

We report a novel method to generate morphologically, antigenically and transcriptionally dependable microglia-like cells in vivo.
Abstract
Recent evidence indicates that hematopoietic stem and progenitor cells (HSPCs) can serve as vehicles for therapeutic molecular delivery to the brain by contributing to the turnover of resident myeloid cell populations. However, such engraftment needs to be fast and efficient to exert its therapeutic potential for diseases affecting the central nervous system. Moreover, the nature of the cells reconstituted after transplantation and whether they could comprise bona fide microglia remain to be assessed. We demonstrate that transplantation of HSPCs in the cerebral lateral ventricles provides rapid engraftment of morphologically, antigenically, and transcriptionally dependable microglia-like cells. We show that the cells comprised within the hematopoietic stem cell compartment and enriched early progenitor fractions generate this microglia-like population when injected in the brain ventricles in the absence of engraftment in the bone marrow. This delivery route has therapeutic relevance because it increases the delivery of therapeutic molecules to the brain, as shown in a humanized animal model of a prototypical lysosomal storage disease affecting the central nervous system.
INTRODUCTION
In the last three decades, hematopoietic cell transplantation (HCT) and hematopoietic stem cell (HSC)–based gene therapy (GT) have been applied, with benefit to patients affected with diseases involving the nervous system such as peroxisomal disorders (1, 2), lysosomal storage diseases (LSDs) (3, 4), and neurodegenerative conditions (5, 6). The clinical evidence generated in these settings, as well as supporting preclinical data, suggests that the transplanted hematopoietic stem and progenitor cells (HSPCs) and/or their progeny could engraft within the central nervous system (CNS) and progressively contribute to resident myeloid populations, possibly including microglia (7–11). These transplant-derived cells could serve as vehicles for the delivery of therapeutic molecules across the blood-brain barrier (BBB) (3, 4). Moreover, renewing myeloid cells and microglia, as well as modulating their phenotype and function, could contribute to the mitigation of key pathologic events in the diseased brain. Myeloid cells and microglia play a crucial role in many of the pathogenic processes contributing to tissue damage in neurodegenerative disorders (12–18), such as neuroinflammation and oxidative stress. However, the therapeutic potential of HSPC-based approaches to treating neurodegenerative conditions is still limited by the turnover rate and slow kinetics of brain myeloid cell renewal after transplant. Proper selection of a pretransplant chemotherapy conditioning capable of ablating functionally defined brain-resident microglia precursors was shown to increase the extent of donor cell chimerism after transplant (10, 19). However, further improvements and the development of novel strategies to anticipate the time and enhance the potential for clinical benefit in the transplanted patients are warranted and would represent an important breakthrough for the field. Moreover, the differentiation and functional characteristics of the transplant-derived cells in murine and human brains are the subject of extensive debate, and in particular, whether bona fide microglia could be reconstituted by the donor cell progeny after HCT remains to be demonstrated (7, 10, 20). Microglia cells have a developmental origin distinct from that of bone marrow (BM)–derived myelomonocytes (21). They are also the most abundant and functionally heterogeneous cells within myeloid brain populations, and their functional replacement with metabolically competent and inactivated cells upon transplantation would be particularly relevant for therapeutic purposes. We and others demonstrated that, under specific experimental conditions, cells of donor origin showing a morphology and expressing surface markers consistent with those of microglia could be successfully generated in the brain of mice transplanted with donor HSPCs. However, whether the functional characteristics of these donor-derived cells in the transplanted brain are consistent with those of bona fide microglia is not known.
Here, we first demonstrate that new myeloid cells of donor origin can be efficiently generated upon administration of HSPCs in the brain lateral ventricles of conditioned mice, in the absence of donor cell engraftment in the BM. Of note, this novel intracerebroventricular route of HSPC delivery allows for a more rapid and robust engraftment of donor cells in the brain compared to the standard intravenous HSPC administration. The donor-derived myeloid cells isolated from the brain of transplanted mice share not only the morphology and the surface markers but also a similar transcriptional profile with adult naïve microglia. These microglia-like cells derive from the transplanted early hematopoietic progenitors and long-term HSCs (LT-HSCs) in the intracerebroventricular setting and mostly from LT-HSCs in the case of intravenous delivery. This intracerebroventricular cell delivery approach has a unique therapeutic relevance, as shown in a murine model of metachromatic leukodystrophy (MLD). Overall, these data demonstrate that intraventricularly injected human HSPCs can contribute to myeloid brain cell populations and increase therapeutic protein delivery to the CNS of diseased animals.
RESULTS
Rapid and robust myeloid cell engraftment in the brain occurs following intracerebroventricular injection of HSPCs
Our recent results (10) propose that engraftment and persistent donor chimerism in the BM hematopoietic niche might not be necessary for obtaining myeloid cell reconstitution in the brain following HCT. On the basis of these findings, we assessed whether myeloid and microglia-like cell reconstitution could occur upon direct transplantation of HSPCs into the cerebroventricular space in conditioned mice. Murine HSPCs were labeled with green fluorescent protein (GFP)–encoding lentiviral vectors (LVs) and transplanted by intracerebroventricular injection in mice after exposure to a myeloablative busulfan (BU) dose or lethal irradiation (fig. S1A). By short-term flow cytometry monitoring, we could demonstrate the presence, persistence, and moderate expansion of the intracerebroventricularly delivered cells in the brain of the recipient animals (Fig. 1A). Conversely, we only detected negligible amounts of GFP+ cells in the BM of the intracerebroventricularly transplanted mice (<1%) (Fig. 1B). The GFP+ cells transiently up-regulated early hematopoietic markers (Fig. 1C) and, afterward, the CD11b, CX3CR1, and CD115 microglia markers up to levels similar to the endogenous microglia (Fig. 1D). The GFP−CD45+ endogenous cells transiently and slightly down-regulated CD115, as a possible effect of the BU treatment (Fig. 1D).
Fig. 1. Myeloid cell reconstitution in the brain after intracerebroventricular injection of HSPCs.
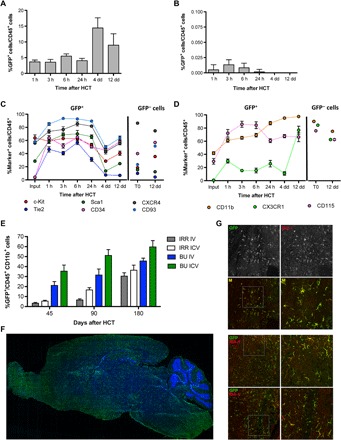
(A and B) Frequency of GFP+ cells identified within CD45+ cells of the brain (A) and BM (B) of BU_TX (BU-treated and transplanted) mice at the indicated time points after intracerebroventricular (ICV) injection of HSPCs transduced with GFP-encoding LVs. n ≥ 3 mice each time point; average and SD are shown. Analyzed by one-way analysis of variance (ANOVA) with Bonferroni post hoc test, 4 days at comparison with 1, 3, 6, and 24 hours shows P < 0.001. (C and D) Expression of the indicated hematopoietic (C) and myeloid/microglia (D) markers by GFP+ (donor) and GFP− (recipient) CD45+ cells retrieved from the brain of BU_TX mice at different time points after intracerebroventricular injection of transduced HSPCs (input represents the HSPCs at time of infusion). n ≥ 3 mice each time point; average and SD are shown. Two-way ANOVA showed a significant effect of the markers and time (P < 0.0001). (E) Frequency of GFP+ cells in the total myeloid (CD45+CD11b+) brain compartment at different time points after intracerebroventricular and intravenous (IV) HSPC transplantation in BU-treated (BU) and irradiated (IRR) mice. n ≥ 5 mice per time point and group; average and SD are shown. Two-way ANOVA showed a significant effect of the route of cell administration and time in BU_TX and IRR mice (intracerebroventricular versus intravenous and time, P < 0.005). (F) Reconstruction of a sagittal brain section of a representative intracerebroventricularly transplanted BU-treated mouse, showing widespread distribution of GFP+ cells at 90 days from GFP-transduced HSPC intracerebroventricular injection. GFP (green) and Topro III (TPIII; blue) for nuclei are shown. Images were acquired via DeltaVision Olympus at 20× magnification and processed using Soft Work 3.5.0. Reconstruction was performed with Adobe Photoshop CS 8.0 software. (G) Immunofluorescence analysis for GFP (green) and IBA-1 (red) on brain sections from BU_TX mice at 90 days after intracerebroventricular transplantation of GFP-transduced HSPCs. M, merge. Magnifications (20× and 40×) of the relative dashed box are shown. Images were acquired using the confocal microscope Radiance 2100 (Bio-Rad) IX70 and processed using Soft Work 3.5.0.
