

Abstract
Background
Hyperphosphatemia is a major factor promoting the formation of arterial medial calcification in chronic kidney disease (CKD). However, arterial medial calcification begins to occur during the early stages of CKD, when hyperphosphatemia is not yet apparent. It is predicted that other factors also play a role. The aim of the present study was to determine the role of pro‐inflammatory nuclear factor‐κB (NF‐κB) signaling in smooth muscle cells (SMCs) for phosphate‐induced arterial medial calcification in CKD mice.
Methods and Results
We first sought to establish a novel mouse model of CKD with arterial medial calcification. CKD was induced in DBA/2 mice by feeding them a low concentration of adenine, and these mice were fed a normal or high‐phosphorus diet. Severe calcification was seen in CKD mice fed the high‐phosphorus diet, while it was undetectable in CKD mice fed the normal phosphorus diet or control mice fed the high‐phosphorus diet. Arterial medial calcification was accompanied by phenotypic switching of SMCs into osteogenic cells. Interestingly, NF‐κB inhibitors, tempol and triptolide, both reduced arterial medial calcification in CKD mice fed the high‐phosphorus diet. Moreover, formation of arterial medial calcification, as well as SMC phenotypic switching, was also markedly attenuated in transgenic mice, in which the NF‐κB activity was inhibited selectively in SMCs. Mechanistic studies revealed that Krüppel‐like factor 4 was involved in NF‐κB‐induced SMC phenotypic switching and calcification.
Conclusions
Results of the present studies suggest that the NF‐κB signaling in SMCs plays an important role in high phosphate‐induced arterial medial calcification in CKD.
Keywords: calcification, chronic kidney disease, nuclear factor‐κB, smooth muscle cell
Subject Categories: Smooth Muscle Proliferation and Differentiation, Vascular Biology
Clinical Perspective
What Is New?
Smooth muscle cell–selective inhibition of nuclear factor‐κB signaling reduced high phosphate‐induced arterial medial calcification in chronic kidney disease mice.
What Are the Clinical Implications?
Multiple factors including hyperphosphatemia and nuclear factor‐κB activation cooperatively contribute to arterial medial calcification in chronic kidney disease.
The present study raises a possibility that reagents that selectively inhibit nuclear factor‐κB signaling in smooth muscle cells will be a novel candidate for the treatment and prevention of arterial medial calcification in chronic kidney disease.
Arterial medial calcification, also known as Mönckeberg's arteriosclerosis, is highly prevalent among patients with chronic kidney disease (CKD) and is associated with an increased risk of cardiovascular disease and mortality.1, 2, 3 Calcification in the arteries results in increased vessel wall stiffness, decreased compliance, increased pulse‐wave velocity, and elevated systolic blood pressure.3 These alterations in mechanical and hemodynamic properties lead to left ventricular hypertrophy, decreased coronary artery perfusion, and heart failure.3 Identification of the molecular and cellular mechanisms underlying the formation of arterial medial calcification is, therefore, required to improve the prognosis of CKD patients.
Hyperphosphatemia is a major factor promoting the formation of arterial medial calcification.1, 2, 3 The results of previous studies by our laboratory and others have shown that high phosphate concentration induces the phenotypic switching of vascular smooth muscle cells (SMCs) into osteogenic cells by decreasing the expression of SMC differentiation marker genes, including smooth muscle α‐actin (Acta2) and SM22α (Tagln), and by increasing the expression of osteogenic genes, such as Runx2, osteopontin (Spp1), and alkaline phosphatase.4, 5 In addition, a number of clinical studies have demonstrated that an elevation in serum phosphate levels is significantly related to vascular calcification in end‐stage CKD patients.6, 7 Moreover, the administration of phosphate binders has been shown to attenuate the progression of vascular calcification in CKD patients,8, 9 as well as in experimental animal models.10, 11, 12, 13 As such, the results of preceding studies provide compelling evidence that high phosphate concentration is a strong factor in the promotion of arterial medial calcification. However, arterial medial calcification, as well as an increase in cardiovascular mortality, begins to occur during the early stages of CKD, when hyperphosphatemia is not yet apparent.14, 15 These findings suggest that factors other than high phosphate concentration also contribute to the formation and the progression of arterial medial calcification in CKD.
The nuclear factor‐κB (NF‐κB) family of transcription factors participates in the inflammatory process in a variety of cells.16, 17, 18 In resting cells, NF‐κB exists in the cytoplasm as an inactive dimer by association with the inhibitory protein, IκB. Upon cell stimulation by inflammatory cytokines, such as tumor necrosis factor‐α (TNF) and interleukin‐1β, IκB is phosphorylated on specific serine residues, serines 32 and 36, leading to its ubiquitination and consecutive proteasomal degradation. Upon its release from IκB, NF‐κB is free to translocate into the nucleus, engage DNA, and initiate the transcription of many genes including cytokines, chemokines, adhesion molecules, and anti‐oxidant proteins. The results of previous studies have shown that serum concentrations of inflammatory cytokines, including TNF and interleukin‐1β, are elevated in CKD patients.19, 20 In addition, NF‐κB has been shown to be activated in calcified vessels.21 These results suggest that NF‐κB is a potential therapeutic target for arterial medial calcification in CKD. Indeed, the administration of the anti‐oxidant tempol, which is known to inhibit multiple signaling pathways including the NF‐κB pathway, ameliorated arterial medial calcification in uremic rats.22 However, the precise mechanisms, including the cell‐autonomous role of NF‐κB in SMCs in vivo, remain to be determined.
Recently, we generated mice expressing a truncated form of IκB (IκBΔN) in a SMC‐selective manner by crossing mice expressing Cre recombinase under the control of the Tagln promoter (Tagln‐Cre mice)23 with IκBΔN mice24 on a C57BL/6 background, and these mice were designated as SM‐IκBΔN mice.24 SM‐IκBΔN mice contain the IκBΔN transgene separated from a universal CAG promoter by a floxed STOP sequence. Following the activation of Cre recombinase in SMCs, they cell‐specifically express IκBΔN, which lacks its N‐terminal 54 amino acids including 2 phosphorylation sites at serines 32 and 36, resulting in the continuous inhibition of NF‐κB activation as a super‐repressor. Using SM‐IκBΔN mice, we previously showed that the inhibition of NF‐κB signaling within SMCs attenuated neointimal formation following vascular injury.24 These mice are suitable for investigating the effect of SMC‐selective NF‐κB inhibition on the development of vascular diseases. However, C57BL/6 mice are calcification‐resistant, whereas DBA/2 mice are susceptible to calcification.25 Thus, we changed the mouse genetic background from C57BL/6 to DBA/2 in the present studies. We then sought to establish a novel mouse model of arterial medial calcification in CKD using DBA/2 mice and to determine whether NF‐κB signaling within SMCs contributed to the formation of arterial medial calcification in CKD.
Materials and Methods
The data, analytic methods, and study materials will be made available on request to other researchers for purposes of reproducing the results or replicating the procedure.
Generation of SM‐IκBΔN Mice
Animal protocols, including the number of animals used, were approved by the Keio University Animal Care and Use Committee, and the procedures followed were in accordance with institutional guidelines. Tagln‐Cre mice23 and IκBΔN mice24 were, respectively, backcrossed more than 8 times to DBA/2 mice (Sankyo Labo Service, Tokyo, Japan). Heterozygous Tagln‐Cre mice and heterozygous IκBΔN mice, both of which had a DBA/2 background, were crossed to generate SMC‐selective IκBΔN transgenic (Tagln‐Cre+/−/IκBΔN+/−; described as SM‐IκBΔN) mice and control (Tagln‐Cre+/−/IκBΔN−/−, Tagln‐Cre−/−/IκBΔN+/−, or Tagln‐Cre−/−/IκBΔN−/−) mice. Tagln‐Cre−/−/IκBΔN−/− control mice were also used as DBA/2 mice. Genotyping was performed using polymerase chain reaction as described previously.24, 26
Arterial Medial Calcification Model
Female mice were used in the calcification studies, because they have shown higher susceptibility to calcification than male mice.25 Fourteen‐week‐old control mice and SM‐IκBΔN mice were fed a normal phosphorus diet (NPD: 1.0% calcium and 1.0% phosphorus; CLEA Japan, Tokyo, Japan) or a high phosphorus diet (HPD: 1.0% calcium and 2.0% phosphorus, CLEA Japan) ad libitum for 10 weeks. Some of these mice were also fed a diet containing 0.25% adenine (Sigma, St. Louis, MO) for 5 weeks (from 14 to 19 weeks of age) to induce CKD. A subset of the HPD‐fed/adenine‐fed CKD mice were treated for 10 weeks with one of the following procedures: 3% CaCO3 in dietary chow, 3 mmol/L tempol (4‐hydroxy‐TEMPO, Sigma) in drinking water, or intraperitoneal injections of triptolide (70 μg/kg body weight, Sigma) every other day. The tempol‐containing water was changed every other day. At the end of the experiment, tissues including the aorta and the heart, as well as blood samples, were collected. Tissues from some mice were directly fixed in 4% paraformaldehyde to obtain whole cardiovascular images using Alizarin red staining, while other tissues were cut into multiple pieces for histology, immunostaining, RNA analysis, and measurement of the calcium content.
