

Abstract
Background
There is increasing interest in the concept of atrial cardiomyopathy, but the underlying molecular and mechanistic determinants remain poorly defined. We identified a family with heritable atrial cardiomyopathy manifesting as progressive atrial‐selective electromechanical dysfunction, tachyarrhythmias, and bradyarrhythmias requiring pacemaker implantation. Myosin light‐chain 4 (MYL4), encoding the atrial‐selective essential myosin light chain, was identified as a candidate gene. We used genetically modified rat models to investigate the role of MYL4 in atrial cardiomyopathy.
Methods and Results
Exome sequencing and systematic bioinformatic analyses identified a rare missense variant of MYL4 (c.31G>A [p.E11K]) in a large multiplex atrial cardiomyopathy family pedigree. The mutation cosegregated with atrial standstill (selected as the principal presenting trait) with a logarithm of the odds score of 5.3. The phenotype of rats with MYL4 mutation knock‐in confirmed the causative role of the mutation. MYL4 knockout rats showed a similar atrial cardiomyopathy phenotype, whereas rats with an adjacent 4‐amino‐acid deletion showed no phenotype. Both MYL4 p.E11K knock‐in rats and MYL4 knockout rats showed progressive atrial electrophysiological, contractile, and fibrotic abnormalities, similar to affected patients. Biochemical analyses of MYL4 p.E11K mutation rats showed activation of proapoptotic and profibrotic signaling, along with increased atrial‐cardiomyocyte terminal deoxynucleotidyl transferase dUTP nick end labeling staining, suggesting enhanced apoptotic cell death, findings that were mimicked by in vitro adenoviral transfer of the mutant gene to neonatal‐rat cardiomyocytes.
Conclusions
Loss‐of‐function MYL4 gene variants cause progressive atrial cardiomyopathy in humans and rats. Our findings identify MYL4 as a key gene required for atrial contractile, electrical and structural integrity. These results improve our understanding of the molecular basis of atrial cardiomyopathy and introduce new models for further mechanistic analysis.
Keywords: animal models, arrhythmia, atrial fibrillation, cardiomyopathy, fibrosis, genetics
Subject Categories: Atrial Fibrillation, Arrhythmias, Genetically Altered and Transgenic Models, Fibrosis
Clinical Perspective
What Is New?
Here, we study a kindred with several members showing a severe atrial cardiomyopathy phenotype and identify a myosin light‐chain 4 (MYL4) loss‐of‐function mutation as the probable cause.
Rat models, created by knocking out the MYL4 gene as well as by knocking in the human MYL4 p.E11K mutation, reproduce the clinical phenotype and point to enhanced apoptotic and profibrotic signaling as a central mechanism, providing new insights into the molecular basis of atrial cardiomyopathy.
What Are the Clinical Implications?
MYL4 is essential for atrial electrical, functional, and structural integrity, and MYL4 dysfunction can produce severe inherited atrial cardiomyopathy.
Introduction
Whereas ventricular cardiomyopathy is a well‐established entity with clear diagnostic and therapeutic implications, atrial cardiomyopathy represents a relatively new concept in active development.1 A wide range of factors contributes to the development of atrial cardiomyopathy, most commonly in association with ventricular pathology.1 To date, few primary cardiomyopathies with highly atrial‐selective manifestations have been recognized, but these may be very instructive about the mechanisms underlying the broad range of atrial cardiomyopathies.2 Genetic analyses of atrial cardiomyopathy are, so far, limited.2 Mutations in the natriuretic peptide precursor A gene are associated with familial atrial cardiomyopathy; long‐term follow‐up of natriuretic peptide precursor A–mutant patients shows progressive fibrosis associated with atrial standstill, a severe clinical scenario characterized by complete loss of atrial excitability.3
Myosin light‐chain 4 (MYL4) encodes an essential myosin light chain that is specifically expressed in human atria after birth.4 The essential light chain forms part of the actomyosin cross‐bridge, providing structural support for the α‐helical neck region of the myosin heavy chain and modulating actin‐activated ATPase activity.5 The ventricular analog, a protein encoded by MYL3, is downregulated in failing or hypertrophied hearts and partly replaced by re‐expression of MYL4 in the context of the fetal‐pattern remodeling typical of ventricular pathology.4 Recently, Gudbjartsson et al reported that MYL4 is linked to atrial cardiomyopathy,6, 7 with a frameshift mutation in MYL4 associated with early‐onset familial atrial fibrillation (AF) and bradyarrhythmias requiring pacemaker insertion. Orr et al8 also reported that heterozygous MYL4 p.Glu11Lys mutation causes familial AF. They found that expression of the analogous mutation in zebrafish leads to atrial enlargement and electrical abnormalities, without AF per se. This work presents the only animal‐model data available regarding the pathophysiology associated with MYL4‐dysfunction, and is limited by the fact that the zebrafish has a primitive heart tube with only 1 atrial and ventricular chamber, with (unlike humans) equal atrial and ventricular expression of the orthologous gene.
We identified a Chinese family with atrial tachyarrhythmias, a high incidence of atrial standstill, and severe bradyarrhythmias. Based on exome sequencing of this family, we identified a rare missense variant of MYL4 (p.E11K), which had perfect cosegregation with atrial standstill in a large multiplex pedigree. Because these studies suggested that the MYL4 was crucial for atrial functional integrity, we generated a series of rat models using CRISPR/Cas9‐mediated genome editing to define the function of MYL4, as well as to validate the ability of MYL4 dysfunction to cause the atrial abnormalities we found in patients and to define underlying mechanisms.
Methods
Additional methodological details are available in Data S1.
Large Atrial Standstill Pedigree and Clinical Features
We collected a large pedigree in which 6 family members were diagnosed as showing atrial standstill. The inheritance pattern was very likely to be autosomal dominant (Figure 1A). The proband and her family members provided written informed consent for the study according to a clinical research protocol approved by the Ethics Committee of Shanghai Tenth People's Hospital.
Figure 1.

Pedigree and clinical data of the atrial standstill family. A, Pedigree of the atrial standstill family, 44 family members were clinically evaluated and 6 were diagnosed as having atrial standstill. B, The ECG records of the 3 patients (I‐7, II‐1, and II‐3) with total atrial standstill: The proband (I‐7) had atrial flutter (indicated by arrows) with junctional escape rhythm and atrial fibrillation at different ages and finally developed atrial standstill and required pacemaker implantation. Another 2 patients (II‐1, II‐3) also required pacemaker implantation; no P waves were found after atrial pacing artifacts (indicated by arrows). C, II‐19, II‐21, and III‐10 were diagnosed as having partial atrial standstill. III‐10, aged 14, had a normal ECG and no symptoms of palpitations or syncope. Though P waves were found in the surface ECGs of these patients, the A wave was absent on TTE. Atrial flutter, atrial fibrillation (II‐19), and sinus arrest (pauses up to 2.3 seconds in II‐19) occurred when they aged. ECG indicates standard 12‐lead electrocardiography; TTE, transthoracic ultrasound.