In the long term, a high and progressively increasing GFP chimerism was observed in the CD45+CD11b+ brain myeloid compartment of the intracerebroventricularly transplanted mice, conceivably derived from the local proliferation of the transplanted cells (Fig. 1E). For each time point and condition, control mice transplanted intravenously with GFP+ HSPCs were used as terms of comparison. Notably, the kinetics of microglia reconstitution was faster, and the extent of GFP chimerism was higher when the GFP+ HSPCs were transplanted intracerebroventricularly as compared to intravenously (Fig. 1E). As in the case of intravenous injection (10), also upon intracerebroventricular cell transplantation, recipient mice pretreated with BU showed a higher brain myeloid donor chimerism as compared to irradiated animals.
Immunofluorescence analysis of brain sections of intracerebroventricularly transplanted mice consistently demonstrated abundant GFP-expressing, donor-derived cells distributed throughout the recipient mice brain (Fig. 1F). GFP+IBA-1+–ramified parenchymal cells were frequently grouped in small clusters, with the highest GFP+ cell frequencies retrieved in the olfactory bulb, hypothalamic area, basal nuclei, subventricular zone and surrounding regions, striatum, and pons (Fig. 1G). GFP+ cell morphology resembled that of intraparenchymal microglia cells, with ramifications and thin processes departing from the cell body already at relatively short term (45 to 60 days) after transplant.
Posttransplant brain myeloid cells derive from early HSPCs
We previously demonstrated that the c-kit+, Sca-1+, Lin− (KSLs) fraction of HSPCs is able to give rise to new myeloid cells in the brain of mice upon intravenous transplantation, whereas c-kit−, Sca-1−, Lin− (non-KSL) progenitor cells are not able to do so (10). We here further explored this initial finding also in the intracerebroventricular route setting using a stringent competitive design by cotransplanting differentially labeled KSL and non-KSL cells in individual animals (Fig. 2, A to C) intravenously (Fig. 2B) and intracerebroventricularly (Fig. 2C). When injected intravenously with non-KSLs, KSL cells almost exclusively contributed to brain myeloid CD45+CD11b+ cells (Fig. 2B) and their subpopulations (10). On the contrary, KSL and non-KSL cells contributed, to a similar extent, to brain myeloid cell reconstitution when injected intracerebroventricularly (Fig. 2C). We next addressed the ability of differentially labeled KSL subpopulations, identified by differential expression of the signaling lymphocytic activation molecule (SLAM) markers CD150 and CD48, to contribute to brain myeloid cell engraftment upon transplantation (Fig. 2, A, D, and E). Upon competitive intravenous transplantation, CD48−/CD150+ LT-HSCs and CD48−/CD150− multipotent progenitors (MPPs) showed a greater ability to reconstitute the brain myeloid compartment over the other injected populations (Fig. 2D and fig. S2A). In contrast, the more committed CD48+CD150− cells also contributed to the reconstitution of brain myeloid populations in the mice transplanted intracerebroventricularly (Fig. 2E and fig. S2A). Hematopoietic reconstitution of the transplanted mice is shown in fig. S2B. Histology on cryostat brain slices from intravenously and intracerebroventricularly transplanted mice confirmed these results (Fig. 2, F and G). Consistent with previous findings, also in these settings, donor-derived cells showed a ramified morphology with thin processes departing from the cell body and expressed the myeloid markers IBA-1 and CD11b (Fig. 2, F and G, and fig. S2, C and D). To interpret these findings, we analyzed the expression of the CXCR4 receptor, which is well known to be involved in HSC recruitment and homing to the BM (22–24), on KSLs, non-KSL cells, and four KSL subpopulations described above before transplantation. The cells enriched in brain myeloid cell reconstitution potential in the intravenous delivery setting, namely, KSL, LT-HSCs, and MPPs, expressed CXCR4 at higher levels as compared to non-KSLs and HPC-1 (hematopoietic progenitor cell) and HPC-2 (Fig. 2H) and could thus be favored in early recruitment to the brain upon intravenous delivery.
Fig. 2. Posttransplant brain myeloid cells derived from early HSPCs.
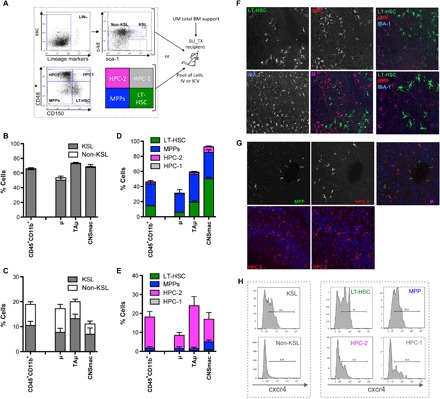
(A) Experimental scheme. Prospectively isolated LT-HSCs and progenitors within the HSPC pool as per c-kit, Sca-1, and lineage− staining and use of the SLAM receptor markers CD150 and CD48. The indicated sorted populations were differentially transduced with LVs encoding GFP (KSL) and deleted nerve growth factor receptor (ΔNGFR) (non-KSL), as well as GFP (LT-HSCs), ΔNGFR (MPP), Tag-blue fluorescent protein (BFP) (HPC-1), and Cherry (HPC-2), and subsequently transplanted intravenously (B and D) or intracerebroventricularly (C and E) in competitive fashion at their original ratio into BU-myeloablated mice. Animals transplanted intracerebroventricularly also received unmanipulated (UM) total BM cells for hematopoietic rescue at day 5 after transplant. (B and C) Frequency of cells derived from intravenously (B) or intracerebroventricularly (C) transplanted KSL and non-KSL within total brain myeloid (CD45+CD11b+) cells, μ and TAμ of BU_TX mice at 90 days after transplant. n = 10 mice per group; average and SD are shown. CNSmac, CNS-associated macrophages. (D and E) Frequency of cells derived from each of the transplanted KSL subpopulations within total brain myeloid cells, μ and TAμ of BU-myeloablated mice transplanted intravenously (D) or intracerebroventricularly (E), at different time points after HCT. n = 10 mice per time point and group; average and SD are shown. (F and G) Immunofluorescence analysis of brain slices of BU-treated mice transplanted mice transplanted intravenously (F) or intracerebroventricularly (G) with KSL subpopulations at 90 days after transplant. In (F), the progeny cells of LT-HSC are GFP+ and those of MPPs are ΔNGFR+ (in red). IBA-1 staining is in the blue channel. Magnification, 20×. M, merge. In the right panels, other representative merged pictures at 20× (top) and their 40× magnifications (bottom) are shown. In (G), the progeny cells of HPC-2 are Cherry+ and those of MPPs are ΔNGFR+ (in green). No GFP+ staining was detected in the absence of ΔNGFR immunofluorescence. TPIII (blue) for nuclei is shown. Magnification, 20× (top). In the bottom panels, other representative merged pictures at 20× (top) and their 40× magnifications (bottom) are shown. Images were acquired using the confocal microscope Radiance 2100 (Bio-Rad) and processed using Soft Work 3.5.0.100. (H) Histogram plots showing the differential level of cxcr4 expression in KSL and non-KSL cells, and KSL subpopulations at the time of transplant.
