

Abstract
Background
Cardiac fibrosis occurs because of disruption of the extracellular matrix network leading to myocardial dysfunction. Angiotensin II has been implicated in the development of cardiac fibrosis. Recently, microRNAs have been identified as an attractive target for therapeutic intervention in cardiac pathologies; however, the underlying mechanism of microRNAs in cardiac fibrosis remains unclear. MicroRNA‐130a (miR‐130a) has been shown to participate in angiogenesis and cardiac arrhythmia; however, its role in cardiac fibrosis is unknown.
Methods and Results
In this study, we found that miR‐130a was significantly upregulated in angiotensin II‐infused mice. The in vivo inhibition of miR‐130a by locked nucleic acid– based anti‐miR‐130a in mice significantly reduced angiotensin II‐induced cardiac fibrosis. Upregulation of miR‐130a was confirmed in failing human hearts. Overexpressing miR‐130a in cardiac fibroblasts promoted profibrotic gene expression and myofibroblasts differentiation, and the inhibition of miR‐130a reversed the processes. Using the constitutive and dominant negative constructs of peroxisome proliferator‐activated receptor γ 3‐′untranslated region (UTR), data revealed that the protective mechanism was associated with restoration of peroxisome proliferator‐activated receptor γ level leading to the inhibition of angiotensin II‐induced cardiac fibrosis.
Conclusions
Our findings provide evidence that miR‐130a plays a critical role in cardiac fibrosis by directly targeting peroxisome proliferator‐activated receptor γ. We conclude that inhibition of miR‐130a would be a promising strategy for the treatment of cardiac fibrosis.
Keywords: angiotensin II, cardiac fibrosis, miR‐130a, myofibroblast, peroxisome proliferator‐activated receptor γ
Subject Categories: Basic Science Research, Fibrosis, Cell Signalling/Signal Transduction, Myocardial Biology
Clinical Perspective
What Is New?
Our study highlights the potential importance of micro RNA‐130a in the progression of cardiac fibrosis and offers a possible therapeutic target (microRNA itself or the target gene) for future intervention.
What Are the Clinical Implications?
Future studies are warranted to determine the effect of microRNA‐130a in the cardiac remodeling process in conditionally overexpressed microRNA‐130a transgenic mice or knock‐out model for therapeutic evaluation in cardiac remodeling and fibrosis.
Introduction
Cardiac fibrosis is a severe pathologic manifestation resulting from an injury to the myocardium because of pressure overload, ischemic insult, or metabolic stress.1, 2, 3 The underlying molecular and morphological correlate of cardiac fibrosis is the maladaptive changes in myocardial structure via uncontrolled deposition of extracellular matrix (ECM) proteins in the interstitium and perivascular region of the heart.3, 4, 5, 6, 7 As a result, myocardial stiffness occurs that alters the mechanics of the heart and impairs its function. Cardiac fibroblasts represent the most abundant cell type in the mammalian heart and play a dominant role in cardiac fibrosis.8, 9, 10 A critical event in cardiac fibrosis is the transformation of cardiac fibroblasts to myofibroblasts.11 Myofibroblasts are the specialized cardiac fibroblasts formed by irreversible acquisition of expression of α‐smooth muscle actin (α‐SMA).11, 12, 13, 14 The excessive deposition of ECM proteins is thought to be produced by myofibroblasts,12, 15, 16, 17, 18 and inhibition of myofibroblasts may be effective to prevent cardiac fibrosis.
MicroRNAs (miRNAs) are short, highly conserved RNA sequences that control gene expression either by degradation of mRNA or repressing translational process.19 Recently, miRNAs have been shown to be key modulators of diverse cardiac diseases including cardiac fibrosis,20, 21, 22, 23, 24, 25, 26, 27 but the role of miRNAs in cardiac fibroblasts activation, which includes myofibroblasts differentiation, remains unknown. Angiotensin II (Ang II), the major component of the renin‐angiotensin system, plays a critical role in cardiac fibrosis via inducing production of collagens (type I and type III) and other ECM constituents.12, 28, 29 Ang II further activates transforming growth factor β (TGFβ) signaling to induce pro‐fibrogenic cascade.30, 31 In the context that Ang II is directly involved in the pathogenesis of cardiac fibrosis through altering an array of gene expression,32 whether miRNAs contribute to the process may provide a new insight.
To date, little is known about the contribution of miR‐130a in cardiac pathology. The miR‐130a is pro‐angiogenic; promoting endothelial cell migration implicated the role in the angiogenic process33, 34, 35 and has also been shown to be upregulated in patients with heart failure.36, 37 Interestingly, miR‐130a has been reported to play a role in smooth cell proliferation,38 liver steatosis,39 or breast cancer40; however, its role in cardiac fibrosis is unknown, albeit a recent study has demonstrated its role in cardiac arrhythmia.41
In this study, we investigated the role of miR‐130a in regulating cardiac fibrosis–related gene expression in an Ang II‐infused mouse model. We have presented for the first time a mechanistic role of miR‐130a in cardiac fibrosis by modulating myofibroblasts phenotype and restoring the peroxisome proliferator‐activated receptors (PPAR) γ level. Our result indicated that miR‐130a was significantly upregulated in the mouse heart starting from day 3 to day 14 post Ang II infusion. Sufficient inhibition of miR‐130a attenuated cardiac fibrosis through directly restoring its target molecule PPARγ along with reducing the expression of collagens and myofibroblasts differentiation. Our results clearly demonstrate that miR‐130a contributes a critical role in Ang II‐induced cardiac fibrosis.
Materials and Methods
Infusion of Ang II and Administration of Locked Nucleic Acid (LNA)‐Anti‐miR‐130a in Mice
Animal care and all experimental procedures were conducted with the approval of the Institutional Animal Care and Use Committee at the Texas A&M Health Science Center and Baylor Scott & White Health, and the Ethics Committee of Animal Research at Peking University Health Science Center. Male 9‐ to 10‐week‐old FVB/N mice received a single intraperitoneal injection of LNA‐anti‐miR‐130a at the dose of 5, 15, and 25 mg/kg or scrambled LNA (25 mg/kg). Three days after injection, mice were euthanized and the hearts were excised in order to detect miR‐130a level. For Ang II infusion, male 9‐ to 10‐week‐old FVB/N mice were implanted with Alzet mini‐osmotic pumps (Alza Pharmaceutics, Palo Alto, CA) containing either normal saline or Ang II (1.4 mg/kg per day for 14 days) under sterile conditions. At 1, 3, and 7 days after surgery, Ang II‐infused mice received an intraperitoneal injection of normal saline, LNA‐anti‐miR‐130a (25 mg/kg/injection), or scrambled LNA (25 mg/kg/injection). Systolic blood pressure was measured in conscious mice by the noninvasive tail‐cuff method using the CODA blood pressure system (Kent Scientific, Torrington, CT) according to the manufacturer's instruction. After 2 weeks, echocardiography was performed and the data were recorded. The hearts were excised for biochemical, histological, and molecular analysis.
Echocardiography
Transthoracic 2‐dimensional echocardiography was performed as described previously.42, 43, 44
Collection of Human Tissues
Nonfailing human hearts were obtained from organ donors whose hearts were not suitable for transplantation but who had no history of cardiac disease. The hearts were from victims of motor vehicle accidents, gunshot wounds, or cerebral vascular accidents without any known hemodynamic abnormalities. Failing human hearts were obtained from transplanted patients who had been diagnosed with hypertrophic cardiomyopathy. Tissues from failing human hearts were collected at the time of transplantation. Ventricular tissues were frozen in liquid nitrogen for storage at −80°C. For determination of miR‐130a expression, left ventricular tissues were used. Collection of human tissues was in accordance with institutional guidelines and was approved by the institutional review board of the Cleveland Clinic. All human sample collections were completed at the Cleveland Clinic, Cleveland, OH.
Synthesis of LNA
The antisense sequence of miR‐130a (LNA‐anti‐miR‐130a) was synthesized by Exiqon (Denmark) and 5 nucleotides or deoxynucleotides at both ends of the antisense molecules were locked. The sequence of LNA‐anti‐miR‐130a and the scramble are as follows:
mmu‐miR‐130a‐3p T*T*T*T*A*A*C*A*T*T*G*C*A*C*T
Scrambled A*C*G*T*C*T*A*T*A*C*G*C*C*C*A
(*=phosphorothioate)
The scrambled LNA consists of a sequence directed against a Caenorhabditis elegans–specific miRNA with a comparable LNA/DNA content.
Preparation of Neonatal Rat Cardiac Fibroblasts
Primary neonatal rat cardiac fibroblasts were prepared as described.23
Transfection of miRNA Mimetic and Inhibitor
The miRNA mimetic and inhibitor of miR‐130a were designed and purchased from Dharmacon (Pittsburgh, PA, USA). We used miRIDIAN mimic mouse mmu‐miR‐130a (C‐310399‐05) and miRIDIAN hairpin inhibitor mmu‐miR‐130a (IH‐310399‐07). Cardiac fibroblasts were transfected with a final concentration of 50 nmol/L for miR‐130a mimetic and inhibitor using DharmaFECT Duo Transfection Reagent according to the manufacturer's instruction (Thermo Scientific, Lafayette, CO, USA).
