

Abstract
Background
Intracoronary stent restenosis, characterized by excessive smooth muscle cell (SMC) proliferation and myointimal hyperplasia, remains a clinical challenge. Mitochondrial membrane potential has been linked to the proliferative rate of SMCs. This study aimed to screen a critical gene regulating mitochondrial potential and to confirm its effects on myointimal formation in preclinical animal models.
Methods and Results
We performed transcriptome screening for genes differentially expressed in ligated versus unligated mouse carotid arteries. We observed that uncoupling protein 2 gene (Ucp2) mRNA, encoding UCP2, was transiently upregulated during the first 3 days after ligation and then significantly downregulated from day 7 through day 21, during which time neointima formed remarkably. The UCP2 protein level also declined after day 7 of ligation. In ligated carotid arteries, Ucp2 −/− mice, compared with wild‐type littermates, exhibited accelerated myointimal formation, which was associated with increased superoxide production and can be attenuated by treatment with antioxidant 4‐hydroxy‐2,2,6,6‐tetramethyl‐piperidinoxyl (TEMPOL). Knockdown of UCP2 enhanced human aortic SMC migration and proliferation that can also be attenuated by TEMPOL, whereas UCP2 overexpression inhibited SMC migration and proliferation, along with decreased activity of nuclear factor‐κB. Moreover, nuclear factor‐κB inhibitor attenuated UCP2 knockdown‐enhanced SMC proliferation. Adenovirus‐mediated overexpression of UCP2 inhibited myointimal formation in balloon‐injured carotid arteries of rats and rabbits and in‐stent stenosis of porcine coronary arteries. Moreover, UCP2 overexpression also suppressed neointimal hyperplasia in cultured human saphenous vein ex vivo.
Conclusions
UCP2 inhibits myointimal hyperplasia after vascular injury, probably through suppressing nuclear factor‐κB–dependent SMC proliferation and migration, rendering UCP2 a potential therapeutic target against restenosis.
Keywords: neointimal hyperplasia, restenosis, smooth muscle cell
Subject Categories: Gene Therapy, Translational Studies
Clinical Perspective
What Is New?
Uncoupling protein 2 (UCP2) is downregulated during myointimal hyperplasia after vascular injury and proliferation of vascular smooth muscle cells.
Overexpression of UCP2 inhibits myointimal hyperplasia in balloon‐injured artery and in‐stent stenosis of coronary artery.
UCP2 inhibits vascular smooth muscle cell proliferation and migration by decreasing the production of superoxide and inhibiting the nuclear factor ‐κB pathway.
What Are the Clinical Implications?
UCP2 is a novel target for the prevention and treatment of in‐stent restenosis.
Agents that increase the expression of UCP2 could be helpful in preventing the myointimal hyperplasia after percutaneous coronary interventions.
Introduction
Restenosis after angioplasty and stent implantation has been historically considered a limitation in percutaneous coronary interventions.1 The rates of in‐stent restenosis in current clinical practice remain >10%, despite the introduction of drug‐eluting stents in complex lesions.2 A recent report demonstrated that 5.3% of patients need target‐lesion revascularization at 6 years after drug‐eluting stent implantation.3 Vascular smooth muscle cells (SMCs), essential components of the vascular wall, are able to dedifferentiate to a proliferative or synthetic phenotype in response to vessel injury and play a critical role in vascular repair.4 Excessive proliferation of vascular SMCs is a critical step for myointimal hyperplasia, which has been viewed as the pathologic basis of restenosis.5, 6 In addition to in‐stent restenosis, neointima hyperplasia is also a major contributor to arteriovenous fistula failure in patients undergoing hemodialysis, which is still a huge clinical problem.7
Mitochondria, the powerhouse of cells, gain a central role in the regulation of cell proliferation during evolution.8 Mitochondrial electron transport chain plays an essential role in cell proliferation, and its inhibition arrests the cell cycle.9 During electron transfer, the electron transport chain pumps protons into the intermembrane space of mitochondria and regulates reactive oxygen species (ROS) production and mitochondrial membrane potential (ΔΨm).10 It has been reported that SMCs with hyperpolarization of ΔΨm temporally acquire a high proliferative rate and resistance to apoptosis, whereas prevention of ΔΨm hyperpolarization facilitates apoptosis and suppresses SMC proliferation.11 This evidence suggests that targeting ΔΨm may be a novel strategy for the prevention of restenosis.