To more stringently assess whether bona fide HSCs could generate new microglia-like cells upon intracerebroventricular transplantation in the absence of competition, we isolated LT-HSCs with alternative markers and the functional signature of Fgd5 expression in Fgd5-Zsgreen animals (25). The Fgd5-green reporter strain allows faithfully isolating cells that are highly enriched in HSC activity. Bona fide HSCs were isolated from CD45.2 Fgd5-Zsgreen donors as Zsgreen+, lineage−, c-kit+, Sca1+, Flkt-2−, and CD34− cells and transplanted intravenously or intracerebroventricularly into BU-conditioned or lethally irradiated CD45.1 recipient mice, along with BM CD45.1 support (Fig. 3A). Intravenous cell delivery resulted in a robust hematopoietic donor chimerism, whereas intracerebroventricularly injected cells did not contribute to hematopoiesis in the peripheral blood (fig. S2E). Although the intracerebroventricularly transplanted animals did not show peripheral blood chimerism, donor-derived CD45.2+ cells were identified in the brain in both transplant settings, indicating that bona fide HSCs can generate a myeloid cell progeny in the brain also upon intracerebroventricular transplantation (Fig. 3, B and C). Reconstitution of brain myeloid cells was confirmed to be less efficient in irradiated than in BU-ablated recipients, as described by Capotondo et al. (10). The donor-derived cells expressed CD45 and CD11b and were mostly part of the transiently amplifying microglia (TAμ) cell compartment (Fig. 3, D and E).
Fig. 3. Fgd5+ HSCs generate a microglia-like progeny in the brain upon both intracerebroventricular and intravenous transplantation.
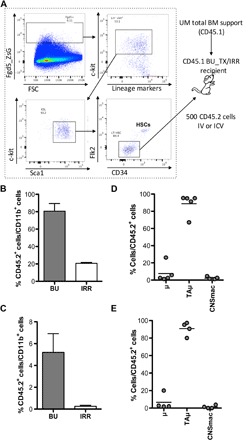
(A) Experimental scheme. Fgd5+ HSCs (Lin− ckit+ Sca-1+ Flk2− CD34−) were isolated from CD45.2 Fdg5-green donor mice. Five hundred Fgd5+ HSCs were transplanted intravenously or intracerebroventricularly into BU-myeloablated or lethally IRR CD45.1 recipient mice. Transplanted animals also received UM CD45.1 total BM cells for hematopoietic rescue at day 5 after transplant. FSC, forward side scatter. (B and C) Frequency of donor cells (CD45.2+) within brain myeloid CD11b+ cells of mice transplanted intravenously (B) and intracerebroventricularly (C) with Fgd5 cells after BU and irradiation conditioning. n ≥ 4 per group. (D and E) Frequency of μ, TAμ, and CNSmac populations within donor-derived cells in intravenously (D) and intracerebroventricularly (E) transplanted BU-conditioned mice. n ≥ 4 per group; average and SD are shown. Student’s t test, P < 0.001 for BU versus irradiation in (B) and (C).
A microglia signature is present in myeloid cells retrieved from the brain of intracerebroventricularly transplanted mice
We then analyzed the brain myeloid cell progeny of the intracerebroventricularly versus intravenously transplanted HSPCs. First, by flow cytometry, we analyzed brain myeloid cell subpopulations (namely, CD45+CD11bhigh μ cells, CD45+CD11b+/low TAμ cells, and CD45highCD11bhigh CNSmac), and we observed that the μ progeny of the HSPCs injected intracerebroventricularly (labeled with GFP) was more abundant than the μ progeny of intravenously injected cells (GFP−), in the presence of a similar but overall higher contribution of the former to TAμ cells (Fig. 4A). To interpret these findings and further characterize the donor-derived cells, we sorted endogenous and donor-derived μ and TAμ cells (fig. S3) from mice that were intracerebroventricularly or intravenously transplanted 90 days earlier and from adult and p10 control animals using fluorescence-activated cell sorting (FACS). We then amplified, by real-time PCR, previously identified genes that are highly expressed in microglia as compared to macrophages (Tmem119, Tgfbr1, P2ry13, Mertk, and Olfml3) (20, 26–29) on the sorted brain myeloid cells and on BM macrophages from adult control (ADULT_CT) animals. The cells isolated from the brain of the intracerebroventricularly (and intravenously) transplanted mice showed expression of these genes at levels similar to those of μ cells isolated from control mice, rather than of macrophages (Fig. 4B). In addition, within both the μ and TAμ fractions, the GFP+ progeny of the intracerebroventricularly transplanted HSPCs showed expression levels very similar to those of ADULT_CT μ and of (GFP− and GFP+) μ from intravenous transplanted mice (table S1). The selected genes were expressed at slightly lower levels in the TAμ populations of intravenously transplanted mice (particularly, GFP+) and in TAμ isolated from p10 mice (table S1).
Fig. 4. Microglia signature is present in myeloid cells retrieved from the brain of transplanted mice.
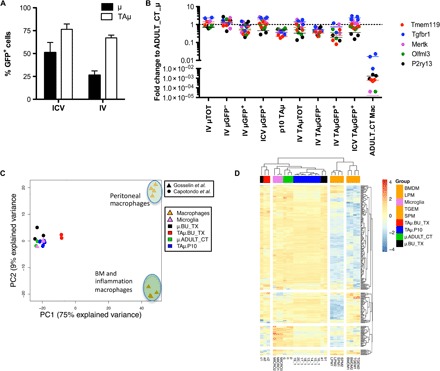
(A) Frequency of μ and TAμ cells within GFP+ cells in the brain of mice at 90 days after transplantation of GFP+ HSPCs intravenously or intracerebroventricularly. n = 5 per group; average and SD are shown. Two-way ANOVA showed a significant effect of the populations analyzed (P < 0.0001). (B) Fold change expression (calculated as 2−DDCT) of selected microglia genes, obtained by real-time polymerase chain reaction (PCR) in each indicated population retrieved from the brain of BU-treated, intravenously and intracerebroventricularly transplanted mice or from P10 mice, calculated on expression of the same genes in ADULT_CT_μ cells. Mean values are shown. For statistical tests, refer to table S1. (C and D) Principal components analysis (PCA) (C) and heat map (D) showing expression analysis of the genes identified as microglia signature by Butovsky et al. within our samples (μ and TAμ retrieved from naïve P10 and ADULT_CT, and HCT animals) and samples reported in Gosselin et al. (30), including microglia and macrophages. LPM, large peritoneal macrophages; SPM, small peritoneal macrophages; BMDM, BM-derived macrophages; TGEM, thioglycollate-elicited peritoneal macrophages.