Standard 12‐lead ECG and transthoracic echocardiography (TTE) were used to analyze atrial electrical and mechanical activity, respectively. TTE parameters of the affected patients are shown in Table S1. The diagnosis of atrial standstill was established according to the following criteria: absence of P wave on the surface 12‐lead ECG; inability to stimulate the atria on invasive electrophysiological study9; and/or loss of the A wave on TTE. Examples of illustrative ECG and TTE data are shown in Figure 1B and 1C.
Exome Sequencing
DNA were extracted from peripheral blood. After excluding mutations in SCN5A and GJA5 known to cause atrial standstill by Sanger sequencing, we performed high‐throughput exome capture sequencing for 2 affected family members, II1 and II3, at the Beijing Genomics Institute in Shenzhen China (for details, see Data S1).
Calling and Prioritizing Exome Sequence Variants
The paired‐end short reads from exome sequencing were mapped onto the UCSC human reference genome by BWA.10 The Genome analysis toolkit (GATK)11 was used to recalibrate the alignments and to call single‐nucleotide variants and short insertion‐deletions (Indels). We prioritized the sequence variants by multilayer resources and functions using KGGSeq (V0.8), (http://grass.cgs.hku.hk/limx/kggseq/).12 For details, see Data S1.
Validation of Candidate Mutations and Genes
A short list of sequence variants prioritized by KGGSeq (V0.8) was validated for all available family members by Sanger sequencing. In addition, for validated sequence variants showing perfect cosegregation with disease, we examined the frequency of mutant alleles in 200 unrelated controls without history of heart disease by Sanger sequencing. In the genomic region perfectly cosegregating with disease, we genotyped 47 sequence variants (covering around 38 Mbp) in all family members by Sequenom MassARRAY system (Sequenom, San Diego, CA). SimWalk2 was used to calculate the logarithm of the odds scores of multipoint parametric linkage analysis.13 We then ranked all genes in the significant linkage region according to their pathogenic potential with 2 widely used bioinformatics tools, Endeavour14 and GeneWanderer.15 Based on this information and the literature, we identified an MYL4 point mutation (31 G→A), causing a glutamic acid to lysine mutation at position 11, as the most likely causative candidate.
Generation of MYL4 p.E11K Rats, MYL4 KO Rats, and MYL4 Deletion Rat Models
All the animal procedures conformed to NIH guidelines (Guide for the Care and Use of Laboratory Animals) and were approved by the Animal Care and Use Committees of Shanghai Tenth People's Hospital.
MYL4 mutant rat strains were generated by CRISPR/Cas‐mediated genome editing. In brief, Cas9 mRNA and sgRNA targeting exon 1 of MYL4 gene was in vitro synthesized as previously described.16, 17 One‐cell rat embryos were obtained by superovulation of females the next morning after mating. The zygotes were cultured in KSOM embryo culture medium (Millipore, Billerica, MA) before injection. TE solution containing 12.5 ng/μL of gRNA and ≈25 ng/μL of Cas9 mRNA was injected into the cytoplasm of 1‐cell embryos through the microinjection needle. Injections were performed using an Eppendorf transferMan NK2 micromanipulator. Injected embryos were transferred into pseudopregnant female rats immediately after injection. Tail genomic DNA of F0 rats was extracted and amplified for sequencing.
Surface ECG and TTE
Surface ECGs were recorded with a biological signal acquisition amplifier (BL‐420S; Taimeng Co, Chengdu, China). For TTE, transthoracic 2‐dimensional and M‐mode images were obtained with a Visualsonics high resolution VEVO 2100 system (VisualSonics, Toronto, Ontario, Canada) with a 20‐MHz transducer. For details, see Data S1.
Immunohistochemistry
Serial 5‐mm‐thick atrial cryosections were stained with Masson trichrome, counterstained with hematoxylin, and quantified with Image‐Pro Plus 6.0 software (version 6.0; Media Cybernetics, Inc, Rockville, MD). Fibrosis was quantified and expressed as percent cross‐sectional area, excluding blood vessels and perivascular tissue. Terminal deoxynucleotidyl transferase dUTP nick end labeling staining (Roche 11684817910 kit; F. Hoffmann‐La Roche AG, Basel, Switzerland) was used as an index of apoptosis. Fluorescence images were obtained with a DMI6000 fluorescent microscope (Leica Microsystems Wetzlar GmbH, Wetzlar, Germany). The sources of the primary antibodies are listed in Table S2.
Statistical Analysis
Continuous variables were described as mean±SEM. Normal distribution was verified with the Shapiro–Wilk test. If data were normally distributed, differences were analyzed by 1‐way ANOVA, following by post hoc analysis (t test with Bonferroni correction) to evaluate statistical significance. For data that were not normally distributed, a nonparametric multiple‐group comparison (Kruskal–Wallis) was performed, followed by a Wilcoxon test with Bonferroni post hoc correction to compare individual group means. All P values are 2‐sided. The null hypothesis was rejected at P<0.05.
Results
Clinical Profile
We collected an unusually large pedigree of a family with inherited atrial cardiomyopathy and atrial standstill as a prominent feature, affecting 6 individuals of 44 family members distributed into 2 branches, with a typical dominant inheritance pattern (Figure 1A). The proband (I‐7) initially visited our hospital for bradycardia (Figure 1B, left) and lower extremity edema. Atrial flutter with a slow ventricular response was followed approximately 4 years later by atrial standstill, neither right nor left atria could be captured during invasive electrophysiological (Figure S1A). II‐1 and II‐3 also developed total atrial standstill (Figure 1B, right): Although they were implanted with dual chamber pacemakers, their atria could not be captured by atrial pacing. These patients had very similar clinical histories, all suffering from bradycardia at around 20 years old, followed by palpitations and reduced exercise tolerance between 30 and 40 years of age. After 40, all patients developed symptoms of heart failure.
II‐19, II‐21, and III‐10 showed partial atrial standstill (Figure 1C). II‐19 and II‐21 had a history of paroxysmal AF with junctional escape rhythm, along with low‐amplitude P waves and sinus‐node dysfunction (Figure 1C and Figure S1). III‐10, aged 14, had a normal ECG and no symptoms of palpitation or syncope (Figure 1C), but markedly diminished atrial contractility on TTE. Despite the P wave in the surface ECG of these patients, the A wave was markedly reduced or absent on TTE.
No atrial or ventricular septal defects or valvular abnormalities were found in these patients. Details of ECG evolution as a function of age can be found in Figure S1. Detailed ECG and TTE data are provided in Tables S1 and S3, respectively.
