

Abstract
Background
Hyperuricemia is associated with hypertension, with elevated serum uric acid levels postulated to have a causal role in the development of hypertension. Consequently, serum uric acid reduction may help lower blood pressure (BP). A Phase 2, double‐blind, placebo‐controlled trial was conducted to assess the potential BP‐lowering effects of the xanthine oxidase inhibitor febuxostat in subjects with hypertension and hyperuricemia (serum uric acid ≥0.42 mmol/L [≥7.0 mg/dL]).
Methods and Results
Subjects (n=121) were randomized 1:1 to febuxostat 80 mg once daily or to placebo. The primary end point was change from baseline to Week 6 in 24‐hour mean ambulatory systolic BP (SBP). Additional end points included the following: change from baseline to Week 3 in 24‐hour mean SBP and changes from baseline to Weeks 3 and 6 in 24‐hour mean ambulatory diastolic BP, serum uric acid, mean daytime and nighttime ambulatory SBP/diastolic BP, and clinic SBP/diastolic BP. For the overall study population, there were no significant differences between febuxostat and placebo for changes from baseline to Weeks 3 or 6 in ambulatory, daytime or nighttime, or clinic SBP or diastolic BP. However, in a preplanned subgroup analysis, there was a significant decrease in SBP from baseline to Week 6 in subjects with normal renal function (estimated glomerular filtration rate ≥90 mL/min) treated with febuxostat versus placebo; least squares mean difference, −6.7; 95% confidence interval −13.3 to −0.0; P=0.049.
Conclusions
This study suggests that febuxostat may lower BP in hyperuricemic patients with hypertension and normal renal function; further studies should be conducted to confirm this finding.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT01496469.
Keywords: ambulatory blood pressure monitoring, febuxostat, hypertension, hyperuricemia, serum urate, uric acid
Subject Categories: Clinical Studies, Hypertension, Blood Pressure
Clinical Perspective
What Is New?
The present study demonstrated that intervention with febuxostat in hyperuricemic subjects with normal renal function could have additional benefit related to the blood pressure response to urate‐lowering therapy.
However, febuxostat appeared to have less effect on blood pressure–lowering in subjects with impaired renal function.
What Are the Clinical Implications?
Patients with kidney disease and long‐standing hypertension may not be as responsive to urate‐lowering therapy because of the association of hypertension and uric acid with vascular damage.
Therefore, early intervention with xanthine oxidase therapy in patients with elevated serum uric acid, before the onset of kidney disease, may be beneficial based on the current evidence.
The key precursor to gout is hyperuricemia, which leads to the deposition of monosodium urate crystals in the joints or soft tissues. Xanthine oxidase inhibitors effectively treat gout through the inhibition of urate synthesis, and by impairing the conversion of hypoxanthine and xanthine to uric acid.
Febuxostat is a xanthine oxidase inhibitor indicated for the long‐term management of hyperuricemia in patients with gout.1 Febuxostat can be used in patients with reduced renal function, as mild‐to‐moderate renal impairment does not reduce its effect on serum uric acid (sUA).2
Experimental evidence suggests that sUA could be important in the pathogenesis and progression of renal disease.3 Preclinical and clinical data show that hyperuricemia induces endothelial dysfunction, which is associated with end‐stage renal disease.4, 5 Febuxostat contributed to improvements in endothelial dysfunction by attenuating oxidative stress in a diabetic rat model.6 In addition, preclinical data suggest that febuxostat may also protect the kidneys from acute renal ischemic reperfusion injury and contribute to preservation of kidney function.7 Febuxostat has also been shown to slow the decline in estimated glomerular filtration rate in subjects with chronic kidney disease stages 3 and 4 compared with placebo, although the sample was small and follow‐up was limited to 6 months.8 Furthermore, an analysis of subjects receiving febuxostat in Phase 3 studies demonstrated that greater sustained decreases in sUA levels were associated with less renal function decline.9
There is a growing interest in sUA as an independent risk factor for hypertension because of the strong association between hyperuricemia and arterial hypertension, which appears to have a complex association with endothelial dysfunction.10 A meta‐analysis of 25 studies (n=97 824) demonstrated that hyperuricemia was associated with a higher risk of incident hypertension.11 Furthermore, a review of several epidemiologic studies reported a relationship between sUA levels and a wide variety of cardiovascular conditions.12
The mechanism for the association between hyperuricemia and arterial hypertension remains unclear. It is also unclear whether endothelial dysfunction is a cause or an effect of hypertension. Soluble urate may have a direct pathogenic role.13, 14 Small clinical trials have demonstrated that urate‐lowering therapy with allopurinol and probenecid lowers blood pressure (BP) in adolescents with hypertension.15, 16 In a retrospective analysis of febuxostat Phase 3 studies, febuxostat significantly improved systolic (SBP) and diastolic BP (DBP) over 28 weeks in subjects with untreated hypertension and gout.17 The reductions in SBP and DBP were ≈10 and 8 mm Hg, respectively.
The potential benefit of urate‐lowering therapy on BP in subjects with hyperuricemia and stable hypertension remains to be fully elucidated, as does the impact of renal impairment on these treatment effects. Consequently, the present study was conducted to test the hypothesis that febuxostat significantly reduced BP compared with placebo in patients with hyperuricemia and stable hypertension, and evaluate any treatment effects across patient subgroups based on baseline renal function. To our knowledge, this was the first prospective placebo‐controlled study to investigate the effect of febuxostat therapy on the BP of adult patients with hyperuricemia and stable hypertension.
Methods
Study Design
This was a Phase 2 double‐blind, placebo‐controlled, multicenter, proof‐of‐concept study conducted at 29 sites in North America (NCT01496469). The study consisted of a screening visit (Day −21), a 2‐week single‐blind placebo run‐in period (Days −14 to −1), and a 6‐week treatment period (Figure S1). Subjects were required to complete 3 clinic visits for BP measurements during the placebo run‐in on Days −14, −7, and −1 to ensure that only subjects who met the criteria for stable BP were randomized into the study. The study also included a safety follow‐up visit 1 week following the last dose of study drug.