Myeloid cells from the brain of transplanted mice display functional features similar to maturing microglia
To analyze the transcriptomic differences between μ and TAμ cells from transplanted mice and mature μ retrieved from control naïve animals, we performed a genome-wide expression analysis by means of an Illumina RNA sequencing (RNA-seq) platform on sorted μ and TAμ populations from mice transplanted 3 months earlier with GFP-expressing HSPCs and from adult and p10 control naïve mice (fig. S3 and table S2). To examine their differential gene expression, we analyzed our data set together with that from Gosselin and colleagues (30), specifically focusing on the 239 genes identified by Butovsky et al. (Fig. 4, C and D) (26). All microglia samples included in this analysis clustered closely to each other (Fig. 4, C and D), confirming that μ and TAμ cells reconstituted after transplant share a pattern of gene expression consistent with that of microglia. We then performed a differential gene expression coupled to gene set enrichment analysis (GSEA) preranked analysis (31) on our RNA-seq data (Fig. 5, A to F). The resulting gene ontology (GO) biological processes enriched in μ from ADULT_CTs versus μ (Fig. 5A and table S3) and TAμ (Fig. 5C and table S3) from the transplanted mice were related to immune cell differentiation, immune responses, DNA/RNA processes, and DNA methylation, pointing to the mature immune function of control μ. On the other hand, the processes enriched in μ cells from the transplanted mice (Fig. 5B and table S3) covered neuronal related processes, such as neuron migration, differentiation, and regulation of synaptic plasticity (32, 33). Among the processes enriched, we also found glial cell differentiation, gliosis, and metabolism and cellular respiration, supporting the idea that transplant-derived μ cells are more oriented to interact with or affect the neuronal environment, a process consuming a considerable amount of energy (34). Posttransplant TAμ-enriched processes (Fig. 5D and table S3) presented a more intense neural function signature underlying a putative different stage of maturation as compared to μ cells, in agreement with the concept that microglia acquire different functions according to their maturation states (35). μ and TAμ cells from transplanted mice expressed the genes that were shown to be robustly modulated during microglia development (35). Notably, μ from transplanted animals matched control μ for the levels of expression of four of the five genes associated to mature microglia function, whereas a different path was observed in TAμ from transplanted mice (Fig. 5E and table S4) and in genes associated to neonatal stage (Fig. 5F and table S4), possibly because of a different and dynamic maturation stage.
Fig. 5. Myeloid cells from the brain of transplanted mice display functions of maturing microglia.
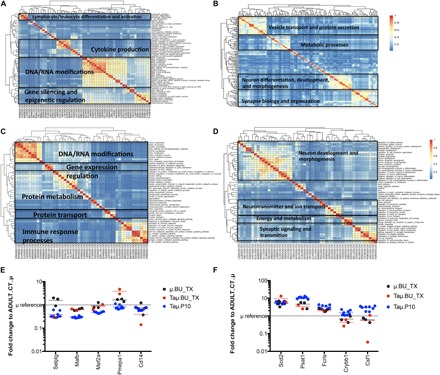
Functional enrichment of differentially up-regulated (A) and down-regulated (B) genes in μCT cells versus μ transplanted cells. Functional enrichment of differentially up-regulated (C) and down-regulated (D) genes in μCT cells versus TAμ transplanted cells. GSEA preranked analysis was performed using our RNA-seq differential gene expression data on GO biological processes (http://software.broadinstitute.org/gsea/msigdb/collections.jsp) with default parameters. Semantic similarity of GOs (GOSemSim) was used to cluster significantly enriched GOs [GOs with false discovery rate (FDR) < 0.05 for up-regulation and FDR < 0.001 for down-regulation were chosen to enhance representation clarity]. A complete list of GO enrichments is presented in table S3. (E and F). Fold change of RNA-seq normalized expression values of genes selected from Matcovitch-Natan et al. (35) in the indicated populations retrieved from the brain of BU_TX mice or P10 mice versus ADULT_CT_μ cells. In (E), genes whose expression is up-regulated in adult mice and, in (F), genes whose expression is up-regulated in p10 mice are shown, as for Matcovitch-Natan et al. (35) For statistical tests, refer to table S4.
Intracerebroventricular delivery of HSPCs has therapeutic relevance
To get insight into the clinical relevance of the establishment of brain myeloic donor chimerism by intracerebroventricular delivery of HSPCs, we tested the ability of human HSPCs to generate microglia-like cells upon intracerebroventricular delivery in conditioned immunodeficient animal models. We first infused intracerebroventricularly or intravenously human GFP+ CD34+ cells into NOD.Cg-PrkdcscidIl2rgtm1WjI/SzJ (NSG) mice (fig. S1B). Moreover, to determine the actual role of intracerebroventricular cell transplantation in augmenting the potential of the transplant to deliver therapeutic molecules to the brain, we used a newly generated mouse model, the Rag−/−γ−chain−/−As2−/, that reproduces the lysosomal disease MLD because of arylsulfatase A (ARSA) deficiency on an immunodeficient background. These mice received human CD34+ cells transduced with an ARSA-encoding LV (3, 4) by intravenous-only or intracerebroventricular-only injection, or by a combination of the intravenous and intracerebroventricular routes (fig. S1C).
A clearly defined human myeloid (CD45+CD11b+) cell progeny was identified in the brain of the transplanted mice long term after intravenous and intracerebroventricular transplant (Fig. 6A and fig. S4, B and C). Intracerebroventricular cell delivery either alone or, even more consistently, in combination with intravenous delivery resulted in a greater human cell engraftment in the brain as compared to intravenous delivery alone (Fig. 6A). In all the tested transplant settings, the human cells largely expressed the microglia marker CX3CR1 (Fig. 6B) at flow cytometry. Cells were distributed within the brain parenchyma, displayed the morphological features of microglia cells (Fig. 6C), and also expressed the IBA-1 and CD11b (Fig. 6C and fig. S4D) markers but not CD68 and CD163 (fig. S4E) that are mostly associated to macrophages. In the case of intracerebroventricular delivery, the progeny cells were identified to be typically grouped in small clusters in the same regions where progeny cells of the intracerebroventricularly transplanted murine HSPCs were identified, that is, in the subventricular zone. The increased contribution of the intracerebroventricularly transplanted human HSPCs to brain myeloid chimerism resulted in a greater ARSA enzyme delivery to the brain of the transplanted mice (Fig. 6D), in which ARSA activity levels indistinguishable with As2+/+ controls were measured. Cotransplantation did not result in increased enzyme activity in the hematopoietic system, confirming that the contribution of intracerebroventricularly delivered cells is mostly restricted to the brain.
Fig. 6. Intracerebroventricular delivery of HSPCs has therapeutic relevance.
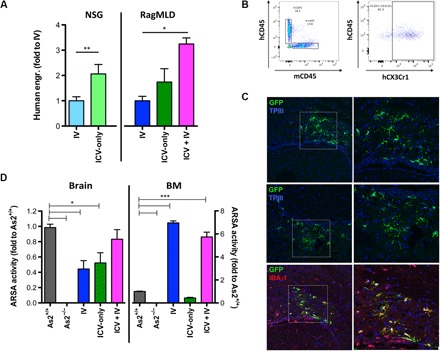
(A) Frequency of human CD45+CD11b+ cells retrieved from the brain of NSG and Rag−/−γ−chain−/−As2−/− (RagMLD) mice transplanted intravenously or intracerebroventricularly with umbilical cord blood (CB)–derived CD34+ cells after BU treatment or sublethal irradiation (Rag−/−γ−chain−/−As2−/−), 12 (NSG) and 5 (Rag−/−γ-chain−/−As2−/−) weeks after transplant. Values are expressed as fold to intravenous transplantation, with intravenous transplantation equal to 3 ± 1.3 in NSG mice, and to 2.9 ± 0.7 in RagMLD. n ≥ 5 mice per group; average and SD are shown. P < 0.001 by Student’s t test in NSG mice; P < 0.05 by one-way ANOVA with Bonferroni post hoc test in RagMLD mice. (B) Representative box plots showing human cell engraftment (human CD45) in the brain of NSG mice that received human CD34+ HSPCs, and expression of the human CX3CR1 marker in the human cells identified in the transplanted mice brains. (C) Immunofluorescence analysis for GFP (green) and IBA-1 (red) on brain sections from NSG mice at 90 days after intracerebroventricular transplantation of GFP-transduced CD34+ cells. In blue, nuclei stained by TPIII. Magnifications (20× and 40×) of the relative dashed box are shown. M, merge. (D) ARSA activity (expressed as fold to the value measured in Rag−/−γ−chain−/−As2+/+ wild-type mice tissues) measured in the brain and BM of Rag−/−γ−chain−/−As2−/− mice transplanted with ARSA-transduced cells intracerebroventricularly or intravenously, as indicated. n = 3 mice per group; average and SD are shown. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA with Bonferroni post hoc test.