In this study, we aimed to screen a critical gene for vascular SMC proliferation among the genes involved in mitochondrial energy metabolism and to test its myointima‐suppressing effects in a variety of preclinical animal models of vascular injury.
Methods
Mouse Carotid Artery Ligation Model
All experimental procedures were approved by the Institutional Animal Care and Use Committee and the Ethic Committee of Chengdu Military General Hospital (Chengdu, Sichuan, China). Uncoupling protein 2 gene knockout (Ucp2 −/−) mice were purchased. Littermate wild‐type (WT) mice (C57BL/6 background) were used as controls. Eight‐week‐old male mice, housed under a 12‐hour/12‐hour day/night cycle, with ad libitum food and water, were used for experiments. Complete left common carotid artery ligation was performed using aseptic techniques, as previously described.12 Mice were given either normal drinking water or water supplemented with 1 mmol/L 4‐hydroxy‐2,2,6,6‐tetramethyl‐piperidinoxyl (TEMPOL). At the end of experiments, mice were deeply anesthetized with pentobarbital (100 mg/kg) and perfused with saline. For histological analysis, mice were fixed with perfusion.
Rat and Rabbit Carotid Artery Balloon Injury Model
Sprague‐Dawley rats, weighing 250 to 300 g, and New Zealand rabbits, weighing ≈2 kg, were purchased, and left carotid arteries were injured using a 2F (for rat) or 3F (for rabbit) Fogarty balloon catheter.13, 14 For gene transfer, 30 μL (for rat) or 50 μL (for rabbit) adenovirus, with a titer of 1011 plaque‐forming units/mL, expressing UCP2 and green fluorescent protein (GFP; Ad‐UCP2) or GFP alone (Ad‐GFP), was injected into the balloon‐injured common carotid artery via the external carotid artery immediately after injury and incubated for 30 minutes.
Minipig Coronary Artery In‐Stent Stenosis Model
Male Guizhou minipigs (40–50 kg) were used. Pig coronary stenting was performed, as previously described.15 After stent implantation, a local drug infusion balloon catheter (ClearWay RX)16 was inserted into the stented segment. A total of 500 μL of Ad‐UCP2 or Ad‐GFP (1011 plaque‐forming units/mL) diluted to 1 mL in saline was infused to inflate the balloon and held for 90 seconds. Pigs received follow‐up coronary angiography via carotid artery at 3 months after procedure.
Transcriptome Array
Mice at 3 days after procedure were euthanized, and the ligated and contralateral unligated carotid arteries were isolated and stored in RNA‐later at −80°C. Total RNA was transcribed to cDNA, then synthesized into cRNA and labeled with cyanine‐3‐CTP. The microarray (SurePrint G3 Mouse GE 8×60K Microarrays) was performed, and the data were processed with Feature Extraction software (version 10.7.1.1). The total data set included 3 biological replicates for both groups. Each sample consisted of 3 mouse carotid arteries.
Pathological Characteristics
Carotid arteries, saphenous veins, and segments distal to the stented porcine coronary arteries were dehydrated, embedded, divided into sections, and affixed to slides. Six serial sections from each animal were used for staining. Stented porcine coronary arteries were embedded in methyl methacrylate and cut at a thickness of 80 μm using a hard tissue slicer with a rotating diamond‐coated saw (SP1600). Sections were rehydrated and stained with hematoxylin and eosin.
Immunohistochemistry
Six serial sections from each animal were used for staining. Tissue sections and chamber slides with a monolayer of cells were subjected to citric acid antigen retrieval, and endogenous peroxidase activity was inhibited by incubation with 3% hydrogen peroxide. After being blocked, sections were incubated overnight at 4°C with primary antibodies against SM α‐actin, Ki67, nuclear factor‐κB (NF‐κB) p65, and UCP2. Sections were then incubated with horseradish peroxidase–conjugated secondary antibody. Optimal visualization of staining was achieved using diaminobenzidine or aminoethyl carbazole detection kits. Sections were counterstained with Mayer hematoxylin.
Immunofluorescence
Frozen sections of vessel or chamber slides with a monolayer of cells were fixed, blocked, and incubated at 4°C with rabbit anti–NF‐kB p65 overnight. The p65 was visualized with Alexa Fluor 594–conjugated secondary antibody. Cell nuclei were stained with 4’,6‐diamidino‐2‐phenylindole.