Novel Candidate Genes Suggested by Exome Sequencing and Bioinformatic Analysis
We then searched for candidate causal gene variants in this atrial cardiomyopathy pedigree. Because of failure to find any promising mutations in the 2 genes previously associated with atrial standstill, SCN5A and GJA5,18 we performed exome sequencing in 2 patients, II‐1 and II‐3. In total, 62 960 single‐nucleotide variants and 4192 insertions/deletions (Indels) were detected after stringent quality control using KGGSeq (V0.8),12 and we chose the top 60 single‐nucleotide variants (listed in Table S4) for validation according to their predicted pathogenic probability.19 It turned out that 2 missense single‐nucleotide variants (c. 610G>C of TUBG2 and c. 31G>A of MYL4) in 17q21 perfectly cosegregated with the dominant inheritance mode in the pedigree. To further map the specific linkage regions in 17q21, the multipoint parametric linkage analysis in 17q21 provided a maximal logarithm of the odds score of 5.3 (Figure 2A). Haplotype analysis showed a ≈6 Mbp (or ≈5 cM) region exclusively shared by patients.
Figure 2.
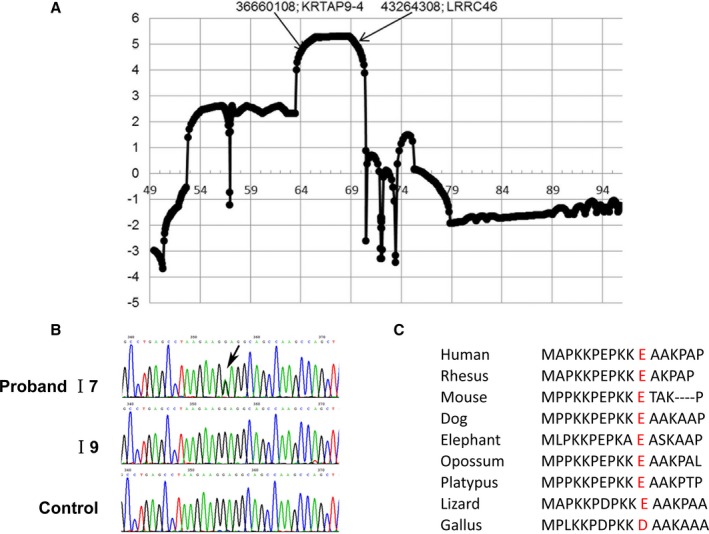
Genetic analysis. A, The multiple linkage analysis logarithm of the odds score in 17q21. B, Mutation (31G>A) in MYL4 confirmed by Sanger sequencing. C, The mutated locus is highly conserved across species. MYL4 indicates myosin light‐chain 4.
Based on bioinformatic analysis, both GeneWanderer and Endeavour ranked MYL4 in the top 2 genes in terms of its functional relationship to SCN5A and GJA5 (listed in Tables S5 and S6). Importantly, the identified missense mutation c. 31G>A in MYL4 changed the negatively charged residue (glutamic acid) to a positively charged 1 (lysine), p.E11K. This mutation was predicted to be pathogenic by KGGSeq (V0.8) under a combined prediction model19 of multiple deleteriousness and conservation scores (including SIFT,20 MutationAssessor,21 and GERP++22). The MYL4 mutation was confirmed in the entire pedigree by Sanger sequencing (Figure 2B). The mutation corresponded positionally to a negatively charged amino acid that is highly conserved across species (Figure 2C).
Of note, all 6 affected individuals carried the MYL4 mutation, and none of 38 nonaffected family members that were sequenced showed the mutation. There were 3 additional family members (I‐1, I‐5, and the father of I‐7), who also had a history of bradycardia, but for whom neither DNA for sequencing nor sufficient clinical data to assess the precise phenotype were available.
MYL4 p.E11K Rats Show an Atrial Cardiomyopathy Phenotype
To assess the pathogenic contribution of the MYL4 p.E11K mutation, we constructed a knock‐in MYL4 p.E11K rat model with CRISPR/Cas‐mediated genome editing (Figure 3A and 3B). The P‐wave amplitude of MYL4 E11K +/− and MYL4 E11K +/+ rats decreased progressively, accompanied by prolongation of the PR interval, from 15 days of age. The phenotype was more severe in homozygous rats, with 40% of MYL4 E11K +/+ rats losing their P wave by 3 months of age and 80% at 6 months. Nevertheless, heterozygote (MYL4 E11K +/−) rats also showed a clear phenotype, albeit less dramatic compared with homozygotes, with reduced P‐wave amplitude (Table S7) and left atrial dilation (Table S8) versus wild type (WT). The QRS duration was similar among all groups, indicating atrial‐specific electrical pathology (Figure 3C through 3E; Table S7). No spontaneous atrial arrhythmias were observed in knock‐in rats during routine ECG recording.
Figure 3.

MYL4 p.E11K rat targeting strategies and atrial cardiomyopathic phenotype. A and B, The MYL4 p.E11K rat model was produced using CRISPR/Cas‐mediated genome editing. The genotypes were confirmed by Sanger sequencing. C, Representative ECGs from WT, heterozygous, and homozygous MYL4 E11K rats at different ages. D and E, Mean data for P‐wave amplitude and PR interval. P‐wave amplitude of MYL4 E11K +/− rats progressively decreased, with prolongation of the PR interval, from 15 days of age. WT Rats: N=9; MYL4 E11K +/− rats: N=15; MYL4 E11K +/+ rats: N=10. F, Representative echocardiographic images from 3 rat genotypes. G and H, Mean data for left atrial dimension and left ventricular ejection fraction. The left atria progressively enlarged in both MYL4 E11K +/− and MYL4 E11K +/+ rat hearts, starting at 3 months of age. For TTE, 4 to 5 rats/group were averaged. Data are mean±SEM. *P<0.05; **P<0.01; ***P<0.001 vs WT, # P<0.05 among 15 days, 1 month, 3 months, and 6 months rats. ECG indicates standard 12‐lead electrocardiography; MYL4, myosin light‐chain 4; N, number; TTE, transthoracic ultrasound; WT, wild type.
Echocardiography showed left atrial dilation (Figure 3F) in both MYL4 E11K +/− and MYL4 E11K +/+ rats at 3 months and progressive increases with aging (Table S8). For example, at 6 months, left atrial diameters were 4.5±0.3 mm in MYL4 E11K +/+ and 3.8±0.1 mm in MYL4 E11K +/− rats, compared with 3.2±0.1 mm in WT (P<0.001; Figure 3F through 3H; Table S8).
In contrast to the clear atrial changes, right and left ventricular chamber sizes and ejection fraction were similar among the 3 genotypes, with no deviation greater than 20% and a small change of borderline statistical significance only at 3 months in MYL4 E11K +/+ rats. There were no differences in growth parameters among the 3 genotypes in either male or female rats (Figure S2).