DISCUSSION
This work identifies a new strategy to improve brain myeloid cell turnover with the donor after HSPC transplantation and first shows that this process can result in robust and rapid engraftment of microglia-like cells. On the basis of our previous observations on the clonal independence of microglia and peripheral hematopoietic cells in repopulated transplant recipients (10), we challenged standard hematopoietic transplantation paradigms and directly infused HSPCs into the brain ventricular space of mice, after proper conditioning administration. Intracerebroventricular HSPC transplantation was associated with extensive and exclusive reconstitution of the brain myeloid cell pool, confirming our hypothesis that donor cell engraftment in the hematopoietic tissue is not necessary for microglia replacement (10). Rather, HSPCs delivered into the brain could generate new bona fide microglia-like cells by local engraftment and, conceivably, proliferation and differentiation. This transplantation route was associated with a more rapid reconstitution of the myeloid brain compartment and, in particular, of the μ pool, as compared to intravenous transplantation. The great and unique contribution of intracerebroventricularly delivered HSPCs to brain myeloid cell populations was also confirmed in humanized mice. Intracerebroventricular transplantation of human HSPCs in two different immunodeficient mouse models resulted in a robust engraftment of the injected cells in the brain, and engraftment was even more pronounced when human HSPCs were codelivered intracerebroventricularly and intravenously. The use of immunodeficient mice affected by the LSD MLD, characterized by extensive CNS involvement, allowed determining the therapeutic relevance of this delivery route. In this setting, intracerebroventricular transplantation (and cotransplantation) of human HSPCs transduced with a therapeutic ARSA-expressing LV resulted in a more effective and abundant ARSA enzyme delivery to the brain of the affected mice as compared to a standard intravenous transplantation of the transduced cells. Because of the limitations intrinsic to the mouse model we have used, we could not assess rescue of disease manifestations other than enzyme deficiency in this experimental setting. However, we and others have documented in both mice models and patients a dose-effect relationship in HSC GT for LSDs and, in particular, that the higher is the enzyme quantity delivered to the brain by HSC GT, the greater is the benefit exerted by the transplant on CNS disease manifestations (9, 36–38) (3, 4). Thus, these data strongly support the therapeutic potential of this novel transplantation modality for MLD and other lysosomal disorders. Given the most favorable kinetics of myeloid cell reconstitution in the brain of intracerebroventricularly transplanted animals, this strategy could address the need to anticipate and increase the clinical benefit of the therapy in those cases characterized by a rapid progression of the pathology.
The use of prospectively isolated populations from the HSPC pool that have increased potential for renewal of microglia-like cells upon transplantation may further enhance timing and extent of the therapeutic benefit of HSC-based approaches for CNS diseases. Our data indicate that fractions enriched in LT-HSCs may enhance the ability of intravenous transplantation to generate a brain myeloid progeny, whereas the use of committed MPPs within the HSPC pool could favor a rapid myeloid cell reconstitution upon intracerebroventricular cell delivery. Upon competitive transplantation of differently labeled HSPC subpopulations, LT-HSCs and MPPs showed the highest potential not only to reconstitute the hematopoietic system but also to give rise to extensive myeloid progeny in the brain. This ability correlates with the levels of CXCR4 expression on the cell surface, a finding that may allow hypothesizing a role of SDF1-CXCR4 signaling in homing of HSPCs not only to the BM niche but also to the brain. Similarly, prospectively isolated Fdg5-expressing HSCs efficiently generated new microglia-like cells. Differently, cells greatly contributing to new myeloid brain populations in the intracerebroventricular setting are represented by the more committed CD48+CD150− cells. This differential contribution could be due to the brain microenvironment that could create conditions favoring the engraftment of the more committed progenitors. In support of this hypothesis, we reported an up-regulation of typical microglia markers (CSF1-R/CD115 and CX3CR1) early after transplant (possibly induced by the brain environment after conditioning) that could influence the fate of the transplanted cells by favoring their engraftment. Moreover, upon intracerebroventricular injection, cells that are quantitatively more represented among the others (HPC-2) could be advantaged and favored in engrafting locally and expanding. In support of this hypothesis, in the absence of competition, purified Fdg5-green HSCs could generate a quantitatively modest but clearly detectable myeloid cell progeny upon intracerebroventricular injection. Finally, the more committed cells, if transplanted intravenously, could be disfavored because of their intrinsically lower ability to migrate to the brain as compared to LT-HSCs and MPPs, as per our CXCR4 analysis. Nevertheless, we could not exclude the idea that, upon intracerebroventricular cell injection, the transplanted cells could respond to a different signaling pathway.
An extensive debate exists in the literature regarding the actual phenotype and differentiation stage of hematopoietic cells appearing in the brain of mice after HSPC transplantation (7, 10, 11, 20, 36, 39). Most authors suggest that transplant-derived cells retain a macrophage-like identity. To contribute to this debate and assess the functional identity of the transplant-derived cells in our setting, we then studied the gene expression signature of newly formed myeloid cells (identified as μ and TAμ cells) retrieved from the brain of chimeric mice. This analysis revealed that donor-derived brain myeloid cells expressed typical microglia markers such as Tmem119, Tgfbr1, P2ry13, Olfml3, and Mertk (20, 26–29) at levels comparable to those of microglia isolated from ADULT_CT mice. This was more evident for the progeny of intracerebroventricularly transplanted HSPCs but also applied to the intravenous HSPC progeny. On the contrary, brain cells reconstituted after the transplant greatly differed in the expression levels of these markers from BM-resident or circulating macrophages (30). These results were also confirmed by genome-wide expression analysis of our samples. In particular, the combination of our data with a data set from Gosselin and colleagues (30) confirmed that μ and TAμ cells reconstituted after transplant share a pattern of gene expression consistent with that of microglia. These results may appear in conflict with other data, such as those of Bennett and colleagues (20), showing that cells in the adult CNS derived from total BM transplantation do not express such markers. This apparent conflict could be explained by the use of different experimental conditions. The use of purified HSPCs instead of total BM cells may affect the outcome of the transplant procedure because the former are highly enriched in the ability to give rise to new microglia-like cells, as per our data. Moreover, the use of intense irradiation in addition to BU for mice conditioning (20) could have induced the recruitment of circulating myeloid cells across a damaged BBB, which is not instead affected by BU alone, which we used (10). In support of these observations, μ from transplanted animals expressed at levels similar to ADULT_CT μ genes associated with mature microglia function by Matcovitch-Natan and colleagues (35). A less homogenous path was observed in TAμ from transplanted mice. Of note, an additional analysis evaluating the transcriptomic differences between μ and TAμ cells showed that newly formed cells after transplant, particularly TAμ, were characterized by up-regulation of processes related to neuronal function, neuron migration, differentiation, and regulation of synaptic plasticity, as expected in microglia under maturation (32, 33). In comparison with the transplant progeny, control μ cells showed up-regulation of mature immune function processes, among others. Assessing whether these observations could indicate that TAμ cells could be more immature than μ, as also suggested by the kinetics of donor cell appearance in these two subsets after transplant (earlier and more abundant in TAμ and late appearing and progressively increasing in μ), will require dedicated fate mapping in vivo studies.