Biochemical Parameters
Serum concentrations of urea nitrogen, creatinine, calcium, and inorganic phosphate were measured as described previously.27, 28 Serum concentrations of cystatin‐C and TNF were determined using the mouse cystatin‐C ELISA kit (BioVendor, Modrice, Czech Republic) and the Quantikine ELISA mouse TNF immunoassay kit (R&D Systems, Minneapolis, MN), respectively.
Calcium Measurements
The calcium contents in the abdominal aorta were determined using the o‐cresolphthalein complexone method, as described previously.27
Histology and Immunostaining Studies
The arteries and the hearts for histology and immunostaining were fixed in 4% paraformaldehyde and embedded into OCT compound. The 6‐μm cross‐sections were prepared and subjected to von Kossa staining, Alizarin red staining, Masson trichrome staining, and immunostaining, as described previously.4, 26, 27, 28, 29 A morphometric analysis of the thoracic aorta was performed using 3 Alizarin red‐stained sections per artery. These sections were located at ≈10.0 mm proximal to the diaphragm, and each section was located 300 μm apart (ie, at 9700, 10 000, and 10 300 μm proximal to the diaphragm). The calcified areas were measured using Image‐Pro Plus software (Media Cybernetics, Silver Spring, MD). Cardiac fibrosis was evaluated semiquantitatively using Masson trichrome–stained samples, as described previously.29
Immunohistochemistry was performed with antibodies for Acta2 (1A4, Sigma), Tagln (Abcam, Cambridge, MA), p65 (F6, Santa‐Cruz Biotechnology, Santa Cruz, CA), and Krüppel‐like factor 4 (Klf4).30 Staining for Acta2 and Tagln was visualized using the Vector Red Alkaline Phosphatase Substrate kit (Vector Laboratories, Burlingame, CA), and the sections were counterstained with hematoxylin. Staining for p65 and Klf4 was visualized using diaminobenzidine, and the sections were counterstained with hematoxylin. Immunofluorescence studies were performed using antibodies for Runx2 (Santa‐Cruz Biotechnology) and Spp1 (Abcam), and the sections were counterstained by 4′,6‐diamidino‐2‐phenylindole.
Real‐Time Reverse Transcription–Polymerase Chain Reaction
Total RNA prepared from the thoracic aorta was used for real‐time reverse transcription–polymerase chain reaction. The primer and probe sequences for Acta2, Tagln, Runx2, Spp1, Klf4, and 18S rRNA have been described previously.4, 26, 31
Western Blotting
Western blotting was performed, as described previously.26, 30 Antibodies for IκB (Santa‐Cruz Biotechnology) and GAPDH (6C5; Millipore, Billerica, MA) were used.
Cell Cultures
Rat aortic SMCs were cultured as described previously.4, 27, 31 One day after plating at 10 000 cells/cm2, SMCs were incubated in Dulbecco's modified Eagle medium (glucose: 4.5 g/L)/5% fetal bovine serum (Invitrogen, Carlsbad, CA) with normal (0.9 mmol/L) or high (2.7 mmol/L) phosphate concentration in the presence or absence of 10 ng/mL of mouse TNF (R&D Systems) and/or 1 μmol/L of BAY11‐7082 (Calbiochem, Darmstadt, Germany) for 8 days. The medium containing the reagents was changed every 2 days. Measurement of the calcium contents, von Kossa staining, RNA analysis, immunoprecipitation assays, and quantitative chromatin immunoprecipitation assays were performed as described previously.4, 26, 27, 30, 32
Statistical Analyses
Data are presented as the mean±SEM. Statistical analyses were done by SigmaPlot/SigmaStat9 (Systat Software Inc, San Jose, CA). After confirming that the data passed the normality test for parametric analyses, unpaired t test, 1‐way factorial ANOVA with a post‐hoc Fisher protected least significant difference test, or 2‐way factorial ANOVA with a post‐hoc Fisher protected least significant difference test were performed. When the data failed to pass the normality test, Kruskal–Wallis nonparametric test was performed, as appropriate. P<0.05 were considered significant.
Results
High Phosphate Induced Arterial Medial Calcification in Adenine‐Fed CKD Mice
The results of previous studies showed that high phosphate induced arterial medial calcification in 5/6‐nephrectomized CKD mice on a DBA/2 background.25 We sought to establish another CKD mouse model with arterial medial calcification. An adenine‐induced CKD rat model is widely used to study arterial medial calcification.4, 10, 11, 12, 13, 21, 22, 27 Thus, we first fed a diet containing 0.75% adenine, the same concentration of adenine as that used in the rat model, to DBA/2 mice to induce CKD. However, all the mice fed the diet containing 0.75% adenine died within 1 week (Figure 1). The concentration of adenine in the diet was adjusted, and we found that a diet containing 0.25% adenine was appropriate for the induction of CKD in DBA/2 mice.
Figure 1.
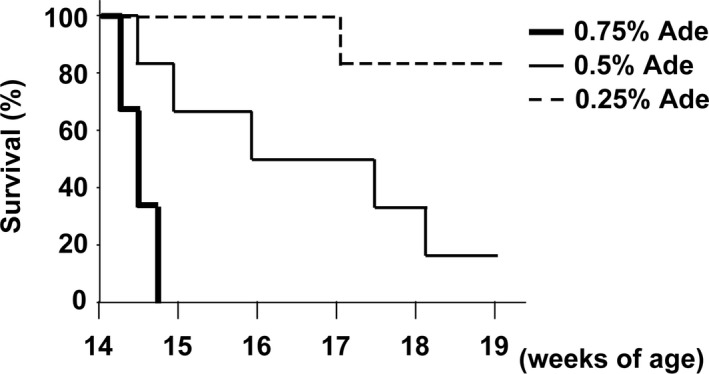
DBA/2 mice could not survive when fed with 0.75% adenine. DBA/2 mice were fed a normal phosphorus diet containing 0.75% adenine (Ade), 0.5% adenine, or 0.25% adenine for 5 wk (n=6 per group). Survival rates are shown.
DBA/2 mice were divided into 5 groups: (1) mice fed a normal phosphorus diet (NPD), (2) mice fed an NPD containing 0.25% adenine (NPD+Ade), (3) mice fed a high phosphorus diet (HPD), (4) mice fed an HPD containing 0.25% adenine (HPD+Ade), and (5) mice fed an HPD containing 0.25% adenine and 3% CaCO3 (HPD+Ade+CaCO3). Adenine was given for 5 weeks (from 14 to 19 weeks of age), whereas the NPD or HPD was fed throughout the entire experimental period of 10 weeks (Figure 2A). The results showed that the body weight was reduced by either the 0.25% adenine or the HPD, compared with the results in the NPD‐fed mice. Serum concentrations of urea nitrogen, creatinine, and cystatin‐C were elevated in mice fed a diet containing 0.25% adenine (Figure 2B through 2D). Serum concentrations of calcium decreased in mice fed an adenine‐containing diet, although treatment with CaCO3 restored the calcium concentration (Figure 2E). Elevation in serum phosphate concentrations was seen in the HPD+Ade‐fed mice (Figure 2F).
Figure 2.
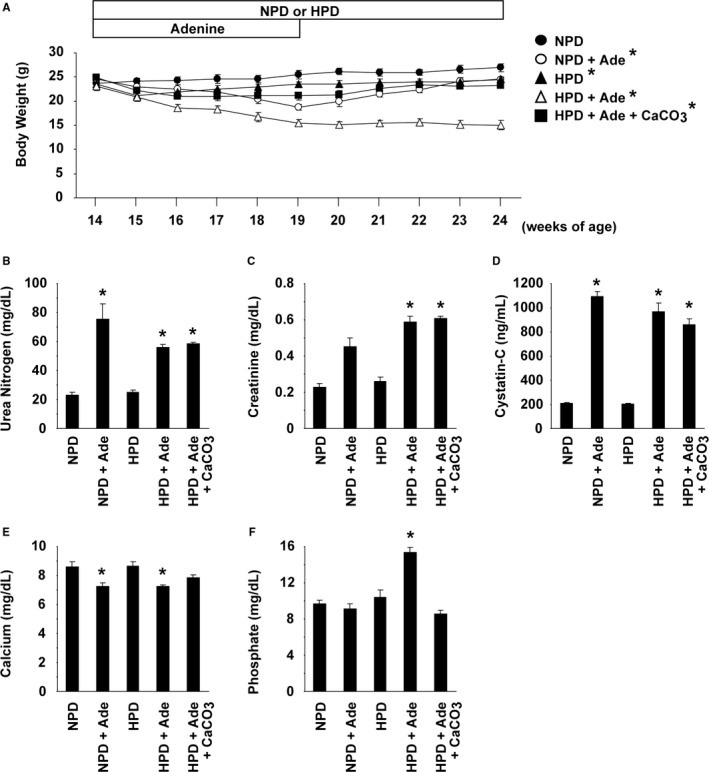
Adenine and high phosphorus diet (HPD)‐fed DBA/2 mice exhibited chronic kidney disease and hyperphosphatemia. DBA/2 mice were fed a normal phosphorus diet (NPD) (n=9), an NPD containing 0.25% adenine (NPD+Ade) (n=9), an HPD (n=8), an HPD containing 0.25% adenine (HPD+Ade) (n=7), or an HPD containing 0.25% adenine and 3% CaCO 3 (HPD+Ade+CaCO 3) (n=9). Adenine was administered for 5 wk. The animals were fed the NPD or HPD throughout the entire experimental period of 10 wk. A, Changes in body weight are shown. B through F, Serum levels of urea nitrogen (B), creatinine (C), cystatin‐C (D), calcium (E), and phosphate (F) were measured at the end of the experimental period. *P<0.05 compared with NPD‐fed mice.
