

Abstract
Background
Atherosclerosis is characterized by the presence of activated immune‐competent cells including dendritic cells (DCs) and T cells, dead cells, and oxidized low‐density lipoprotein. HSP60 (Heat shock protein 60) has been implicated in atherosclerosis. A plasma protein, Annexin A5, has atheroprotective properties.
Methods and Results
Human DCs differentiated from peripheral blood monocytes were treated with human HSP60 or HSP90 and autologous T cells were cocultured with these pretreated DCs (mDCs). HSP60 induced mDCs and T‐cell activation as determined by FACScan (Fluorescence associated cell scan), gene‐activation, and cytokine production. HSP60‐induced T‐cell activation was partly major histocompatibility complex class II–dependent. T cells exposed to HSP60‐treated mDCs produced interferon‐γ, interleukin‐17, but not transforming growth factor‐β. HSP60 did not promote expression of Toll‐like receptors 2 or 4. HSP90 promoted mDCs maturation but had no effect on T‐cell activation. Annexin A5 inhibited HSP60‐proinflammatory Th1/Th17 effects on mDCs and T cells, and partly bound HSP60. Further, Annexin A5 inhibited HSP‐induced activation of mDCs and also oxidized low‐density lipoprotein–induced HSP‐production from mDCs. Experiments on mDCs and T cells derived from carotid atherosclerotic plaques from patients with symptomatic carotid disease gave similar results as from blood donors.
Conclusions
HSP60 induces mDCs activation and partly major histocompatibility complex class II–dependent activation of blood‐ and plaque‐derived T cells, which is mostly of Th1/Th17 type. HSP60 could thus be an important T‐cell antigen in plaques, and also mediate oxidized low‐density lipoproteins immunogenic effects on DC‐T‐cell activation, promoting plaque rupture and clinical manifestations of cardiovascular disease. Annexin A5 inhibits both oxidized low‐density lipoprotein–induced HSP60, and HSP60‐mediated immune activation, which suggests a potential therapeutic role.
Keywords: Annexin A5, atherosclerosis, autoimmunity, dendritic cells, heat shock protein 60, immune system, T cells
Subject Categories: Inflammation, Lipids and Cholesterol, Mechanisms, Vascular Biology
Clinical Perspective
What Is New?
Macrophages, foam cells, dead cells, oxidized low‐density lipoprotein, and also specialized antigen‐presenting cells, dendritic cells, and T cells are characteristic of atherosclerotic lesions.
Heat shock protein 60 has been implicated as proatherogenic.
A plasma protein, Annexin A5, has atheroprotective properties.
What Are the Clinical Implications?
Human heat shock protein 60 is induced by oxidized low‐density lipoprotein and induces dendritic cell activation and dendritic cell–mediated activation of T cells from human atherosclerotic plaques, and it could thus be argued that atherosclerosis has autoimmune characteristics.
Annexin A5 inhibits this immune activation, and could be a potential therapy.
Introduction
Atherosclerotic plaques are characterized by the presence of activated immune‐competent cells, and a dominant Th1 pro‐inflammatory type of immune activation, typically in areas of plaques prone to rupture or undergoing rupture, which could lead to cardiovascular disease (CVD) manifestations as stroke and myocardial infarction.1 Other major components of atherosclerotic lesions are dead cells and oxidized low‐density lipoprotein (OxLDL).2
The nature and cause of the T‐cell activation in plaques is still not clarified in detail, and different antigens could play a role, simultaneously. OxLDL is a major candidate since it activates human T cells,3 also from human atherosclerotic plaques and also monocytes/macrophages and dendritic cells (DCs).4, 5, 6
Another candidate (non–mutually exclusive) is HSP60 (heat shock protein 60). The hypothesis that HSP60 is involved in the immunity of atherosclerosis was originally proposed in the early 1990s by G. Wick and coworkers, who demonstrated that immunization with HSP60 increases atherosclerosis in animal models.7 Since then this concept has continued to be developed, with more supporting data from animal models, and also from human studies, where antibodies against HSP60/65 are associated with CVD and/or atherosclerosis and a pathogenic role of these antibodies has been suggested as an underlying mechanism.8, 9
HSPs, sometimes called stress proteins, are conserved in prokaryotes and eukaryotes and share the sequence homology at both protein and DNA level. Mammalian HSP60 and microbial HSP65 share >90% amino acid sequences.2, 10, 11 It is possible that a side effect of immunity against microbial HSP65 also involves immune reactions to human HSP60 at sites in the arterial wall, exposed to increased stress as in hypertension, which could then promote atherosclerosis. Such a concept could also provide an explanation for the propensity of atherosclerotic lesions to certain sites in the arterial tree, as in bifurcations prone to sheer stress.9, 10 OxLDL induces HSP60 in monocytes/macrophages and DC, and we proposed that such induced HSP could be involved in immune effects of OxLDL.6, 12 However, the mechanisms by which human HSP60 could promote immune activation are not fully known. Interestingly, circulating HSP60 also is a risk marker for CVD.13 Less is known about HSP90 in atherosclerosis, though it also has been implicated as proatherogenic.14
We recently reported that Annexin A5 (ANXA5), a plasma protein with antithrombotic properties, reduces inflammation and improves vascular function in apolipoprotein E−/− mice,15 ameliorates OxLDL‐induced T‐cell and DC activation, and promotes a T‐cell regulatory response.6 In this report we study DC and T‐cell activation in relation to human atherosclerosis with focus on HSP60 as an antigen and a modulatory role of ANXA5.
Materials and Methods
DC Differentiation and Treatment
Monocytes were isolated by human monocyte enrichment cocktail (Stemcell Technologies, France). Isolated monocytes were cultured with recombinant human granulocyte macrophage colony‐stimulating factor and interleukin‐4 (IL‐4) (Immunotools, Germany), 50 ng/mL of each in RPMI complete media. At day 3, half of the media was replaced with fresh RPMI complete media with granulocyte macrophage colony‐stimulating factor and IL‐4. At day 6, 2×106/mL cells were cultured with or without HSP60 (MyBiosource) or HSP90 (HSPß90, Sigma Aldrich, MO) at the concentrations described. Cells were collected after 24 hours and analyzed with >90% cell viability. DC stained with CD11C‐PE, CD86‐Percp/Cy5.5, CD83‐APC, CD40‐fluorescein isothiocyanate (BD Bioscience), and HLA‐II‐fluorescein isothiocyanate (Biolegend) antibodies.
The endotoxin concentration in HSP60 was checked by LAL Chromogenic Endotoxin Quantitation Kit (Thermofisher Scientific, IL). The level of endotoxin in the HSP60 was ≤1 EU/mg, according to manufacturer information, which is equal to ≤0.1 ng/mg. Our working concentration 5 μg/mL of HSP60 contains ≤0.0005 ng endotoxin. A similar concentration of endotoxin was determined in HSP90.
Plaque T‐Cell‐DC Coculture
Atherosclerotic plaques were collected from patients who were undergoing carotid endarterectomy (3) or femoral arterial surgery (3) at the Department of Surgery, Vascular Surgery, Södersjukhuset, Stockholm, Sweden. The study was approved by the Karolinska Institutet research ethics committee and is in accordance with the Declaration of Helsinki. All subjects gave written informed consent before entering the study. Information about clinical characteristics is given in Table.
Table 1.