Experimental conditions associated with the murine transplant setting, as well as artifacts related to the artificial chimeric nature of the human-into-mouse experiments, constitute intrinsic biases of our study that might affect the ability to predict reproducibility of our findings in humans. However, the consistent favorable outcome of HCT-transplant approaches, including HSC GT, and related biochemical findings in murine models and humans suggest that the described phenomena could occur in humans similar to what was observed in animal models.
In summary, the present data provide strong evidence that reconstitution of cells with microglia features occurs upon HSPC transplantation. Generation of these cells is significantly enhanced by delivery (and codelivery) of cells into the lateral cerebral ventricles. Proper selection and dosing of the cells to be administered may further enhance this process for clinical translation. We demonstrated that intracerebroventricular cell delivery is endowed with a curative potential for LSDs and potentially other neurodegenerative conditions for which timely and sustained protein delivery to the brain is presumed to be therapeutic. The demonstration of the feasibility and safety of the intracerebroventricular injection of HSPCs will be the objective of a follow-up work intended at the clinical translation of this approach.
MATERIALS AND METHODS
Study design
The objectives of this study were to:
(1) Study the myeloid cell engraftment in the brain after intracerebroventricular injection of HSPCs;
(2) Assess the contribution of HSPC cell fractions to microglia-like cell reconstitution in the intracerebroventricular (and intravenous) transplant setting;
(3) Characterize the gene expression signature of intracerebroventricularly (and intravenously) transplanted cell progeny;
(4) Study the potential therapeutic impact of intracerebroventricular HSPC delivery.
For each of these objectives, specific experimental designs were planned and then executed in the context of controlled laboratory experiments grouped for thematic relevance. For each of the indicated experiments, multiple (minimum of two) experimental replicates were performed to generate an adequate number of animals in each group. In each of these experimental replicates, animals from all experimental cohorts were generated. Experimental cohort size was determined on the basis of previously collected data in the same experimental settings and by interim analysis of results. No data were excluded from the analysis. Investigators conducting the analyses were blinded to mice code up to the stage of data analysis. For details on gene expression data analysis, please refer to the dedicated sections below and in the Supplementary Materials.
Study the myeloid cell engraftment in brain after intracerebroventricular injection of HSPCs
Murine lineage− HSPCs were transplanted into BU-myeloablated recipients after labeling with GFP-encoding LVs. The short- and long-term fate of the transplanted cells were analyzed by flow cytometry and histology into recipient wild-type mice.
Assess the contribution of HSPC cell fractions to microglia-like cell reconstitution in the intracerebroventricular (and intravenous) transplant setting
Subpopulations at different stages of maturity identified within the HSPC pool by previously validated markers were differentially labeled and transplanted intravenously or intracerebroventricularly (as detailed below) into myeloablated recipients to identify the contribution of each subpopulation to new myeloid cells in the brain. This contribution was assessed by flow cytometry and histology.
Characterize the gene expression signature of intracerebroventricularly (and intravenously) transplanted cell progeny
Myeloid cells (differentiating μ and TAμ and, whenever feasible, GFP+ and GFP− cells) were sorted from the brain of intravenously and intracerebroventricularly transplanted mice and control mice 3 months after transplant. These cells were then subjected to RNA extraction, measurement of the expression of specific genes (see Results and Materials and Methods for details), and gene expression analysis. Control cells were retrieved from naïve adult mice (μ) and p10 untreated mice (TAμ).
Study the potential therapeutic impact of intracerebroventricular HSPC delivery
Human CB CD34+ cells were transduced with either GFP- or ARSA-encoding LVs and then transplanted intravenously, intracerebroventricularly, or intravenously + intracerebroventricularly into myeloablated immunodeficient recipients, also carrying mutations in the murine ortholog of the human ARSA gene in the case of ARSA_LV transduction, to assess (i) the reproducibility of the previous results using therapeutically relevant human HSPCs and (ii) the therapeutic relevance of the intracerebroventricular route used in enhancing the ability of HCT/HSC GT to deliver lysosomal enzyme to the brain of a prototypical LSD animal model. Transplanted mice were then monitored for the appearance of a human myeloid progeny in the brain by flow cytometry and histology. Moreover, ARSA activity was measured in the brain of transplanted Rag−/−γ−chain−/−As2−/− mice. The different experimental groups were compared to each other and to controls.
Mice studies
C57BL6/J and C57BL/6-Ly5.1 mice were provided by Charles River. NSG mice were purchased by The Jackson Laboratory. Rag−/−γ−chain−/−As2−/− and Rag−/−γ-chain−/−As2+/+ mice were generated in the animal facility at San Raffaele Scientific Institute (40). Fgd5ZsGr·ZsGr/+ (Fgd5-ZsGreen) (Stock no. 027788, The Jackson Laboratory) was provided by Derrick J. Rossi’s laboratory, Harvard University/Boston Children’s Hospital (25).
All procedures were approved by the Animal Care and Use Committee of the Fondazione San Raffaele del Monte Tabor [Institutional Animal Care and Use Committee (IACUC) 573] and communicated to the Ministry of Health and local authorities according to Italian law and by Boston Children’s Hospital and Dana-Farber Cancer Institute Committees on Animals (IACUC 3198 and 15-031).
Isolation, transduction, and transplantation of murine hematopoietic cells
Young adult mice (5 to 8 weeks) were killed with CO2, and BM was harvested by flushing the femurs and tibias. Murine HSPCs were purified by lineage− selection, transduced with LV, and transplanted by tail vein injection as previously described (10). See the Supplementary Materials for additional details on subpopulation isolation, transduction, transplantation, and intracerebroventricular injection.
Isolation, transduction, and transplantation of human CD34+ cells
Human CB-HSPCs were purchased from Lonza (2C-101). After transduction, 5 × 105 cells were infused into the tail vein or into the lateral brain ventricle of preconditioned 7- to 9-week-old female NSG mice or postnatal day 2 neonatal Rag−/−γ−chain−/−As2−/−. Supplementary Materials report additional details on subpopulation isolation, transduction, and transplantation.
Flow cytometric analysis
Cells from BM and brain were analyzed by FACS (LSR Fortessa, Becton Dickinson) upon resuspension in blocking solution (phosphate-buffered saline, 5% fetal bovine serum, 1% bovine serum albumin) and labeling at 4°C for 15 min with different antibodies.
Immunofluorescence analysis
Brains were serially cut in the sagittal planes on a cryostat in 15-μm sections. Brain sections were obtained from the contralateral side of cell injection of intracerebroventricularly transplanted mice. A detailed description of immunofluorescence is available in the Supplementary Materials.
RNA extraction and gene expression analysis by real-time PCR
Total RNA was isolated for gene expression analysis from total CD45+CD11b+, μ, and TAμ [sorted according to the expression of CD45, CD11b, and GFP (only HCT mice)] of adult or P10 naïve control and transplanted mice. Moreover, we extracted RNA from macrophages (sorted according to the expression of CD45, CD11b, F4/80, Ly6C, and GFP) of HCT mice. A detailed description of RNA extraction, quantitation, and retrotranscription is available in the Supplementary Materials. A custom-designed TaqMan-based microfluidic card gene expression assay (Applied Biosystems) was used to measure the expression of 16 selected genes and fold change expression of selected microglia genes in μ and Taμ cells retrieved from intracerebroventricularly or intravenously transplanted mice, or P10naïve _ versus ADULT CT_μ was calculated using the 2−ΔΔCt method (41). See details in the Supplementary Materials.