At the end of the experimental periods, the formation of arterial medial calcification was examined using Alizarin red staining and von Kossa staining (Figure 3A). Calcification was not seen in the NPD‐fed mice, the NPD+Ade‐fed mice, or the HPD‐fed mice. However, severe calcification was seen in mice fed the HPD and adenine. Calcification was detectable in the medial layer of various sizes of arteries, including the thoracic aorta, abdominal aorta, carotid arteries, subclavian arteries, renal arteries, and iliac arteries, in the HPD+Ade‐fed mice (Figure 3A through 3C and data not shown). By contrast, treatment with CaCO3 decreased the formation of arterial medial calcification in mice fed the HPD and adenine. Indeed, the calcified area in the thoracic aorta, as well as the calcium content in the abdominal aorta, was significantly increased in the HPD+Ade‐fed mice, but not in the HPD+Ade+CaCO3‐fed mice (Figure 3D and 3E). Moreover, the expression of SMC differentiation markers, such as Acta2 and Tagln, was decreased in the thoracic aorta of the HPD+Ade‐fed mice, whereas it was restored by CaCO3 treatment (Figure 4A). Conversely, the expression of osteogenic markers, including Runx2 and Spp1, was induced in the thoracic aorta of the HPD+Ade‐fed mice, whereas it was not induced in the HPD+Ade+CaCO3‐fed mice. Phenotypic switching of SMCs into osteogenic cells in the HPD+Ade‐fed mice was also confirmed at the mRNA level (Figure 4B through 4E). In summary, these results demonstrate that arterial medial calcification, as well as the phenotypic switching of SMCs into osteogenic cells, was induced in DBA/2 mice treated with the HPD and adenine, in which hyperphosphatemia and CKD were remarkable.
Figure 3.
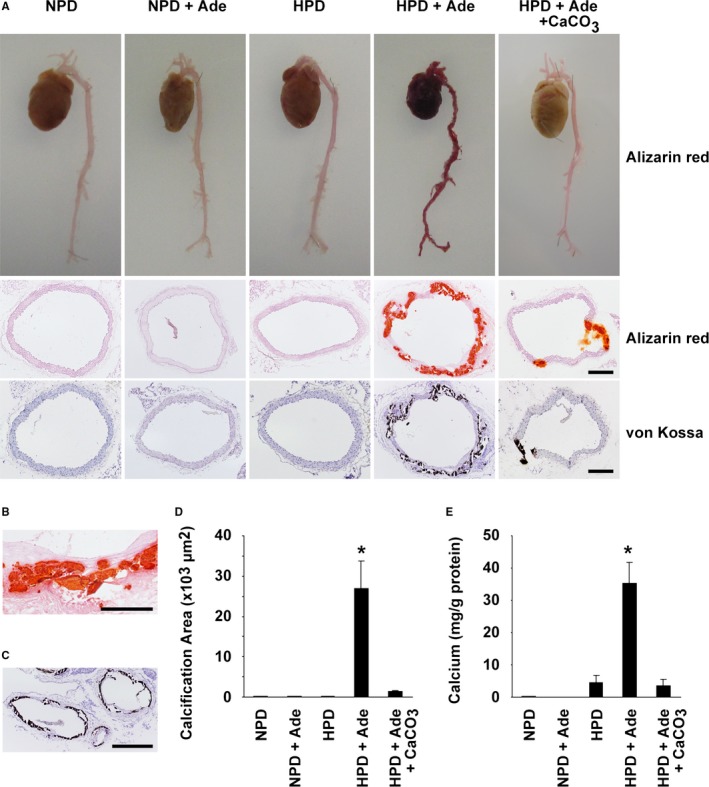
Arterial medial calcification was remarkable in high phosphorus diet (HPD)–fed mice with chronic kidney disease. DBA/2 mice were fed a normal phosphorus diet (NPD), an NPD containing 0.25% adenine (NPD+Ade), an HPD, an HPD containing 0.25% adenine (HPD+Ade), or an HPD containing 0.25% adenine and 3% CaCO 3 (HPD+Ade+CaCO 3). Adenine was administered for 5 wk. The animals were fed the NPD or HPD throughout the entire experimental period of 10 wk. A through D, Formation of arterial medial calcification was examined using Alizarin red staining and von Kossa staining. A, Top: Alizarin red staining for whole cardiovascular images (n=3 per group). Middle and bottom: Alizarin red staining (middle) and von Kossa staining (bottom) in the thoracic aorta are shown (n=5–9 per group). Bar: 200 μm. B, Magnified image of Alizarin red staining in the thoracic aorta of the HPD+Ade‐fed mice is shown. Bar: 100 μm. C, von Kossa staining was performed in the carotid artery and the subclavian artery of the HPD+Ade‐fed mice. Bar: 200 μm. D, Calcified areas in the thoracic aorta were quantified using Alizarin red–stained samples (n=5–9 per group). E, Calcium contents in the abdominal aorta were measured using the o‐cresolphthalein complexone method (n=5–9 per group). *P<0.05 compared with NPD‐fed mice.
Figure 4.
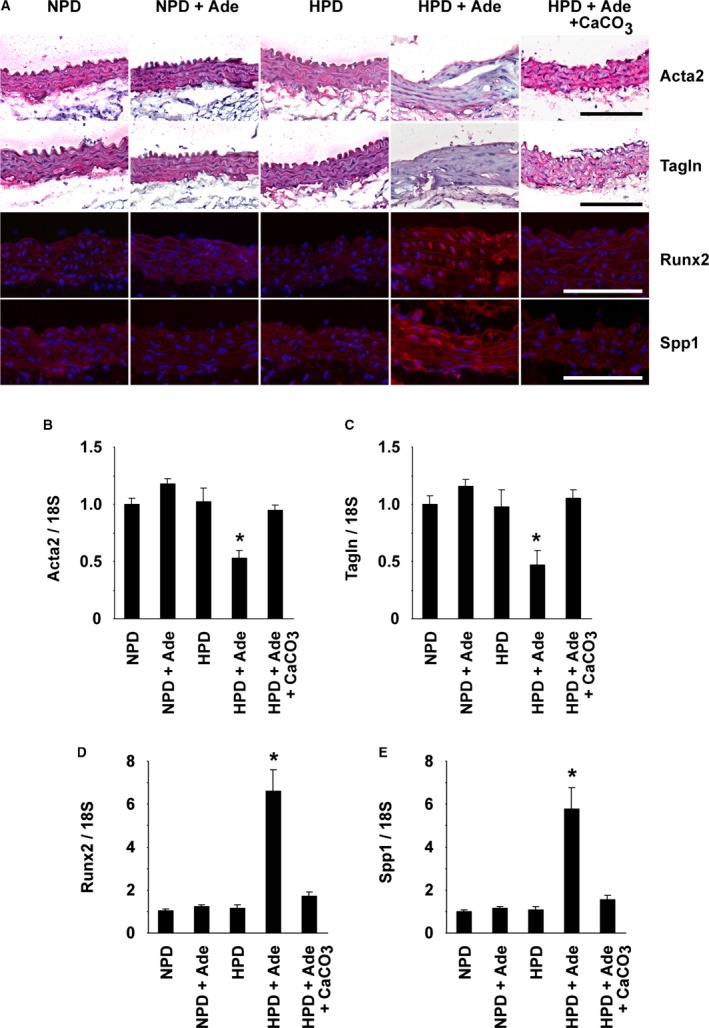
Phenotypic switching of smooth muscle cells into osteogenic cells occurred in the aorta of high phosphorus diet (HPD)–fed mice with chronic kidney disease. DBA/2 mice were fed a normal phosphorus diet (NPD), an NPD containing 0.25% adenine (NPD+Ade), an HPD, an HPD containing 0.25% adenine (HPD+Ade), or an HPD containing 0.25% adenine and 3% CaCO 3 (HPD+Ade+CaCO 3). Adenine was administered for 5 wk. The animals were fed the NPD or HPD throughout the entire experimental period of 10 wk. n=5 to 9 per group. A, Expression of Acta2, Tagln, Runx2, and Spp1 in the thoracic aorta was examined by immunohistochemistry and immunofluorescence studies. Staining for Acta2 and Tagln was visualized using the Vector Red Alkaline Phosphatase Substrate kit, and the sections were counterstained with hematoxylin. Runx2 and Spp1 were detected by Alexa‐Fluor 594‐conjugated secondary antibodies (red), and the sections were counterstained with 4′,6‐diamidino‐2‐phenylindole (blue). Bar: 100 μm. B through E, Expression of Acta2 (B), Tagln (C), Runx2 (D), and Spp1 (E) in the thoracic aorta was determined by real‐time reverse transcription–polymerase chain reaction. *P<0.05 compared with NPD‐fed mice.