Superoxide Assay
Fresh‐frozen sections of vessel or chamber slides with a monolayer of cells were incubated in the dark with dihydroethidium. Images were acquired by using an inverted fluorescence microscope outfitted with a rhodamine filter set.17
Cell Culture
Human aortic SMCs (HA‐SMCs) were grown in medium 231 with SM growth supplement. Cells in passages 4 to 10 were used. For the knockdown of UCP2, cells were transfected with 20 nmol/L of UCP2‐specific siRNA or negative control siRNA by using Lipofectamine RNAiMAX Transfection Reagent. For overexpression of UCP2, HA‐SMCs were infected with Ad‐UCP2 or Ad‐GFP (1011 plaque‐forming units/mL). For specific purposes, 20 ng/mL of recombined human platelet‐derived growth factor (PDGF) or 10 μmol/L of BAY 11‐7085 was used to treat cells. The phenotype of SMCs passaged >5 times may be switched from contractile to synthetic form, which may be a limitation of this study.
Cell Migration and Proliferation Assay
Cell migration assay was performed using a scratch wound healing assay. Cell proliferation was measured using a cell counting kit.
Real‐Time Polymerase Chain Reaction
Real‐time polymerase chain reaction was performed using One Step SYBR Prime Scrip RT‐PCR Kit II. The relative amount of mRNA was calculated by 2−ΔΔCT and was normalized to a housekeeping gene, 18s rRNA. Each sample was run and analyzed in triplicate.
Antibody Array
Antibody array was performed with Human PDGF Phospho Antibody Array, according to the manufacturer's instructions. Three biological replicates were included in each group.
Western Blotting
Extracted protein (40 µg) was separated and transferred to polyvinylidene difluoride membranes. Membranes were blocked and incubated with primary antibodies against UCP2, PDGF, IκB, phosphorylated IκB, IκB kinase (IKK), phosphorylated IKK, p65, p50, SM myosin heavy chain, calponin, SM α‐actin, GAPDH, and histone H3. After incubation with horseradish peroxidase–conjugated secondary antibodies, chemiluminescence detection reagent was added onto the membranes. The luminescent signal was detected by exposure to x‐ray film.
ΔΨm Measurement
A 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolylcarbocyanine iodide (JC‐1) assay kit was used to measure ΔΨm, according to the manufacturer's instructions. The increases in the ratios between fluorescence intensity in the red and green channels were interpreted as an increase in ΔΨm.
NF‐κB p65 Transcription Factor Assay
Nuclear fractions of SMCs were isolated, and the bound NF‐κB was detected using NF‐κB p65 Transcription Factor Assay Kit.
Human Saphenous Vein Culture Ex Vivo
The experiment conformed to the principles outlined in the Declaration of Helsinki. Written informed consent and approval from the Ethics Committee of Chengdu Military General Hospital were obtained before tissue collection. Leftover human saphenous vein tissue from surgery was collected and cultured, as previously described.12 After incubation for 2 weeks, tissue explants were fixed and embedded in paraffin, and the paraffin sections were stained with hematoxylin and eosin and Verhoeff–Van Gieson stain.
Statistical Analysis
Continuous data are presented as mean±SEM. Normality was tested with the Kolmogorov‐Smirnov test. Comparisons among groups were determined by analysis of variance with post hoc Tukey honestly significant difference test, whereas statistically significant differences between 2 groups were determined by using the Student t test. The nonparametric Mann‐Whitney U test was used if data were not normally distributed. To compare means that involve 2 factors, we performed 2‐way ANOVA. P<0.05 was considered statistically significant. See Data S1 for further details.