Enhanced Green Fluorescent Protein Reporters Containing PPARγ mRNA Sequences
Enhanced green fluorescent protein (EGFP) reporters containing wild‐type and mutant PPARγ 3′‐UTR were kindly provided by Dr M. Gorospe. The reporter assay was performed as described previously.23
Western Blot Analysis
Myocardial tissues or cells were lysed using RIPA lysis buffer containing protease inhibitors and Western blotting along with quantification of images were performed as described previously.44 The antibodies for PPARγ, α‐SMA, and GAPDH were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody for GFP was from Proteintech Group, Inc (Chicago, IL, USA).
RNA Isolation and Quantitative Reverse Transcriptase Polymerase Chain Reaction Analysis
The quantitative reverse transcriptase polymerase chain reaction for miR‐130a, collagen I (Col I), collagen III (Col III), connective tissue growth factor (CTGF), fibronectin (Fn), matrix metalloproteinase 9, α‐SMA, and PPARγ was performed using either human/mouse gene‐specific primers as described previously.43, 44, 45
Morphological Examination
Masson's trichrome‐staining, fluorescein isothiocyanate conjugate–wheat germ agglutinin staining, and microscopy images analysis were performed as described previously.43, 44
Immunohistochemistry
Cardiac fibroblasts were fixed in 4% paraformaldehyde and permeabilized in 0.2% Triton X‐100 in PBS. Frozen sections were fixed with pre‐cooled acetone. Cardiac fibroblasts or sections were stained with anti‐α‐SMA antibody overnight at 4°C, then secondary antibody for 2 hours at 37°C. Nuclei were stained with 4, 6‐diamidino‐2‐phenylindole (DAPI, Sigma‐Aldrich). Fluorescence images were captured by use of the Leica TCS SP5 confocal system as described previously.45
Cross sections from paraffin‐embedded ventricles were deparaffinized and stained with the antibody for CD11b at 4°C overnight, then incubated with the horseradish peroxidase–conjugated secondary antibody at 37°C for 2 hours, and reacted with diaminobenzidine substrate. Images were captured by using the Leica 550IW system (Leica, Mannheim, Germany).
Statistical Analysis
Data are expressed as means±SEM. Data were analyzed using Prism 5.0 GraphPad software (GraphPad, San Diego, CA). Data from animal experiments that passed the Shapiro–Wilk normality test were analyzed with 1‐way ANOVA for multiple groups followed by Tukey–Kramer post‐tests. Data from animal experiments that did not pass the Shapiro–Wilk normality test were analyzed with the Kruskal–Wallis test followed by Dunn's multiple comparisons test. Data from cell experiments were compared by Student t test for 2 groups and 1‐way ANOVA for multiple groups followed by Tukey–Kramer post‐tests. P<0.05 was considered statistically significant.
Results
Selective Inhibition of miR‐130a Rescues Ang II‐Induced Cardiac Damage
MiR‐130a expression is upregulated in Ang II‐infused mice hearts
First, we detected the expression of miR‐130a in heart tissues of Ang II‐infused mice. MiR‐130a expression was increased to 1.87‐, 2.34‐, and 2.45‐ (P<0.05) fold, respectively, at 3, 7, and 14 days after Ang II infusion (Figure 1A). Similar expression trend was observed in miR‐130b (Figure 1B).
Figure 1.
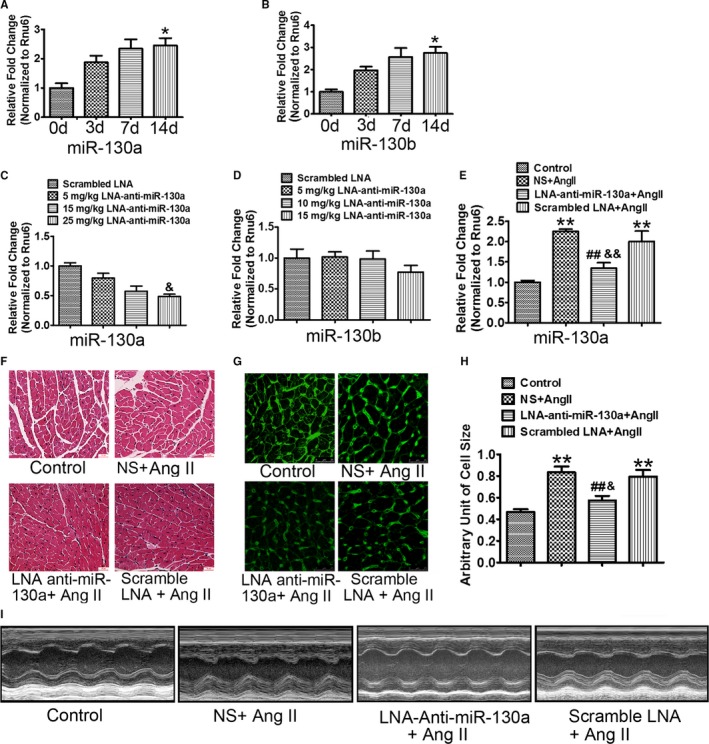
Selective inhibition of miR‐130a rescues Ang II‐induced cardiac damage. Wild‐type mice were treated with Ang II for 3, 7, and 14 d and myocardial expression of miR‐130a (A) and miR‐130b (B) was determined by qRT‐PCR. Rnu6 was used as an internal control. Cardiac miR‐130a (C) and miR‐130b (D) were determined by qRT‐PCR in mice injected with the scrambled LNA (locked nucleic acid) at 25 mg/kg of body weight or LNA‐anti‐miR‐130a at 5, 15, and 25 mg/kg of body weight for 3 d. Rnu6 was used as an internal control. A through D, Data are expressed as means±SE from 3 independent mice. *P<0.05 vs 0 d; & P<0.05 vs scrambled LNA. E, Cardiac miR‐130a was determined by qRT‐PCR in control, NS+Ang II, LNA‐anti‐miR‐130a+AngII, and scrambled LNA+Ang II groups. Rnu6 was used as an internal control. F, Representative H&E staining images in control, NS+Ang II, LNA‐anti‐miR‐130a+AngII, and scrambled LNA+Ang II groups. Scale bars indicate 30 μm. G and H, Representative WGA (wheat germ agglutinin) staining images of left ventricles (G) and histogram of cardiac myocyte cross‐sectional area derived from WGA staining (H) (100 myocytes in each section were scanned and averaged for each group). Scale bars indicate 25 μm (G). E and H, Data are expressed as means±SE from 6 independent mice. **P<0.01 vs control; ## P<0.01 vs NS+Ang II; && P<0.01 vs scrambled LNA+Ang II. I, Representative LV (left ventricular) trace images showing the LV thickness and LV free wall for control, NS+Ang II, LNA‐anti‐miR‐130a+AngII, and scrambled LNA+Ang II groups, respectively. Ang II indicates angiotensin II; mIR‐130a, microRNA‐130a; NS, normal saline; qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction.
Delivery of LNA‐anti‐miR‐130a inhibits expression of miR‐130a
We assessed the inhibitory effect of LNA‐anti‐miR‐130a. LNA‐anti‐miR‐130a at 5, 15, and 25 mg/kg of body weight was injected intraperitoneally into mice. The miR‐130a and miR‐130b expression in the ventricles was determined at 3 days after injection. Expression of miR‐130a was decreased by 20.39%, 42.40%, and 51.69% (P<0.05), respectively, at the dose of 5, 15, and 25 mg/kg of body weight (Figure 1C). LNA‐anti‐miR‐130a had no effect on the myocardial expression of miR‐130b, suggesting that the LNA anti‐miR‐130a is specific for miR‐130a but not miR‐130b (Figure 1D).
We then examined the potential effect of LNA‐anti‐miR‐130a on miR‐130a expression in Ang II‐treated mice. Compared with the normal saline (NS)‐ and scrambled LNA‐treated mice, myocardial expression of miR‐130a was reduced significantly by LNA‐anti‐miR‐130a treatment (NS+Ang II versus LNA‐anti‐ miR‐130a+Ang II, 2.25±0.06 versus 1.34±0.13, P<0.01; scrambled LNA+Ang II versus LNA‐anti‐miR‐130a+Ang II, 2.00±0.26 versus 1.34±0.13, P<0.01) (Figure 1E).
