib showed limited effects on the metabolic activity of GBM cells at moderate concentrations (2 and 10 μM). When concentrations are increased to 100 μM, a cytotoxic response (increased dead cells seen in live/dead staining) and a significant reduction in metabolic activity with increasing local HA content becomes evident. While a reduction in metabolic activity for EGFR+ GBM12 may seem intuitive, the reduction in metabolic activity with increasing matrix-bound HA content for EGFRwt GBM10 may be associated with the erlotinib acting to abrogate effects of significant local upregulation of HAS-3 as well as CD44-associated upregulation of ERK1/2, which is known to enhance glioma activity.[38,43] Hence, the decrease in metabolic activity of GBM10 may be driven by TKI disruption of amplified GBM cell activity in HA microenvironments.
Figure 3.

Metabolic activity (MTT) and cell viability (live/dead images) of GBM10 (a) and GBM12 (b) within GelMA/HAMA hydrogel gradients exposed to first dose of erlotinib. Gradient hydrogels were exposed to single doses of erlotinib of 2 (bottom images), 10 (middle), and 100 μM (top). Scale bar 200 μm.
*P < 0.05 compared with section 1.
Acquired resistance to erlotinib of GBM cells in HA microenvironments
Cancer cells exhibit multiple mechanisms of resistance to chemotherapeutic agents, such as enhanced drug efflux via the ATP-binding cassette family transporters.[44] However, in addition to genetic resistance-promoting mutations, it is suspected that EGFR-dependent cancers may escape targeted therapy by developing dependence on other non-amplified, non-mutated RTKs. Interactions between CD44 and hyaluronan may increase resistance to numerous drugs and stimulate expression of drug transporters.[45] While glioma cells can be characterized by intrinsic resistance to EGFR inhibitors or acquired mechanisms that allow them to escape from EGFR-targeted treatment,[14] we examine the use of our gradient hydrogel platform to rapidly examine such phenomena in the context of the tumor microenvironment.
We subsequently employed the gradient hydrogel to define recovery and acquired resistance parameters for GBM specimens exposed to erlotinib. After 3 days of exposure to erlotinib, the media surrounding the hydrogels was replaced with fresh (erlotinib-free) media for 4 days; subsequently, the media was replaced with erlotinib-containing media for an additional 3 days. We report relative changes in metabolic activity of GBM PDX cells in each hydrogel region for each treatment and recovery cycle. Shifts in local metabolic activity of the cells at the end of each stage of this culture describes recovery (day 7/day 3 metabolic activity ratio) and second dose (day 10/ day 7 metabolic activity ratio) responses. The complete data set of “recovery and second dose” response (compared with control hydrogel not exposed to erlotinib) for all erlotinib doses and local positions across GelMA hydrogels containing margin-inspired gradients of matrix-immobilized HA are reported in Supplementary Fig. S1. To more rapidly compare the effect of matrix HA gradients, we also report explicit comparison of “recovery” and “second dose” response for the regions of the hydrogel with the lowest (region 1) and highest (region 4) matrix-immobilized HA content (Fig. 4). Both EGFRwt GBM10 and EGFR+ GBM12 showed significant recovery of metabolic activity in a matrix-immobilized HA dose-dependent manner. When exposed to a second dose of erlotinib, GBM10 cells show a dose-dependent response more sensitive than in response to a first dose. However, margin-inspired gradient in HA do not seem to have any effect on response of GBM10 cells to this second dose. Interestingly, EGFR+ GBM12 specimens show patterns of “recovery” and “second dose” response strongly influenced by local matrix-immobilized HA content (Supplementary Fig. S1). Hence, acquired resistance to erlotinib seems to be higher in EGFR+/ PTEN+ GBM12 cells and enhanced by margin-inspired gradients of hyaluronan. Unexpectedly, the second dose of erlotinib has a larger overall effect in GBM10 than GBM12. The mechanistic origin of this behavior is under investigation, but highlights the importance of dosage concentration, number of doses, and timing between those doses are for every GBM tumor type. It also supports the idea of HA as a protective environment for EGFR+ cells against TK inhibitors.