Results
UCP2 Is Decreased During Myointimal Formation
Myointimal formation was induced by ligating the mouse left carotid artery. Three days after the procedure, the ligated carotid artery and the contralateral unligated artery were harvested for transcriptome array. Among 84 genes involved in the regulation of mitochondrial function, 6 were significantly upregulated, whereas 9 genes were downregulated in ligated carotid arteries compared with unligated arteries (Figure 1A). Notably, Ucp2 was the most significantly increased gene on day 3 after carotid artery ligation (Figure 1A). To confirm the change of Ucp2 expression in ligated arteries, we measured the mRNA by quantitative polymerase chain reaction. Interestingly, the mRNA of Ucp2 was transiently increased during the first 3 days after procedure. However, it was significantly decreased to lower than the baseline level after 7 days of ligation (Figure 1B), during which time myointima developed remarkably. Likewise, the protein level of UCP2 was also decreased at days 7, 14, and 21 after ligation (Figure 1C). The transient increase of Ucp2 at day 3 after ligation was able to be inhibited by treatment with antioxidant TEMPOL (Figure S1). The downregulation of Ucp2 at day 14 after ligation was also confirmed by comparing with carotid arteries from mice that received sham surgery rather than contralateral artery surgery (Figure S2). As shown by immunohistochemistry, the upregulation of UCP2 at day 3 and the downregulation at day 14 primarily occurred in the medial layer of carotid artery (Figure S3). It has been recognized that PDGF is a key mediator for medial SMC migration and proliferation after vessel injury. Because PDGF was significantly upregulated in ligated arteries (Figure 1D), the recombined PDGF was used to stimulate cultured SMCs in vitro. Both the mRNA (Figure 1E) and protein (Figure 1F) levels of UCP2 in cultured HA‐SMCs were gradually decreased after treatment with recombined PDGF, without an initial, temporary upregulation that occurred in vivo. The decrease of UCP2 level in PDGF‐treated cells was confirmed by immunohistochemistry staining (Figure S4A). To evaluate the basic function of UCP2 (decreasing ΔΨm), we measured the ΔΨm of SMCs by staining with JC‐1, which revealed a significant increase in the red (high ΔΨm) to green (low ΔΨm) ratio in PDGF‐treated SMCs when compared with control cells (Figure 1G and 1H). Moreover, superoxide was increased in PDGF‐treated SMCs, indicating the function of UCP2 was decreased along with the reduced expression (Figure S4B). Because UCP2 was substantially changed, along with the vessel injury and proliferation of SMCs, it is worth testing the effects of UCP2 on myointimal hyperplasia.
Figure 1.
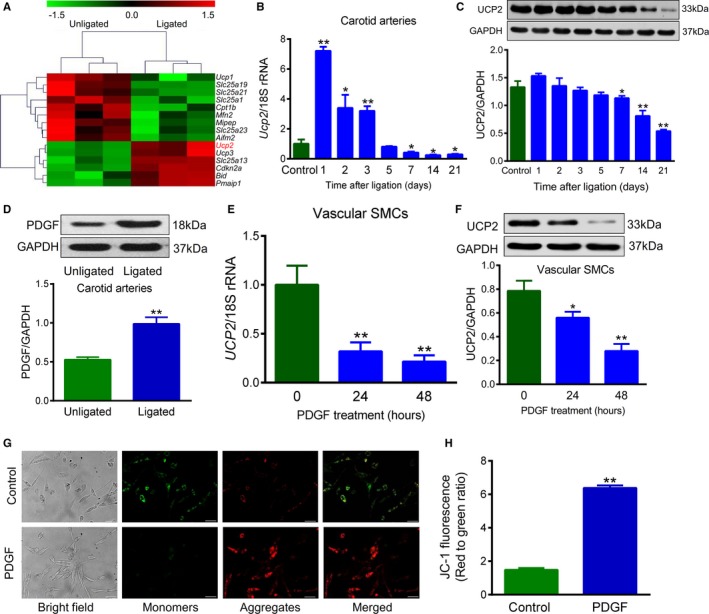
Uncoupling protein 2 (UCP2) is involved in myointimal hyperplasia and vascular smooth muscle cell (SMC) proliferation. A, Heat map of transcriptome array comparing expression of genes involved in the regulation of mitochondrial function between ligated and contralateral unligated mouse carotid arteries harvested 3 days after the procedure. n=3. B and C, mRNA and protein expression levels of UCP2 in unligated and ligated mouse carotid arteries at different time points after the procedure were measured by quantitative real‐time polymerase chain reaction and Western blot, respectively. n=5 for mRNA, n=3 for protein. *P<0.05, **P<0.01 vs control. D, Protein expression of platelet‐derived growth factor (PDGF) in unligated and ligated mouse carotid arteries 3 days after procedure. n=3. **P<0.01 vs unligated. E and F, mRNA and protein expression of UCP2 in human aortic SMCs treated with or without human recombined PDGF for 24 and 48 hours. n=6 for mRNA, n=4 for protein. *P<0.05, **P<0.01 vs control (0 hour). G, JC‐1 staining of SMCs treated with or without PDGF for 48 hours. JC‐1 monomers emit green fluorescence, whereas J‐aggregates emit orange‐red fluorescence. Bar=50 μm. H, Mitochondrial membrane potential (ΔΨm) was evaluated with green (low ΔΨm) to red (high ΔΨm) fluorescence. n=6 independent experiments. **P<0.01 vs control.