RNA-seq analysis
Total RNA was extracted using the RNeasy Plus Micro Kit (Qiagen), and RNA-seq libraries were prepared with Ovation RNA-seq System V2 (NuGen). Barcoded complementary DNA fragments of total RNA were then sequenced at IGA Technologies on a HiSeq 2500 instrument from Illumina with 1 × 50 base pair SE (single end) chemistry. Reads were mapped against the reference genome mm10 using STAR version 2.3.0e_r291 (42), supplied with the gene annotation of the Ensembl database (v72). Data were normalized to library size using the trimmed mean of M values method (43). Normalized counts were log2-transformed using the voom function in limma (linear models for microarray data) (44) before test for differential expression analysis. PCA was made in R with mixOmics. Raw reads of the previously published RNA-seq data in Gosselin et al. (30) were downloaded from the Gene Expression Omnibus (GEO) (GSE62826) and analyzed as detailed in Supplementary Materials and Methods. RNA-seq data have been submitted to the GEO database (accession no. GSE87799). Although the presence of a batch effect could be expected to affect our analysis, we could not apply the available statistical tools to remove it because the removal depends heavily on the presence of the same type of cells in both studies and our data set did not contain macrophages. Despite not removing it, we found that, for the subset of microglial genes published by Butovsky et al. (26), the batch effect was minor.
To assess functional associations of μCT, μBU_TX, and TAμBU_TX cells, enrichment analysis was performed by GSEA preranked on GO biological processes (31). Significant enrichments were clustered on the basis of semantic similarities with GoSemSim (45).
Statistical analysis
All statistical tests were two-sided. For comparisons other than RNA-seq results, Student’s t test was used for two-group comparisons. For comparisons with more than two groups, one-way ANOVA with Bonferroni post hoc test was used; for multiple variable comparisons, two-way ANOVA was applied. Differences were considered statistically significant at a value of *P < 0.05 (*0). **P < 0.01, ***P < 0.001. In all figures with error bars, the graphs depict means ± SD.
Supplementary Material
Acknowledgments
We acknowledge FRACTAL (Flow cytometry Resource and Advanced Cytometry Technical Applications Laboratory) and ALEMBIC (Advanced Light and Electron Microscopy BioImaging Center) at San Raffaele Scientific Institute and L. Sergi Sergi for technical support and S. Martino, F. Cecere, L. S. Sergi, A. Lombardo, E. Zonari, B. Gentner, and G. Tonon for their support. Funding: This study was funded by the European Community (Consolidator European Research Council 617162), the Italian Ministry of Health (GR-2011-02347261), Telethon Foundation (TGT_B01), and Boston Children’s Hospital (to A.B.). Author contributions: Conceptualization: A.B. and A.C.; methodology: A.B., A.C., R.M., J.M.G.-M., E.C., M.P., D.J.R., and B.S.G.; investigation: A.C., R.M., J.M.G.-M., E.C., A.M., M.P., D.J.R., and B.S.G.; writing (original draft): A.B., A.C., R.M, and J.M.G.-M.; writing (review and editing): A.B., A.C., J.M.G.-M., and D.J.R.; funding acquisition, resources, and supervision: A.B. Competing interests: A.C., R.M., M.P., and A.B. are authors on a patent application related to this work filed by Boston Children’s Hospital, Ospedal San Raffaele, and Politecnico of Milano (application no. PCT/US17/56774, filed 16 October 2017). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. RNA-seq data are available under accession code GSE87799, and data reanalyzed from Gosselin et al. (30) are available under accession code GSE62826.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/12/e1701211/DC1
Supplementary Materials and Methods
fig. S1. Experimental schemes for the different transplantation settings described in the paper, using both mouse and human HSPCs.
fig. S2. Analysis of the engraftment of murine HSPC subpopulations in BU_TX or irradiated mice.
fig. S3. FACS plots of cell populations sorted for gene expression analysis.
fig. S4. Evaluation of hCD34+ cell engraftment in BM and brain of transplanted NSG mice.
table S1. ANOVA P values with Tukey’s post hoc test of contrasts between cells in Fig. 4B.
table S2. Differential expression of RNA-seq profiles of μ and TAμ cell populations.
table S3A. Genes up-regulated in μCT versus μBU_TX as per Fig. 5A.
table S3B. Genes down-regulated in μCT versus μBU_TX as per Fig. 5B.
table S3C. Genes up-regulated in μCT versus TAμBU_TX as per Fig. 5C.
table S3D. Genes down-regulated in μCT versus TAμBU_TX as per Fig. 5D.
table S4. Moderated t test (limma) after FDR (46) adjustment for the different contrast of interest.
REFERENCES AND NOTES
- 1.Cartier N., Hacein-Bey-Abina S., Bartholomae C. C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L., Mahlaoui N., Kiermer V., Mittelstaedt D., Bellesme C., Lahlou N., Lefrère F., Blanche S., Audit M., Payen E., Leboulch P., l’Homme B., Bougnères P., Von Kalle C., Fischer A., Cavazzana-Calvo M., Aubourg P., Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326, 818–823 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Beam D., Poe M. D., Provenzale J. M., Szabolcs P., Martin P. L., Prasad V., Parikh S., Driscoll T., Mukundan S., Kurtzberg J., Escolar M. L., Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol. Blood Marrow Transplant. 13, 665–674 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S., Benedicenti F., Vallanti G., Biasco L., Leo S., Kabbara N., Zanetti G., Rizzo W. B., Mehta N. A. L., Cicalese M. P., Casiraghi M., Boelens J. J., Del Carro U., Dow D. J., Schmidt M., Assanelli A., Neduva V., Di Serio C., Stupka E., Gardner J., von Kalle C., Bordignon C., Ciceri F., Rovelli A., Roncarolo M. G., Aiuti A., Sessa M., Naldini L., Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341, 1233158 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Sessa M., Lorioli L., Fumagalli F., Acquati S., Redaelli D., Baldoli C., Canale S., Lopez I. D., Morena F., Calabria A., Fiori R., Silvani P., Rancoita P. M. V., Gabaldo M., Benedicenti F., Antonioli G., Assanelli A., Cicalese M. P., del Carro U., Sora M. G. N., Martino S., Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 388, 476–487 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Corti S., Locatelli F., Donadoni C., Guglieri M., Papadimitriou D., Strazzer S., Del Bo R., Comi G. P., Wild-type bone marrow cells ameliorate the phenotype of SOD1-G93A ALS mice and contribute to CNS, heart and skeletal muscle tissues. Brain 127, 2518–2532 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Simard A. R., Soulet D., Gowing G., Julien J.-P., Rivest S., Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Ajami B., Bennett J. L., Krieger C., Tetzlaff W., Rossi F. M. V., Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–1543 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Ajami B., Bennett J. L., Krieger C., McNagny K. M., Rossi F. M. V., Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 14, 1142–1149 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Biffi A., Capotondo A., Fasano S., del Carro U., Marchesini S., Azuma H., Malaguti M. C., Amadio S., Brambilla R., Grompe M., Bordignon C., Quattrini A., Naldini L., Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Invest. 116, 3070–3082 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capotondo A., Milazzo R., Politi L. S., Quattrini A., Palini A., Plati T., Merella S., Nonis A., di Serio C., Montini E., Naldini L., Biffi A., Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. U.S.A. 109, 15018–15023 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mildner A., Schmidt H., Nitsche M., Merkler D., Hanisch U.