Characterization of Patients Operated on Because of Atherosclerotic Disease
| Patient No. | Anatomic Region | Macroscopic Appearance | Indication for Surgery |
|---|---|---|---|
| 1 | Right femoral artery | Heavily calcified plaque, subtotal occlusion | Severe claudication |
| 2 | Right carotid artery | Tight stenosis, fragile, ruptured plaque | Stroke right cerebral hemisphere 6 d before surgery, and yet new symptoms 5 d before surgery |
| 3 | Left carotid artery | Tight stenosis, fragile plaque with hematoma | TIA with symptoms from left hemisphere: dysphasia, right‐side hemiplegia (transitory weakness of right arm and leg). The patient had initial symptoms 3 d preop and even on the morning at the day of surgery |
| 4 | Left carotid artery | Very tight stenosis with fragile plaque | TIA 3 d preop with transitory right arm paresis, facial paresis, and dysphasia |
| 5 | Right femoral artery | Occlusion of the common femoral artery. Heavily calcified plaque with fresh thrombus | Severe claudication, walking distance 10 m, rest pain |
| 6 | Right femoral artery | Occlusive calcified plaque | Severe claudication |
TIA indicates transient ischemic attack.
According to previously reported methodology,6 plaques were dissected into small pieces for mononuclear cells separation. In short, small pieces of plaques were incubated in enzyme mixture containing 1.25 mg/mL collagenase IV (Life Technologies Europe BV, Sweden), 25 μg/mL Liberase DL (Roche Applied Science, Sweden), and 0.2 mg/mL DNase I (Roche Applied Science, Sweden) for 1 hour at 37°C. The released cells from plaques were strained through a 100‐μm Celltrics filter (Millipore AB, Sweden) to remove unwanted tissue fat or debris. Filtered cells were collected and T cells were purified by easysep T‐cell enrichment kit (Stemcell Technologies, France). DCs generated from peripheral blood of the plaque donors were cultured with or without 5 μg/mL of HSP60. After overnight culture, cells were washed, resuspended in complete RPMI media, and were cocultured with plaque T cells for 48 hours. Cells were collected and stained with anti‐CD3‐Percp/Cy5.5, anti‐CD 25‐PE/fluorescein isothiocyanate, anti‐CD69‐APC/fluorescein isothiocyanate and anti‐CD71‐BV421 (BD Bioscience). Cell supernatants were frozen down for cytokine measurement. Details of composition of plaque T cells have been published before using similar technique as herein.5, 6
DC‐T‐Cell Cocultures
CD3 T cells were isolated from buffy coat of healthy donors by human T‐cell enrichment cocktail (Stemcell Technologies, France) according to the manufacturer's instruction. Monocyte‐derived DCs (mDCs) were cultured with or without HSP60 or HSP90 as indicated and with or without addition of 10 μg/mL ANXA5. To investigate concentration‐dependent effect on T‐cell activation, mDCs were stimulated with 2.5, 5, or 10 μg/mL of HSP60. As mentioned above, after overnight incubation, autologous T cells 4×105 were cocultured with 1×105 DCs in 400 μL complete medium. In case of HLA‐II blocking, low endotoxin azide‐free purified anti‐HLA‐II antibodies (Biolegend), 8 μg/mL, were added into mDCs before T‐cell addition. Cells were collected after 48 hours and stained as mentioned for plaque cells.
Cell Proliferation Assay
Cell proliferation assays were determined by BrDu (colorimetric assay kit) assays according to manufacturer protocol (Sigma Aldrich). Briefly, mDCs with or without 5 μg/mL of HSP60 were cultured for 12 hours. After washing and resuspending in complete RPMI media, 0.5×105 DCs were cocultured with 2×105 autologous T cells in a 96‐well round‐bottom plate (Becton Dickinson). After 72 hours of incubation, cells were labeled with BrDu and incubated for 20 hours. Cells were collected, centrifuged, and dried at 60°C for 1 hour. Dried cells were fixed with FixDenat solution before incubation with anti‐BrDu POD (Peroxidase) antibodies. After 2 hours of incubation, cells were washed and substrate solution was added for color development. To stop the reaction, 1M H2SO4 was added into substrate solution and read the reaction plate at 450‐nm wavelength with 690 reference wavelength.
Trans‐Well Assay
mDCs were stimulated with HSP60 and as mentioned above, cultured with T cells in trans‐well plates (Sigma Aldrich). T cells 4×105 in 200 μL culture media was cultured in an upper chamber of a 24‐well trans‐well plate while autologous HSP60‐stimulated mDCs 2×105 in 500 μL were in the bottom chamber. In parallel, T cells and mDCs were in the same wells of the plate for comparison. Cells were collected after 48 hours and stained as mentioned.
Cytokine Quantification
The amount of interferon (IFN)‐γ, IL‐6, tumor necrosis factor‐α, IL‐1β, IL‐12.p70, IL‐10, and transforming growth factor‐β cytokines in cell supernatants were detected by ELISA (R&D Systems, UK). Supernatants of mDCs cultures were collected after 24 hours, whereas mDC‐T‐cell coculture supernatants were collected after 48 hours. All the measurement of cytokines was performed according to manufacturer protocol.
Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was extracted with a Qiagen mini or micro kit (Qiagen, Germany) and cDNA was synthesized from total RNA using a cDNA synthesis high‐capacity kit (Applied Biosystem). One microliter of cDNA was used in each reaction of quantitative real‐time polymerase chain reaction. and TBET and GATA3 genes were analyzed for Th1 and Th2 transcription factor, respectively. T‐reg transcription factor FoxP3 and RORC for IL‐17‐producing T cell was also analyzed. Housekeeping gene GAPDH was used as reference gene or to normalize the difference. PCR reagent TaqMan master mix was used to run the reaction (Applied Biosystems, CA) in 7500 Real‐Time PCR system (Applied Biosystems, CA). ΔΔCT method was used to calculate the relative difference in gene expression.
HSP60 Induction
LDL (Sigma Aldrich) was oxidized by CuSO4 as previously reported.3 Briefly, 0.5 mg/mL LDL in PBS was incubated with 20 μmol/L CuSO4 at 37°C. After 24 hours of incubation, the solution was dialyzed in PBS overnight. Lipid oxidation was confirmed by thiobarnituric acid assay.
mDCs were cultured with or without 20 μg/mL of oxidized LDL in presence or absence of 10 μg/mL of ANXA5 as indicated. After 6 hours of culture condition, cell supernatant was collected and HSP60 was measured by human HSP60 Duoset ELISA kit (R&D systems, UK). In similar experimental condition, cells were collected for RNA extraction. As mentioned above, cDNA synthesized from RNA was used for mRNA level in quantitative real‐time polymerase chain reaction.
Competition/Binding Assay (Protein–Protein Interaction Assay)
HSP60 and/or ANXA5 was resuspended in PBS. The same concentration of HSP60 with different concentration of ANXA5 as presented in Figure 6F was incubated for 2 hours. After incubation, HSP60 was measured from HSP60 in PBS or HSP60 with ANXA5 in PBS by Human HSP60 duoset ELISA kit (R&D systems, UK).
Statistical Analysis
Statistical analysis was performed by Student t test, where P≤0.05 is considered statistically significant. P≤0.05, ≤0.01, and ≤0.001 are expressed as *, **, and, ***, respectively. The bar diagrams are expressed as mean±SD and the line plot is expressed as mean±SEM.