Study investigators were selected based on qualifications and experience, the adequacy of the facilities to perform study‐related procedures, and the adequacy of the site to manage study‐related activities and requirements. Each investigator conducted the study according to the Declaration of Helsinki and the International Conference on Harmonisation Harmonised Tripartite Guideline for good clinical practices and Food and Drug Administration regulatory requirements. The study protocol was approved by the Institutional Review Boards of each participating site. All subjects provided written informed consent.
Subjects
Male or female subjects ≥18 years old who were taking ≤2 baseline (Day 1) BP medications were eligible for enrollment. Subjects enrolled also had to have documented and stable hypertension, defined as average clinic SBP of ≥145 and ≤165 mm Hg or average clinic DBP of ≥90 and ≤105 mm Hg, at the Day −21 screening visit.
Subjects also had to demonstrate SBP ≥130 and ≤165 mm Hg, as measured by 24‐hour ambulatory blood pressure monitoring (ABPM). Baseline ABPM measurements were conducted for 24 hours starting at 8:00 AM (±2 hours) on Day −1 until Day 1. If the ABPM measurement was unsuccessful (documented equipment failure or failed quality reading), it was to be repeated within 72 hours of the completion of the unsuccessful ABPM measurement, and the subject continued taking the single‐blind placebo medication until the day of the repeat ABPM. If the ABPM was unsuccessful the second time, the subject was considered a run‐in failure. In addition, subjects were required to have a sUA level of ≥0.42 mmol/L not associated with gout at the screening visit.
Subjects were excluded if they had an average clinic SBP >165 mm Hg or DBP >105 mm Hg at 1 or more visits during the placebo run‐in period (Days −14 to −1). Similarly, subjects with secondary causes of high BP or high sUA, a history within 6 months before screening of cardiovascular events, type 1 or inadequately controlled type 2 diabetes mellitus, or other major medical conditions that may have interfered with treatment safety or compliance were excluded.
Subjects who met the BP criteria described above at 2 of the 3 placebo run‐in visits, and who did not have increases or decreases of >10 mm Hg in either clinic SBP or DBP measurements between visits (Day −14 to −7, Day −7 to −1, or Day −14 to −1), were considered to have stable BP and were then eligible for randomization in the study.
Subjects were recruited via a central advertising campaign conducted by Blue Chip Marketing Worldwide (Chicago, IL) and local advertising campaigns carried out by individual participating sites. In addition, individual sites screened and included subjects from their own databases, where appropriate.
Randomization
Subjects were randomized to 1 of 2 arms in a 1:1 ratio at Day 1 (baseline randomization visit) via an interactive voice response system in the order in which they were enrolled. Randomization was stratified using 2 strata: subjects taking an angiotensin‐converting enzyme inhibitor (ACEi) or an angiotensin receptor blocker (ARB) at baseline versus subjects not taking these drugs at baseline. Randomization was stratified according to baseline ACEi/ARB use in case the BP benefit of urate lowering was limited to subjects with an intact renin–angiotensin–aldosterone system, as suggested by prior clinical and nonclinical data.14 At least 50% of subjects enrolled were not to be taking an ACEi or an ARB. Once 60 subjects taking an ACEi or an ARB were randomized, no additional subjects taking these drugs were allowed to enroll.
Treatments
During the 14‐day placebo run‐in period, subjects orally self‐administered 1 placebo capsule every day. Medication was dispensed and managed via the interactive voice response system. After completion of the single‐blind placebo run‐in period, subjects with stable clinic BP, and a mean 24‐hour ambulatory SBP of ≥130 and ≤165 mm Hg at the Day 1 visit and who continued to meet the inclusion criteria, received either febuxostat 80 mg once daily or placebo on Day 1. Concomitant medications, including up to 2 previous BP medications, were continued at stable doses throughout the study. Treatment compliance (%) was calculated as (number of capsules dispensed−number of capsules returned)/(number of days on drug)×100.
Efficacy Assessments
The primary efficacy end point was change from baseline to Week 6 in 24‐hour mean ambulatory SBP. Secondary efficacy end points were change from baseline to Week 6 in 24‐hour mean ambulatory DBP and sUA. Additional BP end points were change from baseline to Week 3 in 24‐hour mean ambulatory SBP and DBP, and change from baseline to Weeks 3 and 6 in mean daytime and nighttime ambulatory SBP and DBP, and clinic SBP and DBP.
Twenty‐four‐hour ABPM was recorded using an automated, lightweight device (Spacelabs 90207 ambulatory blood pressure monitor, Snoqualmie, WA) that did not interfere with daily activities and was preprogrammed with a 24‐hour cuff‐inflation schedule. Twenty‐four‐hour ABPM was measured at baseline (between Days −1 and 1) and from the end of the Weeks 3 and 6 visits (Figure S1). The daytime and nighttime periods for ABPM were defined as 06:00 to 22:00 and 22:01 to 05:59, respectively.
Planned subgroup analyses for the primary efficacy end point were summarized according to baseline renal function. Categorically, renal function was defined as moderately impaired (estimated glomerular filtration rate 30 to <60 mL/min), mildly impaired (60 to <90 mL/min), or normal (≥90 mL/min) renal function. The estimated glomerular filtration rate was calculated using the Modification of Diet in Renal Disease formula as follows: when serum creatinine (Scr) is in μmol/L, glomerular filtration rate =30 849×standardized Scr −1.154×age−0.203×1.212 (if black)×0.742 (if female).18, 19
Additional planned analyses included BP response in subgroups according to use of a concomitant ACEi or ARB.
Analyses of soluble and expression‐based inflammation and cardiovascular biomarkers were exploratory end points that will be presented separately.
Safety Assessments
Safety assessments included a physical examination, 12‐lead ECG, vital signs, hematologic/serum chemistries, and urinalysis at baseline and at the end of Weeks 3 and/or 6. A safety follow‐up visit was scheduled ≈1 week after the last dose of study medication for all subjects, including those who had discontinued prematurely.