Figure 4.

Recovery of GBM 10 and GBM12 cells metabolic activity (MTT) within GelMA/HAMA hydrogel gradients after 4 days in fresh media (a), results are compared with MTT after 3 days in erlotinib (1st dose). (b) MTT after second dose of erlotinib (day 10) compared with metabolic activity recovery (day 7). MTT results are normalized to control for every erlotinib dose. *P < 0.05 compared with section 1.
These results insinuate that transitions in ECM properties of the TME, specifically matrix-bound HA, may strongly influence compensatory pathways and gene expression changes of GBM cells, but that they may be largely dependent on the unique molecular signature associated with each patient (e.g., GBM12: CD44 expression; GBM10: HAS-3). Analysis of metabolic activity data over a course of erlotinib dosages (Supplementary Fig. S1) suggests a protective environment established by matrix-bound HA that alters PDX GBM cells response to the model TK inhibitor erlotinib. PDX specimens showed enhanced recovery in HA-rich regions of the hydrogel, and their response to a second dose was strongly influenced by local matrix HA content in only EGFR+/PTEN+ GBM12 specimens rather than EGFRwt/PTEN−GBM10. The ability to create hydrogel environments containing localized gradients in ECM content, inspired by features of the native GBM TME, then employ these cultures to rapidly profile changes in phenotype (metabolic activity, gene expression) suggests the potential value for this platform to screen for potential compensatory pathways and multi-drug combination therapies for expanded libraries of GBM specimens ahead of costly in vivo trials using xenograft models.
Conclusions
We highlight the application of ex vivo biomaterial platforms that recreate transitions in the native GBM tumor microenvironment to rapidly profile cell response to therapeutic inhibitors. Rather than cell lines, we employed patient-derived GBM specimens that maintain patient morphological and molecular signatures. We demonstrate the capacity of this hydrogel platform to classify tumors response to local gradient in ECM properties and to multiple rounds of exposure to a model therapeutic agent via changes in gene expression and metabolic activity. These gradient hydrogels facilitate analysis of cell response to small-molecule inhibitors such as erlotinib in the context of the local ECM. We show the influence of extra-cellular HA in the efficiency of erlotinib to treat GBM tumors. Moreover, we demonstrate that this biomaterial tumor model can recreate relevant features of the GBM tumor microenvironment, and be used as a valuable tool in the mechanistic studies of tumor development, and prediction of TKIs efficacy and resistance.
Supplementary Material
Acknowledgments
The authors are grateful for seed funding provided by the Illini 4000 as well as the by Mayo Clinic—University of Illinois Alliance for Technology-Based Healthcare. Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under Award Number R01 CA197488. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors are also grateful for additional funding provided by the Illinoi4000, the Department of Chemical and Biomolecular Engineering, and the Carl R. Woese Institute for Genomic Biology at the University of Illinois at Urbana-Champaign.
Footnotes
The supplementary material for this article can be found at https://doi.org/10.1557/mrc.2017.85.