Results
HSP60 and DC Maturation
First, we investigated the effect of HSP60 in DCs. mDCs were treated with or without 5 μg/mL of HSP60 at day 6. After 24 hours of incubation, cells were stained with CD11C, CD86, CD83, CD40, and HLA‐II surface marker antibodies. HSP60‐treated mDCs induced activation markers and expression of several costimulatory proteins (Figure 1A). Inflammatory cytokines IL‐6, tumor necrosis factor‐α, IL‐12.p70 and IL‐1β, also IL‐10 but not transforming growth factor‐β significantly increased in HSP60‐treated mDCs (Figure 1B). A similar pattern of mDCs activation was observed by 5 μg/mL of HSP90 (Figure 1C).
Figure 1.
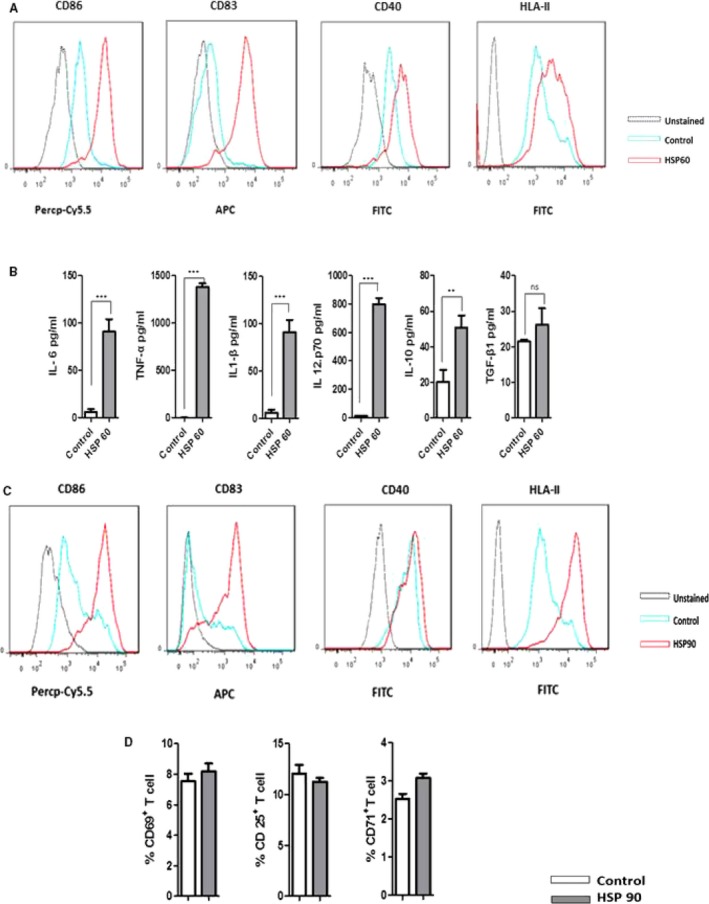
Heat shock protein 60 (HSP60) induces dendritic cell (DC) activation/maturation; 1 of 3 individual experiments is presented here. DCs at the concentration of 2×106/mL were cultured with or without HSP60. A, Expression of CD86, CD83, CD40, and HLA‐II was analyzed by flow cytometry after 24 h of stimulation with 5 μg/mL of HSP60. Figure in histogram shows higher induction of these markers by HSP60. Percentage of histogram shift or mean fluorescence intensity was calculated from triplicate samples and P values are CD86 ≤0.001, CD83 ≤0.001, HLA‐II ≤0.05, and CD40 ≤0.001. B, Cytokine profile of DC against HSP60 is listed. DCs were stimulated as mentioned, cultured for 24 h, and cell supernatant was collected for measurement of cytokines. Mostly pro‐inflammatory cytokines are highly increased by HSP60. C, In similar condition, DCs activation was observed by HSP90 (5 μg/mL). P value from triplicates samples CD86 ≤0.0001, CD83 ≤0.001, CD40 ≤0.01, and HLA‐II ≤0.001. D, HSP90 induced‐DCs were cocultured with T cells but no activation of T cells was observed. APC indicates antigen‐presenting cells; FITC, fluorescein isothiocyanate; IL‐6, interleukin‐6; Percp‐Cy5.5, Peridinin Chlorophyll Protein‐Cyanine 5.5; TGF‐β1, transforming growth factor‐β1.
HSP60‐Stimulated T‐Cell Activation Through DCs
Mature DCs have a major role in T‐cell stimulation. We therefore investigated T‐cell activation and proliferation in HSP60‐stimulated mDC‐T‐cell coculture. We induced DCs with 2.5, 5, or 10 μg/mL of HSP60 and cocultured with T cells. All the concentrations induced T‐cell activation but 5 or 10 μg/mL showed a strong effect in comparison to 2.5 μg/mL as determined by CD25 expression (Figure 2A). In the next experiments we continued with 5 μg/mL. We observed that HSP60 induced DC‐mediated T‐cell activation, as determined by surface expression of T‐cell early/intermediate/late activation markers (Figure 2B), and T‐cell proliferation by BrDu incorporation assays (Figure 2C). Interestingly, HSP90‐stimulated mDCs could not induce T cells in DCs‐T‐cell culture (Figure 1D). In further studies we focused on HSP60.
Figure 2.
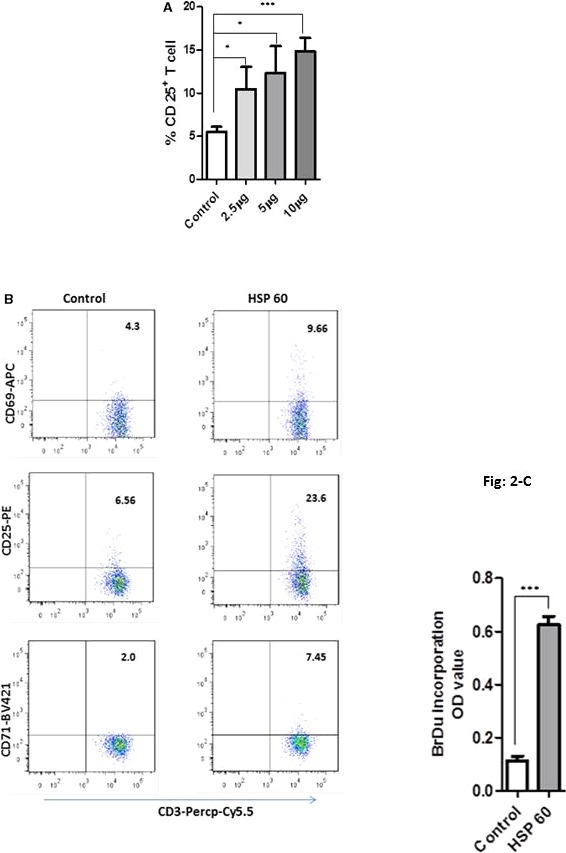
T‐cell activation and proliferation in dendritic cell (DC)+T‐cell coculture. A, DCs were stimulated with heat shock protein 60 (HSP60) at the concentration of 2.5, 5, or 10 μg/mL. After overnight incubation, autologous T cells 4×105 were cocultured with 1×105 DCs. All the concentrations of HSP60 induced T‐cell activation, where 5 or 10 μg/mL were a little stronger, which was tested by CD25 expression in CD3 T cells. B, One representative of minimum 3 experiments of T‐cell activation, which was determined by CD69 early activation, CD25 and CD71 intermediate/late activation markers. DCs were stimulated with 5 μg/mL of HSP60 and cocultured with CD3+ T cells. For analysis, CD3+ cells were gated, then percentage of CD3+CD69/CD25/CD71+ cells was shown in the upper right of each gate. HSP60‐induced DCs activated all of 3 activation markers in CD3+ T cells, P≤0.0001 from triplicate samples. C, In response to HSP60, DC+T cells show a high proliferation rate; 1 representative of 3 individual experiments is shown here. APC Allophycocyanine; BrDu, 5‐brom‐2‐deoxiuridin; OD, Optical density; Percp‐Cy5.5, Peridinin Chlorophyll Protein‐Cyanine 5.5. *P≤0.05; ***P≤0.0001.