Statistical Analyses
The full analysis set was used for efficacy analyses and included all subjects who were randomized and received at least 1 dose of double‐blind study medication. A subject was included in the analysis of a specific variable only when there was both a baseline value and at least 1 value during the double‐blind treatment period; missing data for all efficacy analyses were imputed using the last available postbaseline observation carried forward. Subjects were analyzed according to the treatment group to which they were randomized. The safety analysis set included all subjects who received at least 1 dose of double‐blind study medication. All routine safety analyses were based on the safety analysis set.
The HP‐Unix‐UX SAS v9.2 operating system was used for statistical analyses. Unless otherwise specified, all statistical tests and confidence intervals (CIs) were 2‐sided and conducted at the 0.05 significance level with no adjustment for multiple comparisons. The primary analysis was based on an ANCOVA model for change from baseline to Week 6 (or last on‐treatment visit) in 24‐hour mean ambulatory SBP. The model included treatment and prior use of an ACEi or ARB (yes/no) as factors and baseline 24‐hour mean ambulatory SBP as a covariate. The least squares (LS) mean, P value, and 2‐sided 95% CI of treatment differences were calculated. Similar ANCOVA analyses were performed on the 2 secondary end points: change from baseline to Week 6 in 24‐hour mean ambulatory DBP and sUA. Additional end points analyzed by ANCOVA included the following: change from baseline to Weeks 3 and 6 in mean daytime and nighttime ambulatory SBP and DBP and clinic SBP and DBP; change from baseline to Week 3 in 24‐hour mean ambulatory SBP, DBP, and sUA; and post hoc analysis of change from baseline to Week 6 in body weight. The Cochran–Mantel–Haenszel test was used to compare the percentage of subjects in each treatment group who demonstrated a decrease from baseline to Weeks 3 and 6 in mean ambulatory SBP ≥4 mm Hg and DBP ≥3 mm Hg. Prior use of an ACEi or ARB at baseline was a stratification variable. Safety and tolerability were assessed by evaluating the incidence of treatment‐emergent adverse events (TEAEs), cardiovascular adverse events, 12‐lead ECG findings, clinical laboratory tests, and vital signs. A TEAE was defined as any adverse event, regardless of relationship to study drug, which occurred from Day 1 through to 30 days after the last dose of the double‐blind study drug and summarized using the Medical Dictionary for Regulatory Activities (Version 17.0) terminology. No statistical inference was performed for safety end points.
Sample size was based on a total of 120 enrolled subjects (60 subjects per treatment group) and was sufficient to achieve 80% power to detect a difference of 6.0 mm Hg between the placebo and febuxostat treatment groups. This calculation assumed a dropout rate of 20% and a common SD of 10 mm Hg for the change from baseline in 24‐hour mean ambulatory SBP for both treatment groups.
Results
Subjects
Overall, 1034 subjects were screened and 121 subjects (placebo, n=60; febuxostat, n=61) were randomized. The majority of screened subjects did not meet entry criteria (Figure 1). The most common reason for discontinuation was voluntary withdrawal of consent and included 4 subjects in each treatment group who did not want to perform study procedures (ie, ABPM) or cited work/job conflicts, and 1 subject who withdrew at the request of his physician for a required surgical procedure. The mean treatment compliance over the double‐blind treatment period in the placebo and febuxostat groups was 100% and 99%, respectively, and the proportion of subjects who were ≥90% compliant was 97% and 95%, respectively.
Figure 1.
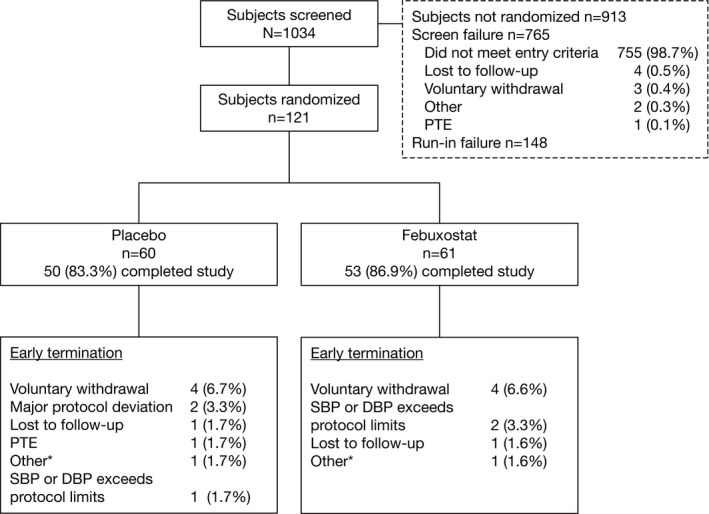
Disposition of subjects. *Reasons for other included subject who did not want to perform study procedures. DBP indicates diastolic blood pressure; PTE, pretreatment adverse event; SBP, systolic blood pressure.
The baseline demographic characteristics for the febuxostat and placebo treatment groups (and overall population) are presented in the Table (please see Table S1 for baseline demographic characteristics of treatment groups stratified by baseline renal function); baseline demographics were well balanced across treatment groups. The study population was predominantly male, white, and balanced for racial origin between treatment groups; mean age was 54 years. Incidences of concurrent medical conditions were similar between treatment groups, with a low concurrent incidence of clinically relevant conditions such as cardiac conditions (n=2) and stroke (n=0). The mean baseline sUA was similar between the placebo and febuxostat treatment groups (0.46 and 0.45 mmol/L, respectively); 71.9% (87/121) of subjects had sUA <0.48 mmol/L and 28.1% (34/121) had sUA ≥0.48 mmol/L. A total of 37.2% (45/121) of subjects had normal renal function and 62.8% (76/121) had mild‐to‐moderate renal impairment. Among the overall population, 42.1% (51/121) were treated with an ACEi/ARB; other concomitant BP medications included β‐blockers (12.4%), calcium channel blockers (12.4%), and diuretics (13.2%). ACEi/ARB treatment in the renal function subgroups was 44.4% (20/45), 39.7% (25/63), and 46.2% (6/13) in subjects with normal, mildly impaired, and moderately impaired renal function, respectively. Overall, 28.1% (34/121) of subjects were not treated with any BP medication: 26.7% (12/45), 31.7% (20/63), and 15.4% (2/13) in the subgroups with normal, mildly impaired, and moderately impaired renal function, respectively.
Table 1.