Contributor Information
S. Pedron, Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West Gregory Drive, Urbana, IL 61801, USA
H. Polishetty, Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West Gregory Drive, Urbana, IL 61801, USA
A.M. Pritchard, Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West Gregory Drive, Urbana, IL 61801, USA
B.P. Mahadik, Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West Gregory Drive, Urbana, IL 61801, USA
J.N. Sarkaria, Department of Radiation Oncology, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA
B.A.C. Harley, Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West Gregory Drive, Urbana, IL 61801, USA; Department of Chemical and Biomolecular Engineering, Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 110 Roger Adams Lab., 600 S. Mathews Avenue, Urbana, IL 61801, USA
References
- 1.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 2.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 3.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012;107:359. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 4.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59:1169. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 5.Jackson C, Ruzevick J, Phallen J, Belcaid Z, Lim M. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. Clin Dev Immunol. 2011;2011:20. doi: 10.1155/2011/732413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 7.Okada M, Saio M, Kito Y, Ohe N, Yano H, Yoshimura S, Iwama T, Takami T. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int J Oncol. 2009;34:1621. doi: 10.3892/ijo_00000292. [DOI] [PubMed] [Google Scholar]
- 8.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thaker NG, Pollack IF. Molecularly targeted therapies for malignant glioma: rationale for combinatorial strategies. Expert Rev Neurother. 2009;9:1815. doi: 10.1586/ern.09.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang TT, Sarkaria SM, Cloughesy TF, Mischel PS. Targeted therapy for malignant glioma patients: lessons learned and the road ahead. Neurotherapeutics. 2009;6:500. doi: 10.1016/j.nurt.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misra S, Toole BP, Ghatak S. Hyaluronan constitutively regulates activation of multiple receptor tyrosine kinases in epithelial and carcinoma cells. J Biol Chem. 2006;281:34936. doi: 10.1074/jbc.C600138200. [DOI] [PubMed] [Google Scholar]
- 12.Rape A, Ananthanarayanan B, Kumar S. Engineering strategies to mimic the glioblastoma microenvironment. Adv Drug Delivery Rev. 2014;79–80:172. doi: 10.1016/j.addr.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roth P, Weller M. Challenges to targeting epidermal growth factor receptor in glioblastoma: escape mechanisms and combinatorial treatment strategies. Neuro Oncol. 2014;16:viii14. doi: 10.1093/neuonc/nou222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets. 2012;12:197. doi: 10.2174/156800912799277557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulte A, Liffers K, Kathagen A, Riethdorf S, Zapf S, Merlo A, Kolbe K, Westphal M, Lamszus K. Erlotinib resistance in EGFR-amplified glioblastoma cells is associated with upregulation of EGFRvIII and PI3Kp110δ. Neuro Oncol. 2013;15:1289. doi: 10.1093/neuonc/not093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slomiany MG, Dai L, Bomar PA, Knackstedt TJ, Kranc DA, Tolliver L, Maria BL, Toole BP. Abrogating drug resistance in malignant peripheral nerve sheath tumors by disrupting hyaluronan-CD44 interactions with small hyaluronan oligosaccharides. Cancer Res. 2009;69:4992. doi: 10.1158/0008-5472.CAN-09-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedron S, Becka E, Harley BA. Spatially gradated hydrogel platform as a 3D engineered tumor microenvironment. Adv Mater. 2015;27:1567. doi: 10.1002/adma.201404896. [DOI] [PubMed] [Google Scholar]
- 18.Heddleston JM, Hitomi M, Venere M, Flavahan WA, Yan K, Kim Y, Minhas S, Rich JN, Hjelmeland AB. Glioma stem cell maintenance: the role of the microenvironment. Curr Pharm Des. 2011;17:2386. doi: 10.2174/138161211797249260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN Canc Genome Atlas Res N. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westermark B. Glioblastoma—a moving target. Ups J Med Sci. 2012;117:251. doi: 10.3109/03009734.2012.676574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hambardzumyan D, Cheng Y-K, Haeno H, Holland EC, Michor F. The probable cell of origin of NF1- and PDGF-driven glioblastomas. PLoS ONE. 2011;6:e24454. doi: 10.1371/journal.pone.0024454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labussiere M, Sanson M, Idbaih A, Delattre JY. IDH1 gene mutations: a new paradigm in glioma prognosis and therapy? Oncologist. 2010;15:196. doi: 10.1634/theoncologist.2009-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rich JN, Hans C, Jones B, Iversen ES, McLendon RE, Rasheed BK, Dobra A, Dressman HK, Bigner DD, Nevins JR, West M. Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res. 2005;65:4051. doi: 10.1158/0008-5472.CAN-04-3936. [DOI] [PubMed] [Google Scholar]