HSP60‐Stimulated DCs Activate Plaque T Cell
To elucidate mechanisms behind T‐cell activation in plaques, we tested effects of HSP60 on activation of T cells from atherosclerotic plaques, by peripheral monocyte–derived DCs (from patients) and thus cocultured with each patient's plaque T cells. We determined activation of the classical T‐cell activation marker CD25 in T cells from 3 carotid and 3 femoral plaques, cocultured with HSP60‐stimulated mDCs from the same individual (Figure 3). T cells from 4 atherosclerotic plaques (where 2, 3, 4 were carotid and 1, 5, 6 were femoral) were available for further testing of T‐cell activation markers. As indicated in Figure 3, CD69 (early activation) and CD71 (intermediate/late activation) were also induced by HSP60. There was no material available for triplicate testing, in all samples, as indicated in Figure 3, but only duplicates. When different experiments were pooled, the increase in CD25 and CD71 was significant (P≤0.05) and there was a nonsignificant increase in CD69 (P≤0.1).
Figure 3.
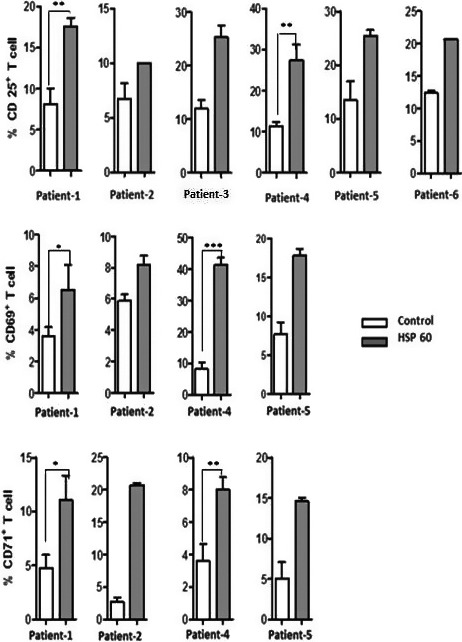
Plaque T‐cell activation. To investigate the plaque T‐cell activation, dendritic cells (DCs) were generated from blood monocytes of atherosclerotic patients; DCs were stimulated with 5 μg/mL of heat shock protein 60 (HSP60) and coculture with the plaque T cells of the same patients. First, we have determined classic T‐cell activation marker CD25 (intermediate activation) from 6 patients, where we observed CD25 induction against HSP60. Next, early activation marker CD69 and late activation marker CD71 from 4 patients also tested against HSP60. The induction of CD69 and CD71 also was similar to CD25 induction. Cells were cultured in triplicates (with determination of statistical significance) or duplicates (where material from plaques was not sufficient for triplicates) from each patient. *P≤0.05; **P≤0.01; ***P≤0.0001.
Characterization of T‐Cell Subset Against HSP60
To investigate the effects of HSP60 on Th‐cell differentiation, we quantified cytokines IL‐6, tumor necrosis factor‐α, IFN‐γ, IL‐17, and transforming growth factor‐β. In mDCs‐T‐cell cocultures, a higher amount of IFN‐γ, tumor necrosis factor‐α, and IL‐6 were observed (Figure 4A). IL‐5 and IL‐4 were undetected in mDC‐T‐cell co‐culture supernatant. In addition, IL‐17 also increased in HSP60‐stimulated cocultures, but there was no increase in transforming growth factor‐β1 (Figure 4A). Further, quantitative real‐time polymerase chain reaction analysis quantified a higher level of Th1 transcription factor expression in HSP60‐stimulated cocultures, whereas there was no significant increase in Th2 transcription factor expression (Figure 4B). HSP60‐stimulated mDCs mainly regulate Th1 differentiation, where little increase in Th2 differentiation was observed. In addition, RORC transcription factor for IL‐17‐producing T‐cell and interestingly Treg transcription factor Fox P3 also increased by HSP60 in similar culture condition (Figure 4B).
Figure 4.
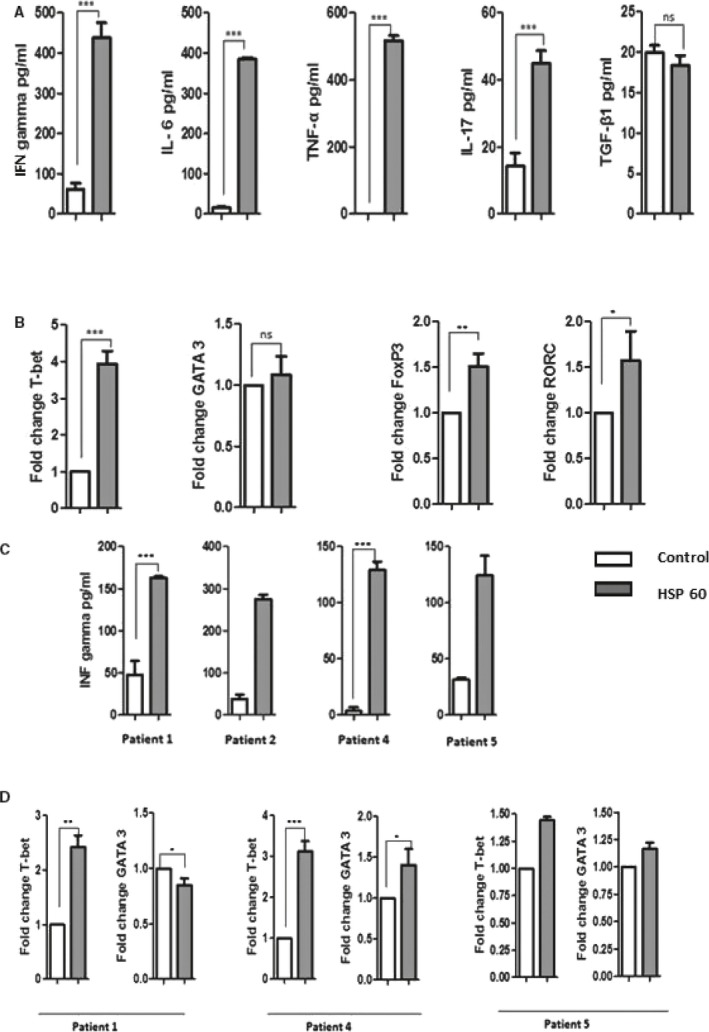
T helper (Th) cells differentiation: 1 representative experiment of 3 individual experiments. A, Heat shock protein 60 (HSP60) (5 μg/mL) induced DCs were cocultured with CD3+ T cells. Cytokines were measured from coculture supernatants. In HSP60 treated coculture, high level of pro‐inflammatory cytokines was observed. B, Next, Th cell differentiating transcription factors were analyzed by reverse transcriptase quantitative polymerase chain reaction. Th1 transcription factor T‐bet was increased more than 3‐fold whereas Th2 transcription factor GATA‐3 increase was negligible. In addition, FoxP3 and RORC transcription factor for T regulatory cells and IL‐17 producing T cells, respectively, also increased. C, Cell culture supernatants from the patients were used only for IFN‐γ measurement. IFN‐γ detection from 4 patients shows higher induction by HSP60. Patients’ samples were in duplicates. D, mRNA level or transcription factor was analyzed from 3 patients, where mostly Th1 transcription factor was increased. DCs indicates dendritic cells; FoxP3, Forkhead box P3; GATA3, GATA binding protein 3; IFN‐γ, interferon‐γ; IL‐6, interleukin‐6; ns, not significant; RORC, RAR Related Orphan Receptor C; T‐bet, T‐box expressed in‐T‐cells; TNF‐α, tumor necrosis factor α; TGF‐β1, transforming growth factor‐β1. *P≤0.05; **P≤0.01; ***P≤0.0001.