NF‐κB Signaling Was Involved in High Phosphate‐Induced Arterial Medial Calcification in CKD Mice
Inflammatory cytokines, such as TNF, are known to activate NF‐κB signaling and have been shown to be elevated in CKD patients.16, 19, 20 We sought to determine the role of inflammatory signaling in arterial medial calcification in CKD. First, serum levels of TNF were measured in mice fed the HPD and/or adenine. The results showed that the TNF levels were significantly higher in the HPD+Ade‐fed mice, compared with the NPD‐fed mice (Figure 5A). Tempol and triptolide have been shown to inhibit NF‐κB signaling, although they also block other intracellular signaling pathways. The HPD+Ade‐fed CKD mice were treated with either 3% tempol in drinking water or intraperitoneal injections of triptolide for 10 weeks. Serum levels of TNF were not reduced by treatment with either tempol or triptolide (Figure 5A), suggesting that TNF elevation is not attributed to the activation of NF‐κB signaling in the HPD+Ade‐fed CKD mice. Indeed, biosynthesis of TNF is under the control of multiple and complex regulatory mechanisms, including gene transcription, mRNA turnover, and translation.33 Moreover, TNF gene transcription is regulated by multiple factors, such as nuclear factor of activated T cells, Ets, interferon‐regulating factor, CCAAT/enhancer‐binding protein α, as well as NF‐κB. TNF levels in the HPD+Ade‐fed CKD mice are likely to be induced by NF‐κB‐independent mechanisms. Of interest, tempol and triptolide both decreased arterial medial calcification in mice fed the HPD and adenine (Figure 5B and 6). Indeed, treatment with either tempol or triptolide decreased the calcium contents in the abdominal aorta by 75% and 60%, respectively (Figure 6G). Moreover, the phenotypic switching of SMCs into osteogenic cells was also blunted by these reagents. These results suggest that NF‐κB signaling contributes to high phosphate‐induced arterial medial calcification in CKD mice.
Figure 5.
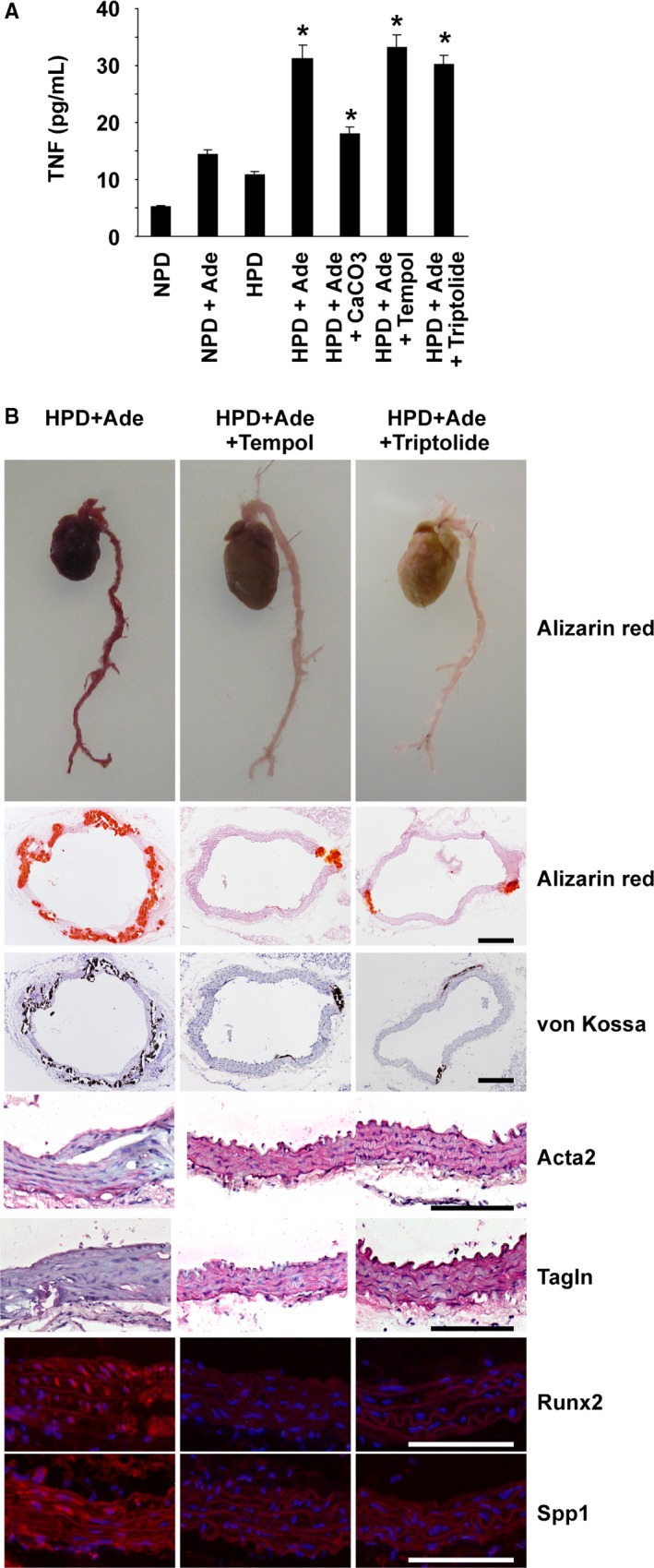
Nuclear factor‐κB inhibitors attenuated arterial medial calcification in high phosphorus diet (HPD)–fed mice with chronic kidney disease. DBA/2 mice were fed a normal phosphorus diet (NPD), an NPD containing 0.25% adenine (NPD+Ade), an HPD, an HPD containing 0.25% adenine (HPD+Ade), or an HPD containing 0.25% adenine and 3% CaCO 3 (HPD+Ade+CaCO 3). Adenine was administered for 5 wk. The animals were fed the NPD or HPD throughout the entire experimental period of 10 wk. Some of the mice fed the HPD and adenine were treated with either 3% tempol in drinking water (HPD+Ade+tempol) or intraperitoneal injections of triptolide (HPD+Ade+triptolide) for 10 wk. A, Serum tumor necrosis factor‐α (TNF) levels were measured at the end of the experimental period (n=7–9 per group). *P<0.05 compared with NPD‐fed mice. B, Formation of arterial medial calcification and expression of smooth muscle cell differentiation markers and osteogenic markers were examined. Top: Alizarin red staining for whole cardiovascular images (n=3 per group). Second and third rows: Alizarin red staining and von Kossa staining in the thoracic aorta are shown (n=5–7 per group). Bar: 200 μm. Fourth to seventh rows: Expression of Acta2, Tagln, Runx2, and Spp1 in the thoracic aorta was examined by immunohistochemistry and immunofluorescence studies (n=5–7 per group). Staining for Acta2 and Tagln was visualized using the Vector Red Alkaline Phosphatase Substrate kit, and the sections were counterstained with hematoxylin. Runx2 and Spp1 were detected by Alexa‐Fluor 594‐conjugated secondary antibodies (red), and the sections were counterstained with 4′,6‐diamidino‐2‐phenylindole (blue). Bar: 100 μm.
Figure 6.
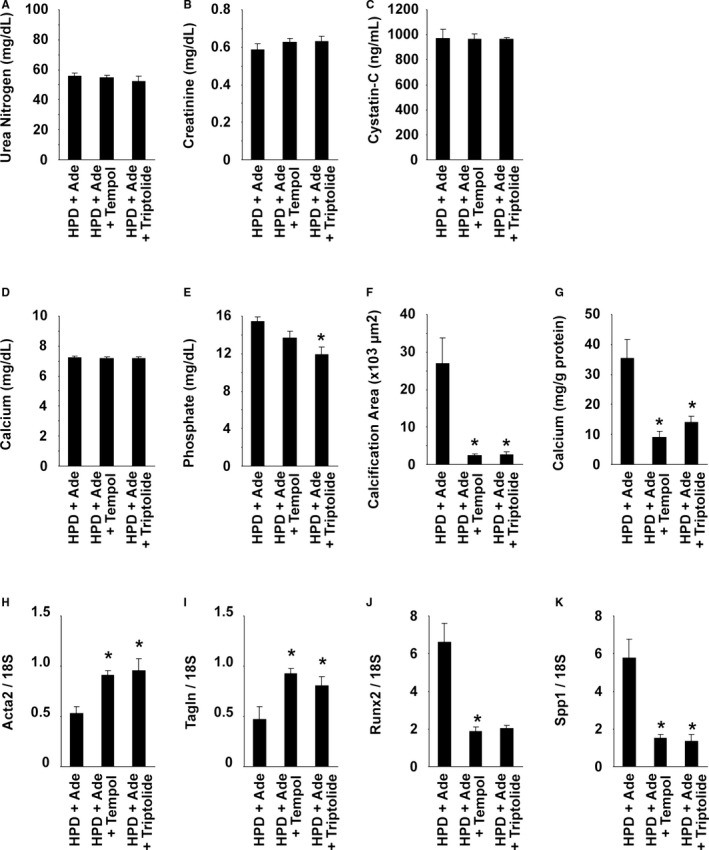
Nuclear factor‐κB inhibitors attenuated arterial medial calcification in high phosphorus diet (HPD)–fed mice with chronic kidney disease. DBA/2 mice were fed an HPD containing 0.25% adenine (HPD+Ade). Adenine was administered for 5 wk. The animals were fed the HPD throughout the entire experimental period of 10 wk. Some mice were treated with either 3% tempol in drinking water (HPD+Ade+tempol) or intraperitoneal injections of triptolide (HPD+Ade+triptolide) for 10 wk. n=7 to 9 per group. A through E, Serum levels of urea nitrogen (A), creatinine (B), cystatin‐C (C), calcium (D), and phosphate (E) were measured at the end of the experimental period. F, Calcified areas in the thoracic aorta were quantified using Alizarin red–stained samples. G, Calcium contents in the abdominal aorta were measured using the o‐cresolphthalein complexone method. H through K, Expression of Acta2 (H), Tagln (I), Runx2 (J), and Spp1 (K) in the thoracic aorta was determined by real‐time reverse transcription–polymerase chain reaction. *P<0.05 compared with mice fed the HPD and adenine.