Atrial Fibrosis and Cardiomyocyte Apoptosis in MYL4 p.E11K Rats
To further assess the atrial remodeling caused by p.E11K mutation, we collected atrial tissues from WT, MYL4 E11K +/− and MYL4 E11K +/+ rats at ages of 2 days, 15 days, 1 month, 3 months, and 6 months. Left atrial fibrosis was quantified with Masson trichrome staining. MYL4 E11K +/− and MYL4 E11K +/+ rats developed progressive left atrial fibrosis compared with WT rats, with statistically significant increases noted as early as 2 days of age (Figure 4A). At 2 days, 16.4±0.7% fibrous tissue content was observed in MYL4 E11K +/+ rats and 8.4±1.5% in MYL4 E11K +/− rats, compared with 5.6±0.6% in WT (P<0.001). At 6 months old, 27.6±5.8% fibrosis was observed in MYL4 E11K +/+ rats and 12.0±1.0% in MYL4 E11K +/− rats versus 8.7±0.6% in WT (P=0.016; Figure 4A and 4B).
Figure 4.

MYL4 p.E11K causes atrial fibrosis and cardiomyocytes apoptosis. A and B, Atrial fibrosis obtained by Masson staining was averaged for 4 to 5 hearts per group (6–10 fields per heart for each group). C and D, Representative images and mean data for TUNEL staining of atrial sections (4–5 hearts per group, 6–10 fields per heart for each group) from WT,MYL4 E11K +/−, and MYL4 E11K +/+rats. E, Confocal immunofluorescence showed that TUNEL‐positive cells costained with α‐actin, indicating that they were cardiomyocytes. TUNEL‐positive staining is green. Nuclei are stained with DAPI (blue). Myocytes are stained for α‐actin (red). Arrows indicate TUNEL‐positive nuclei. Values are mean±SEM. *P<0.05; **P<0.01; ***P<0.001 vs WT;, # P<0.05 among 15 days, 1 month, 3 months, and 6 months rats. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; LA, left atrial; MYL4, myosin light‐chain 4; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; WT, wild type.
Reparative fibrosis typically replaces cardiomyocytes that have been lost through apoptosis or necrosis.23, 24 The proportion of terminal deoxynucleotidyl transferase dUTP nick end labeling–positive nuclei was significantly increased in atrial tissue of heterozygous and homozygous MYL4 p.E11K rats compared with WT, even at 2 days of age (Figure 4C and 4D). Confocal immunofluorescence showed that terminal deoxynucleotidyl transferase dUTP nick end labeling–positive cells appeared to also costain with α‐actin (Figure 4E). We also performed proliferating cell nuclear antigen staining to obtain an index of fibroblast numbers. The percentage of proliferating cell nuclear antigen–positive nuclei increased significantly in atrial tissue for both MYL4 E11K +/− and MYL4 E11K +/+ rats at day 15 (P<0.001; Figure S3A and S3B).
There were no differences among WT, MYL4 E11K +/− rat and MYL4 E11K +/+ rats in atrial or ventricular cardiomyocyte size, based on FITC‐conjugated wheat germ agglutinin staining (Figure S4). Similarly, there were no differences in ventricular fibrous tissue content, apoptosis, or proliferating cell nuclear antigen–positive cell content among genotypes (Figures S3C, S3D, and S5).
Activation of Profibrotic and Proapoptotic Signals in the p.E11K Cardiomyocytes In Vivo and In Vitro
Atrial fibrosis can result from the activation of a variety of fibroproliferative signaling pathways.25, 26 Both small mothers against decapentaplegic and c‐Jun N‐terminal kinase phosphorylation was significantly increased in MYL4 E11K +/+ (but not MYL4 E11K +/−) atria (Figure 5A through 5C). Both MYL4 E11K +/+ and MYL4 E11K +/− atria expressed significantly stronger expression of proapoptotic proteins B‐cell lymphoma 2–associated X protein and Caspase‐3 compared with WT (Figure 5D through 5G).
Figure 5.
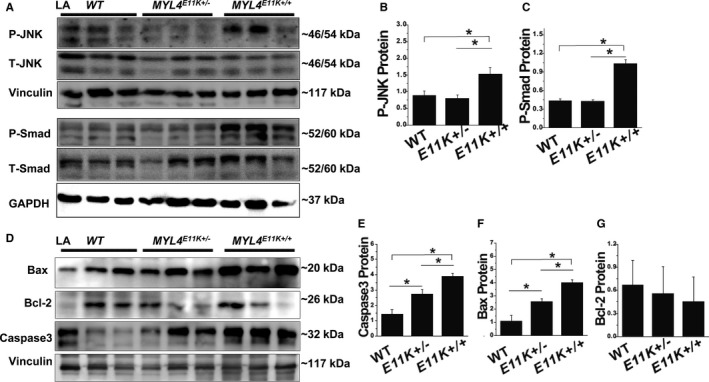
Profibrotic and proaptotic signaling. A through C, Western blots and mean data for JNK and Smad signaling in atria from WT,MYL4 E11K +/−, and MYL4 E11K +/+rats. D through G, Western blots and mean data for Bax and Caspase‐3 expression in atria from WT,MYL4 E11K +/−, and MYL4 E11K +/+rats. Data are mean±SEM. Protein from 3 independent rat atrial samples were subjected to immunoblot for each groups. *P<0.05. Bax indicates B‐cell lymphoma 2–associated X protein; Bcl‐2, B‐cell lymphoma 2; JNK, c‐Jun N‐terminal kinase; LA, left atrial; MYL4, myosin light‐chain 4; Smad, small mothers against decapentaplegic; WT, wild type.
To independently assess the direct effects of the MYL4 mutation on cardiomyocytes, we exposed cultured neonatal rat cardiomyocytes to WT MYL4 or MYL4E11K overexpression by adenovirus‐mediated gene transfer in vitro (Figure 6A through 6C). Neonatal rat cardiomyocytes overexpressing MYL4E11K showed increased apoptosis compared with WT (Figure 6D and 6E), along with upregulation of the proapoptotic protein, caspase‐3, and downregulation of the antiapoptotic protein, B‐cell lymphoma 2 (Figure 6F through 6I). In addition, MYL4E11K overexpression also increased small mothers against decapentaplegic and c‐Jun N‐terminal kinase phosphorylation relative to MYL4‐WT (Figure 6F, 6J, and 6K). These data suggest that MYL4 p.E11K directly induces atrial profibrotic and proapoptotic signaling.
Figure 6.