Because of the low volume of cell supernatants from the patient's mDC‐T‐cells coculture, we measured only IFN‐γ (Figure 4C). In addition, transcription factors were tested from 3 patients, where mainly Th1 transcription factor was increased (Figure 4D). When different experiments were pooled, the increase in IFN‐γ and T‐box expressed in‐T‐cells (T‐bet) was significant (P≤0.05).
HSP60‐Induced DCs Secreted Cytokines in T‐Cell Activation
We then determined that HSP60 induces mDCs to secrete different cytokines. We studied whether HSP60‐induced mDC‐secreted cytokines play any role in T‐cell activation. For this purpose, we cocultured HSP60‐induced mDCs and T cells in a trans‐well culture system, where we observed a relatively weak role of cytokines for T‐cell activation. Cytokine‐induced T‐cell activation was very low in comparison to T‐cell activation in mix coculture (Figure 5A).
Figure 5.
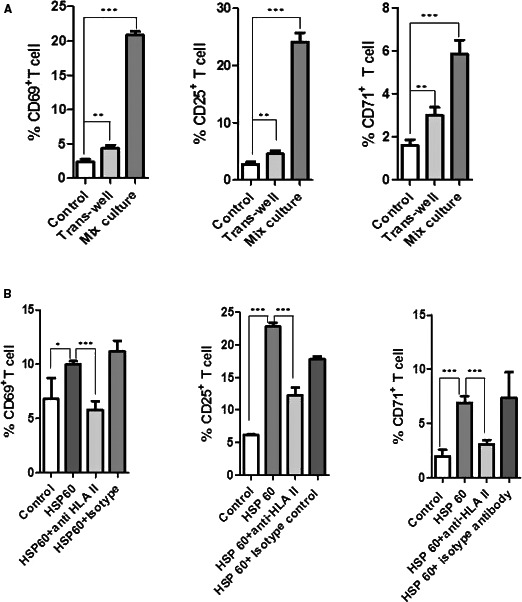
Heat shock protein 60 (HSP60; 5 μg/mL)‐induced dendritic cells (DCs) were cocultured with T cells in 24‐well trans‐well culture plates. T cells 4×105 (upper well) in 200 μL were cultured in the upper chamber while autologous HSP60‐stimulated DCs 2×105 (bottom well) in 500 μL were in the bottom chamber. In parallel, DCs induced in the same condition were cocultured with T cell in same well as mix coculture. T‐cell activation from trans‐well culture was weak in comparison to mix culture. One of 2 individual experiments is represented here (A). HSP60‐induced DCs were cocultured with CD3+ T cells in presence or absence of 8 μg/mL anti‐HLA II antibodies (HLA‐II‐blocking antibodies). DCs‐mediated T‐cell activation was inhibited in presence of HLA II antibodies (B). Experiments were performed 3 times and 1 of them is shown as a representative, except for CD69 where inhibition by HLA‐II‐blocking antibodies was nonsignificant in some experiments. *P≤0.05; **P≤0.01; ***P≤0.0001.
HSP60 Induces HLA‐II‐Mediated T‐Cell Activation
In order to further elucidate details of HSP‐induced mDC‐mediated T‐cell activation, we studied effects of antibodies to MHC class II. HSP60 (5 μg/mL) stimulated mDCs and T‐cell culture with HLA‐II‐blocking antibodies (8 μg/mL), attenuating the T‐cell activation (Figure 5B). There was some variability in inhibition of CD69, since nonsignificant inhibition also was determined in some experiments.
ANXA5 Inhibits/Attenuates HSP60 Effect and oxLDL‐Induced HSP60
To study the anti‐inflammatory or inhibitory effect of ANXA5 on HSP60‐induced T‐cell activation, we stimulated mDCs with HSP60 (5 μg/mL) in presence or absence of ANXA5 (10 μg/mL). Stimulated mDCs were resuspended in fresh medium and cocultured with T cells. T‐cell activation was inhibited in HSP60+ANXA5‐stimulated mDCs in comparison to HSP60‐induced mDC‐mediated T‐cell activation (Figure 6A).
Figure 6.
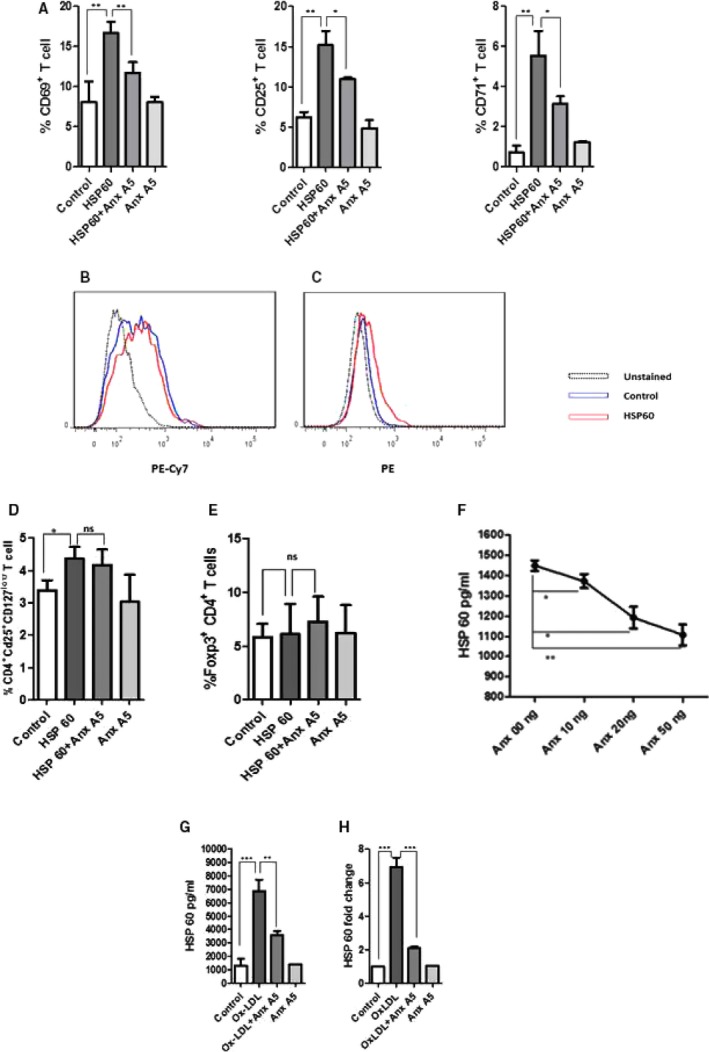
Annexin A5 (ANXA5) inhibited heat shock protein 60 (HSP60)‐induced immune response. Dendritic cells (DCs) at the concentration of 2×106 were stimulated with HSP60: 5 μg/mL in presence or absence of ANXA5 (10 μg/mL) and cocultured with CD3+ T cells (DC: T‐cell concentration was same as mentioned in Figure 2). ANXA5 attenuated HSP60‐induced T‐cell activation. One of 3 individual experiments is represented (A). HSP60 induced P38 MAPK activation, mean fluorescence intensity was calculated and P≤0.05 (C) but there was no effect on NFκB (B) activation. ANXA5 did not induce T regulatory cells (T‐reg cells) in HSP60‐stimulated DC+T‐cell coculture, whereas little induction of T‐reg by HSP60 was observed. The experiments were performed 3 times and 1 of them is presented (D). FoxP3‐positive CD4 T cells did not show significant increase in response to ANX A5 or HSP60. One of 2 individual experiments is shown (E). Protein–protein interaction assay shows HSP60‐ANX A5 have transient or weak interaction where HSP60‐ANXA5 incubation reduced HSP60 detection in ELISA measurement. One of 3 individual experiments is presented. F, Ox‐LDL induced HSP60 production in DCs and it was inhibited by ANX A5 in both protein (1 representative experiment from 3 individual experiments) (G) and mRNA level (H). Foxp3, Forkhead box P3; MAPK, Mitogen activated protein kinase; Ox‐LDL, oxidized low‐density lipoprotein; NFκB, nuclear factor kappa B; PE, Phicoerythrin; PE‐Cy7, Phicoerythrin‐cyanine 7. *P≤0.05; **P≤0.01; ***P≤0.0001.