-K., Mack M., Heikenwalder M., Brück W., Priller J., Prinz M., Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 10, 1544–1553 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Jeyakumar M., Thomas R., Elliot-Smith E., Smith D. A., van der Spoel A. C., d’Azzo A., Perry V. H., Butters T. D., Dwek R. A., Platt F. M., Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 126, 974–987 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Wada R., Tifft C. J., Proia R. L., Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. U.S.A. 97, 10954–10959 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohmi K., Greenberg D. S., Rajavel K. S., Ryazantsev S., Li H. H., Neufeld E. F., Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. U.S.A. 100, 1902–1907 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eichler F. S., Ren J.-Q., Cossoy M., Rietsch A. M., Nagpal S., Moser A. B., Frosch M. P., Ransohoff R. M., Is microglial apoptosis an early pathogenic change in cerebral X-linked adrenoleukodystrophy? Ann. Neurol. 63, 729–742 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Boillee S., Yamanaka K., Lobsiger C. S., Copeland N. G., Jenkins N. A., Kassiotis G., Kollias G., Cleveland D. W., Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Jack C. S., Arbour N., Manusow J., Montgrain V., Blain M., McCrea E., Shapiro A., Antel J. P., TLR signaling tailors innate immune responses in human microglia and astrocytes. J. Immunol. 175, 4320–4330 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Cartier N., Lewis C.-A., Zhang R., Rossi F. M. V., The role of microglia in human disease: Therapeutic tool or target? Acta Neuropathol. 128, 363–380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkinson F. L., Sergijenko A., Langford-Smith K. J., Malinowska M., Wynn R. F., Bigger B. W., Busulfan conditioning enhances engraftment of hematopoietic donor-derived cells in the brain compared with irradiation. Mol. Ther. 21, 868–876 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett M. L., Bennett F. C., Liddelow S. A., Ajami B., Zamanian J. L., Fernhoff N. B., Mulinyawe S. B., Bohlen C. J., Adil A., Tucker A., Weissman I. L., Chang E. F., Li G., Grant G. A., Hayden Gephart M. G., Barres B. A., New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. U.S.A. 113, E1738–E1746 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ginhoux F., Greter M., Leboeuf M., Nandi S., See P., Gokhan S., Mehler M. F., Conway S. J., Ng L. G., Stanley E. R., Samokhvalov I. M., Merad M., Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dar A., Kollet O., Lapidot T., Mutual, reciprocal SDF-1/CXCR4 interactions between hematopoietic and bone marrow stromal cells regulate human stem cell migration and development in NOD/SCID chimeric mice. Exp. Hematol. 34, 967–975 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Rettig M. P., Ansstas G., DiPersio J. F., Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia 26, 34–53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugiyama T., Kohara H., Noda M., Nagasawa T., Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Gazit R., Mandal P. K., Ebina W., Ben-Zvi A., Nombela-Arrieta C., Silberstein L. E., Rossi D. J., Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J. Exp. Med. 211, 1315–1331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butovsky O., Jedrychowski M. P., Moore C. S., Cialic R., Lanser A. J., Gabriely G., Koeglsperger T., Dake B., Wu P. M., Doykan C. E., Fanek Z., Liu L., Chen Z., Rothstein J. D., Ransohoff R. M., Gygi S. P., Antel J. P., Weiner H. L., Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiu I. M., Morimoto E. T. A., Goodarzi H., Liao J. T., O’Keeffe S., Phatnani H. P., Muratet M., Carroll M. C., Levy S., Tavazoie S., Myers R. M., Maniatis T., A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385–401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hickman S. E., Kingery N. D., Ohsumi T. K., Borowsky M. L., Wang L.-c., Means T. K., El Khoury J., The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grommes C., Lee C. Y. D., Wilkinson B. L., Jiang Q., Koenigsknecht-Talboo J. L., Varnum B., Landreth G. E., Regulation of microglial phagocytosis and inflammatory gene expression by Gas6 acting on the Axl/Mer family of tyrosine kinases. J. Neuroimmune Pharmacol. 3, 130–140 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gosselin D., Link V. M., Romanoski C. E., Fonseca G. J., Eichenfield D. Z., Spann N. J., Stender J. D., Chun H. B., Garner H., Geissmann F., Glass C. K., Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colonna M., Butovsky O., Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tay T. L., Savage J. C., Hui C. W., Bisht K., Tremblay M.-È., Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J. Physiol. 595, 1929–1945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyamoto A., Wake H., Moorhouse A. J., Nabekura J., Microglia and synapse interactions: Fine tuning neural circuits and candidate molecules. Front. Cell. Neurosci. 7, 70 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matcovitch-Natan O., Winter D. R., Giladi A., Vargas Aguilar S., Spinrad A., Sarrazin S., Ben-Yehuda H., David E., Zelada González F., Perrin P., Keren-Shaul H., Gury M., Lara-Astaiso D., Thaiss C. A., Cohen M., Bahar Halpern K., Baruch K., Deczkowska A., Lorenzo-Vivas E., Itzkovitz S., Elinav E., Sieweke M. H., Schwartz M., Amit I., Microglia development follows a stepwise program to regulate brain homeostasis. Science 353, aad8670 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Biffi A., De Palma M., Quattrini A., Del Carro U., Amadio S., Visigalli I., Sessa M., Fasano S., Brambilla R., Marchesini S., Bordignon C., Naldini L., Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Invest. 113, 1118–1129 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visigalli I., Delai S., Politi L. S., Di Domenico C., Cerri F., Mrak E., D’Isa R., Ungaro D., Stok M., Sanvito F., Mariani E., Staszewsky L., Godi C., Russo I., Cecere F., del Carro U., Rubinacci A., Brambilla R., Quattrini A., Di Natale P., Ponder K., Naldini L., Biffi A., Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 116, 5130–5139 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gentner B., Visigalli I., Hiramatsu H., Lechman E., Ungari S., Giustacchini A., Schira G., Amendola M., Quattrini A., Martino S., Orlacchio A., Dick J. E., Biffi A., Naldini L., Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci. Transl. Med. 2, 58ra84 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Simard A. R., Rivest S., Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 18, 998–1000 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Meneghini V., Frati G., Sala D., De Cicco S., Luciani M., Cavazzin C., Paulis M., Mentzen W., Morena F., Giannelli S., Sanvito F., Villa A., Bulfone A., Broccoli V., Martino S., Gritti A., Generation of human induced pluripotent stem cell-derived bona fide neural stem cells for ex vivo gene therapy of metachromatic leukodystrophy. Stem Cells Transl. Med. 6, 352–368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Livak K. J., Schmittgen T. D., Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson M. D., Oshlack A., A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu G., Li F., Qin Y., Bo X., Wu Y., Wang S., GOSemSim: An R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26, 976–978 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/12/e1701211/DC1
Supplementary Materials and Methods
fig. S1. Experimental schemes for the different transplantation settings described in the paper, using both mouse and human HSPCs.
fig. S2. Analysis of the engraftment of murine HSPC subpopulations in BU_TX or irradiated mice.
fig. S3. FACS plots of cell populations sorted for gene expression analysis.
fig. S4. Evaluation of hCD34+ cell engraftment in BM and brain of transplanted NSG mice.
table S1. ANOVA P values with Tukey’s post hoc test of contrasts between cells in Fig. 4B.
table S2. Differential expression of RNA-seq profiles of μ and TAμ cell populations.
table S3A. Genes up-regulated in μCT versus μBU_TX as per Fig. 5A.
table S3B. Genes down-regulated in μCT versus μBU_TX as per Fig. 5B.
table S3C. Genes up-regulated in μCT versus TAμBU_TX as per Fig. 5C.
table S3D. Genes down-regulated in μCT versus TAμBU_TX as per Fig. 5D.
table S4. Moderated t test (limma) after FDR (46) adjustment for the different contrast of interest.