Baseline Characteristics of Study Population
| Characteristic | Overall Study Population (N=121) | Placebo (n=60) | Febuxostat (n=61) |
|---|---|---|---|
| Male sex, n (%) | 98 (81.0) | 48 (80.0) | 50 (82.0) |
| Age, y | |||
| Mean (SD) | 53.6 (10.6) | 55.1 (10.6) | 52.2 (10.5) |
| Range | 26 to 77 | 26 to 77 | 27 to 76 |
| Race, n (%) | |||
| American Indian or Alaska Native | 1 (0.8) | 0 | 1 (1.6)a |
| Asian | 15 (12.4) | 7 (11.7) | 8 (13.1) |
| Black | 21 (17.4) | 11 (18.3) | 10 (16.4) |
| White | 83 (68.6) | 42 (70.0) | 41 (67.2) |
| Other | 1 (0.8) | 0 | 1 (1.6) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 23 (19.0) | 9 (15.0) | 14 (23.0) |
| Not Hispanic or Latino | 98 (81.0) | 51 (85.0) | 47 (77.0) |
| Height (cm), mean (SD) | 172.72 (8.88) | 172.10 (8.56) | 173.33 (9.21) |
| Weight (kg), mean (SD) | 98.14 (20.46) | 95.35 (21.20) | 100.88 (19.49) |
| BMI (kg/m2), mean (SD) | 32.78 (5.57) | 31.99 (5.13) | 33.55 (5.91) |
| Smoking history, n (%) | |||
| Never smoked | 62 (51.2) | 32 (53.3) | 30 (49.2) |
| Ex‐smoker | 41 (33.9) | 20 (33.3) | 21 (34.4) |
| Current smoker | 18 (14.9) | 8 (13.3) | 10 (16.4) |
| Alcohol history, n (%) | |||
| Never drank | 46 (38.0) | 26 (43.3)a | 20 (32.8)a |
| Ex‐drinker | 6 (5.0) | 2 (3.3) | 4 (6.6) |
| Current drinker | 69 (57.0) | 32 (53.3) | 37 (60.7) |
| Renal function,b n (%) | |||
| Moderately impaired | 13 (10.7) | 8 (13.3) | 5 (8.2) |
| Mildly impaired | 63 (52.1) | 33 (55.0) | 30 (49.2) |
| Normal | 45 (37.2) | 19 (31.7) | 26 (42.6) |
| Baseline sUA, n (%) | |||
| <0.48 mmol/L | 87 (71.9) | 40 (66.7) | 47 (77.0) |
| ≥0.48 mmol/L | 34 (28.1) | 20 (33.3) | 14 (23.0) |
| BP medication, n (%) | |||
| None | 34 (28.1) | 16 (26.7)a | 18 (29.5) |
| 1 BP medication | 76 (62.8) | 37 (61.7) | 39 (63.9) |
| 2 BP medications | 11 (9.1) | 7 (11.7) | 4 (6.6) |
| BP medication type, n (%) | |||
| ACEi/ARB | 51 (42.1) | 25 (41.7) | 26 (42.6) |
| β‐blockers | 15 (12.4) | 8 (13.3) | 7 (11.5) |
| Ca++ channel blockers | 15 (12.4) | 7 (11.7) | 8 (13.1) |
| Diuretics | 16 (13.2) | 10 (16.7) | 6 (9.8) |
| Other | 1 (0.8) | 1 (1.7) | 0 |
ACEi indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; BMI, body mass index; BP, blood pressure; eGFR, estimated glomerular filtration rate; sUA, serum uric acid.
Total proportion does not add up to 100% because of rounding.
Renal function was defined as moderately impaired (eGFR 30 to <60 mL/min), mildly impaired (60 to <90 mL/min), or normal (≥90 mL/min); eGFR was calculated using the Modification of Diet in Renal Disease formula.
Efficacy
Analysis of BP
For the primary 24‐hour ABPM end point, there was no statistically significant difference between placebo and febuxostat in change from baseline to Week 6 SBP (Figure 2A). Results were similar for secondary (Week 6) and additional (Week 3) 24‐hour ABPM end points, with no significant differences observed between placebo and febuxostat for change from baseline to Weeks 3 or 6 DBP or Week 3 SBP. There was no significant difference between placebo and febuxostat in the analysis of daytime or nighttime mean SBP or DBP (Figure 2B). Clinic BP assessments also showed no significant difference between placebo and febuxostat at Weeks 3 or 6 (Figure 2C).
Figure 2.
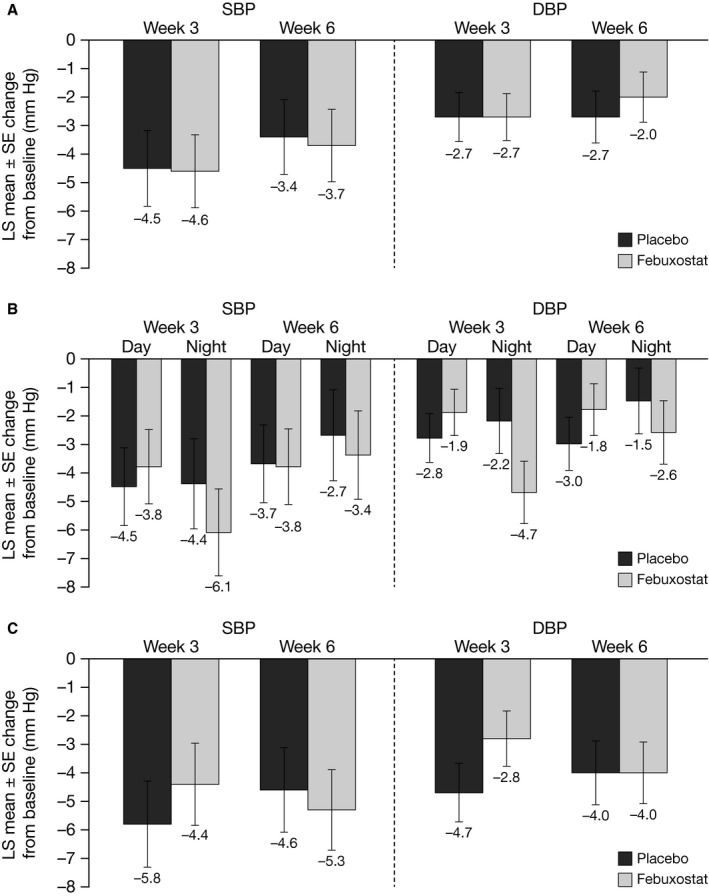
Mean change from baseline in (A) overall, (B) daytime and nighttime, and (C) clinic ambulatory SBP and DBP at Wks 3 and 6. DBP indicates diastolic blood pressure; LS, least squares; SBP, systolic blood pressure.