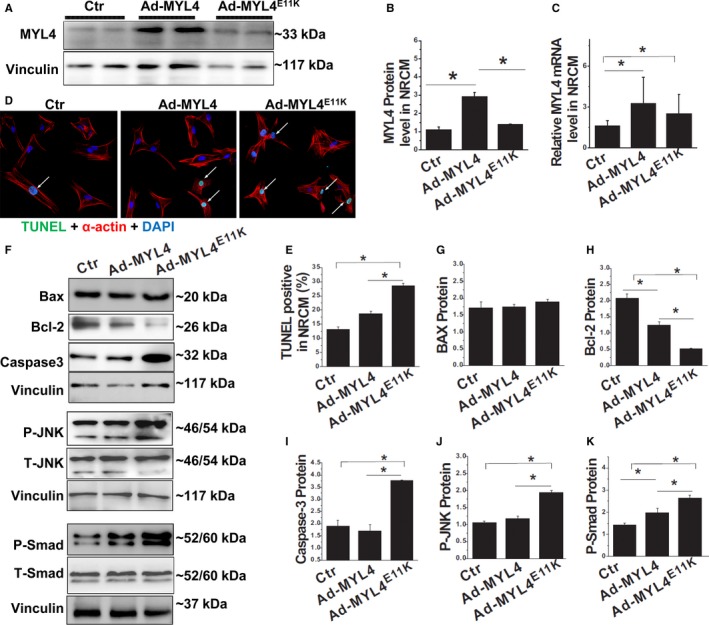
Acute MYL4 p.E11K gene transfer causes NRCM apoptosis and activates profibrotic signaling. A through C, Western blot and quantitative RT‐PCR analysis of MYL4 expression in NRCM exposed to gene transfer with different adenoviral constructs. D and E, Cardiomyocytes exposed to MYL4 E11K adenovirus showed significantly greater TUNEL‐positivity compared with WT. TUNEL‐positive staining is FITC (green). Nuclei are stained with DAPI (blue). Myocytes are stained for α‐actin (red). Arrows indicate TUNEL‐positive nuclei. F through K, Western blot analysis for proapoptotic protein caspase‐3 and antiapoptotic protein Bcl‐2 (F through I) and Smad/JNK signaling. Data are mean±SEM. The sample sizes were N=3 NRCM isolations per group with n=3 replicates from each isolate for each experiment. *P<0.05. Bax indicates B‐cell lymphoma 2–associated X protein; Bcl‐2, B‐cell lymphoma 2; Ctr indicates control; DAPI, 4′,6‐diamidino‐2‐phenylindole; JNK, c‐Jun N‐terminal kinase; MYL4, myosin light‐chain 4; N, number; NRCM, neonatal rat cardiomyocytes; Smad, small mothers against decapentaplegic; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; WT, wild type.
MYL4 protein expression decreased in MYL4 p.E11K knock‐in rats compared to WT rats (Figure S6), and E11K protein overexpression in cultured neonatal rat cardiomyocytes reduced MYL4 protein levels (Figure 6A), suggesting that MYL4 p.E11K causes a loss of MYL4 protein stability and that this effect contributes to loss of function.
Atrial Arrhythmia and Heart Function in MYL4 KO Rats
To further assess the functional contribution of MYL4, we constructed a MYL4 knockout rat model (for gene editing strategy, see Figure S6). MYL4 knockout eliminated detectable MYL4 protein on western blot. Note that MYL4 p.E11K knock‐in similarly reduced MYL4 protein expression, pointing to loss of protein stability (Figure S6). MYL4 KO rats demonstrated a similar phenotype to MYL4 p.E11K rats, but the phenotype manifested earlier and was more severe. Some homozygous MYL4 KO lost their P wave as early as 1 month of age. Overall, P‐wave amplitudes decreased significantly in homozygous KO rats at the age of 1 month, whereas the PR interval increased (see Figure 7A through 7C; Table S9). On routine ECG recording, homozygous rats also showed spontaneous atrial arrhythmias, including atrial tachycardias, AF, and atrial standstill (Figure 7D; Table S9). Heterozygous KO rats (MYL4 HT) also showed atrial electrical conduction disorders as they aged (Figure 7A through 7D; Table S9). Echocardiography showed left atrial dilation in KO rats (8.7±2.2 mm in KO rats versus 5.4±0.1 mm in WT; P<0.001; Figure 7E and 7F; Table S10). KO rats also showed progressive atrial fibrosis (Figure 7H and 7I) and lost almost all contractile force generation (Figure S7). In vitro, MYL4 knockdown in cultured myocytes led to a similar myocyte death phenotype to MYL4 p.E11K overexpression (Figure S8), suggesting that loss of MYL4 function causes cell death that likely contributes to the progressive atrial cardiomyopathy phenotype.
Figure 7.

Atrial cardiomyopathy phenotype in MYL4 KO rats. A, Representative ECGs for WT, heterozygous, and homozygous MYL4 KO rats at different ages. B and C, Mean data for P‐wave amplitude and PR interval. WT Rats: N=9; MYL4 HT rats: N=12; MYL4 KO rats: N=5. D, Typical spontaneous atrial arrhythmias. E through G, Echocardiography showed left atrial enlargement in KO rats. WT Rats: N=3; MYL4 HT rats: N=5; MYL4 KO rats: N=3. H and I, KO rats developed atrial fibrosis (4–5 hearts per group, 6–10 fields per heart for each group). Data are mean±SEM. *P<0.05; **P<0.01; ***P<0.001 vs WT; # P<0.05 among 1 month, 3 months, and ≥6 months rats. ECG indicates standard 12‐lead electrocardiography; HT, heterozygous; KO, knockout; MYL4, myosin light‐chain 4; N, number; WT, wild type.
Though there was no difference in ventricular fibrosis among WT, MYL4 HT, and MYL4 KO rats (Figure S9A and S9B), moderate statistically significant decreases in left ventricular function were noted in 6‐month‐old MYL4 KO rats, which may have been attributed to atrial arrhythmia and/or atrial structural and contractile remodeling (Figure 7G; Table S10).
Effects on Myosin ATPase Activity
In order to assess the molecular signature of MYL4 dysfunction, we constructed 3 types of recombinant plasmids: (1) pcDNA3.1‐MYL4‐GG (WT); (2) pcDNA3.1‐MYL4‐AA (the E11K mutation); and (3) pcDNA3.1‐MYL4‐12bp del (the Del, with 4 consecutive amino acids deleted near the MYL4 p.E11 site), then expressed and purified the corresponding proteins in yeast. MYL4 modulates actin‐activated ATPase activity,5 and the ATPase activities of different proteins were analyzed based on the Km and Vmax of myosin. Both Km and Vmax were significantly smaller in the presence of MYL4‐E11K protein than MYL4‐WT protein (Figure S10). MYL4‐Del protein did not affect myosin ATPase activity (Figure S10). These results indicate that the MYL4‐E11K mutant protein specifically reduces the activity of myosin ATPase.
To relate effects on myosin ATPase activity to atrial remodeling and obtain an additional control for the specificity of the MYL4 p.E11K mutation, we constructed an additional rat model, with the same 4 consecutive amino‐acid deletion near the MYL4 p.E11K site (Figure S6A and S6C). Neither MYL4 del+/− nor MYL4 del+/+ rats showed atrial or ventricular fibrosis, arrhythmia, or structural remodeling (Figure 8 and Figure S9C and S9D; Tables S11 and S12), indicating that mutations in the gene that do not affect myosin function do not cause atrial cardiomyopathy.
Figure 8.