UCP2 Ablation Exacerbates Myointimal Hyperplasia
The Ucp2 −/− mice and WT littermates underwent ligation of left carotid arteries to await myointima development. Histological measurement was performed on days 7, 14, 21, and 28 after ligation on the basis of hematoxylin and eosin staining, which showed that the overt neointima was developed after 14 days in both strains of mice (Figure 2A). Interestingly, neointima hyperplasia was exacerbated in Ucp2 −/− mice when compared with WT littermates, whereas the medial layer thickness was similar between the 2 strains of mice (Figure 2A and 2B, Figure S5A through S5E). Because UCP2 ablation causes vascular remodeling by excessively producing ROS,18 we treated another cohort of mice with antioxidant TEMPOL in drinking water. The ligated arteries from Ucp2 −/− mice displayed a higher level of ROS than WT mice (Figure 2C), which is in good correspondence with our previous study indicating increased ROS in aortas from high‐salt diet–fed Ucp2 −/− mice.17 As expected, ROS production was suppressed by TEMPOL treatment in both WT and Ucp2 −/− mice (Figure 2C and 2D). More important, oral administration with TEMPOL abolished the difference in neointima formation between Ucp2 −/− and WT mice (Figure 2E and 2F, Figure S6A through S6E), indicating that knockout of UCP2 exacerbates neointimal hyperplasia by overproducing ROS. The immunostaining of Ki67, which indicates cell proliferation, in ligated carotid arteries was concentrated in the neointimal area, closer to the lumen (Figure 2G). The number of Ki67‐positive cells was more abundant in Ucp2 −/− mice than WT littermates (Figure 2G and 2H), which is in correspondence with the degree of neointimal hyperplasia. Moreover, TEMPOL treatment significantly decreased the number of Ki67‐positive cells in both strains (Figure 2G and 2H). In contrast, the terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling–positive cells, which depict apoptosis, were few within the intima and similar between WT and Ucp2 −/− mice (Figure S7A and S7B). Because endothelial function plays an important role in neointimal development, we tested acetylcholine‐induced relaxation of ligated carotid arteries. The results showed that endothelium‐dependent relaxation in ligated arteries was significantly impaired 3 days after ligation when compared with unligated groups; it was almost recovered on days 7 and 14 (Figure S8A through S8C). However, there was no difference in the vasorelaxation between Ucp2 −/− mice and WT littermates (Figure S8A through S8C). These findings suggest that UCP2 deficiency likely enhances myointimal hyperplasia through ROS‐dependent SMC proliferation, rather than improving endothelial function.
Figure 2.
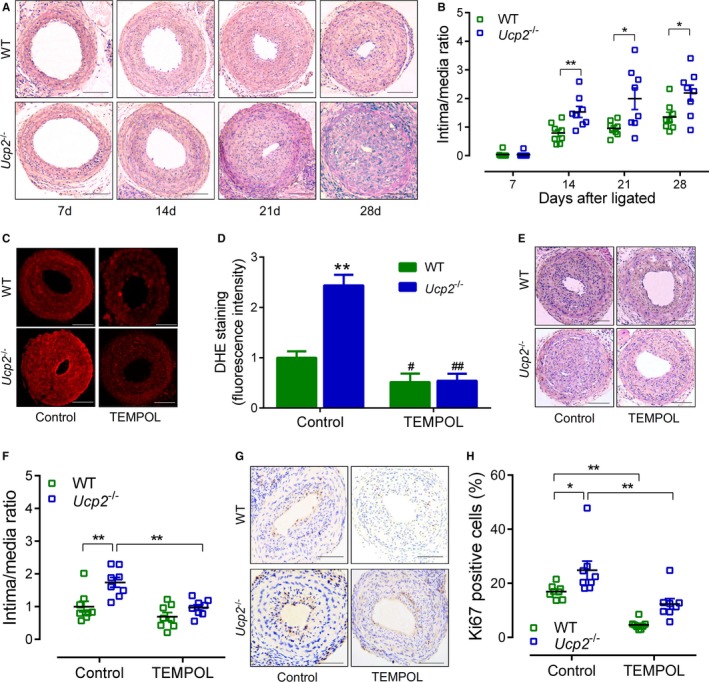
Uncoupling protein 2 (UCP2) ablation exacerbates mouse carotid artery ligation‐induced myointimal hyperplasia. A, Hematoxylin and eosin (H&E)‐stained sections of ligated carotid arteries from Ucp2 −/− mice and wild‐type (WT) littermates 7, 14, 21, and 28 days after ligation. Bar=50 μm. B, Intima/media ratio of ligated arteries. n=8. *P<0.05, **P<0.01. C, Dihydroethidium (DHE)‐stained frozen sections of ligated carotid arteries from Ucp2 −/− and WT mice treated with or without 4‐hydroxy‐2,2,6,6‐tetramethyl‐piperidinoxyl (TEMPOL) in drinking water (1 mmol/L) for 21 days. Bar=50 μm. D, Quantification of DHE fluorescence intensity. n=6. **P<0.01 vs WT‐control; # P<0.05, ## P<0.01 vs isogenic mice treated without TEMPOL. E, H&E‐stained sections of ligated carotid arteries from mice treated with or without TEMPOL. Bar=50 μm. F, Intima/media ratio of ligated arteries. n=8 in each group. **P<0.01. G, Immunohistochemistry staining of Ki67 (brown) in sections of ligated carotid arteries from mice treated with or without TEMPOL. Bar=50 μm. H, Percentage of Ki67‐positive cells within neointima. n=8. *P<0.05, **P<0.01.