MiR‐130a inhibition improves Ang II‐induced cardiac dysfunction
We investigated whether LNA‐anti‐miR‐130a exerts a protective effect in in vivo conditions. Ang II infusion in the NS‐ and scrambled LNA‐treated mice resulted in cardiac hypertrophy as revealed by the increase in heart weight:body weight ratio (Table 1) and cardiac myocyte cross‐section area (NS+Ang II versus control, 0.83±0.05 versus 0.47±0.03, P<0.01; Scrambled LNA+Ang II versus control, 0.80±0.06 versus 0.47±0.03, P<0.01) (Figure 1F through 1H). LNA‐anti‐miR‐130a treatment significantly reduced Ang II‐induced cardiac hypertrophy, compared with the NS‐ and scrambled LNA‐treated mice (Table 1 and Figure 1F through 1H) (cardiac myocyte cross‐section area: LNA‐anti‐miR‐130a+Ang II versus NS+Ang II, 0.57±0.04 versus 0.83±0.05, P<0.01; LNA‐anti‐miR‐130a+Ang II versus Scrambled LNA+Ang II, 0.57±0.04 versus 0.80±0.06, P<0.05). Echocardiography examination showed that Ang II infusion increased the left ventricular posterior wall in diastole and systole and reduced the E/A ratio in the NS‐treated mice. These parameters were reversed by inhibition of miR‐130a. Scrambled LNA produced no effect on Ang II‐induced cardiac dysfunction (Figure 1I and Table 2).
Table 1.
Heart Weight/Body Weight Ratio of Ang II‐Infusion Mice Treated With LNA‐Anti‐miR‐130a or Scrambled LNA
| Control (n=6) | NS+Ang II (n=6) | LNA‐Anti‐miR‐130a+Ang II (n=6) | Scrambled LNA+Ang II (n=6) | |
|---|---|---|---|---|
| HW, mg | 115.90±6.00 | 134.30±2.05* | 124.30±2.58 | 131.80±5.76 |
| BW, g | 27.36±0.47 | 26.40±0.65 | 27.83±0.22 | 26.67±0.99 |
| HW/BW ratio, mg/g | 4.23±0.17 | 5.10±0.07† | 4.47±0.11‡ , § | 4.94±0.08† |
Data are expressed as means±SE. Ang II indicates angiotensin II; BW, body weight; HW, heart weight; LNA, locked nucleic acid; miR‐130a, microRNA‐130‐a; NS, normal saline.
* P<0.05, † P<0.01 vs control.
‡ P<0.01 vs NS+Ang II.
§ P<0.05 vs scrambled LNA+Ang II.
Table 2.
Echocardiography of Ang II‐Infusion Mice Treated With LNA‐Anti‐miR‐130a or Scrambled LNA
| Control (n=6) | Ang II (n=6) | LNA‐Anti‐miR‐130a+Ang II (n=6) | Scrambled LNA+Ang II (n=6) | |
|---|---|---|---|---|
| LVIDd | 3.65±0.14 | 3.07±0.20 | 3.33±0.41 | 3.15±0.23 |
| LVIDs | 2.58±0.16 | 2.07±0.24 | 2.41±0.15 | 2.18±0.20 |
| LVPWd | 0.63±0.03 | 0.79±0.03† | 0.69±0.02‡ | 0.77±0.02† |
| LVPWs | 0.61±0.01 | 0.72±0.02† | 0.64±0.03‡ | 0.70±0.02* |
| E/A ratio | 1.70±0.06 | 0.99±0.14† | 1.43±0.11‡ , § | 0.94±0.10† |
| FS% | 30.31±2.34 | 33.65±3.89 | 29.13±2.98 | 32.11±3.60 |
Data are expressed as means±SE. E/A ratio indicates ratio of early to late peak left ventricular filling velocities; FS, fractional shortening; LNA, locked nucleic acid; LVIDd, left ventricular (LV) internal diameter in diastole; LVIDs, LV internal diameter in systole; LVPWd, LV posterior wall diameter in diastole; LVPWs, LV posterior wall diameter in systole; miR‐130a, microRNA‐130‐a.
*P<0.05, † P<0.01 vs control.
‡ P<0.05 vs Ang II.
§ P<0.05 vs scrambled LNA+Ang II.
MiR‐130a inhibition marginally reduces Ang II‐induced blood pressure
Ang II is a potent stimulus for increasing blood pressure level. In this study, we also determined whether inhibition of miR‐130a reduced the blood pressure. Ang II infusion into NS‐ and scrambled LNA‐treated mice for 2 weeks resulted in significant increase in systolic blood pressure to 167.61±2.86 and 168.50±2.16 mm Hg, respectively (P<0.01), compared with the control mice in 2 weeks of treatment. LNA‐anti‐miR‐130a treatment marginally reduced the blood pressure level to 155.11±2.83 (P<0.05) compared with Ang II‐infused mice (Table 3).
Table 3.
Systolic Blood Pressure of Ang II‐Infusion Mice Treated With LNA‐Anti‐miR‐130a or Scrambled LNA
| Control (n=6) | NS+Ang II (n=6) | LNA‐Anti‐miR‐130a+Ang II (n=6) | Scrambled LNA+Ang II (n=6) | |
|---|---|---|---|---|
| 0 wk | 92.61±1.69 | 94.17±1.73 | 96.94±2.53 | 98.61±2.12 |
| 1 wk | 100.72±3.86 | 144.83±1.66* | 142.44±2.48* | 148.78±2.82* |
| 2 wk | 102.39±3.61 | 167.61±2.86* | 155.11±2.83* , † , ‡ | 168.50±2.16* |
Data are expressed as means±SE. Ang II indicates angiotensin II; LNA, locked nucleic acid; miR‐130a, microRNA‐130‐a.
*P<0.01 compared with the mice at 0‐wk time point in each group.
† P<0.05 vs NS+Ang II.
‡ P<0.05 vs scrambled LNA+Ang II.
Inhibition of miR‐130a Reduces Cardiac Fibrosis and Myofibroblasts Differentiation In Vivo
Compared with the control group, the NS‐ and scrambled LNA‐treated mice subjected to continuous Ang II infusion showed a greater degree of cardiac fibrosis. Delivery of LNA‐anti‐miR‐130a significantly decreased collagen deposition as determined by Masson's Trichrome staining (NS+Ang II versus LNA‐anti‐miR‐130a+Ang II, 11.28±1.27 versus 5.20±1.08, P<0.05; scrambled LNA+Ang II versus LNA‐anti‐miR‐130a+Ang II, 10.82±1.36 versus 5.20±1.08, P<0.05) (Figure 2A and 2B). Upregulation of collagen I (Col I), collagen III (Col III), connective tissue growth factor (CTGF), fibronectin (Fn), and matrix metalloproteinase 9 in the hearts of Ang II‐infused mice was attenuated by LNA‐anti‐miR‐130a delivery, as demonstrated by quantitative reverse transcriptase polymerase chain reaction, whereas scrambled LNA produced no such effects (Figure 2C). Immunohistochemical analysis showed rare expression of α‐SMA in the control group but increased α‐SMA‐positive staining cells in the NS‐ or scrambled LNA‐treated group infused with Ang II. Mice treated with LNA‐anti‐miR‐130a possessed fewer α‐SMA‐positive myofibroblasts than the NS‐ or scrambled LNA‐treated mice (Figure 2D). The quantification of α‐SMA‐positive myofibroblasts is shown in Figure 2E. In addition, our data showed an upregulation of α‐SMA gene expression in the hearts of Ang II‐infused mice and was significantly attenuated by LNA‐anti‐miR‐130a delivery (Figure 2F).
Figure 2.
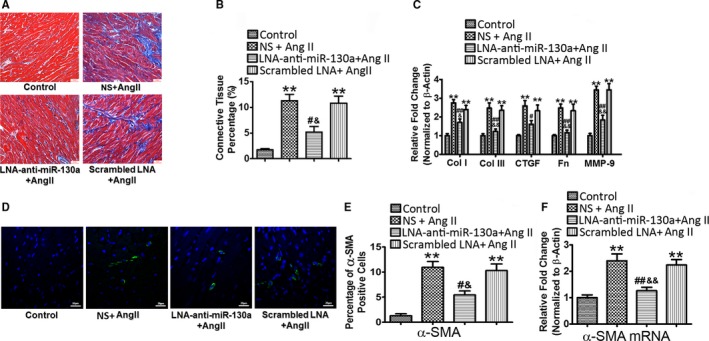
Inhibition of miR‐130a reduces cardiac fibrosis and myofibroblast differentiation in vivo. Representative image of Masson's trichrome staining (A) and histogram of connective tissue percentage of hearts (B). Blue staining indicates connective tissue (n=6); 6 fields in each sample were scanned and averaged. Scale bars indicate 30 μm. C, Cardiac gene expression of Col I, Col III, CTGF (connective tissue growth factor), Fn (fibronectin), and MMP‐9 was determined by qRT‐PCR. β‐Actin was used as an internal control. D, Representative immunofluorescence images of α‐SMA in control, NS+Ang II, LNA‐anti‐miR‐130a+Ang II, and scrambled LNA+Ang II groups. Green signals represent α‐SMA protein and blue signals represent nuclei. Scale bars indicate 20 μm. E, Quantification of α‐SMA‐positive myofibroblasts from the immunofluorescence images of (D). Three fields in each sample were counted and calculated, n=6 in each group. F, Cardiac gene expression of α‐SMA was determined by qRT‐PCR. β‐Actin was used as an internal control. Data are expressed as means±SE from 6 independent mice. **P<0.01 vs control; # P<0.05, ## P<0.01 vs NS+Ang II; & P<0.05, && P<0.01 vs scrambled LNA+Ang II. Ang II indicates angiotensin II; Col I, Col III, collagen I, collagen III; CTGF, connective tissue growth factor; LNA, locked nucleic acid; miR‐130‐a, microRNA‐130‐a; MMP‐9, matrix metalloproteinase 9; NS, normal saline; qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction; α‐SMA, α‐smooth muscle actin.