Masson trichrome staining was performed in the hearts. The results showed that cardiac fibrosis was induced in the hearts of mice fed the HPD and/or adenine, compared with the NPD‐fed mice (Figure 7). In addition, treatment with CaCO3 or NF‐κB inhibitors reduced the severity of cardiac fibrosis in HPD+Ade‐fed CKD mice. These results raise a possibility that cardiac phenotype affects the formation of arterial medial calcification in the HPD+Ade‐fed CKD mice, although further studies are required to confirm this hypothesis.
Figure 7.
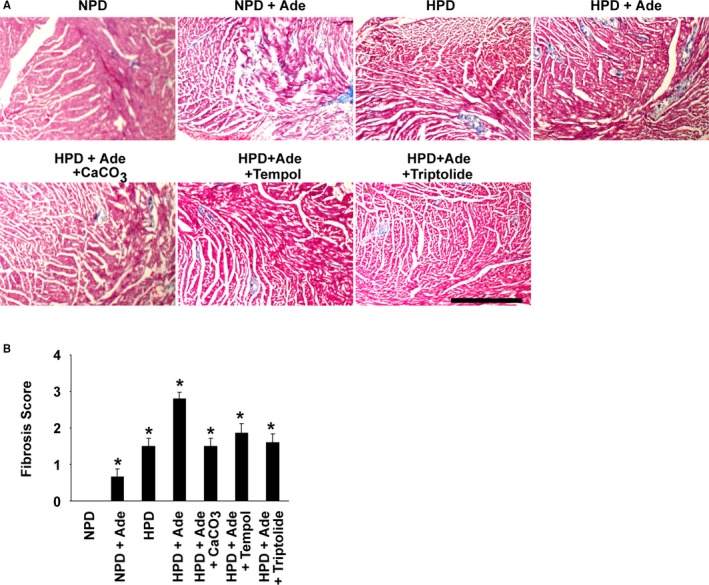
Cardiac fibrosis was induced in the hearts of high phosphorus diet (HPD)–fed mice with chronic kidney disease. DBA/2 mice were fed a normal phosphorus diet (NPD), an NPD containing 0.25% adenine (NPD+Ade), an HPD, an HPD containing 0.25% adenine (HPD+Ade), or an HPD containing 0.25% adenine and 3% CaCO 3 (HPD+Ade+CaCO 3). Adenine was administered for 5 wk. The animals were fed the NPD or HPD throughout the entire experimental period of 10 wk. Some of the mice fed the HPD and adenine were treated with either 3% tempol in drinking water (HPD+Ade+tempol) or intraperitoneal injections of triptolide (HPD+Ade+triptolide) for 10 wk. A, Cardiac fibrosis was examined using Masson trichrome staining (n=5–7 per group). Bar: 500 μm. B, The levels of cardiac fibrosis were graded. *P<0.05 compared with NPD‐fed mice.
Inhibition of NF‐κB Within SMCs Attenuated Arterial Medial Calcification in CKD Mice
The cell‐autonomous role of NF‐κB signaling in SMCs for arterial medial calcification was examined using SM‐IκBΔN mice, in which the NF‐κB activity was inhibited in a SMC‐selective manner (Figure 8A). SM‐IκBΔN mice on a DBA/2 background were born at the expected Mendelian ratio and were grown to adulthood without exhibiting any differences in visible appearance, compared with controls (data not shown). The body weight (24.6±1.1 g in SM‐IκBΔN mice versus 23.7±0.8 g in control mice), systolic blood pressure (97±2 mm Hg in SM‐IκBΔN mice versus 101±3 mm Hg in control mice), diastolic blood pressure (58±3 mm Hg in SM‐IκBΔN mice versus 63±4 mm Hg in control mice), and heart rate (659±15 beats/min in SM‐IκBΔN mice versus 616±23 beats/min in control mice) at 14 weeks of age were similar between SM‐IκBΔN and control mice (n=8 per group). Overexpression of IκBΔN in the aorta, but not in the brain or liver, was confirmed in SM‐IκBΔN mice (Figure 8B).
Figure 8.
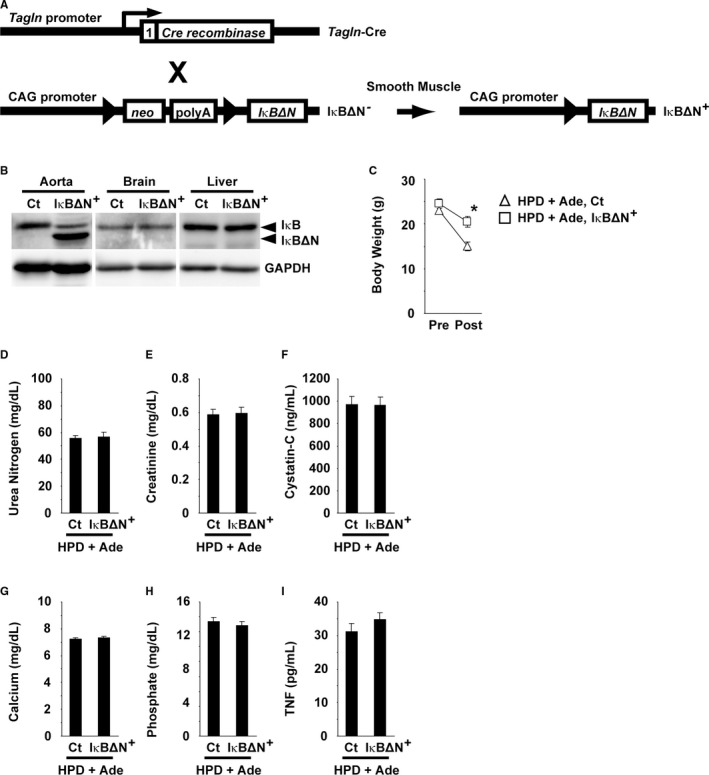
IκBΔN was predominantly expressed in the aorta of SM‐IκBΔN mice. A, Schematic representation of smooth muscle cell–selective expression of IκBΔN is shown. Tagln‐Cre mice were bred with IκBΔN mice to generate SM‐IκBΔN and control mice. Triangles represent the loxP sites. The number 1 represents exon 1 of the Tagln gene. X, breeding; neo, the neomycin‐resistant gene; polyA, polyadenylation signal. B, Expression of IκBΔN and GAPDH in the aorta, brain, and liver of SM‐IκBΔN and control mice was examined using Western blotting. C through I, SM‐IκBΔN and control (Ct) mice were fed a high phosphorus diet (HPD) containing 0.25% adenine (HPD+Ade). Adenine was administered for 5 wk. The animals were fed the HPD throughout the entire experimental period of 10 wk. n=8 to 9 per group. The body weight before and after the experimental period of 10 wk is shown (C). Serum levels of urea nitrogen (D), creatinine (E), cystatin‐C (F), calcium (G), phosphate (H), and tumor necrosis factor‐α (TNF) (I) were measured at the end of the experimental period. *P<0.05 compared with control mice fed the HPD and adenine.