UCP2 Suppresses SMC Proliferation and Migration
Next, we asked whether UCP2 influences SMC proliferation and migration in vitro. The UCP2 expression was knocked down in HA‐SMCs using specific siRNA (Figure 3A). The SMC migration was characterized by scratch assay and expressed as recovered area. UCP2 knockdown exacerbated PDGF‐induced SMC migration, which was attenuated by treatment with TEMPOL (Figure 3B and 3C). PDGF‐induced cell proliferation was also enhanced by UCP2 knockdown, whereas TEMPOL significantly inhibited the proliferation of SMCs treated with or without UCP2‐siRNA (Figure 3D). We used a second siRNA probe to knock down the UCP2, which yielded similar results (Figures S9 and S10). Then, the UCP2 was overexpressed by adenovirus‐mediated gene transfer (Figure 3E). As expected, both the migration and proliferation of HA‐SMCs were suppressed by overexpression of UCP2 (Figure 3F and 3G). The expression levels of SMC contractile markers, including SM myosin heavy chain, SM α‐actin, and calponin, were decreased in primarily cultured aortic SMCs from Ucp2 −/− mice when compared with those from WT mice, which can be partly reversed by overexpression of UCP2 with Ad‐UCP2 (Figure S11).
Figure 3.
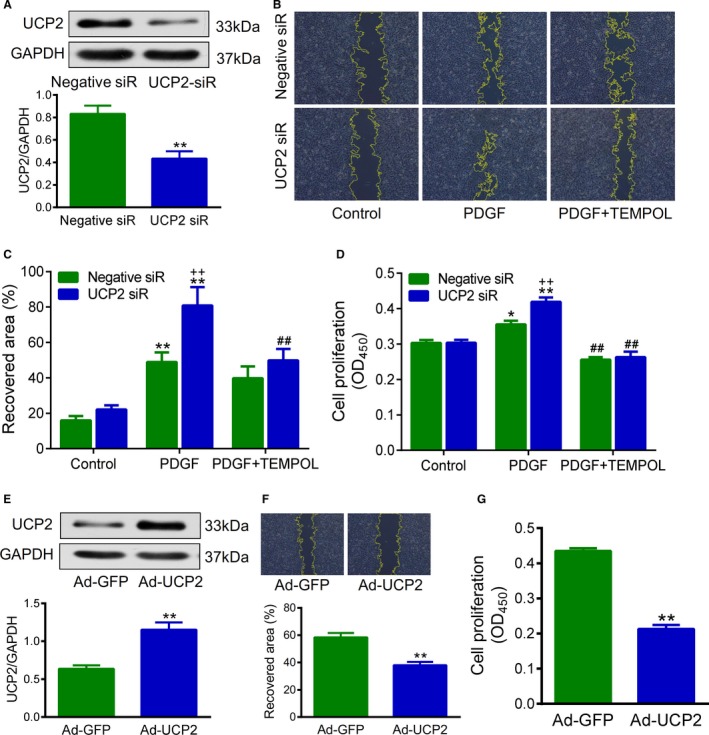
Uncoupling protein 2 (UCP2) suppresses smooth muscle cell (SMC) proliferation and migration. A, Protein expression of UCP2 in cultured human aortic SMCs transfected with UCP2‐specific and control‐negative siRNA. n=3 independent experiments. **P<0.01 vs negative siRNA. B, SMC migration was evaluated by scratch assay. UCP2‐specific and negative siRNA‐transfected SMCs were cultured in monolayer to confluency, scratched with a sterile 200‐μL pipette tip, and treated with or without 4‐hydroxy‐2,2,6,6‐tetramethyl‐piperidinoxyl (TEMPOL; 1 mmol/L). Images were taken after 24 hours. Yellow lines outline the area without cells. C, Recovered area was calculated based on B. **P<0.01 vs control cells transfected with the same siRNA; ## P<0.01 vs platelet‐derived growth factor (PDGF)‐BB–treated cells transfected with UCP2‐specific siRNA; ++ P<0.01 vs PDGF‐BB–treated cells transfected with negative siRNA. n=6 independent experiments. D, SMC proliferation was measured using a colorimetric assay kit (CCK‐8). UCP2‐specific and negative siRNA‐transfected SMCs were treated with or without TEMPOL and incubated with CCK‐8 reagent