MiR‐130a Is Upregulated in Failing Human Hearts
We compared the expression of mature miR‐130a in nonfailing (normal) and failing human hearts (hypertrophic cardiomyopathy). Our results demonstrated that mature miR‐130a expression was significantly upregulated in failing hearts compared with nonfailing human hearts (3.80‐fold over healthy controls, P<0.05) (Figure 3A). To further determine whether these failing hearts evinced alteration of fibrotic genes, we analyzed the mRNA expression of Col I, Col III, and CTGF. Our data showed that both Col I and Col III (2.29‐ and 2.98‐fold, respectively, P<0.05) and CTGF (2.55‐fold, P<0.05) were significantly increased in failing human hearts compared with the nonfailing hearts (Figure 3B). Together, our result indicates a strong association between miR‐130a and cardiac fibrosis.
Figure 3.
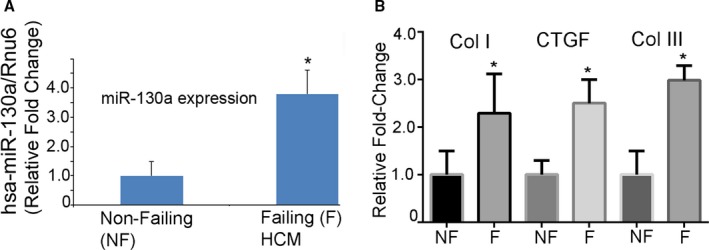
MiR‐130a is upregulated in failing human hearts. A, Mature miR‐130a expression was determined by qRT‐PCR in failing and nonfailing human hearts. Rnu6 was used as an internal control. B, The mRNA expression of Col I, Col III, and CTGF were determined by qRT‐PCR in failing and nonfailing human hearts. GAPDH was used as an internal control. *P<0.05, vs nonfailing hearts (n=8 for nonfailing and n=12 for failing hearts). Col I, Col III, collagen I indicate collagen III; CTGF, connective tissue growth factor; HCM, hypertrophic cardiomyopathy; miR‐130‐a, microRNA‐130‐a; qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction.
MiR‐130a Inhibits Ang II‐Induced Myofibroblasts Differentiation and Profibrotic Response in Cardiac Fibroblasts
First, we assessed the efficacy of miR‐130a mimic and inhibitor transfection in cardiac fibroblasts stimulated with Ang II. Data presented in Figure 4A show that Ang II treatment significantly upregulated miR‐130a expression. The mimic transfection showed 757‐fold miR‐130a expression; however, we did not observe any further cumulative effect in the presence of Ang II. The miR‐130a inhibitor showed significant reduction of miR‐130a in both unstimulated‐ and Ang II‐treated cells (Figure 4A). Together, our data indicated that the mimic and inhibitor transfections are optimum. To define the role of miR‐130a in Ang II‐triggered profibrotic response, cardiac fibroblasts were transfected with the miR‐130a mimetic or inhibitor in the presence or absence of Ang II. Gene expression of Col I, Col III, CTGF, and Fn was increased to 2.14‐ (P<0.01), 2.18‐ (P<0.01), 1.76‐ (P<0.05), and 2.57‐ (P<0.01) fold, respectively, in cells treated with Ang II, compared with the unstimulated cells. Cardiac fibroblasts transfected with miR‐130a mimetic in the presence of Ang II showed further increased expression of these fibrotic genes (Col I, 2.14±0.17 versus 2.92±0.12, P<0.05; Col III, 2.18±0.11 versus 3.17±0.34, P<0.05; CTGF, 1.76±0.13 versus 2.48±0.14, P<0.05; Fn, 2.57±0.21 versus 3.61±0.25, P<0.05), compared with the Ang II‐treated cells. The miR‐130a inhibitor transfection in the presence of Ang II resulted in significant attenuation of mRNA expression of Col I (2.14±0.17 versus 1.38±0.11, P<0.05), Col III (2.18±0.11 versus 1.32±0.09, P<0.05), CTGF (1.76±0.13 versus 1.18±0.10, P<0.05), and Fn (2.57±0.21 versus 1.73±0.14, P<0.05), compared with the Ang II‐stimulated cells (Figure 4B through 4E).
Figure 4.
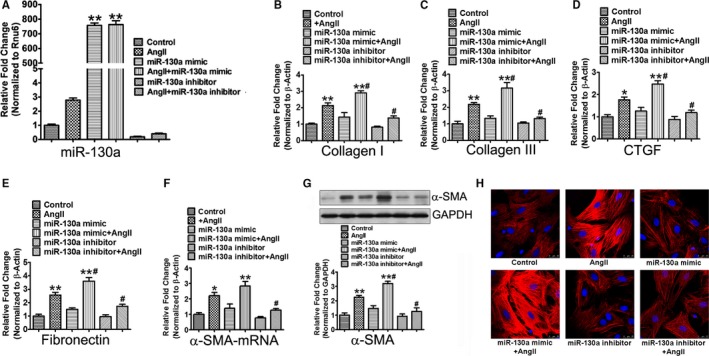
MiR‐130a inhibits Ang II‐induced myofibroblasts differentiation and profibrotic response in cardiac fibroblasts. A, Cardiac fibroblasts were transfected with 50 nmol/L miR‐130a mimetic or inhibitor in the presence and absence of 1 μmol/L Ang II. The mature miR‐130a expression was determined by qRT‐PCR. Rnu6 was used as an internal control. B through E, Cardiac fibroblasts were transfected with 50 nmol/L mimetic or inhibitor of miR‐130a for 12 h, and then stimulated with 1 μmol/L Ang II for 48 h. Gene expression of Col I (B), Col III (C), CTGF (D), Fn (E), and α‐SMA (F) was determined by qRT‐PCR. β‐Actin was used as an internal control. G, Protein expression of α‐SMA was determined by Western blot analysis. GAPDH was used as an internal loading control. Data are expressed as means±SE from 3 independent experiments. *P<0.05, **P<0.01 vs control; # P<0.05 vs Ang II. H, Representative immunofluorescence images of α‐SMA. Red signals represent α‐SMA protein and blue signals represent nuclei. Scale bars indicate 25 μm. Ang II indicates angiotensin II; Col I, Col III, collagen I, collagen III; CTGF, connective tissue growth factor; Fn, fibronectin; miR‐130‐a, microRNA‐130‐a; qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction.
Many effects of cardiac fibroblasts are mediated through differentiation into the myofibroblasts phenotype that expresses contractile proteins and exhibits increased migratory, proliferative, and secretory properties. We detected the effect of miR‐130a on myofibroblasts differentiation in cultured cardiac fibroblasts. Ang II treatment increased the mRNA and protein expression of α‐SMA, compared with the unstimulated cells. The miR‐130a mimetic further elevated the expression of α‐SMA induced by Ang II, whereas miR‐130a inhibitor restored the level of α‐SMA (Figure 4F and 4G). Immunofluorescence microscopy for α‐SMA indicated that miR‐130a mimetic further facilitated whereas miR‐130a inhibitor impeded the Ang II‐induced myofibroblasts differentiation (Figure 4H).
MiR‐130a Attenuates Ang II‐Induced Macrophages Infiltration and Myocardial Inflammation
Activated macrophages are capable of secreting pro‐inflammatory mediators and are essential for cardiac fibrotic response triggered by Ang II. Immunohistochemical analysis showed a marked increase in CD11b+ macrophages in the NS‐ (25.67±3.40, P<0.01) or scrambled LNA‐treated mice (22.11±3.00, P<0.01) infused with Ang II, compared with the control mice (1.78±0.46). Mice treated with LNA‐anti‐miR‐130a showed a significantly lower content in CD11b+ macrophages than the NS‐ or scrambled LNA‐treated mice (9.67±1.35, P<0.01) (Figure 5A and 5B).
Figure 5.
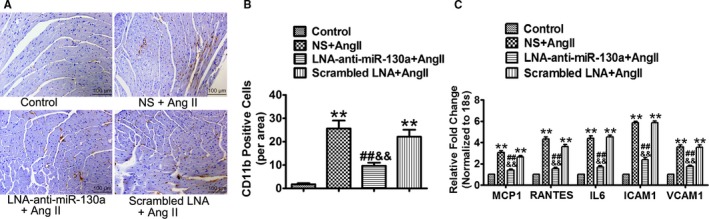
MiR‐130a attenuates Ang II‐induced macrophages infiltration and myocardial inflammation. A, Representative images of CD11b+ staining cells in control, NS+Ang II, LNA‐anti‐miR‐130a+ Ang II, and scrambled LNA+Ang II groups. B, Averaged bar graphs of numbers of CD11b+ cells per field (3 fields in each sample were counted, n=6 in each group). C, Gene expression of MCP1, RANTES, IL6, ICAM1, and VCAM1 was determined by qRT‐PCR. 18s was used as an internal control. **P<0.01 vs control; ## P<0.01 vs NS+Ang II; && P<0.01 vs scrambled LNA+Ang II. Ang II indicates angiotensin II; ICAM1, intercellular adhesion molecule 1; IL6, interleukin 6; LNA, locked nucleic acid; miR‐130a, microRNA‐130a; MCP1, macrophage chemoattractant protein 1; NS, normal saline; qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction.