SM‐IκBΔN mice were fed the HPD and adenine, and the biochemical parameters and the formation of arterial medial calcification were compared with those in HPD+Ade‐fed control mice. As shown in Figure 8C, the decrease in body weight was blunted in SM‐IκBΔN mice, even though they were fed the HPD and adenine. The elevations in serum levels of urea nitrogen, creatinine, and cystatin‐C were similar between SM‐IκBΔN mice and control mice after feeding them the HPD and adenine (Figure 8D through 8F). Serum concentrations of calcium and phosphate, as well as serum TNF levels, were also not significantly different between SM‐IκBΔN and control mice (Figure 8G through 8I). However, of significant interest, the formation of arterial medial calcification was markedly decreased in SM‐IκBΔN mice, compared with control mice (Figure 9A through 9C). The calcified area in the thoracic aorta was significantly smaller by 87% in SM‐IκBΔN mice fed the HPD and adenine, compared with the HPD+Ade‐fed control mice (Figure 9B). The calcium content in the abdominal aorta of SM‐IκBΔN mice fed the HPD and adenine (9.6 μg/mg protein) was also markedly lower, compared with that in the HPD+Ade‐fed control mice (35.3 μg/mg protein) (Figure 9C). In addition, phenotypic switching of SMCs into osteogenic cells was attenuated in SM‐IκBΔN mice (Figure 9D through 9H). Indeed, the reduction in the expression of Acta2 and Tagln, and the induction in the expression of Runx2 and Spp1, were not obvious in SM‐IκBΔN mice. Moreover, the expression of p65, a subunit of NF‐κB, was detectable in the nuclei of SMCs in the aorta of the HPD+Ade‐fed control mice, whereas it was not seen in those of SM‐IκBΔN mice (Figure 9D), indicating that NF‐κB was selectively inhibited in SMCs in SM‐IκBΔN mice. The expression of p65 was undetectable in control mice fed the NPD, or the HPD, or adenine (data not shown). These results provide novel evidence that the NF‐κB activity within SMCs plays a key role in high phosphate‐induced arterial medial calcification in CKD mice.
Figure 9.
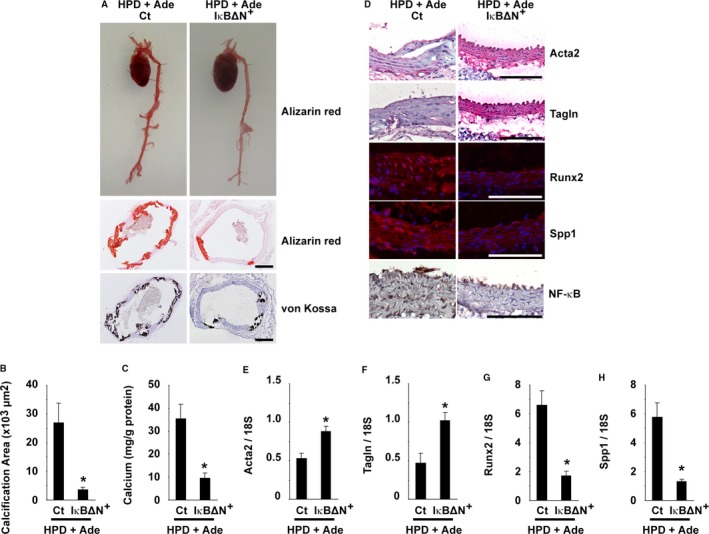
Nuclear factor‐κB (NF‐κB) inhibition in smooth muscle cells reduced arterial medial calcification in high phosphorus diet (HPD)–fed mice with chronic kidney disease. SM‐IκBΔN and control mice were fed an HPD containing 0.25% adenine (HPD+Ade). Adenine was administered for 5 wk. The animals were fed the HPD throughout the entire experimental period of 10 wk. A, Formation of arterial medial calcification was examined. Top: Alizarin red staining for whole cardiovascular images (n=3 per group). Middle and bottom: Alizarin red staining and von Kossa staining in the thoracic aorta are shown (n=5–6 per group). Bar: 200 μm. B, Calcified areas in the thoracic aorta were quantified using Alizarin red–stained samples (n=5–6 per group). C, Calcium contents in the abdominal aorta were measured using the o‐cresolphthalein complexone method (n=5–6 per group). D, Expression of Acta2, Tagln, Runx2, Spp1, and NF‐κB (p65) in the thoracic aorta was examined by immunohistochemistry and immunofluorescence studies (n=5–6 per group). Staining for Acta2 and Tagln was visualized using the Vector Red Alkaline Phosphatase Substrate kit, and the sections were counterstained with hematoxylin. Runx2 and Spp1 were detected by Alexa‐Fluor 594‐conjugated secondary antibodies (red), and the sections were counterstained with 4′,6‐diamidino‐2‐phenylindole (blue). Staining for p65 was visualized using diaminobenzidine, and the sections were counterstained with hematoxylin. Bar: 100 μm. E through H, Expression of Acta2 (E), Tagln (F), Runx2 (G), and Spp1 (H) in the thoracic aorta was determined by real‐time reverse transcription–polymerase chain reaction. *P<0.05 compared with control mice fed the HPD and adenine. Ct indicates control.
NF‐κB Contributed to High Phosphate‐Induced SMC Calcification by Interacting With Klf4
To determine the mechanisms by which the activation of NF‐κB signaling in SMCs contributes to high phosphate‐induced arterial medial calcification and SMC phenotypic switching, cultured rat aortic SMCs were treated with high phosphate and TNF. As shown in Figure 10A, a combination of high phosphate (2.7 mmol/L) and TNF (10 ng/mL) together induced calcification in cultured SMCs. Inhibition of the NF‐κB signaling by BAY11‐7082 at 1 μmol/L diminished SMC calcification induced by high phosphate and TNF. These results were also confirmed by the measurement of the calcium contents in cultured SMCs (Figure 10B). Consistently, treatment with high phosphate and TNF reduced the expression of Acta2, whereas it induced Spp1 expression in cultured SMCs (Figure 10C and 10D). These results suggest that cultured SMCs treated with high phosphate and TNF are suitable for investigations of what happens in HPD‐induced arterial medial calcification in CKD mice.
Figure 10.
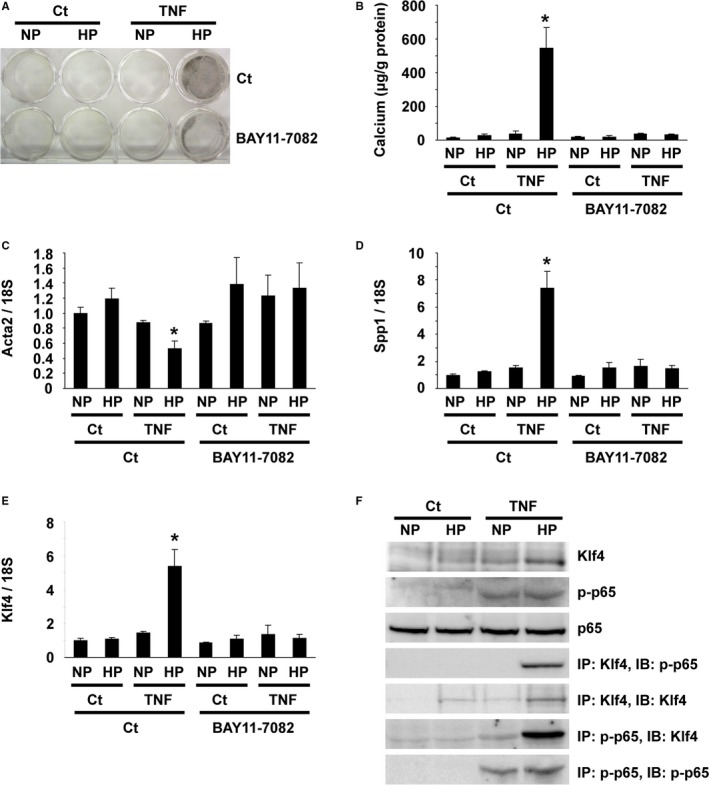
NF‐κB contributed to high phosphate‐induced smooth muscle cell (SMC) calcification by interacting with Krüppel‐like factor (Klf4). A through E, Cultured rat aortic SMCs were incubated with normal (NP: 0.9 mmol/L) or high (HP: 2.7 mmol/L) phosphate concentration in the presence or absence of 10 ng/mL of tumor necrosis factor‐α (TNF) and/or 1 μmol/L of BAY11‐7082 for 8 d (n=4). A representative picture of von Kossa staining is shown (A). Calcium contents were measured using the o‐cresolphthalein complexone method (B). Expression of Acta2 (C), Spp1 (D), and Klf4 (E) was determined by real‐time RT‐PCR. *P<0.05 compared with SMCs incubated with normal phosphate concentration. F, Physical interaction between p65 and Klf4 was examined by co‐immunoprecipitation assays. Cultured rat aortic SMCs were incubated with normal or high phosphate concentration in the presence or absence of 10 ng/mL of TNF for 48 h (n=4). Expression of Klf4, p65, and phospho‐p65 was examined by Western blotting. Cell lysates from rat aortic SMCs were immunoprecipitated (IP) with anti‐Klf4 antibody or anti‐phospho‐p65 antibody and were immunoblotted (IB) with anti‐Klf4 antibody or anti‐phospho‐p65 antibody. Ct indicates control; RT‐PCR, reverse transcription–polymerase chain reaction.