Analysis of change from baseline in sUA and weight
As expected, febuxostat significantly reduced sUA compared with placebo over the treatment period. The LS mean change from baseline to Week 3 in sUA was 0.0 and −0.19 mmol/L for placebo and febuxostat, respectively (LS mean difference −0.19; 95% CI, −0.22 to −0.16; P<0.001). At Week 6, the LS mean change from baseline was 0.01 and −0.19 mmol/L for placebo and febuxostat, respectively (LS mean difference −0.20; 95% CI, −0.23 to −0.17; P<0.001). An exploratory, post hoc analysis of LS mean (SE) change in weight from baseline to Week 6 showed that there was no weight change (0.0 [0.49] kg) in the placebo group and a reduction of −0.9 (0.47) kg in the febuxostat group. The difference between the treatment groups was not significant (LS mean difference −0.8; 95% CI, −2.2 to 0.5; P=0.223).
Planned subgroup analyses by renal function
The 24‐hour ABPM change from baseline to Week 3 or 6 by renal function subgroups is shown in Figure 3. There was a small, statistically significant difference between placebo and febuxostat in change from baseline SBP in the subgroup with normal renal function at Week 6 (LS mean difference −6.7; 95% CI, −13.3 to 0.0; P=0.049). No significant differences between placebo and febuxostat in change from baseline SBP to Weeks 3 or 6 were observed in the subgroups with mildly or moderately impaired renal function. Irrespective of renal function, febuxostat significantly reduced sUA versus placebo at Weeks 3 and 6 (Tables S2 and S3).
Figure 3.
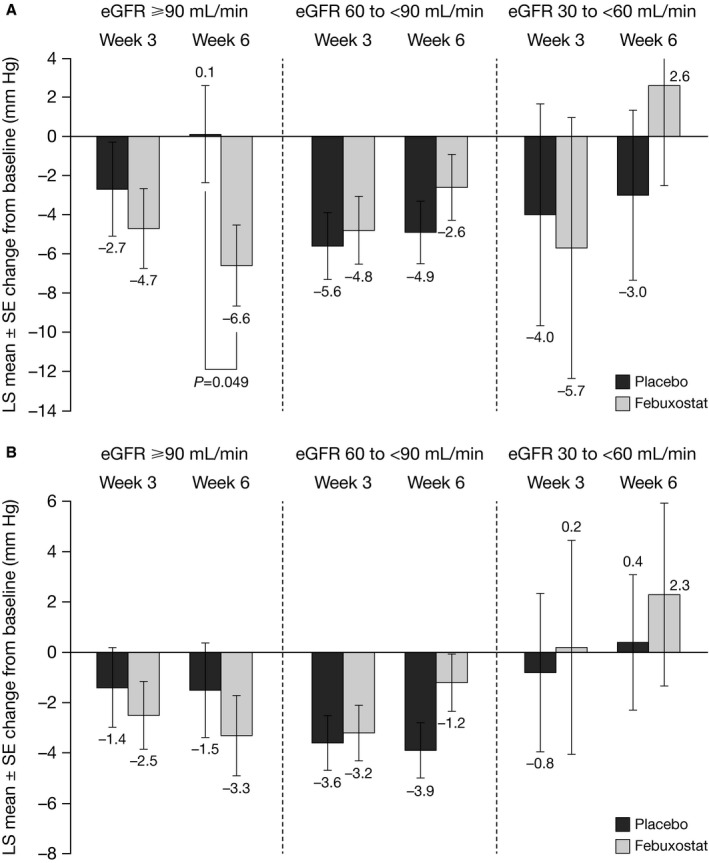
Mean change from baseline in ambulatory (A) SBP and (B) DBP at Wks 3 and 6 by renal function subgroups. DBP indicates diastolic blood pressure; eGFR, estimated glomerular filtration rate; LS, least squares; SBP, systolic blood pressure.
Additional subgroup and BP analyses
Tables S4 and S5 summarize the analysis of change from baseline at Week 6 in 24‐hour ambulatory SBP and DBP, respectively, by use of an ACEi/ARB. There was no significant difference between placebo and febuxostat in change from baseline SBP or DBP by use of an ACEi/ARB. However, febuxostat significantly reduced sUA versus placebo at Weeks 3 and 6 (P<0.001 for both time points), irrespective of ACEi/ARB use at baseline (Tables S6 and S7).
The analysis of the percentage of subjects who demonstrated a change from baseline to Week 3 or 6 in mean ambulatory SBP of ≥4 mm Hg and mean ambulatory DBP of ≥3 mm Hg is shown in Table S8. No significant differences between placebo and febuxostat were observed in the percentage of subjects with changes in ambulatory SBP ≥4 mm Hg or DBP ≥3 mm Hg at Week 3 or 6.
Safety
Treatment with febuxostat was well tolerated. The incidence of TEAEs was similar between the febuxostat and placebo groups (30.0% versus 24.6%, respectively). There were 2 nonfatal serious TEAEs during the study: 1 patient in the febuxostat group who was a current consumer of alcohol, had a body mass index of 35.3, and had a history of hypertension, hyperlipidemia, diabetes mellitus, and asthma, experienced a serious TEAE of coronary artery insufficiency and required hospitalization; 1 patient in the placebo group who had a body mass index of 28.9 and a history of hypertension experienced a serious TEAE of increased BP that was considered life threatening and required hospitalization.