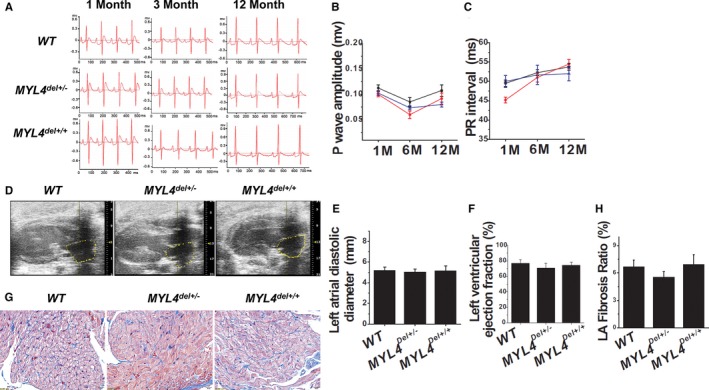
MYL4 del mutation rats do not show atrial cardiomyopathy. A through C, ECG data for WT (N=4), heterozygous, and homozygous MYL4 deletion mutation rats at different ages. Neither the MYL4 del+/− (N=5) nor MYL4 del+/+ (N=4) rats had significant arrhythmia. D through F, Echocardiography showed the absence of structural remodeling in MYL4 del+/− and MYL4 del+/+ rats. WT Rats: N=3; MYL4 del+/− rats: N=5; MYL4 del+/+ rats: N=3. G and H, Neither the MYL4 del+/− nor MYL4 del+/+ rats had significant atrial fibrosis (4–5 hearts per group, 6–10 fields per heart for each group). ECG indicates standard 12‐lead electrocardiography; MYL4, myosin light‐chain 4; N, number; WT, wild type.
Discussion
In this study, we first identified the missense mutation, c.31G>A (p.E11K), as a candidate causal gene variant for atrial cardiomyopathy through genetic analysis of a large family with atrial standstill. We then confirmed the role of this mutation with the use of a series of novel rat models generated by CRISPR/Cas‐mediated genome editing. We found that normal MYL4 function is essential for atrial electrical, contractile, and structural integrity. MYL4 dysfunction, whether as a result of knockout or targeted knock‐in of the human mutation, causes early atrial fibrosis associated with enhanced proaptotoic and profibrotic signaling, associated with an atrial cardiomyopathy featuring atrial arrhythmia, atrial contractile failure, and atrial enlargement.
Relationship to Past Studies of MYL4 Gene‐Variant–Associated Atrial Disease
Orr et al recently described a family with early‐onset AF and an apparently causal MYL4 mutation, p.Glu11Lys.8 Interestingly, this mutation results in the same E11K amino‐acid alteration that we observed in our kindred. This is a highly nonconservative mutation of an acidic to a basic amino acid, with the potential to strongly alter protein function. Affected individuals had early‐onset AF, evidence of atrial contractile dysfunction and conduction abnormalities requiring pacemaker insertion, a very similar clinical presentation to our patients. Gudbjartsson et al very recently identified a frameshift deletion in MYL4 causing AF at an early age, pacemaker requirement, and a high incidence of stroke.7 The clinical features were predominantly atrial, but signs of mild ventricular dysfunction eventually developed, primarily in the fifties and sixties. The clinical features of affected patients in the reports by Orr et al and Gudbardtsson et al were similar overall to those of our patients. As in our patients, inheritance appeared to be dominant in the Orr kindred,8 whereas it was recessive in the Gudbjartsson study.7
The only previous animal‐model data relating to these MYL4 gene variants come from the Orr study,8 in which the investigators reproduced the orthologous mutation (E17K in cMLC‐1) in zebrafish. They noted abnormal sinoatrial impulse generation and bradycardia, along with prolonged P‐wave duration and atrial enlargement. They noted marked disruption of sarcomere structure as early as 5 days postfertilization with loss of Z‐discs.
Here, we created novel rat models of MYL4 dysfunction with the use of CRISPR/Cas gene editing. The affected rats confirmed the ability of MYL4 dysfunction, whether by knockout or by knock‐in of a gene variant reproducing the clinical E11K mutation, to produce the cardinal clinical features of atrial electrical dysfunction, loss of atrial contractility, and atrial dilation constituting a highly atrial‐selective cardiomyopathy. Proapoptotic and profibrotic signaling were enhanced in knock‐in rats, and this appeared to be a direct result of the mutation rather than secondary to the atrial cardiomyopathy phenotype, because in vitro gene transfer of the mutation into neonatal rat cardiomyocytes reproduced the signaling changes. Interestingly, evidence of apoptosis and atrial fibrosis was present in vivo as early as 2 days of age (Figure 4), well before substantial loss of P‐wave amplitude or increase in left atrial dimension (Figure 3). These observations are consistent with the notion that Z‐disc disruption affects the many macromolecular complexes involved in intracellular signaling that are located at the Z‐disc,8, 27 producing profound atrial cardiomyocyte remodeling. Unlike the zebrafish, in which the orthologous gene is expressed in both the atria and ventricles, in rats and humans MYL4 shows strong atrial‐selective expression, consistent with the atrial‐selective phenotype noted.
Both in clinical reports7 and in our rats, some evidence of ventricular dysfunction appeared later in life. Ventricular dysfunction may be a secondary consequence of atrial arrhythmias, bradycardia, and dyssynchronous contraction caused by the primary atrial cardiomyopathy, or may be attributed to ventricular consequences of the mutation related to a return of ventricular MYL4 expression because of cardiac dysfunction.4
Insights Into Atrial Cardiomyopathy
The term “atrial cardiomyopathy” first appeared in the literature in 1972.28 Kottkamp has argued that many cases of AF may be attributed to an underlying “atrial fibrotic cardiomyopathy”.2, 29 Recently, a consensus has emerged, defining and characterizing the concept of atrial cardiomyopathy.1 Insights from genetic paradigms greatly contributed to our understanding of ventricular cardiomyopathies,30 but very little information has been available to date about genetic causes of atrial cardiomyopathy. Our identification of a pathological MYL4 mutation causing atrial cardiomyopathy and the development of powerful new animal models with which to study its pathophysiology is a step forward in the understanding of atrial cardiomyopathy. Atrial fibrosis appears to be a very early development in the evolution of this model. Along with the direct activation of proapoptotic and profibrotic signaling by gene transfer of the pathogenic mutation in vitro, these results point to this entity as a prototype for fibrotic atrial cardiomyopathy and provide insights into the underlying mechanisms. Prominent pathological features include highly atrial‐selective cardiomyocyte apoptosis and tissue fibrosis (eg, see Figure 4). According to the EHRA/HRS/APHRS/SOLAECE expert consensus, our rat model would be classified as a type III atrial cardiomyopathy, which combines cardiomyocyte pathology and fibrosis.1
Clinical Relevance
Clinically, atrial cardiomyopathy caused by MYL4 mutation progressed gradually in our patients: Despite sinus rhythm at a young age, contractile abnormalities were already evident, followed by atrial arrhythmia, atrioventricular block, and finally total atrial standstill. In the early stage of the condition, the ECG may appear normal and patients may have negligible symptoms. Nevertheless, TTE indicates reduced mechanical activity in the atria, implying that TTE may be a useful technique for the early identification of affected individuals in a disease‐bearing kindred.