In addition, we analyzed whether human HSP60 activates P38 MAPK or nuclear factor kappa B, and effects by ANXA5. Phosphorylation is required for P38 MAPK or nuclear factor kappa B activation. For this purpose, antibody against dual phosphorylated site pT180/pY182 of p38α, β, γ, and δ or phosphorylated serine 529 (pS529) in the transactivation domain of nuclear factor kappa B p65 subunit was tested. There was no activation of the P65 subunit of nuclear factor kappa B (Figure 6B), although p38 MAPK showed little activation by HSP60 (Figure 6C). We were curious to study the T‐reg population in HSP60‐stimulated mDCs‐T coculture in presence or absence of ANXA5. mDCs were thus stimulated with HSP60 in presence or absence of ANXA5 as described and cocultured with T cells. There was a slight increase in T‐reg population by HSP60‐stimulated mDC, which was not affected significantly by ANXA5 (Figure 6D). Next, HSP60‐ or ANXA5‐induced mDC‐T cells coculture was analyzed for FoxP3+ Cd4 T cells. There was no significant change in Fox P3‐positive CD4 T cells in response to HSP60 or ANXA5 (Figure 6E).
As a next step, we investigated whether HSP60 interacts with ANXA5 or inhibits binding of HSP60 to antibodies. We incubated the HSP60 with different concentrations of ANXA5. We measured HSP60 by ELISA in the presence or absence of ANXA5. A lower level of HSP60 was detected in the presence of ANXA5 (Figure 6F). ANXA5 by itself gave no detectable binding in this assay (data not shown).
We then went on to study OxLDL‐induced HSP60 in mDC and influence of ANXA5. We demonstrate that 20 μg/mL of OxLDL promoted a strong induction of HSP60, an effect abolished by 10 μg/mL of ANXA5 at both protein (Figure 6G) and mRNA level (Figure 6H).
Discussion
We here report that HSP60 induces activation of mDCs from both healthy donors and from individuals with symptom‐giving CVD, either from carotid arteries or from femoral arteries. DCs, as specialized antigen‐presenting cells, could be of major importance in initiation, progression, and also late stages of atherosclerosis with CVD complications, orchestrating immune responses and pro‐inflammatory effects.2, 16 Interestingly, DCs are also present in immature forms in the arterial wall in healthy young humans.17 DCs are more prevalent in areas with turbulent flow, which are the same as where atherosclerosis preferentially occurs.17 In human atherosclerotic lesions, DCs are present both at an early stage and late stage (more DCs in vulnerable plaques),17, 18 and may modulate disease development. Further, DCs and T cells co‐localize in plaques,19 suggesting their participation in cellular immune reactions. Also in mice models of atherosclerosis, DCs are present through all stages of atherosclerosis development and are implicated as drivers of atherogenesis.20, 21
Specific mechanisms by which DC could promote atherogenesis in both mouse models and humans have been demonstrated, including the chemokine CCL17, which is present in plaques and blocking of this leads to expansion of Tregs.22 Further, antigen presentation by antigen‐presenting cells in the arterial wall to CD4‐positive T cells in mouse models of atherosclerosis leads to local T‐cell activation and production of proinflammatory cytokines and foam cell formation.23
There is an extensive literature on HSP60 effects on myeloid cells in general. Still, relatively little is known about effects and underlying immune mechanisms of human HSP60 on human DC, especially from patients with manifest CVD as in this report. In a previous study using mouse models, human HSP60 promoted maturation of mouse DCs, also towards a Th1 phenotype.24 In another study, human HSP60 promoted DC activation and pro‐inflammatory cytokines in monocyte‐derived DCs from healthy individuals.25 Our data using human mDCs derived from patients with advanced atherosclerotic disease are thus in line with and extend previous findings. Similar to HSP60, mDCs were activated also by HSP90β, but these stimulated mDCs were unable to induce T‐cell activation. In general, DCs take up protein antigen and present the peptide to T cells and activate them. It might be that intact protein of HSP90 activates the DCs but peptides of HSP90 are not sufficient to activate T cells.
Another finding in this study is that human HSP60 induces activation of human T cells from both healthy donors and from atherosclerotic plaques from patients with symptom‐giving CVD, either from carotid arteries or from femoral arteries, and that the effect on T cells was mediated by mDCs from the same donor. This activation was at least partly MHC class II dependent, and thus represents a “classic” immune response. There was variability in inhibitory effects of MHC class II inhibition of CD69 that did not reach significance in some experiments. However, our findings indicate for the first time that in humans there is an autoimmune DC‐mediated classic protein antigen‐induced T‐cell activation both among healthy individuals and in cells from atherosclerotic plaques derived from patients with symptom‐giving atherosclerosis. Further, this mDC‐T‐cell activation was characterized by a Th1/Th17 cytokine response while Th2‐ or Treg‐related cytokines were not stimulated. These findings also apply to gene activation level.
Involvement of HSP through immunological mechanisms involving T cells has been much discussed in atherogenesis, but DC‐mediated T‐cell activation in humans, and T‐cell activation of T cells from atherosclerotic plaque through direct involvement of DC have not been reported to the best of our knowledge.
There are several other lines of evidence where HSP60 has been reported to be involved in T‐cell activation. HSP60 could function as chaperones, and potentiate MHC class I or II presentation of peptides.26 This type of mechanism could play an important role in atherosclerosis, since there could very well be other antigens that induce T‐cell activation, an effect that could be further promoted by HSP60.
There are reports of HSP60‐reactive T cells in human atherosclerosis, where HSP60 or peptide epitopes derived therefrom are recognized by T cells clonally expanded from atherosclerotic plaques, interestingly, also in early stages of atherogenesis.10, 27 It is well known that HSP60 nonspecifically can activate cells of the innate immune system. This can occur by HSP60 being an adjuvant, promoting activation of T cells specific for other antigens, and even potentiation of mitogen‐induced T‐cell activation is described. HSP60 could have a direct effect on T cells, and could have distinct functions, being pro‐inflammatory, anti‐inflammatory, or a messenger via Toll‐like receptors 2 and 4 between innate and adaptive immunity.28, 29 Previous studies of HSP60‐related T‐cell activation thus do not prove the effect to be mediated by DC–T‐cell interaction and the role of MHC class II.