Klf4 has been shown to be involved in SMC phenotypic switching and neointimal formation in response to vascular injury.26 We examined the role of Klf4 in high phosphate and TNF‐induced SMC calcification. As shown in Figure 10E, Klf4 expression was induced by treatment with high phosphate and TNF in cultured SMCs. In addition, co‐immunoprecipitation assays revealed that Klf4 physically interacted with p65 in cultured SMCs treated with high phosphate and TNF (Figure 10F). Moreover, the results of quantitative chromatin immunoprecipitation assays showed that high phosphate and TNF induced the binding of Klf4 with the promoter region of the Acta2 gene, as well as the Spp1 gene, in cultured SMCs (Figure 11A and 11B). Furthermore, Klf4 expression was induced in the nuclei of SMCs in the aorta of CKD mice fed the HPD and adenine, whereas it was not detectable in the aorta of SM‐IκBΔN mice fed the same diet (Figure 11C). Taken together, these results suggest that the NF‐κB signaling contributes to SMC phenotypic switching and calcification by interacting with Klf4.
Figure 11.
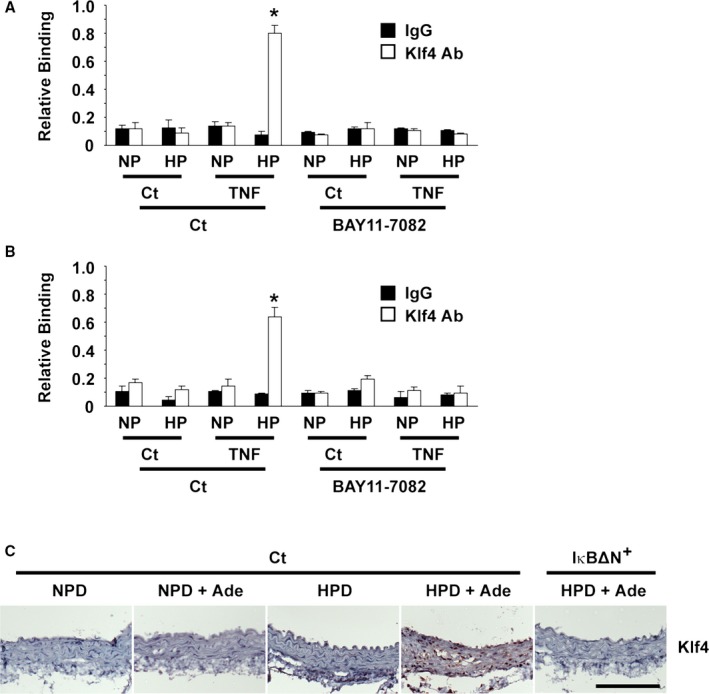
Krüppel‐like factor 4 (Klf4) was involved in the nuclear factor‐κB–mediated inhibition of smooth muscle cell (SMC) phenotypic switching and calcification. A and B, Cultured rat aortic SMCs were incubated with normal (NP: 0.9 mmol/L) or high (HP: 2.7 mmol/L) phosphate concentration in the presence or absence of 10 ng/mL of tumor necrosis factor‐α (TNF) and/or 1 μmol/L of BAY11‐7082 for 48 h (n=4). Association of Klf4 with the Acta2 promoter (A) and the Spp1 promoter (B) was determined by chromatin immunoprecipitation assays. *P<0.05 compared with SMCs incubated with normal phosphate concentration. C, Klf4 expression in mice fed a normal phosphorus diet (NPD), an NPD and adenine, a high phosphorus diet (HPD), or an HPD and adenine, as well as in SM‐IκBΔN mice fed an HPD and adenine, was determined by immunohistochemistry. Staining for Klf4 was visualized using diaminobenzidine, and the sections were counterstained with hematoxylin. Bar: 100 μm. Ade indicates adenine; Ct, control.
Discussion
In the present studies, we demonstrated that the inhibition of NF‐κB signaling in SMCs contributed to high phosphate‐induced arterial medial calcification in CKD mice. Indeed, by using a novel CKD mouse model of arterial medial calcification, we showed that each of the NF‐κB inhibitors that were examined, tempol and triptolide, reduced arterial medial calcification in CKD mice fed the HPD. We also showed that phenotypic switching of SMCs into osteogenic cells was attenuated by treatment with the NF‐κB inhibitors in these mice. In addition, we showed that the formation of arterial medial calcification, as well as SMC phenotypic switching, in HPD‐fed CKD mice was attenuated in SM‐IκBΔN mice, in which the NF‐κB activity was inhibited in a SMC‐selective manner. Moreover, we showed that Klf4 expression was induced in aortic SMCs in HPD‐fed CKD mice and that NF‐κB activation contributed to SMC phenotypic switching and calcification by interacting with Klf4 in cultured SMCs. As such, the results of the present studies provide clear evidence showing that NF‐κB activation in SMCs plays a critical role in SMC phenotypic switching and arterial medial calcification in HPD‐fed CKD mice.
Most studies reported in the field of arterial medial calcification thus far have been performed using an adenine‐fed rat model.4, 10, 11, 12, 13, 21, 22, 27 Unfortunately, this model has limitations in regard to gain‐of‐function experiments and loss‐of‐function experiments because of the use of rats, rather than mice. Recently, DBA/2 mice have been found to be a suitable mouse model for the study of arterial medial calcification, whereas C57BL/6 mice, the most popular strain, are calcification‐resistant.25 Indeed, El‐Abbadi et al25 showed that an HPD induced arterial medial calcification in uremic DBA/2 mice, in which CKD was induced by 5/6 nephrectomy. In the present studies, CKD was induced by feeding the animals a low concentration of adenine in their diet. Compared with 5/6 nephrectomy, the adenine‐feeding procedure has an advantage, in terms of its simplicity and the consistency of its effect on the impairment of renal function. Although we showed that the SMC‐selective inhibition of NF‐κB reduced HPD‐induced arterial medial calcification in adenine‐fed CKD mice in the present studies, it is expected that multiple signaling pathways and factors, such as the RANK‐RANKL system, advanced glycation end products, and fetuin‐A, also affect arterial medial calcification in CKD.1, 2, 3 It would be interesting to determine whether these factors contribute to HPD‐induced arterial medial calcification in CKD using the mouse model system developed in the present studies.
Because NF‐κB has been considered to be a potential therapeutic target of multiple vascular diseases, several studies have been performed to examine the effects of NF‐κB inhibition on arterial medial calcification in CKD. Zhao et al34 showed that siRNA‐induced knockdown of p65 reduced β‐glycerophosphate‐induced SMC calcification in cultured bovine aortic SMCs. In addition, they showed that a superoxide dismutase mimic, MnTMPyP, inhibited the activation of NF‐κB and attenuated arterial medial calcification in adenine‐fed CKD rats. The results of subsequent studies by their group also showed that adenovirus‐mediated overexpression of IκB in the aorta blunted arterial medial calcification in adenine‐fed CKD rats.21 Moreover, Yamada et al22 showed that tempol ameliorated arterial medial calcification in uremic rats. Because they used tempol as an inhibitor of oxidative stress, they showed that the expression of oxidative stress markers, 8‐hydroxy‐2′‐deoxyguanosine and 4‐hydroxy‐2‐nonenal, was decreased by tempol in CKD rats, but they did not examine the NF‐κB activity. Although the results of these studies suggest that NF‐κB inhibition is a potent therapeutic approach for arterial medial calcification, the target cell types were unclear because of the global inhibition of NF‐κB activity in these studies.21, 22, 34 It is possible that paracrine factors secreted by endothelial cells and/or monocytes/macrophages affect the characteristics of SMCs. In this regard, the results of the present studies provide compelling evidence that NF‐κB activation within SMCs is critical for HPD‐induced SMC phenotypic switching and arterial medial calcification in CKD. The NF‐κB activity in SMCs would be a great therapeutic target for arterial medial calcification.
The results of the present studies showed that Klf4 was involved in NF‐κB‐induced SMC phenotypic switching and calcification. Indeed, treatment with high phosphate and TNF induced the expression of Klf4 in cultured SMCs, concomitant with the phosphorylation of p65, a NF‐κB component. In addition, phosphorylated p65 and Klf4 physically interacted with each other, as demonstrated by co‐immunoprecipitation assays using endogenous proteins. Moreover, Klf4 binding to the promoter region of the Acta2 gene and the Spp1 gene was enhanced in response to high phosphate and TNF, as determined by quantitative chromatin immunoprecipitation assays. Furthermore, Klf4 expression was induced in the aorta in HPD‐fed CKD mice, but not in HPD‐fed SM‐IκBΔN mice. These results suggest that Klf4 contributes to the phenotypic switching of differentiated SMCs into calcifying SMCs by cooperating with p65. Consistently, the results of our previous studies showed that siRNA‐induced knockdown of Klf4 attenuated high phosphate‐induced SMC calcification in cultured SMCs.4 It is likely that NF‐κB and Klf4 cooperatively induce SMC phenotypic switching and calcification in response to inflammatory cues. Recently, multiple Klf factors have also been shown to interact with NF‐κB. Klf11 was shown to inhibit TNF‐induced expression of cell adhesion molecules by interacting with p65 in vascular endothelial cells.35 Klf6 was identified as a co‐activator of NF‐κB that mediated p65‐dependent transcription of downstream genes in HEK293 cells.36 As such, Klf family members, including Klf4, play an important role in modulating inflammatory signals by directly interacting with NF‐κB.