To gain mechanistic insight, mRNA levels of a variety of vascular genes, inflammatory cytokines, and chemokines were examined by quantitative reverse transcriptase polymerase chain reaction. Compared with the control group, Ang II‐infused mice treated with NS‐ or scrambled LNA showed markedly increased gene expression of monocyte chemotactic protein 1 (NS+Ang II, 3.05±0.20; Scrambled LNA+Ang II, 2.64±0.13), regulated on activation, normal T cell expressed and secreted (NS+Ang II, 4.33±0.20; Scrambled LNA+Ang II, 3.63±0.17), interleukin 6 (NS+Ang II, 4.41±0.21; Scrambled LNA+Ang II, 4.54±0.15), intercellular adhesion molecule 1 (NS+Ang II, 5.87±0.12; Scrambled LNA+Ang II, 5.87±0.14), and vascular cell adhesion molecule 1 (NS+Ang II, 3.57±0.22; Scrambled LNA+Ang II, 3.54±0.25). Delivery of LNA‐anti‐miR‐130a significantly decreased the gene expression of the above inflammatory mediators (monocyte chemotactic protein 1, 1.40±0.10; regulated on activation, normal T cell expressed and secreted, 1.52±0.15; interleukin 6, 1.70±0.14; intercellular adhesion molecule 1, 2.37±0.21; vascular cell adhesion molecule 1, 1.75±0.11) (Figure 5C).
MiR‐130a Downregulates PPARγ Expression in Ang II‐Treated Cardiac Fibroblasts
First, we detected the expression of miR‐130a in Ang II‐stimulated cardiac fibroblasts. Treatment of cardiac fibroblasts with Ang II for 24 and 48 hours increased miR‐130a to 1.86‐ (P<0.05) and 2.45‐ (P<0.01) fold, respectively (Figure 6A). Gene expression of PPARγ was reduced to 59.35% (P<0.05) and 41.26% (P<0.01), respectively, after treatment with Ang II for 24 and 48 hours (Figure 6B). We also observed significant decrease of PPARγ protein levels with Ang II treatment (Figure 6C). The miR‐130a mimetic transfection in the presence of Ang II showed further reduction of PPARγ expression in both mRNA expression and protein levels, compared with the Ang II‐stimulated cells (P<0.05). Cardiac fibroblasts transfected with miR‐130a inhibitor in the presence of Ang II showed restored expression of PPARγ (Figure 6D and 6E). PPARγ mRNA and protein expression was reduced by 23.52% and 23.47%, respectively, when transfected with miR‐130a mimetic alone, compared with the unstimulated control cells (Figure 6D and 6E).
Figure 6.
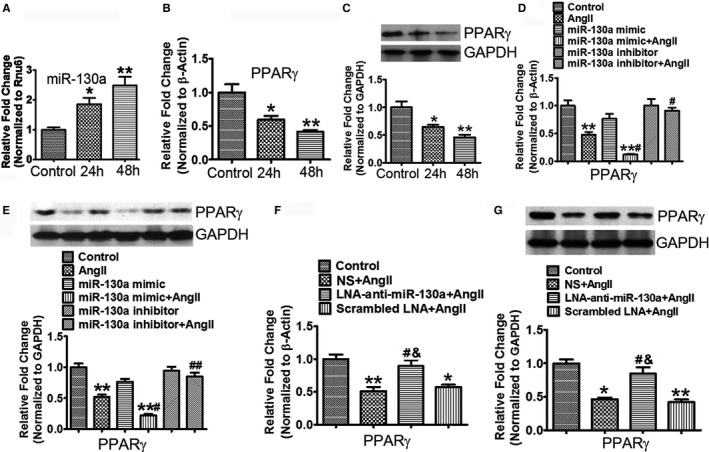
MiR‐130a downregulates PPARγ expression in Ang II‐treated cardiac fibroblasts. A through C, Cardiac fibroblasts were treated with 1 μmol/L Ang II for 24 and 48 h. The mature miR‐130a expression (A) was determined by qRT‐PCR. Rnu6 was used as an internal control. Gene expression of PPARγ (B) was determined by qRT‐PCR. β‐Actin was used as an internal control. Protein expression of PPARγ (C) was determined by Western blot analysis. GAPDH was used as an internal loading control. D and E, Cardiac fibroblasts were transfected with 50 nmol/L mimetic or inhibitor of miR‐130a for 12 h, and then stimulated with 1 μmol/L Ang II for 48 h. Gene expression of PPARγ (D) was determined by qRT‐PCR. β‐Actin was used as an internal control. Protein expression of PPARγ (E) was determined by Western blot analysis. GAPDH was used as an internal loading control. A through E, Data are expressed as means±SE from 3 independent experiments. *P<0.05, **P<0.01 vs control; # P<0.05, ## P<0.01 vs Ang II. F and G, Determination of PPARγ expression in control, NS+Ang II, LNA‐anti‐miR‐130a+Ang II, and scrambled LNA+Ang II‐treated groups. The PPARγ mRNA level (F) was determined by qRT‐PCR and β‐Actin was used as an internal control. The PPARγ protein level (G) was determined by Western blotting. GAPDH was used as an internal loading control. F and G, Data are expressed as means±SE from 6 independent mice. *P<0.05, **P<0.01 vs control; # P<0.05 vs NS+Ang II; & P<0.05 vs scrambled LNA+Ang II. Ang II indicates angiotensin II; LNA, locked nucleic acid; miR‐130a, microRNA‐130a; NS, normal saline; PPARγ, peroxisome proliferator‐activated receptor γ level; qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction.
There was a significant reduction in PPARγ mRNA expression and protein levels in the hearts of the NS‐ and scrambled LNA‐treated mice with Ang II infusion, compared with the control mice (P<0.01). Compared with the NS‐ and scrambled LNA‐treated mice, mice that received LNA‐anti‐miR‐130a injection showed restoration of PPARγ mRNA (NS versus LNA‐anti‐miR‐130a, 0.51±0.06 versus 0.89±0.09, P<0.05; scrambled LNA versus LNA‐anti‐miR‐130a, 0.57±0.04 versus 0.89±0.09, P<0.05) and protein expression (NS versus LNA‐anti‐miR‐130a, 0.46±0.02 versus 0.84±0.10, P<0.05; scrambled LNA versus LNA‐anti‐miR‐130a, 0.42±0.04 versus 0.84±0.10, P<0.05) in heart (Figure 6F and 6G).
MiR‐130a Directly Targets PPARγ
To identify putative binding sites of miR‐130a in the 3′‐UTR of PPARγ transcript, we used the miRNA target predicted search engine, TargetScan 5.2. As shown in miRNA: messenger RNA alignment analysis, 3′‐UTR of the PPARγ gene contained miR‐130a binding sites and were highly conserved among different species (Figure 7A). Based on the above analysis, HEK293 cells were transiently transfected with plasmids pEGFP, pEGFP‐PPARγ UTR, or pEGFP‐ PPARγ UTRmut along with control siRNA or miR‐130a mimetic, respectively. MiR‐130a mimetic potently suppressed EGFP expression from pEGFP‐ PPARγ UTR, but showed no influence on EGFP expression from pEGFP and pEGFP‐PPARγ UTR‐mut (Figure 7B and 7C). These results indicate that miR‐130a targets 3′‐UTR of PPARγ mRNA and thereby inhibits PPARγ mRNA and protein levels.
Figure 7.
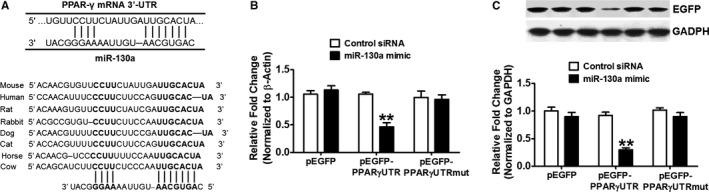
MiR‐130a directly targets PPARγ. A, An alignment between PPARγ and miR‐130a as predicted by TargetScan 6.2 analysis in various animals as shown by vertical bars. The sequence alignment of putative miR‐130a and its targeting site on 3′‐UTR of PPARγ shows a high level of complementarity and sequence conservation among vertebrates. B and C, HEK293 cells were transfected with 100 ng pEGFP, pEGFP‐PPARγ UTR or pEGFP‐PPARγ UTR mut along with control siRNA or miR‐130a mimetic, respectively. After 48 h of transfection, the EGFP gene expression (B) and protein level (C) were determined by qRT‐PCR and Western blotting, respectively. Data are expressed as means±SE from 3 independent experiments. **P<0.01 vs control siRNA. miR‐130a indicates microRNA‐130a; pEGFP, plasmid carrying enhanced green fluorescent protein; PPARγ, peroxisome proliferator‐activated receptor γ level; qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction.