HSP60 could thus induce T‐cell activation by several different mechanisms, and this also includes cross‐reactivity to microbial HSP. Antibodies against HSP60 (or microbial HSP60/65) are not studied herein, but could represent 1 important mechanism by which HSP60 immunity could promote vascular inflammation and atherosclerosis (for example, by activating the endothelium where HSP60 is induced). Several studies support this notion in different aspects of atherosclerosis and CVD.8, 9 It appears likely that such antibodies are T‐cell dependent, though this is not known and this issue deserves further study.
HSP60 could thus promote a pro‐inflammatory immune response by several different mechanisms, non–mutually exclusive.
In previous studies, we suggested that OxLDL, which may be a major pro‐inflammatory factor in atherosclerosis development by inducing HSP60 in monocytes/macrophages, could indirectly induce immune activation.12 We recently added support to this notion by demonstrating that OxLDL (and enzymatically modified LDL)‐induced T‐cell activation is indeed dependent on HSP60, although we could not formally demonstrate that such induced HSP60 was a classic antigen for T cells, presented through MHC class II by antigen‐presenting cells.6
Other risk factors including hypertension and hyperlipidemia could promote atherosclerosis‐related inflammation and immune activation by inducing stress at some sites in the arterial wall that are prone to turbulent flow, inducing HSP, which becomes immunogenic. It is also possible that immune reactivity to HSP from microorganisms is an underlying factor through cross‐reactivity with human HSP60.10
In principle, there could also be other possibilities. For example, we reported that in vitro hyperthermia, as a model of fever, induced a pro‐inflammatory cytokine response, including also the typical pro‐inflammatory Th1 cytokine IFN‐γ, mainly T‐cell derived.30 Of note, infections such as influenza, typically with fever as a prominent symptom, are associated with myocardial infarction.31
The concentration of HSP60 and HSP90 in normal tissue where a role in atherosclerosis is likely to occur is not known, to the best of our knowledge. Since we demonstrate here that HSP60, but not HSP90, induce DC‐mediated T‐cell activation, we are focusing on HSP60, and demonstrate that OxLDL induces HSP60 in DC, in line with our previous findings.12 HSP60 could be immunogenic in the local environment of the plaque. Less is known about HSP90 induction in this context.
Still, both HSP60 and HSP90 are present in human plasma, and have been proposed by us and others as potential risk markers, as discussed. Even though there is variability in studies of circulating HSP60, several studies showed HSP60 concentrations in serum that are in a range comparable to those in our experimental studies.13, 32, 33
HSP90 levels in serum appear to be lower than in this study, ≈300 ng/mL.34 Of note, HSP levels in different stressful conditions such as infections are not well described, and there could be variations, and even though we think the major immune effects of HSPs are local, the possibility of an immune response also occurring systemically cannot be excluded.
One further observation is that ANXA5, a plasma protein that has antithrombotic properties, inhibits HSP60‐induced mDCs maturation and HSP60‐mediated T‐cell activation. We recently demonstrated that ANXA5 has interesting anti‐atherogenic properties both in vivo and ex vivo, by reducing inflammation and improving vascular function in apolipoprotein E−/− mice and by inhibiting OxLDL‐induced DC‐mediated activation of atherosclerotic plaque–derived T cells.6, 15 While OxLDL‐mediated T‐cell activation was not only inhibited by ANXA5, but also polarized into a Treg response, this was not the case with HSP60‐induced T‐cell activation in the present study. This indicates that HSP60 is only 1 part of an explanation of OxLDL‐mediated immune activation, since the effects of ANXA5 differs, while being anti‐inflammatory for both antigens, not promoting a Treg response from HSP60.
ANXA5 showed signs of binding to HSP60, decreasing binding of HSP60 to the anti‐HSP60 antibodies in the ELISA system (while not binding anti‐HSP60 in the assay), and it is thus possible that it could dampen the immune stimulation of HSP60 through inhibition of binding to cellular receptors, including on DCs. Further, ANXA5 could compete for antibody binding with HSP60. It is possible that ANXA5 and HSP60 have weak or transient interaction.
We also extend our previous observation that OxLDL induces HSP60 in monocytes/macrophages and confirm that OxLDL induces HSP60 in mDC and report that this effect was ameliorated by ANXA5, both at the gene activation level and as secreted protein. Of note, HSP60 itself, not only the antibodies against HSP60, are associated with CVD.13
Taken together, our results indicate that HSP60 induced mDC maturation and mDC mediated MHC class II T‐cell activation, mainly of pro‐inflammatory Th1/17 type both in cells from healthy individuals and patients with symptom‐giving atherosclerosis in carotid or femoral arteries. OxLDL‐mediated immune activation could at least partly be explained by HSP60‐related specific immunity. ANXA5 abolished both HSP60‐induced T‐cell activation and OxLDL‐induced HSP60, which adds support to the possibility that this plasma protein could be of therapeutic interest.
Sources of Funding
This study was supported by the Swedish Heart and Lung Foundation, the Swedish Rheumatism Association, AFA, Torsten Söderberg's Foundation, and King Gustav V:s 80‐year Fund.
Disclosures
JF is on the Board of Directors and is a minor shareholder in Annexin Pharmaceuticals. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2017;6:e006778 DOI: 10.1161/JAHA.117.006778.)29151033
Contributor Information
Anquan Liu, Email: anquan.liu@ki.se.
Johan Frostegård, Email: Johan.frostegard@ki.se.