Discussion
This proof‐of‐concept trial was conducted to evaluate the potential for any BP‐lowering effects of febuxostat, using ABPM measurements, in hypertensive subjects with hyperuricemia; most subjects (71.9%) were treated with at least 1 antihypertensive medication and 28.1% were not treated with any antihypertensive medication. The use of 24‐hour ABPM has the advantage of measuring BP during normal activities of daily living. It can be used to assess differences in BP between daytime and nighttime, and avoids the potential “white‐coat effect” of clinic readings.
Overall, treatment with febuxostat in subjects with confirmed hypertension, treated with up to 2 antihypertensive medications, and hyperuricemia showed no significant differences from placebo in change in ambulatory SBP or DBP from baseline to Week 3 or 6. Additionally, no significant differences between placebo and febuxostat were observed for other BP analyses for the overall population: changes in mean ambulatory daytime or nighttime SBP or DBP from baseline to Weeks 3 and 6, clinic BP assessments, and percentage of subjects with changes in ambulatory SBP ≥4 mm Hg or DBP ≥3 mm Hg at Weeks 3 and 6. As expected, febuxostat significantly reduced sUA at Weeks 3 and 6 versus placebo, and was well tolerated over the 6 weeks of treatment.
In the overall study population, 62.8% (76/121) of subjects had mild‐to‐moderate renal impairment; 40.8% (31/76) of these subjects were treated with an ACEi or ARB; 28.9% (22/76) with mild‐to‐moderate renal impairment were not taking any antihypertensive medication. In subgroup analyses, no statistically significant differences between placebo and febuxostat were observed for changes in ambulatory SBP or DBP from baseline to Week 3 or 6 in subgroups with impaired renal function, or those treated with an ACEi or ARB. Compared with placebo, a significant reduction from baseline sUA was observed in the febuxostat group at Weeks 3 and 6, regardless of renal function status or use of an ACEi or ARB. Of note, animal model data suggest that some of the effects of sUA on BP may be mediated through the activation of the renin–angiotensin system.20, 21 The high rate of ACEi/ARB use in the subjects with chronic kidney disease may significantly blunt BP response to urate‐lowering therapy. Alternatively, the animal models also suggest the development, over time, of uric‐acid‐induced changes in vascular geometry and reactivity that do not revert with urate‐lowering therapy.21 Patients with established chronic kidney disease would be expected to have longer‐standing disease and be more likely to have had vascular progression leading to a resistance to sUA reduction.
The key finding of this study was a small, but significant, decrease in SBP from baseline to Week 6 in subjects with normal renal function treated with febuxostat compared with placebo. A progressive improvement in SBP from the beginning of the study to Weeks 3 and 6 in the febuxostat subgroup was observed in subjects with normal renal function. While not reaching statistical significance, the magnitude of change from baseline DBP in the normal renal function group showed the same trend. Furthermore, the baseline to Week 6 changes of SBP 6.6 mm Hg and DBP 3.3 mm Hg in this normal renal function subgroup may be clinically significant.22, 23, 24, 25
There is a strong association between sUA and arterial hypertension, although the mechanism for this remains unclear. Experimental models that have investigated the relationship between uric acid and hypertension suggest 2 phases in the development of hypertension.20, 26 First, uric acid can induce acute vasoconstriction by activation of the renin–angiotensin–aldosterone system. Second, uptake of uric acid into vascular smooth muscle cells leads to cellular proliferation and secondary arteriolosclerosis that impairs compliance of resistance vessels and BP‐induced natriuresis. Recent experimental and clinical studies suggest that uric acid could play a contributory role in the pathogenesis of elevated BP.27 Chronic elevation of sUA not only sustains an elevated BP but also may cause irreversible vascular and glomerular changes that are associated with a form of salt‐sensitive hypertension. Patients with chronic hypertension and chronic kidney disease are likely to fall into this category, and therefore may not respond to urate‐lowering treatment.
The weight of the evidence suggests that uric acid could be a true modifying and possibly causal factor for human primary hypertension.27 Early treatment of hyperuricemia, therefore, might delay the development of essential hypertension, whereas years of hyperuricemia along with the emergence of renal impairment and hypertension might be a setting in which lowering of sUA would be expected to have less effect on BP. The reason for this may be attributed to the association of both hypertension and hyperuricemia with vascular damage. Consequently, sUA in an adult population with established hypertension may differ from sUA in early‐stage hypertension. Although more research is clearly necessary, the available data suggest that in some cases uric acid is the likely cause of early‐onset hypertension.28
The findings of our study provide further support to earlier observations with allopurinol and probenecid in antihypertensive‐treatment‐naïve adolescent subjects; the antihypertensive effect of urate‐lowering therapy was demonstrated in a randomized interventional study in adolescents with newly diagnosed essential hypertension as well as in a study in prehypertensive adolescents treated with allopurinol and probenecid.15, 16 Subjects were not treated with antihypertensive therapy in these studies, and abnormal serum creatinine or renal impairment were the exclusion criteria. Taking these studies and the present one into account, the results suggest that the reduction of sUA may lower BP in the context of normal renal function and in the early stages of hypertension, and that BP lowering may be less urate dependent in later stages of the disease.
While results from the current study suggest that febuxostat may improve SBP in hyperuricemic patients with hypertension and normal renal function, it should be noted that this study did not assess the ability of febuxostat to prevent BP increases. The primary steps in the prevention and management of hypertension are lifestyle measures including cessation of smoking, reduced alcohol consumption, weight loss, regular exercise, and diet modification. The Dietary Approaches to Stop Hypertension (DASH) diet has been shown to effectively reduce BP and substantially reduce sUA levels (compared with a typical American diet) in patients with hyperuricemia.29, 30, 31 Where appropriate, these preventative lifestyle strategies should be incorporated into the management of hypertension in patients with hyperuricemia.