for 24 hours; then, the absorbance at 450 nm was read using a microplate reader. n=6 independent experiments. *P<0.05, **P<0.01 vs control cells transfected with the same siRNA; ## P<0.01 vs PDGF‐BB–treated cells transfected with the same siRNA; ++ P<0.01 vs PDGF‐BB–treated cells transfected with negative siRNA. E, Protein expression of UCP2 in SMCs transfected with adenovirus expressing UCP2 and green fluorescent protein (GFP; Ad‐UCP2) or GFP alone (Ad‐GFP). n=3. **P<0.01 vs Ad‐GFP. F and G, Migration and proliferation of Ad‐UCP2– or Ad‐GFP–transfected SMCs treated with PDGF‐BB for 24 hours. n=8. **P<0.01 vs Ad‐GFP.
UCP2 Inhibits NF‐κB
We attempted to reveal the underlying mechanisms by which UCP2 suppresses SMC proliferation. Because PDGF signaling has been considered the major pathway involved in SMC proliferation, we performed a protein array by detecting the total and phosphorylated proteins related to the PDGF pathway using 195 highly specific antibodies. The results showed that overexpression of UCP2 in HA‐SMCs significantly downregulated 8 phosphorylated and 2 total proteins, among which 3 belong to NF‐κB pathway (Figure 4A). Immunoblotting showed that UCP2 knockdown increased the protein level of both cytoplasmic and nuclear p65 in HA‐SMCs (Figure 4B and 4C). Similarly, immunostaining demonstrated that the PDGF‐induced increase in NF‐κB p65 was augmented by UCP2 knockdown, whereas either TEMPOL or NF‐κB inhibitor BAY 11‐7085 was able to decrease the level of p65 in SMCs treated with UCP2‐specific siRNA or negative control siRNA (Figure 4D and 4E). Western blot confirmed that BAY 11‐7085 decreased the expression of phosphorylated IκB and IKK with or without UCP2 siRNA (Figure S12). The transcription activity of NF‐κB p65 in nuclear extracts was measured by detecting its response element binding ability. This ELISA showed an enhanced transcription activity of p65 in SMCs with UCP2 knockdown when compared with that in control cells; this difference was abolished by treatment with TEMPOL or BAY 11‐7085 (Figure 4F). Interestingly, BAY 11‐7085 also abolished UCP2 knockdown‐induced increase in SMC proliferation (Figure 4G). In addition, the NF‐κB p65 level was increased in Ucp2 −/− neointima when compared with WT artery in vivo (Figure 4H). These results suggest that UCP2 may inhibit SMC proliferation through the NF‐κB pathway.
Figure 4.