- 24.Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB, James CD. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6:1167. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- 25.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A, Pandita A, James CD. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 26.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, James CD. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7:164. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedron S, Becka E, Harley BAC. Regulation of glioma cell phenotype in 3D matrices by hyaluronic acid. Biomaterials. 2013;34:7408. doi: 10.1016/j.biomaterials.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 28.Mahadik BP, Wheeler TD, Skertich LJ, Kenis PJ, Harley BA. Microfluidic generation of gradient hydrogels to modulate hematopoietic stem cell culture environment. Adv Healthc Mater. 2014;3:449. doi: 10.1002/adhm.201300263. [DOI] [PubMed] [Google Scholar]
- 29.Mahadik BP, Pedron Haba S, Skertich LJ, Harley BAC. The use of covalently immobilized stem cell factor to selectively affect hematopoietic stem cell activity within a gelatin hydrogel. Biomaterials. 2015;67:297. doi: 10.1016/j.biomaterials.2015.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 31.Duffy GP, McFadden TM, Byrne EM, Gill SL, Farrell E, O’Brien FJ. Towards in vitro vascularisation of collagen-GAG scaffolds. Eur Cells Mater. 2011;21:15. doi: 10.22203/ecm.v021a02. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(−Delta Delta C) method. Methods. 2001;25:402. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Wiranowska MR, Rojiani MV. Extracellular matrix microenvironment in glioma progression. In: Ghosh A, editor. Glioma—Exploring Its Biology and Practical Relevance. InTech; Rijeka, Croatia: 2011. p. 257. [Google Scholar]
- 34.Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27:5416. doi: 10.1038/onc.2008.239. [DOI] [PubMed] [Google Scholar]
- 35.Perez A, Neskey DM, Wen J, Pereira L, Reategui EP, Goodwin WJ, Carraway KL, Franzmann EJ. CD44 interacts with EGFR and promotes head and neck squamous cell carcinoma initiation and progression. Oral Oncol. 2013;49:306. doi: 10.1016/j.oraloncology.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cha J, Kang S-G, Kim P. Strategies of mesenchymal invasion of patient-derived brain tumors: microenvironmental adaptation. Sci Rep. 2016;6:24912. doi: 10.1038/srep24912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 38.Tsatas D, Kanagasundaram V, Kaye A, Novak U. EGF receptor modifies cellular responses to hyaluronan in glioblastoma cell lines. J Clin Neurosci. 2002;9:282. doi: 10.1054/jocn.2001.1063. [DOI] [PubMed] [Google Scholar]
- 39.Chen J-W, Pedron S, Harley BAC. The combined influence of hydrogel stiffness and matrix-bound hyaluronic acid content on glioblastoma invasion. Macromol Biosci. 2017;17:1616. doi: 10.1002/mabi.201700018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klank RL, Decker Grunke SA, Bangasser BL, Forster CL, Price MA, Odde TJ, SantaCruz KS, Rosenfeld SS, Canoll P, Turley EA, McCarthy JB, Ohlfest JR, Odde DJ. Biphasic dependence of glioma survival and cell migration on CD44 expression level. Cell Rep. 18:23. doi: 10.1016/j.celrep.2016.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003;21:2787. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- 42.Akita RW, Sliwkowski MX. Preclinical studies with erlotinib (Tarceva) Semin Oncol. 2003;30:15. [PubMed] [Google Scholar]
- 43.Wang SJ, Bourguignon LYW. Role of hyaluronan-mediated CD44 signaling in head and neck squamous cell carcinoma progression and chemoresistance. Am J Pathol. 2011;178:956. doi: 10.1016/j.ajpath.2010.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 45.Ohashi R, Takahashi F, Cui R, Yoshioka M, Gu T, Sasaki S, Tominaga S, Nishio K, Tanabe KK, Takahashi K. Interaction between CD44 and hyaluronate induces chemoresistance in non-small cell lung cancer cell. Cancer Lett. 2007;252:225. doi: 10.1016/j.canlet.2006.12.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.