A limitation of the present study is that the results are not generalizable beyond a study population that was not antihypertensive treatment naïve, had established hypertension, and included a subset with renal impairment. The mean (SD) age of the study population (53.6 [10.6] years) should also be taken into consideration; the study findings may not be applicable to subjects with hyperuricemia and hypertension falling outside of this age range. In addition, the BP cutoffs used in this study may have been too restrictive based on the literature; the BP response to febuxostat may have been greater if the upper limit of SBP and/or DBP had been higher. Furthermore, a hyperuricemic, hypertensive, antihypertensive‐treatment‐naïve adult population may have a different BP response profile in the setting of monotherapy with urate‐lowering treatment. Although the definition of hyperuricemia used in this study is in line with that used in the literature, it is important to recognize that the use of such a generic definition does not take into consideration the variable contribution of xanthine oxidase to cumulative levels of sUA seen in individuals and subpopulations. Another study limitation was the use of enrollment criteria that required sUA >0.42 mmol/L not associated with gout, so most subjects had baseline sUA within a relatively narrow band. Serum urate levels >0.36 or >0.42 mmol/L are well‐established cutoffs in the context of gout, but there is no established cutoff for the cardiovascular associations in the “normal” sUA range. Other limitations include the short study duration and small sample size: this was only a 6‐week study, and the low numbers of subjects in the subgroup analyses may be inadequate as the power estimations were for the primary end point. In addition, the small sample size meant that it was not possible to examine any potential differences in study outcomes across the multiple study sites. Furthermore, any missing data for efficacy analyses were imputed using the last available postbaseline observation carried forward, which may have overestimated the treatment effect. However, as <10% of subjects had missing data for the primary end point, the potential impact is considered minimal.
In conclusion, findings from this small Phase 2 study suggest that febuxostat may reduce 24‐hour ambulatory SBP from baseline in subjects with hypertension, hyperuricemia, and normal renal function. Further studies should be conducted to confirm this finding and better characterize the potential antihypertensive effects of febuxostat. It is important to recognize that these beneficial effects are unlikely to be achieved in all subjects with hyperuricemia and hypertension, and more studies are needed to identify those who are most likely to respond to febuxostat therapy.
Sources of Funding
This study was sponsored by Takeda Pharmaceutical International, Inc., Deerfield, IL.
Disclosures
Dr Gunawardhana and Dr McLean are employees of, and own stock/stock options in, Takeda Pharmaceutical Inc. Ms Hunt and Mr Palmer are employees of Takeda Pharmaceutical International, Inc. Dr Punzi has participated in speakers bureaus for Allergan and AstraZeneca and has received study grants from Allergan, Ferrer, Actelion, Boehringer Ingelheim, and National Institutes of Health. Dr Whelton has been a consultant and participated in a speakers bureau for Takeda. Dr Feig has no relevant disclosures.
Supporting information
Table S1. Baseline Characteristics of Patient Subgroups With Normal or Impaired Renal Function
Table S2. Change From Baseline at Wk 3 in sUA (mmol/L) by Renal Function Subgroup
Table S3. Change from Baseline at Wk 6 in sUA (mmol/L) by Renal Function Subgroup
Table S4. Change From Baseline at Wk 6 in 24‐H Ambulatory SBP (mm Hg) by Use of ACEi/ARB Subgroups
Table S5. Change From Baseline at Wk 6 in 24‐H Ambulatory DBP (mm Hg) by Use of ACEi/ARB Subgroups
Table S6. Change in sUA (mmol/L) at Wk 3 by Use of ACEi/ARB Subgroups
Table S7. Change in sUA (mmol/L) at Wk 6 by Use of ACEi/ARB Subgroups
Table S8. The Percentage of Subjects Who Demonstrated a Change From Baseline to Wk 3 and to Wk 6 in Mean Ambulatory SBP of ≥4 mm Hg and Mean Ambulatory DBP of ≥3 mm Hg
Figure S1. Study design.
Acknowledgments
The authors would like to thank all of the investigators and patients who participated in this study. Medical writing assistance was provided by Frank Beebe, PharmD, MBA (Caudex, New York, NY) and Stephen Craig, PhD (Caudex, Oxford, UK) and funded by Takeda Pharmaceutical America, Inc.
(J Am Heart Assoc. 2017;6:e006683 DOI: 10.1161/JAHA.117.006683.)29102979
References
- 1. Takeda Pharmaceuticals . Febuxostat prescribing information 2009. Available at: http://general.takedapharm.com/content/file.aspx?filetypecode=ULORICPI&cacheRandomizer=57f97fb6-d754-4a3f-8ebd-27c184fcb3c0. Accessed May 8, 2017.
- 2. Hosoya T, Ono I. A repeated oral administration study of febuxostat (TMX‐67), a non‐purine‐selective inhibitor of xanthine oxidase, in patients with impaired renal function in Japan: pharmacokinetic and pharmacodynamic study. J Clin Rheumatol. 2011;17(4 suppl 2):S27–S34. [DOI] [PubMed] [Google Scholar]
- 3. Sánchez‐Lozada LG, Lanaspa MA, Cristóbal‐García M, García‐Arroyo F, Soto V, Cruz‐Robles D, Nakagawa T, Yu MA, Kang D‐H, Johnson RJ. Uric acid‐induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol. 2012;121:e71–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ho W‐J, Tsai W‐P, Yu K‐H, Tsay P‐K, Wang C‐L, Hsu T‐S, Kuo C‐T. Association between endothelial dysfunction and hyperuricaemia. Rheumatology. 2010;49:1929–1934. [DOI] [PubMed] [Google Scholar]
- 5. Morris ST, Jardine AG. The vascular endothelium in chronic renal failure. J Nephrol. 2000;13:96–105. [PubMed] [Google Scholar]
- 6. Hwang SJ, Lee KH, Jang HH, Lee SR, Woo JS, Lee HJ, Jung KH, Kim W. Febuxostat contributes to improvement of endothelial dysfunction in an experimental model of streptozocin‐induced diabetic rats. Int J Cardiol. 2014;171:e110–e112. [DOI] [PubMed] [Google Scholar]