Besides the specific rare familial entity we studied, our work can provide broader insights into the development of atrial cardiomyopathy. Signaling events of the type we identified have also been noted in various acquired forms of atrial cardiomyopathy.31 Further study in our rat models might allow for the identification of vulnerable molecular targets in atrial cardiomyopathy development that could produce new therapeutic approaches.
Potential Limitations
The phenotype of MYL4 mutated or KO rats directly paralleled the ECG changes and atrial dysfunction that we observed in our patients. MYL4 p.E11K rats had evidence of apoptosis and atrial fibrosis at an early stage. Although the phenotype of KO rats was similar overall to that of the MYL4 p.E11K rats, it was generally more severe and developed faster. Atrial tachyarrhythmias were easily noted in KO rats, but were not apparent in the MYL4 p.E11K knock‐in animals. The greater susceptibility of KO rats may be attributed to their complete loss of MYL4 function. The absence of atrial tachyarrhythmias in knock‐in rats may be attributed to the fact that the only rhythm recordings available to us were standard ECGs; continuous telemetry might have revealed spontaneous arrhythmias. Although the phenotype of the knock‐in rats was similar to that of our patients, there were some differences, most notably in the occurrence of spontaneous arrhythmias. Some of the discrepancy may be attributed to the absence of extended rhythm recording in the rats. Alternatively, background genetic control may play a role in conditioning the phenotype and promoting atrial arrhythmogenesis in humans. Nevertheless, KO rats did manifest spontaneous atrial tachyarrhythmias, confirming the principle that MYL4 dysfunction is, by itself, sufficient to cause atria arrhythmogenesis.
The underlying molecular mechanism of the myosin ATPase activity reduction is complex. Force production is considered to be associated with the Pi release step and the transition from weakly (non‐force‐generating) to strongly attached (force‐generating) states.32 Both Km and Vmax of myosin head ATPase are lower in the MYL4‐E11K protein than MYL4‐WT protein.
The work provided in this article presents a novel model with which to study the MYL4‐mutation–related atrial cardiomyopathy and characterizes the model in relationship to the clinical presentation. However, the precise mechanisms underlying key processes like apoptotic cell death and atrial fibrosis in this model remain to be determined in future studies.
Conclusions
We have shown, with the use of novel genetically defined rat models, that MYL4 is essential for atrial electrical, functional, and structural integrity, and that MYL4 dysfunction can account for severe inherited atrial cardiomyopathy in an extended kindred. We have obtained evidence that atrial apoptotic cell death and fibrosis are very early findings and are apparent before any functional manifestations of atrial cardiomyopathy. These observations are consistent with an important pathophysiological role for enhanced proapoptotic and profibrotic signaling that is activated rapidly in the presence of the causative mutation. These biological findings have the potential to advance our understanding of the molecular basis of atrial cardiomyopathy and eventually to lead to new diagnosis and therapeutic modalities.
Sources of Funding
This work was supported by Chinese National Natural Science Foundation No. 81270256, 81370391, 81470394, and 81670230; Shanghai Sailing Program No. 15YF1409400; Hong Kong Research Grants Council GRF HKU 776412M, Hong Kong Research Grants Council Theme‐Based Research Scheme T12‐705/11; European Community Seventh Framework Programme Grant on European Network of National Schizophrenia Networks Studying Gene‐Environment Interactions (EU‐GEI); the HKU Seed Funding Programme for Basic Research 201302159006; the Health and Medical Research Fund 01121436; the Canadian Institutes of Health Research; and the Heart and Stroke Foundation of Canada.
Disclosures
None.
Supporting information
Data S1. Supplemental methods.
Table S1. Transthoracic Echocardiographic Parameters in 6 Patients
Table S2. Sources of Primary Antibodies
Table S3. ECG Parameters of Affected Patients
Table S4. Candidate Genes and Mutations Identified by Preliminary Bioinformatic Analysis
Table S5. The Top 10 Gene List Based on GeneWanderer
Table S6. The Top 10 Gene List Based on Endeavour
Table S7. MYL4 E11K Knock‐in Rat ECG Parameters
Table S8. Measurements of MYL4 E11K+/− Mutation Rat TTE
Table S9. MYL4 KO Rat ECG Measurements
Table S10. MYL4 KO Rat TTE Results
Table S11. MYL4 del Rat ECG Measurements
Table S12. MYL4 del Rat TTE Measurements
Figure S1. Clinical data of patients.
Figure S2. MYL4 p.E11K did not affect rat growth or heart tissue weight.
Figure S3. MYL4 p.E11K exacerbated atrial cell (fibroblast) proliferation, but did not affect the cell proliferation in ventricles.
Figure S4. MYL4 E11K does not affect the cell size of the atrial or ventricle cardiomyocytes, or NRCM.
Figure S5. MYL4 E11K does not affect ventricular fibrosis or apoptosis.
Figure S6. MYL4 KO and MYL4 del targeting strategies and identification.
Figure S7. Lost atrial contractile force in MYL4 KO rats.
Figure S8. MYL4 knockdown in cultured myocytes led to a similar myocyte death phenotype.
Figure S9. MYL4 KO or del mutations do not cause ventricular fibrosis.
Figure S10. ATPase activity assays.
Acknowledgments
We thank the patients and family members for taking part in this study.