In summary, we provide novel evidence that NF‐κB activation in SMCs plays a key role in high phosphate‐induced arterial medial calcification in mice with CKD. Further studies are needed to develop novel reagents that selectively inhibit NF‐κB signaling in SMCs for the treatment and prevention of arterial medial calcification in CKD.
Sources of Funding
This study was supported by Grants‐in‐Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science to Yoshida (16K09655) and Hayashi (15K09271) and by grants from the Japanese Association of Dialysis Physicians (JADP grants 2013‐5 and 2016‐9) to Yoshida and the Kidney Foundation, Japan (JFKB14‐33) to Yoshida.
Disclosures
None.
(J Am Heart Assoc. 2017;6:e007248 DOI: 10.1161/JAHA.117.007248.)29146611
References
- 1. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol. 2014;307:F891–F900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hruska KA, Mathew S, Lund R, Qiu P, Pratt R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008;74:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoshida T, Yamashita M, Hayashi M. Krüppel‐like factor 4 contributes to high phosphate‐induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J Biol Chem. 2012;287:25706–25714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: up‐regulation of Cbfa1 and down‐regulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. [DOI] [PubMed] [Google Scholar]
- 6. Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, Wang Y, Chung J, Emerick A, Greaser L, Elashoff RM, Salusky IB. Coronary‐artery calcification in young adults with end‐stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478–1483. [DOI] [PubMed] [Google Scholar]
- 7. Nishizawa Y, Jono S, Ishimura E, Shioi A. Hyperphosphatemia and vascular calcification in end‐stage renal disease. J Ren Nutr. 2005;15:178–182. [DOI] [PubMed] [Google Scholar]
- 8. Chertow GM, Burke SK, Raggi P; for the Treat to Goal Working Group . Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002;62:245–252. [DOI] [PubMed] [Google Scholar]
- 9. Toussaint ND, Lau KK, Polkinghorne KR, Kerr PG. Attenuation of aortic calcification with lanthanum carbonate versus calcium‐based phosphate binders in haemodialysis: a pilot randomized controlled trial. Nephrology. 2011;16:290–298. [DOI] [PubMed] [Google Scholar]
- 10. Katsumata K, Kusano K, Hirata M, Tsunemi K, Nagano N, Burke SK, Fukushima N. Sevelamer hydrochloride prevents ectopic calcification and renal osteodystrophy in chronic renal failure rats. Kidney Int. 2003;64:441–450. [DOI] [PubMed] [Google Scholar]
- 11. Neven E, Dams G, Postnov A, Chen B, De Clerck N, De Broe ME, D'Haese PC, Persy V. Adequate phosphate binding with lanthanum carbonate attenuates arterial calcification in chronic renal failure rats. Nephrol Dial Transplant. 2009;24:1790–1799. [DOI] [PubMed] [Google Scholar]
- 12. Iida A, Kemmochi Y, Kakimoto K, Tanimoto M, Mimura T, Shinozaki Y, Uemura A, Matsuo A, Matsushita M, Miyamoto K. Ferric citrate hydrate, a new phosphate binder, prevents the complications of secondary hyperparathyroidism and vascular calcification. Am J Nephrol. 2013;37:346–358. [DOI] [PubMed] [Google Scholar]
- 13. Phan O, Maillard M, Peregaux C, Mordasini D, Stehle JC, Funk F, Burnier M. PA21, a new iron‐based noncalcium phosphate binder, prevents vascular calcification in chronic renal failure rats. J Pharmacol Exp Ther. 2013;346:281–289. [DOI] [PubMed] [Google Scholar]
- 14. Tonelli M, Wiebe N, Culleton B, House A, Rabbat C, Fok M, McAlister F, Garg AX. Chronic kidney disease and mortality risk: a systematic review. J Am Soc Nephrol. 2006;17:2034–2047. [DOI] [PubMed] [Google Scholar]
- 15. Nakano C, Hamano T, Fujii N, Matsui I, Tomida K, Mikami S, Inoue K, Obi Y, Okada N, Tsubakihara Y, Isaka Y, Rakugi H. Combined use of vitamin D status and FGF23 for risk stratification of renal outcome. Clin J Am Soc Nephrol. 2012;7:810–819. [DOI] [PubMed] [Google Scholar]
- 16. de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor κB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:904–914. [DOI] [PubMed] [Google Scholar]
- 17. Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of IκB‐α proteolysis by site‐specific, signal‐induced phosphorylation. Science. 1995;267:1485–1488. [DOI] [PubMed] [Google Scholar]
- 18. Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human IκB‐α on serine 32 and 36 controls IκB‐α proteolysis and NF‐κB activation in response to diverse stimuli. EMBO J. 1995;14:2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pereira BJ, Shapiro L, King AJ, Falagas ME, Strom JA, Dinarello CA. Plasma levels of IL‐1β, TNFα and their specific inhibitors in undialyzed chronic renal failure, CAPD and hemodialysis patients. Kidney Int. 1994;45:890–896. [DOI] [PubMed] [Google Scholar]
- 20. Descamps‐Latscha B, Herbelin A, Nguyen AT, Roux‐Lombard P, Zingraff J, Moynot A, Verger C, Dahmane D, de Groote D, Jungers P, Dayer JM. Balance between IL‐1β, TNF‐α, and their specific inhibitors in chronic renal failure and maintenance dialysis: relationships with activation markers of T cells, B cells, and monocytes. J Immunol. 1995;154:882–892. [PubMed] [Google Scholar]
- 21. Zhao G, Xu MJ, Zhao MM, Dai XY, Kong W, Wilson GM, Guan Y, Wang CY, Wang X. Activation of nuclear factor‐kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int. 2012;82:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamada S, Taniguchi M, Tokumoto M, Toyonaga J, Fujisaki K, Suehiro T, Noguchi H, Iida M, Tsuruya K, Kitazono T. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J Bone Miner Res. 2012;27:474–485. [DOI] [PubMed] [Google Scholar]
- 23. Zhang J, Zhong W, Cui T, Yang M, Hu X, Xu K, Xie C, Xue C, Gibbons GH, Liu C, Li L, Chen YE. Generation of an adult smooth muscle cell‐targeted Cre recombinase mouse model. Arterioscler Thromb Vasc Biol. 2006;26:e23–e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoshida T, Yamashita M, Horimai C, Hayashi M. Smooth muscle‐selective inhibition of nuclear factor‐κB attenuates smooth muscle phenotypic switching and neointima formation following vascular injury. J Am Heart Assoc. 2013;2:e000230 DOI: 10.1161/JAHA.113.000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. El‐Abbadi MM, Pai AS, Leaf EM, Yang HY, Bartley BA, Quan KK, Ingalls CM, Liao HW, Giachelli CM. Phosphate feeding induces arterial medial calcification in uremic mice: role of serum phosphorus, fibroblast growth factor‐23, and osteopontin. Kidney Int. 2009;75:1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoshida T, Kaestner KH, Owens GK. Conditional deletion of Krüppel‐like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ Res. 2008;102:1548–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoshida T, Yamashita M, Horimai C, Hayashi M. High glucose concentration does not modulate the formation of arterial medial calcification in experimental uremic rats. J Vasc Res. 2013;50:512–520. [DOI] [PubMed] [Google Scholar]
- 28. Yoshida T, Yamashita M, Iwai M, Hayashi M. Endothelial Krüppel‐like factor 4 mediates the protective effect of statins against ischemic AKI. J Am Soc Nephrol. 2016;27:1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshida T, Yamashita M, Horimai C, Hayashi M. Kruppel‐like factor 4 protein regulates isoproterenol‐induced cardiac hypertrophy by modulating myocardin expression and activity. J Biol Chem. 2014;289:26107–26118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshida T, Gan Q, Owens GK. Krüppel‐like factor 4, Elk‐1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol. 2008;295:C1175–C1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoshida T, Sinha S, Dandré F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG‐dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–864. [DOI] [PubMed] [Google Scholar]
- 32. Shang Y, Yoshida T, Amendt BA, Martin JF, Owens GK. Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. J Cell Biol. 2008;181:461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moelants EA, Mortier A, Van Damme J, Proost P. Regulation of TNF‐α with a focus on rheumatoid arthritis. Immunol Cell Biol. 2013;91:393–401. [DOI] [PubMed] [Google Scholar]
- 34. Zhao MM, Xu MJ, Cai Y, Zhao G, Guan Y, Kong W, Tang C, Wang X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high‐phosphate‐induced vascular calcification in vitro and in vivo. Kidney Int. 2011;79:1071–1079. [DOI] [PubMed] [Google Scholar]
- 35. Fan Y, Guo Y, Zhang J, Subramaniam M, Song CZ, Urrutia R, Chen YE. Krüppel‐like factor‐11, a transcription factor involved in diabetes mellitus, suppresses endothelial cell activation via the nuclear factor‐κB signaling pathway. Arterioscler Thromb Vasc Biol. 2012;32:2981–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y, Lei CQ, Hu YH, Xia T, Li M, Zhong B, Shu HB. Krüppel‐like factor 6 is a co‐activator of NF‐κB that mediates p65‐dependent transcription of selected downstream genes. J Biol Chem. 2014;289:12876–12885. [DOI] [PMC free article] [PubMed] [Google Scholar]