- 7. Tsuda H, Kawada N, Kaimori JY, Kitamura H, Moriyama T, Rakugi H, Takahara S, Isaka Y. Febuxostat suppressed renal ischemia reperfusion injury via reduced oxidative stress. Biochem Biophys Res Commun. 2012;427:266–272. [DOI] [PubMed] [Google Scholar]
- 8. Sircar D, Chatterjee S, Waikhom R, Golay V, Raychaudhury A, Chatterjee S, Pandey R. Efficacy of febuxostat for slowing the GFR decline in patients with CKD and asymptomatic hyperuricemia: a 6‐month, double‐blind, randomized, placebo‐controlled trial. Am J Kidney Dis. 2015;66:945–950. [DOI] [PubMed] [Google Scholar]
- 9. Whelton A, MacDonald PA, Chefo S, Gunawardhana L. Preservation of renal function during gout treatment with febuxostat: a quantitative study. Postgrad Med. 2013;125:106–114. [DOI] [PubMed] [Google Scholar]
- 10. Dharmashankar K, Widlansky ME. Vascular endothelial function and hypertension: insights and directions. Curr Hypertens Rep. 2010;12:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J, Qin T, Chen J, Li Y, Wang L, Huang H, Li J. Hyperuricemia and risk of incident hypertension: a systematic review and metaanalysis of observational studies. PLoS One. 2014;9:e114259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feig DI, Kang D‐H, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359:1811–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson RJ, Kang D‐H, Feig D, Kivlighn S, Kanellis J, Watanabe S, Tuttle KR, Rodriguez‐Iturbe B, Herrera‐Acosta J, Mazzali M. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension. 2003;41:1183–1190. [DOI] [PubMed] [Google Scholar]
- 14. Becker MA, Jolly M. Hyperuricemia and associated diseases. Rheum Dis Clin North Am. 2006;32:275–293. [DOI] [PubMed] [Google Scholar]
- 15. Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension. 2012;60:1148–1156. [DOI] [PubMed] [Google Scholar]
- 16. Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA. 2008;300:924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whelton A, MacDonald P, Hunt B, Gunawardhana L. Hypertension in hyperuricemic gout subjects receiving febuxostat or allopurinol [Abstract]. J Clin Hypertens. 2011;13:A57 (PO‐104). [Google Scholar]
- 18. Levey AS, Coresh J, Balk E, Kausz AT, Levin A, Steffes MW, Hogg RJ, Perrone RD, Lau J, Eknoyan G; National Kidney Foundation . National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139:137–147. [DOI] [PubMed] [Google Scholar]
- 19. Levey AS, Coresh J, Greene T, Marsh J, Stevens LA, Kusek JW, Van Lente F; Chronic Kidney Disease Epidemiology Collaboration . Expressing the Modification of Diet in Renal Disease Study equation for estimating glomerular filtration rate with standardized serum creatinine values. Clin Chem. 2007;53:766–772. [DOI] [PubMed] [Google Scholar]
- 20. Mazzali M, Hughes J, Kim Y‐G, Jefferson JA, Kang D‐H, Gordon KL, Lan HY, Kivlighn S, Johnson RJ. Elevated uric acid increases blood pressure in the rat by a novel crystal‐independent mechanism. Hypertension. 2001;38:1101–1106. [DOI] [PubMed] [Google Scholar]
- 21. Feig DI. The role of uric acid in the pathogenesis of hypertension in the young. J Clin Hypertens. 2012;14:346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee . Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–1252. [DOI] [PubMed] [Google Scholar]
- 23. Franklin SS, Lopez VA, Wong ND, Mitchell GF, Larson MG, Vasan RS, Levy D. Single versus combined blood pressure components and risk for cardiovascular disease: the Framingham Heart Study. Circulation. 2009;119:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Franklin SS, Larson MG, Khan SA, Wong ND, Leip EP, Kannel WB, Levy D. Does the relation of blood pressure to coronary heart disease risk change with aging? The Framingham Heart Study. Circulation. 2001;103:1245–1249. [DOI] [PubMed] [Google Scholar]
- 25. Law MR, Morris JK, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta‐analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338:b1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mazzali M, Kanellis J, Han L, Feng L, Xia Y‐Y, Chen Q, Kang D‐H, Gordon KL, Watanabe S, Nakagawa T, Lan HY, Johnson RJ. Hyperuricemia induces primary renal arteriolopathy in rats by a blood pressure‐independent mechanism. Am J Physiol Renal Physiol. 2002;282:F991–F997. [DOI] [PubMed] [Google Scholar]
- 27. Mazzali M, Kanbay M, Segal MS, Shafiu M, Jalal D, Feig DI, Johnson RJ. Uric acid and hypertension: cause or effect? Curr Rheumatol Rep. 2010;12:108–117. [DOI] [PubMed] [Google Scholar]
- 28. Feig DI. Hyperuricemia and hypertension. Adv Chronic Kidney Dis. 2012;19:377–385. [DOI] [PubMed] [Google Scholar]
- 29. Juraschek SP, Gelber AC, Choi HK, Appel LJ, Miller ER III. Effects of the Dietary Approaches to Stop Hypertension (DASH) diet and sodium intake on serum uric acid. Arthritis Rheumatol. 2016;68:3002–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rai SK, Fung TT, Lu N, Keller SF, Curhan GC, Choi HK. The Dietary Approaches to Stop Hypertension (DASH) diet, Western diet, and risk of gout in men: prospective cohort study. BMJ. 2017;357:j1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sacks FM, Campos H. Dietary therapy in hypertension. N Engl J Med. 2010;362:2102–2112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Baseline Characteristics of Patient Subgroups With Normal or Impaired Renal Function
Table S2. Change From Baseline at Wk 3 in sUA (mmol/L) by Renal Function Subgroup
Table S3. Change from Baseline at Wk 6 in sUA (mmol/L) by Renal Function Subgroup
Table S4. Change From Baseline at Wk 6 in 24‐H Ambulatory SBP (mm Hg) by Use of ACEi/ARB Subgroups
Table S5. Change From Baseline at Wk 6 in 24‐H Ambulatory DBP (mm Hg) by Use of ACEi/ARB Subgroups
Table S6. Change in sUA (mmol/L) at Wk 3 by Use of ACEi/ARB Subgroups
Table S7. Change in sUA (mmol/L) at Wk 6 by Use of ACEi/ARB Subgroups
Table S8. The Percentage of Subjects Who Demonstrated a Change From Baseline to Wk 3 and to Wk 6 in Mean Ambulatory SBP of ≥4 mm Hg and Mean Ambulatory DBP of ≥3 mm Hg
Figure S1. Study design.