PPARγ Attenuates miR‐130a‐Mediated Profibrotic Effect in Cardiac Fibroblasts
To assess the impact of PPARγ on miR‐130a‐mediated profibrotic effect in Ang II‐treated cardiac fibroblasts, we transfected cardiac fibroblasts with PPARγ plasmid and/or miR‐130a mimic followed by Ang II treatment for 48 hours. PPARγ protein expression in cardiac fibroblasts transfected with PPARγ plasmid was increased to 3.59‐fold, compared with that in cells transfected with pCDNA (Figure 8A). Compared with cardiac fibroblasts transfected with pCDNA, PPARγ plasmid reduced the increased mRNA expression of Col I (2.64±0.35 versus 1.41±0.15, P<0.05), Col III (2.44±0.13 versus 1.31±0.13, P<0.01), CTGF (2.54±0.12 versus 1.22±0.13, P<0.01), Fn (2.27±0.25 versus 1.24±0.20, P<0.05), and α‐SMA (2.50±0.22 versus 1.46±0.17, P<0.05) in response to Ang II. MiR‐130a resulted in further increased mRNA expression of these profibrotic genes, which was attenuated by PPARγ overexpression (Col I, 3.83±0.16 versus 1.70±0.19, P<0.01; Col III, 3.39±0.17 versus 1.55±0.14, P<0.01; CTGF, 3.70±0.10 versus 1.65±0.10, P<0.01; Fn, 3.86±0.16 versus 1.60±0.24, P<0.01; α‐SMA, 3.67±0.20 versus 1.78±0.19, P<0.01) (Figure 8B through 8F).
Figure 8.
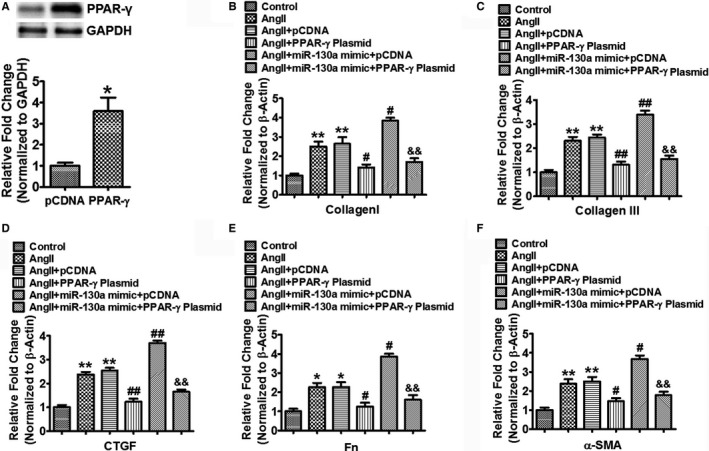
PPARγ attenuates miR‐130a‐mediated profibrotic effect in cardiac fibroblasts. A, Cardiac fibroblasts were transfected with 100 ng pCDNA or PPARγ plasmid for 48 h and protein expression of PPARγ was determined by Western blotting. GAPDH was used as an internal loading control. Data are expressed as means±SE from 3 independent experiments. *P<0.05 vs pCDNA. B through F, Cardiac fibroblasts were transfected with 100 ng pCDNA or PPARγ plasmid with or without 50 nmol/L miR‐130a mimic for 24 h followed by Ang II treatment for another 48 h. Gene expression of Col I (B), Col III (C), CTGF (D), Fn (E), and α‐SMA (F) were determined by qRT‐PCR. β‐Actin was used as an internal control. Data are expressed as means±SE from 3 independent experiments. *P<0.05, **P<0.01 vs control; # P<0.05, ## P<0.01 vs Ang II+pCDNA; && P<0.01 vs Ang II+miR‐130a+pCDNA. Ang II indicates angiotensin II; Col I, Col III, collagen I, collagen III; CTGF, connective tissue growth factor; miR‐130‐a, microRNA‐130‐a; qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction; pCDNA, plasmid carrying control DNA; PPARγ, peroxisome proliferator‐activated receptor γ level.
Discussion
Our study demonstrates that cardiac miR‐130a is upregulated in Ang II‐infused mice and makes a contribution to the pathogenesis of cardiac fibrosis leading to cardiac dysfunction. Although miR‐130a directly targets PPARγ, miR‐130a also influences gene expression associated with cardiac fibrosis. Furthermore, we elucidate for the first time that miR‐130a plays a key role in myofibroblasts differentiation. Overexpression of miR‐130a in cardiac fibroblasts increased expression of several fibrotic genes including collagens, Fn and CTGF, enhanced macrophages (MCP1), vascular (intercellular adhesion molecule 1 and vascular cell adhesion molecule 1), and inflammatory genes (regulated on activation, normal T cell expressed and secreted and interleukin 6), and repressed PPARγ. Importantly, inhibition of miR‐130a in mice hearts significantly reduced Ang II‐induced α‐SMA expression and cardiac fibrosis. Finally, we corroborated our findings with human subjects and provided evidence that miR‐130a is critically involved in the pathogenesis of cardiac fibrosis. Together, these results indicate that miR‐130a plays an important role in Ang II‐induced cardiac fibrosis and inhibition of miR‐130a may offer a therapeutic potential to treat cardiac fibrosis.
Cardiac fibrosis that occurs because of disturbance of the ECM network ultimately leads to myocardial dysfunction.3, 46 Deposition of excessive collagens and other matrix components is the primary determinant of fibrotic myocardium and is thought to be originated from myofibroblasts, the specialized cardiac fibroblasts formed by irreversible acquisition of expressions of α‐SMA.10, 11 However, the molecular mechanism underlying this process remains unknown. Recently it has been determined that miRNAs elicit an important role in many cardiac pathologies and they gained considerable attention. A panel of dysregulated miRNAs has been catalogued in recent years for cardiac fibrosis including miR‐133, miR‐29, miR‐26, miR‐21, or let‐7i by targeting various fibrotic genes with utilizing various mechanisms.21, 23, 47, 48, 49, 50, 51 We identified miR‐130a as a potential candidate in cardiac remodeling. MiR‐130a appears to be widely expressed in diverse cell types and regulates various cell functions through different targets in different cell types. A recent report showed that miR‐130a is critical in cardiac arrhythmia, indicating a potential role in cardiac disorders.41 However, the role of miR‐130a in cardiac pathologies such as cardiac fibrosis remains elusive.
In this study, we clearly demonstrated that miR‐130a is a critical player in the Ang II‐induced cardiac fibrosis in a mouse model. By selective blocking of miR‐130a in Ang II‐infused mice, our results showed the significant reduction in cardiac mass (hypertrophy) and fibrosis compared with the untreated mice. Mechanistically, our data revealed that miR‐130a inhibition selectively restored the PPARγ level and impeded the process of myofibroblasts differentiation. It is also not uncommon that miRNA has multiple targets in a specific biological process.52 While our study identified PPARγ as a target, the other potential targets cannot be ignored. The ability of a single miRNA to control the expression of multiple genes suggests that modulation of a single miRNA may influence several signaling pathways associated with it. While each target gene for miRNA is regulated individually, the additive effect of other mRNA expressions is believed to be the output of downstream signaling events such as fibrotic cascade in this study. In this context, our data revealed that mRNA expression of Col I, Col III, CTGF, and Fn was significantly reduced after blocking of miR‐130a. The PPARγ, a nuclear receptor/transcription factor, has been shown to have the function of antimyocardial fibrosis.53 It has been reported that PPARγ has a wide spectrum of functions in attenuating inflammation, inhibiting apoptosis, and reducing oxidative stress, which may have a significant impact in improving cardiac function from adverse remodeling.54 However, the underling mechanisms of PPARγ modulation by miRNA in the regulation of cardiac fibrosis remain elusive. This study showed for the first time that miR‐130a directly targets PPARγ in controlling cardiac fibrosis. Our data demonstrated that overexpression of miR‐130a repressed the PPARγ level and promoted the process of myofibroblasts differentiation by enhancing α‐SMA level, a critical morphological alteration marker in myofibroblasts differentiation. Inhibition of miR‐130a reversed the process. Our data further elicit that all fibrotic genes including collagens, CTGF, and Fn were significantly attenuated after inhibiting miR‐130a in Ang II‐induced cardiac fibrosis. Altogether, our study underscores a pivotal role of miR‐130a in cardiac fibrosis by targeting PPARγ.