(J Am Heart Assoc. 2017;6:e007030 DOI; 10.1161/JAHA.117.007030.)29080865
References
- 1. Goette A, Kalman JM, Aguinaga L, Akar J, Cabrera JA, Chen SA, Chugh SS, Corradi D, D'Avila A, Dobrev D, Fenelon G, Gonzalez M, Hatem SN, Helm R, Hindricks G, Ho SY, Hoit B, Jalife J, Kim YH, Lip GY, Ma CS, Marcus GM, Murray K, Nogami A, Sanders P, Uribe W, Van Wagoner DR, Nattel S; Document Reviewers . EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: definition, characterization, and clinical implication. Europace. 2016;18:1455–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kottkamp H. Human atrial fibrillation substrate: towards a specific fibrotic atrial cardiomyopathy. Eur Heart J. 2013;34:2731–2738. [DOI] [PubMed] [Google Scholar]
- 3. Disertori M, Mase M, Marini M, Mazzola S, Cristoforetti A, Del Greco M, Kottkamp H, Arbustini E, Ravelli F. Electroanatomic mapping and late gadolinium enhancement MRI in a genetic model of arrhythmogenic atrial cardiomyopathy. J Cardiovasc Electrophysiol. 2014;25:964–970. [DOI] [PubMed] [Google Scholar]
- 4. Hernandez OM, Jones M, Guzman G, Szczesna‐Cordary D. Myosin essential light chain in health and disease. Am J Physiol Heart Circ Physiol. 2007;292:H1643–H1654. [DOI] [PubMed] [Google Scholar]
- 5. Ho G, Chisholm RL. Substitution mutations in the myosin essential light chain lead to reduced actin‐activated ATPase activity despite stoichiometric binding to the heavy chain. J Biol Chem. 1997;272:4522–4527. [DOI] [PubMed] [Google Scholar]
- 6. Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, Besenbacher S, Magnusson G, Halldorsson BV, Hjartarson E, Sigurdsson GT, Stacey SN, Frigge ML, Holm H, Saemundsdottir J, Helgadottir HT, Johannsdottir H, Sigfusson G, Thorgeirsson G, Sverrisson JT, Gretarsdottir S, Walters GB, Rafnar T, Thjodleifsson B, Bjornsson ES, Olafsson S, Thorarinsdottir H, Steingrimsdottir T, Gudmundsdottir TS, Theodors A, Jonasson JG, Sigurdsson A, Bjornsdottir G, Jonsson JJ, Thorarensen O, Ludvigsson P, Gudbjartsson H, Eyjolfsson GI, Sigurdardottir O, Olafsson I, Arnar DO, Magnusson OT, Kong A, Masson G, Thorsteinsdottir U, Helgason A, Sulem P, Stefansson K. Large‐scale whole‐genome sequencing of the Icelandic population. Nat Genet. 2015;47:435–444. [DOI] [PubMed] [Google Scholar]
- 7. Gudbjartsson DF, Holm H, Sulem P, Masson G, Oddsson A, Magnusson OT, Saemundsdottir J, Helgadottir HT, Helgason H, Johannsdottir H, Gretarsdottir S, Gudjonsson SA, Njolstad I, Lochen ML, Baum L, Ma RC, Sigfusson G, Kong A, Thorgeirsson G, Sverrisson JT, Thorsteinsdottir U, Stefansson K, Arnar DO. A frameshift deletion in the sarcomere gene MYL4 causes early‐onset familial atrial fibrillation. Eur Heart J. 2017;38:27–34. [DOI] [PubMed] [Google Scholar]
- 8. Orr N, Arnaout R, Gula LJ, Spears DA, Leong‐Sit P, Li Q, Tarhuni W, Reischauer S, Chauhan VS, Borkovich M, Uppal S, Adler A, Coughlin SR, Stainier DY, Gollob MH. A mutation in the atrial‐specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun. 2016;7:11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Woolliscroft J, Tuna N. Permanent atrial standstill: the clinical spectrum. Am J Cardiol. 1982;49:2037–2041. [DOI] [PubMed] [Google Scholar]
- 10. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li MX, Gui HS, Kwan JS, Bao SY, Sham PC. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012;40:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker‐sharing statistics. Am J Hum Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 14. Tranchevent LC, Barriot R, Yu S, Van Vooren S, Van Loo P, Coessens B, De Moor B, Aerts S, Moreau Y. ENDEAVOUR update: a web resource for gene prioritization in multiple species. Nucleic Acids Res. 2008;36:W377–W384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kohler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82:949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shao Y, Guan Y, Wang L, Qiu Z, Liu M, Chen Y, Wu L, Li Y, Ma X, Liu M, Li D. CRISPR/Cas‐mediated genome editing in the rat via direct injection of one‐cell embryos. Nat Protoc. 2014;9:2493–2512. [DOI] [PubMed] [Google Scholar]
- 17. Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, Zhao Y, Liu M. Heritable gene targeting in the mouse and rat using a CRISPR‐Cas system. Nat Biotechnol. 2013;31:681–683. [DOI] [PubMed] [Google Scholar]
- 18. Groenewegen WA, Firouzi M, Bezzina CR, Vliex S, van Langen IM, Sandkuijl L, Smits JP, Hulsbeek M, Rook MB, Jongsma HJ, Wilde AA. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res. 2003;92:14–22. [DOI] [PubMed] [Google Scholar]
- 19. Li MX, Kwan JS, Bao SY, Yang W, Ho SL, Song YQ, Sham PC. Predicting Mendelian disease‐causing non‐synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013;9:e1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 21. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 2010;6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hanna N, Cardin S, Leung TK, Nattel S. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing‐induced congestive heart failure. Cardiovasc Res. 2004;63:236–244. [DOI] [PubMed] [Google Scholar]
- 24. Cardin S, Li D, Thorin‐Trescases N, Leung TK, Thorin E, Nattel S. Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin‐dependent and ‐independent pathways. Cardiovasc Res. 2003;60:315–325. [DOI] [PubMed] [Google Scholar]
- 25. Nattel S, Dobrev D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol. 2016;13:575–590. [DOI] [PubMed] [Google Scholar]
- 26. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–809. [DOI] [PubMed] [Google Scholar]
- 27. Nattel S. Close connections between contraction and rhythm: a new genetic cause of atrial fibrillation/cardiomyopathy and what it can teach us. Eur Heart J. 2017;38:35–37. [DOI] [PubMed] [Google Scholar]
- 28. Nagle RE, Smith B, Williams DO. Familial atrial cardiomyopathy with heart block. Br Heart J. 1972;34:205. [PubMed] [Google Scholar]
- 29. Kottkamp H. Fibrotic atrial cardiomyopathy: a specific disease/syndrome supplying substrates for atrial fibrillation, atrial tachycardia, sinus node disease, AV node disease, and thromboembolic complications. J Cardiovasc Electrophysiol. 2012;23:797–799. [DOI] [PubMed] [Google Scholar]
- 30. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention . Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. [DOI] [PubMed] [Google Scholar]
- 31. Wakili R, Voigt N, Kääb S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011;121:2955–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siemankowski RF, Wiseman MO, White HD. ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc Natl Acad Sci U S A. 1985;82:658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Table S1. Transthoracic Echocardiographic Parameters in 6 Patients
Table S2. Sources of Primary Antibodies
Table S3. ECG Parameters of Affected Patients
Table S4. Candidate Genes and Mutations Identified by Preliminary Bioinformatic Analysis
Table S5. The Top 10 Gene List Based on GeneWanderer
Table S6. The Top 10 Gene List Based on Endeavour
Table S7. MYL4 E11K Knock‐in Rat ECG Parameters
Table S8. Measurements of MYL4 E11K+/− Mutation Rat TTE
Table S9. MYL4 KO Rat ECG Measurements
Table S10. MYL4 KO Rat TTE Results
Table S11. MYL4 del Rat ECG Measurements
Table S12. MYL4 del Rat TTE Measurements
Figure S1. Clinical data of patients.
Figure S2. MYL4 p.E11K did not affect rat growth or heart tissue weight.
Figure S3. MYL4 p.E11K exacerbated atrial cell (fibroblast) proliferation, but did not affect the cell proliferation in ventricles.
Figure S4. MYL4 E11K does not affect the cell size of the atrial or ventricle cardiomyocytes, or NRCM.
Figure S5. MYL4 E11K does not affect ventricular fibrosis or apoptosis.
Figure S6. MYL4 KO and MYL4 del targeting strategies and identification.
Figure S7. Lost atrial contractile force in MYL4 KO rats.
Figure S8. MYL4 knockdown in cultured myocytes led to a similar myocyte death phenotype.
Figure S9. MYL4 KO or del mutations do not cause ventricular fibrosis.
Figure S10. ATPase activity assays.