References
- 1. Frostegard J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, Hansson GK. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro‐inflammatory (Th1) and macrophage‐stimulating cytokines. Atherosclerosis. 1999;145:33–43. [DOI] [PubMed] [Google Scholar]
- 2. Frostegard J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013;11:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frostegard J, Wu R, Giscombe R, Holm G, Lefvert AK, Nilsson J. Induction of T‐cell activation by oxidized low density lipoprotein. Arterioscler Thromb. 1992;12:461–467. [DOI] [PubMed] [Google Scholar]
- 4. Frostegard J, Nilsson J, Haegerstrand A, Hamsten A, Wigzell H, Gidlund M. Oxidized low density lipoprotein induces differentiation and adhesion of human monocytes and the monocytic cell line U937. Proc Natl Acad Sci USA. 1990;87:904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frostegard J, Zhang Y, Sun J, Yan K, Liu A. Oxidized low‐density lipoprotein (OxLDL)‐treated dendritic cells promote activation of T cells in human atherosclerotic plaque and blood, which is repressed by statins: microRNA let‐7c is integral to the effect. J Am Heart Assoc. 2016;5:e003976 DOI: 10.1161/JAHA.116.003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu A, Ming JY, Fiskesund R, Ninio E, Karabina SA, Bergmark C, Frostegard AG, Frostegard J. Induction of dendritic cell‐mediated T‐cell activation by modified but not native low‐density lipoprotein in humans and inhibition by annexin a5: involvement of heat shock proteins. Arterioscler Thromb Vasc Biol. 2015;35:197–205. [DOI] [PubMed] [Google Scholar]
- 7. Xu Q, Dietrich H, Steiner HJ, Gown AM, Schoel B, Mikuz G, Kaufmann SH, Wick G. Induction of arteriosclerosis in normocholesterolemic rabbits by immunization with heat shock protein 65. Arterioscler Thromb. 1992;12:789–799. [DOI] [PubMed] [Google Scholar]
- 8. Xu Q, Willeit J, Marosi M, Kleindienst R, Oberhollenzer F, Kiechl S, Stulnig T, Luef G, Wick G. Association of serum antibodies to heat‐shock protein 65 with carotid atherosclerosis. Lancet. 1993;341:255–259. [DOI] [PubMed] [Google Scholar]
- 9. Frostegard J, Lemne C, Andersson B, van der Zee R, Kiessling R, de Faire U. Association of serum antibodies to heat‐shock protein 65 with borderline hypertension. Hypertension. 1997;29:40–44. [DOI] [PubMed] [Google Scholar]
- 10. Wick G, Jakic B, Buszko M, Wick MC, Grundtman C. The role of heat shock proteins in atherosclerosis. Nat Rev Cardiol. 2014;11:516–529. [DOI] [PubMed] [Google Scholar]
- 11. Perschinka H, Mayr M, Millonig G, Mayerl C, van der Zee R, Morrison SG, Morrison RP, Xu Q, Wick G. Cross‐reactive B‐cell epitopes of microbial and human heat shock protein 60/65 in atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1060–1065. [DOI] [PubMed] [Google Scholar]
- 12. Frostegard J, Kjellman B, Gidlund M, Andersson B, Jindal S, Kiessling R. Induction of heat shock protein in monocytic cells by oxidized low density lipoprotein. Atherosclerosis. 1996;121:93–103. [DOI] [PubMed] [Google Scholar]
- 13. Pockley AG, Wu R, Lemne C, Kiessling R, de Faire U, Frostegard J. Circulating heat shock protein 60 is associated with early cardiovascular disease. Hypertension. 2000;36:303–307. [DOI] [PubMed] [Google Scholar]
- 14. Madrigal‐Matute J, Lopez‐Franco O, Blanco‐Colio LM, Munoz‐Garcia B, Ramos‐Mozo P, Ortega L, Egido J, Martin‐Ventura JL. Heat shock protein 90 inhibitors attenuate inflammatory responses in atherosclerosis. Cardiovasc Res. 2010;86:330–337. [DOI] [PubMed] [Google Scholar]
- 15. Ewing MM, de Vries MR, Nordzell M, Pettersson K, de Boer HC, van Zonneveld AJ, Frostegard J, Jukema JW, Quax PH. Annexin A5 therapy attenuates vascular inflammation and remodeling and improves endothelial function in mice. Arterioscler Thromb Vasc Biol. 2011;31:95–101. [DOI] [PubMed] [Google Scholar]
- 16. Feig JE, Feig JL. Macrophages, dendritic cells, and regression of atherosclerosis. Front Physiol. 2012;3:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Millonig G, Niederegger H, Rabl W, Hochleitner BW, Hoefer D, Romani N, Wick G. Network of vascular‐associated dendritic cells in intima of healthy young individuals. Arterioscler Thromb Vasc Biol. 2001;21:503–508. [DOI] [PubMed] [Google Scholar]
- 18. Yilmaz A, Lochno M, Traeg F, Cicha I, Reiss C, Stumpf C, Raaz D, Anger T, Amann K, Probst T, Ludwig J, Daniel WG, Garlichs CD. Emergence of dendritic cells in rupture‐prone regions of vulnerable carotid plaques. Atherosclerosis. 2004;176:101–110. [DOI] [PubMed] [Google Scholar]
- 19. Bobryshev YV, Watanabe T. Ultrastructural evidence for association of vascular dendritic cells with T‐lymphocytes and with B‐cells in human atherosclerosis. J Submicrosc Cytol Pathol. 1997;29:209–221. [PubMed] [Google Scholar]
- 20. Jongstra‐Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low‐grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu P, Yu YR, Spencer JA, Johnson AE, Vallanat CT, Fong AM, Patterson C, Patel DD. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler Thromb Vasc Biol. 2008;28:243–250. [DOI] [PubMed] [Google Scholar]
- 22. Weber C, Meiler S, Doring Y, Koch M, Drechsler M, Megens RT, Rowinska Z, Bidzhekov K, Fecher C, Ribechini E, van Zandvoort MA, Binder CJ, Jelinek I, Hristov M, Boon L, Jung S, Korn T, Lutz MB, Forster I, Zenke M, Hieronymus T, Junt T, Zernecke A. CCL17‐expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest. 2011;121:2898–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koltsova EK, Garcia Z, Chodaczek G, Landau M, McArdle S, Scott SR, von Vietinghoff S, Galkina E, Miller YI, Acton ST, Ley K. Dynamic T cell‐APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest. 2012;122:3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flohe SB, Bruggemann J, Lendemans S, Nikulina M, Meierhoff G, Flohe S, Kolb H. Human heat shock protein 60 induces maturation of dendritic cells versus a Th1‐promoting phenotype. J Immunol. 2003;170:2340–2348. [DOI] [PubMed] [Google Scholar]
- 25. Bethke K, Staib F, Distler M, Schmitt U, Jonuleit H, Enk AH, Galle PR, Heike M. Different efficiency of heat shock proteins (HSP) to activate human monocytes and dendritic cells: superiority of HSP60. J Immunol. 2002;169:6141–6148. [DOI] [PubMed] [Google Scholar]
- 26. Murshid A, Gong J, Calderwood SK. The role of heat shock proteins in antigen cross presentation. Front Immunol. 2012;3:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Knoflach M, Kiechl S, Mayrl B, Kind M, Gaston JS, van der Zee R, Faggionato A, Mayr A, Willeit J, Wick G. T‐cell reactivity against HSP60 relates to early but not advanced atherosclerosis. Atherosclerosis. 2007;195:333–338. [DOI] [PubMed] [Google Scholar]
- 28. Quintana FJ, Cohen IR. The HSP60 immune system network. Trends Immunol. 2011;32:89–95. [DOI] [PubMed] [Google Scholar]
- 29. Pockley AG, Muthana M, Calderwood SK. The dual immunoregulatory roles of stress proteins. Trends Biochem Sci. 2008;33:71–79. [DOI] [PubMed] [Google Scholar]
- 30. Huang YH, Haegerstrand A, Frostegard J. Effects of in vitro hyperthermia on proliferative responses and lymphocyte activity. Clin Exp Immunol. 1996;103:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barnes M, Heywood AE, Mahimbo A, Rahman B, Newall AT, Macintyre CR. Acute myocardial infarction and influenza: a meta‐analysis of case‐control studies. Heart. 2015;101:1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rea IM, McNerlan S, Pockley AG. Serum heat shock protein and anti‐heat shock protein antibody levels in aging. Exp Gerontol. 2001;36:341–352. [DOI] [PubMed] [Google Scholar]
- 33. Pockley AG, Bulmer J, Hanks BM, Wright BH. Identification of human heat shock protein 60 (Hsp60) and anti‐Hsp60 antibodies in the peripheral circulation of normal individuals. Cell Stress Chaperones. 1999;4:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fredly H, Reikvam H, Gjertsen BT, Bruserud O. Disease‐stabilizing treatment with all‐trans retinoic acid and valproic acid in acute myeloid leukemia: serum hsp70 and hsp90 levels and serum cytokine profiles are determined by the disease, patient age, and anti‐leukemic treatment. Am J Hematol. 2012;87:368–376. [DOI] [PubMed] [Google Scholar]