Ang II has been demonstrated to promote vascular inflammation, and macrophage infiltration leading to the development of fibrosis55, 56, 57; however, the role of miR‐130a in this signaling is unknown. Data presented in this study showed an increased CD11b+ immunoreactivity in Ang II‐infused hearts along with upregulation of vascular (intercellular adhesion molecule 1 and vascular cell adhesion molecule 1), inflammatory (interleukin 6 and regulated on activation, normal T cell expressed and secreted), and chemotaxis genes (MCP1). The increased patterns of all these genes were significantly reduced by the inhibition of miR‐130a. Thus, the present observation suggests that Ang II‐induced vascular inflammatory signaling and macrophage infiltration cascade are restored by miR‐130a inhibition and, thereby, preventing cardiac fibrosis.
Cardiac fibrosis that occurs as a result of aberrant deposition of ECM protein is a highly debilitating process leading to the development of heart failure. Despite the progress in delineating the mechanisms of fibrosis, antifibrotic therapies never received salutary implementation. The first line of therapies for cardiac fibrosis relies on angiotensin‐converting enzyme inhibitor, β‐blocker, or angiotensin II‐receptor 1 blocker and shows promise in alleviating fibrosis; however, the key effector molecules are now required for more potent therapeutic intervention. In this context, our investigation is providing a new insight for the treatment of cardiac fibrosis. We demonstrated that Ang II infusion in mice showed significant fibrosis in the heart and was accompanied by alteration of fibrotic genes, upregulation of miR‐130a, and compromised cardiac function. This observation is corroborated with human cardiac fibrosis. This increased fibrosis was distinctly reduced by inhibition of miR‐130a compared with the Ang II‐infused group. The magnitude of miR‐130a inhibitor to attenuate the progression of cardiac fibrosis indicated its therapeutic potential in future. It would be interesting to further investigate the effect of ablation of miR‐130a in the heart in adulthood as global knock‐out of miR‐130a is embryonically lethal.58
Recent evidence showing the antifibrotic effect of miR‐130a in bleomycin‐induced lung fibrosis by modulating α‐SMA and procollagen level in mice indicated the pivotal role of miR‐130a in another fibrosis model.59 Besides the report that Su et al demonstrated the downregulation of miR‐130a‐3p in macrophage‐induced lung fibrosis, their identification of PPARγ as a target of miR‐130a‐3p supported our observation. Our study found that miR‐130a was increased significantly in cardiac fibrosis, while inhibition of miR‐130a reduced fibrosis by targeting PPARγ, indicating miR‐130a as a positive regulator for cardiac fibrosis. Furthermore, in myocardial infarction, setting delivery of lentivirus expressing miR‐130a into the mouse heart improves cardiac function, promotes angiogenesis, and attenuates collagen deposition in the infarcted myocardium. The protective effect of miR‐130a appears to be suppression of phosphatase and tensin homolog deleted on chromosome ten (PTEN) expression and activation of PI3K/Akt signaling.60 Both reports support our observation and suggest a critical role for miR‐130a in the fibrotic phenomenon. Interestingly, miR‐130a transgenic mice developed spontaneous atrial arrhythmias by suppressing connexin 43.41 Regional conduction abnormalities or arrhythmia is reported to contribute to the development of both atrial fibrosis and atrial fibrillation.61 Together with the afore mentioned reports, our report underscores an association between miR‐130a and cardiac fibrosis, and PPARγ contributes a potential role in this process.
In conclusion, we have demonstrated a miR‐130a‐mediated novel mechanism of cardiac fibrosis where myofibroblasts differentiation contributes a critical role. Along the way, our study showed that PPARγ is a key player in Ang II‐induced cardiac fibrosis. Restoring the PPARγ level and reversing the process of myofibroblasts differentiation by inhibiting miR‐130a further elicits a fine‐tuning of the activating process of multiple fibrogenic genes.
Sources of Funding
This study was partly supported by American Heart Association‐Grant‐in‐Aid (14GRNT20490320), by start‐up funds from the Texas A & M Health Science Center, College of Medicine, to Gupta, by Scientist Development Grant (13SDG 914630018) to Chatterjee, and by grants from National Natural Science Foundation of China (No. 81370192) to Li.
Disclosures
None.
Acknowledgments
The authors acknowledge Dr Christine Moravec and Ms Wendy Sweet at Kaufman Center for Heart Failure, Department of Cardiovascular Medicine, Cleveland Clinic, Cleveland OH for providing failing and nonfailing human heart samples. The authors acknowledge Dr M. Gorospe for providing wild‐type and mutant PPARγ 3′‐UTR plasmids. The authors also acknowledge Central Texas Veterans Affairs Health Care System for providing the research facility to complete this work. The open access publishing fees for this article have been covered by the Texas A&M University Open Access to Knowledge Fund (OAKFund), supported by the University Libraries and the Office of the Vice President for Research.
(J Am Heart Assoc. 2017;6:e006763 DOI: 10.1161/JAHA.117.006763.)29114000
References
- 1. Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. [DOI] [PubMed] [Google Scholar]
- 2. Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure‐overloaded heart. Cardiovasc Res. 2011;89:265–272. [DOI] [PubMed] [Google Scholar]
- 3. Segura AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev. 2014;19:173–185. [DOI] [PubMed] [Google Scholar]
- 4. Edgley AJ, Krum H, Kelly DJ. Targeting fibrosis for the treatment of heart failure: a role for transforming growth factor‐beta. Cardiovasc Ther. 2012;30:e30–e40. [DOI] [PubMed] [Google Scholar]
- 5. Espira L, Czubryt MP. Emerging concepts in cardiac matrix biology. Can J Physiol Pharmacol. 2009;87:996–1008. [DOI] [PubMed] [Google Scholar]
- 6. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sivakumar P, Gupta S, Sarkar S, Sen S. Upregulation of lysyl oxidase and MMPs during cardiac remodeling in human dilated cardiomyopathy. Mol Cell Biochem. 2008;307:159–167. [DOI] [PubMed] [Google Scholar]
- 8. Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, Conway SJ, Nagai R. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;120:254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–212. [DOI] [PubMed] [Google Scholar]
- 10. Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast‐mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013;10:15–26. [DOI] [PubMed] [Google Scholar]
- 12. Shi Q, Liu X, Bai Y, Cui C, Li J, Li Y, Hu S, Wei Y. In vitro effects of pirfenidone on cardiac fibroblasts: proliferation, myofibroblast differentiation, migration and cytokine secretion. PLoS One. 2011;6:e28134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stempien‐Otero A, Kim DH, Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol. 2016;97:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci USA. 2005;102:437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Daskalopoulos EP, Janssen BJ, Blankesteijn WM. Myofibroblasts in the infarct area: concepts and challenges. Microsc Microanal. 2012;18:35–49. [DOI] [PubMed] [Google Scholar]
- 16. Fan YH, Dong H, Pan Q, Cao YJ, Li H, Wang HC. Notch signaling may negatively regulate neonatal rat cardiac fibroblast‐myofibroblast transformation. Physiol Res. 2011;60:739–748. [DOI] [PubMed] [Google Scholar]
- 17. Lijnen P, Petrov V. Transforming growth factor‐beta 1‐induced collagen production in cultures of cardiac fibroblasts is the result of the appearance of myofibroblasts. Methods Find Exp Clin Pharmacol. 2002;24:333–344. [DOI] [PubMed] [Google Scholar]
- 18. Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. [DOI] [PubMed] [Google Scholar]
- 19. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. [DOI] [PubMed] [Google Scholar]
- 20. Thum T, Catalucci D, Bauersachs J. MicroRNAs: novel regulators in cardiac development and disease. Cardiovasc Res. 2008;79:562–570. [DOI] [PubMed] [Google Scholar]
- 21. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR‐29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du XJ, Lin RC, McMullen JR. Therapeutic inhibition of the miR‐34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci USA. 2012;109:17615–17620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei C, Kim IK, Kumar S, Jayasinghe S, Hong N, Castoldi G, Catalucci D, Jones WK, Gupta S. NF‐kappaB mediated miR‐26a regulation in cardiac fibrosis. J Cell Physiol. 2013;228:1433–1442. [DOI] [PubMed] [Google Scholar]
- 24. Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H, Xu J, Che L, Chen X, Sluijter JP, Das S, Cretoiu D, Xu B, Zhong J, Xiao J, Li X. Crucial role of miR‐433 in regulating cardiac fibrosis. Theranostics. 2016;6:2068–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Du W, Liang H, Gao X, Li X, Zhang Y, Pan Z, Li C, Wang Y, Liu Y, Yuan W, Ma N, Chu W, Shan H, Lu Y. MicroRNA‐328, a potential anti‐fibrotic target in cardiac interstitial fibrosis. Cell Physiol Biochem. 2016;39:827–836. [DOI] [PubMed] [Google Scholar]
- 26. Zhou Y, Deng L, Zhao D, Chen L, Yao Z, Guo X, Liu X, Lv L, Leng B, Xu W, Qiao G, Shan H. MicroRNA‐503 promotes angiotensin II‐induced cardiac fibrosis by targeting Apelin‐13. J Cell Mol Med. 2016;20:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang X, Tsitsiou E, Herrick SE, Lindsay MA. MicroRNAs and the regulation of fibrosis. FEBS J. 2010;277:2015–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simoes ESAC, Teixeira MM. ACE inhibition, ACE2 and angiotensin‐(1‐7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol Res. 2016;107:154–162. [DOI] [PubMed] [Google Scholar]
- 29. Schellings MW, Vanhoutte D, van Almen GC, Swinnen M, Leenders JJ, Kubben N, van Leeuwen RE, Hofstra L, Heymans S, Pinto YM. Syndecan‐1 amplifies angiotensin II‐induced cardiac fibrosis. Hypertension. 2010;55:249–256. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR‐29b as a therapeutic agent for angiotensin II‐induced cardiac fibrosis by targeting TGF‐beta/Smad3 signaling. Mol Ther. 2014;22:974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schneider MD. Serial killer: angiotensin drives cardiac hypertrophy via TGF‐beta1. J Clin Invest. 2002;109:715–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iwata M, Cowling RT, Yeo SJ, Greenberg B. Targeting the ACE2‐ang‐(1‐7) pathway in cardiac fibroblasts to treat cardiac remodeling and heart failure. J Mol Cell Cardiol. 2011;51:542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jakob P, Doerries C, Briand S, Mocharla P, Krankel N, Besler C, Mueller M, Manes C, Templin C, Baltes C, Rudin M, Adams H, Wolfrum M, Noll G, Ruschitzka F, Luscher TF, Landmesser U. Loss of angiomiR‐126 and 130a in angiogenic early outgrowth cells from patients with chronic heart failure: role for impaired in vivo neovascularization and cardiac repair capacity. Circulation. 2012;126:2962–2975. [DOI] [PubMed] [Google Scholar]
- 34. Chen Y, Gorski DH. Regulation of angiogenesis through a microRNA (miR‐130a) that down‐regulates antiangiogenic homeobox genes GAX and HOXA5. Blood. 2008;111:1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meng S, Cao J, Zhang X, Fan Y, Fang L, Wang C, Lv Z, Fu D, Li Y. Downregulation of microRNA‐130a contributes to endothelial progenitor cell dysfunction in diabetic patients via its target Runx3. PLoS One. 2013;8:e68611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matkovich SJ, Van Booven DJ, Youker KA, Torre‐Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB, Dorn GW II. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation. 2009;119:1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. [DOI] [PubMed] [Google Scholar]
- 38. Wu WH, Hu CP, Chen XP, Zhang WF, Li XW, Xiong XM, Li YJ. MicroRNA‐130a mediates proliferation of vascular smooth muscle cells in hypertension. Am J Hypertens. 2011;24:1087–1093. [DOI] [PubMed] [Google Scholar]
- 39. Xiao F, Yu J, Liu B, Guo Y, Li K, Deng J, Zhang J, Wang C, Chen S, Du Y, Lu Y, Xiao Y, Zhang Z, Guo F. A novel function of microRNA 130a‐3p in hepatic insulin sensitivity and liver steatosis. Diabetes. 2014;63:2631–2642. [DOI] [PubMed] [Google Scholar]
- 40. Pan Y, Wang R, Zhang F, Chen Y, Lv Q, Long G, Yang K. MicroRNA‐130a inhibits cell proliferation, invasion and migration in human breast cancer by targeting the RAB5A. Int J Clin Exp Pathol. 2015;8:384–393. [PMC free article] [PubMed] [Google Scholar]
- 41. Osbourne A, Calway T, Broman M, McSharry S, Earley J, Kim GH. Downregulation of connexin43 by microRNA‐130a in cardiomyocytes results in cardiac arrhythmias. J Mol Cell Cardiol. 2014;74:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumar S, Wei C, Thomas CM, Kim IK, Seqqat R, Kumar R, Baker KM, Jones WK, Gupta S. Cardiac‐specific genetic inhibition of nuclear factor‐kappaB prevents right ventricular hypertrophy induced by monocrotaline. Am J Physiol Heart Circ Physiol. 2012;302:H1655–H1666. [DOI] [PubMed] [Google Scholar]
- 43. Li L, Wei C, Kim IK, Janssen‐Heininger Y, Gupta S. Inhibition of nuclear factor‐kappaB in the lungs prevents monocrotaline‐induced pulmonary hypertension in mice. Hypertension. 2014;63:1260–1269. [DOI] [PubMed] [Google Scholar]
- 44. Kumar S, Seqqat R, Chigurupati S, Kumar R, Baker KM, Young D, Sen S, Gupta S. Inhibition of nuclear factor kappaB regresses cardiac hypertrophy by modulating the expression of extracellular matrix and adhesion molecules. Free Radic Biol Med. 2011;50:206–215. [DOI] [PubMed] [Google Scholar]
- 45. Kumar S, Gupta S. Thymosin beta 4 prevents oxidative stress by targeting antioxidant and anti‐apoptotic genes in cardiac fibroblasts. PLoS One. 2011;6:e26912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zannad F, Rossignol P, Iraqi W. Extracellular matrix fibrotic markers in heart failure. Heart Fail Rev. 2010;15:319–329. [DOI] [PubMed] [Google Scholar]
- 47. Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW II. MicroRNA‐133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure‐overloaded adult hearts. Circ Res. 2010;106:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Castoldi G, Di Gioia CR, Bombardi C, Catalucci D, Corradi B, Gualazzi MG, Leopizzi M, Mancini M, Zerbini G, Condorelli G, Stella A. MiR‐133a regulates collagen 1A1: potential role of miR‐133a in myocardial fibrosis in angiotensin II‐dependent hypertension. J Cell Physiol. 2012;227:850–856. [DOI] [PubMed] [Google Scholar]
- 49. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. miR‐133 and miR‐30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–178, 176p following 178. [DOI] [PubMed] [Google Scholar]
- 50. Lorenzen JM, Schauerte C, Hubner A, Kolling M, Martino F, Scherf K, Batkai S, Zimmer K, Foinquinos A, Kaucsar T, Fiedler J, Kumarswamy R, Bang C, Hartmann D, Gupta SK, Kielstein J, Jungmann A, Katus HA, Weidemann F, Muller OJ, Haller H, Thum T. Osteopontin is indispensible for AP1‐mediated angiotensin II‐related miR‐21 transcription during cardiac fibrosis. Eur Heart J. 2015;36:2184–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang X, Wang HX, Li YL, Zhang CC, Zhou CY, Wang L, Xia YL, Du J, Li HH. MicroRNA Let‐7i negatively regulates cardiac inflammation and fibrosis. Hypertension. 2015;66:776–785. [DOI] [PubMed] [Google Scholar]
- 52. Tarang S, Weston MD. Macros in microRNA target identification: a comparative analysis of in silico, in vitro, and in vivo approaches to microRNA target identification. RNA Biol. 2014;11:324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang ZZ, Shang QH, Jin HY, Song B, Oudit GY, Lu L, Zhou T, Xu YL, Gao PJ, Zhu DL, Penninger JM, Zhong JC. Cardiac protective effects of irbesartan via the PPAR‐gamma signaling pathway in angiotensin‐converting enzyme 2‐deficient mice. J Transl Med. 2013;11:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu HJ, Liao HH, Yang Z, Tang QZ. Peroxisome proliferator‐activated receptor‐gamma is critical to cardiac fibrosis. PPAR Res. 2016;2016:2198645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Usui M, Egashira K, Tomita H, Koyanagi M, Katoh M, Shimokawa H, Takeya M, Yoshimura T, Matsushima K, Takeshita A. Important role of local angiotensin II activity mediated via type 1 receptor in the pathogenesis of cardiovascular inflammatory changes induced by chronic blockade of nitric oxide synthesis in rats. Circulation. 2000;101:305–310. [DOI] [PubMed] [Google Scholar]
- 56. Zhao QW, Ishibashi M, Hiasa K, Tan CY, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin II‐induced vascular inflammation and remodeling. Hypertension. 2004;44:264–270. [DOI] [PubMed] [Google Scholar]
- 57. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim GH, Samant SA, Earley JU, Svensson EC. Translational control of FOG‐2 expression in cardiomyocytes by microRNA‐130a. PLoS One. 2009;4:e6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Su SC, Zhao QY, He CH, Huang D, Liu J, Chen F, Chen JN, Liao JY, Cui XY, Zeng YJ, Yao HR, Su FX, Liu Q, Jiang SP, Song EW. miR‐142‐5p and miR‐130a‐3p are regulated by IL‐4 and IL‐13 and control profibrogenic macrophage program. Nat Commun. 2015;6:8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lu C, Wang XH, Ha TZ, Hu YP, Liu L, Zhang X, Yu HH, Miao J, Kao R, Kalbfleisch J, Williams D, Li CF. Attenuation of cardiac dysfunction and remodeling of myocardial infarction by microRNA‐130a are mediated by suppression of PTEN and activation of PI3K dependent signaling. J Mol Cell Cardiol. 2015;89:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lofsjogard J, Persson H, Diez J, Lopez B, Gonzalez A, Edner M, Mejhert M, Kahan T. Atrial fibrillation and biomarkers of myocardial fibrosis in heart failure. Scand Cardiovasc J. 2014;48:299–303. [DOI] [PubMed] [Google Scholar]