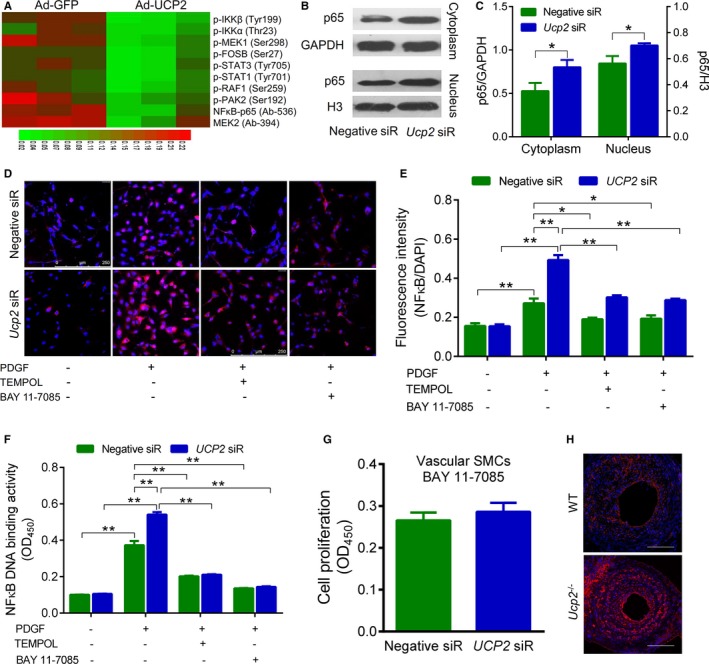
Uncoupling protein 2 (UCP2) inhibits nuclear factor‐κB (NF‐κB). A, Protein phosphorylation array showing the different expression profile related to the platelet‐derived growth factor (PDGF) pathway between PDGF‐stimulated human aortic smooth muscle cells (SMCs) transfected with adenovirus expressing UCP2 (Ad‐UCP2) and green fluorescent protein (GFP) alone (Ad‐GFP). B, Protein expression of NF‐κB p65 in cytoplasmic and nuclear extracts from PDGF‐treated SMCs transfected with Ad‐UCP2 or Ad‐GFP. p65, 65 kDa; GAPDH, 37 kDa; H3, 18 kDa. C, Quantification for the expression of cytoplasmic and nuclear p65 normalized by GAPDH and H3, respectively. n=6 independent experiments. *P<0.05. D, Immunofluorescence staining of p65 (red) in SMCs transfected with UCP2‐specific or negative siRNA. Cells were treated with or without PDGF (20 ng/mL), 4‐hydroxy‐2,2,6,6‐tetramethyl‐piperidinoxyl (TEMPOL; 1 mmol/L), and NF‐κB inhibitor BAY 11‐7085 (10 μmol/L) for 24 hours before staining. Cell nuclei were counterstained with 4’,6‐diamidino‐2‐phenylindole (DAPI; blue). E, Quantification of NF‐κB p65 level was expressed as immunofluorescence intensity normalized by DAPI. n=6 independent experiments. *P<0.05, **P<0.01. F, NF‐κB p65 transcription activity was measured using an ELISA kit. Nuclear extract of cells treated as in D was added into a plate that was immobilized with NF‐κB response element. The p65 was detected with a specific primary antibody and a secondary antibody conjugated to horseradish peroxidase. After adding developing reagents, absorbance at 450 nm was read using a microplate reader. n=6 independent experiments. **P<0.01. G, Proliferation of siRNA‐transfected SMCs treated with PDGF and BAY 11‐7085 for 24 hours was measured using CCK‐8. n=4 independent experiments. H, Immunofluorescence staining of p65 (red) in frozen sections of ligated carotid arteries from Ucp2 −/− and wild‐type (WT) mice. Cell nuclei were counterstained with DAPI (blue). Bar=50 μm.
UCP2 Ameliorates Balloon Injury–Induced Myointima Hyperplasia
We tested the effects of local delivery of adenovirus expressing UCP2 on balloon injury–induced myointima hyperplasia in rat and rabbit carotid arteries. The decrease of UCP2 level was confirmed in rabbit balloon‐injured carotid arteries (Figure S13). Western blot of GFP showed that the overexpression lasted at least 3 weeks and then decreased at week 4 after delivery by adenovirus (Figure S14A). In addition, the GFP was not detected in other major organs, including heart, lung, kidney, and liver (Figure S14B). UCP2 overexpression significantly inhibited the neointima/media ratio in both rat and rabbit injured arteries (Figure 5A through 5C). However, the medial layer was not significantly affected (Figures S15A through S15E and S16A through S16E). Reendothelialization, an important factor for neointimal formation, was similar between these 2 groups, on days 7 and 21 after injury (Figure S17A through S17C), indicating that UCP2 does not affect the endothelial recovery, which is similar to the results from mouse experiments. The SM α‐actin immunostaining highlighted the neointimal and medial layers of vessel wall, and the ratio of SM α‐actin–positive area/neointimal area was similar between Ad‐GFP– and Ad‐UCP2–treated arteries of both rats and rabbits (Figure 5D through 5F). Notably, the percentage of Ki67‐positive proliferating cells in myointima of Ad‐UCP2–treated arteries was less than that in Ad‐GFP–treated vessels (Figure 5G through 5I). In addition, p65 in myointima was decreased in arteries transfected with Ad‐UCP2 (Figure S18A and S18B).
Figure 5.
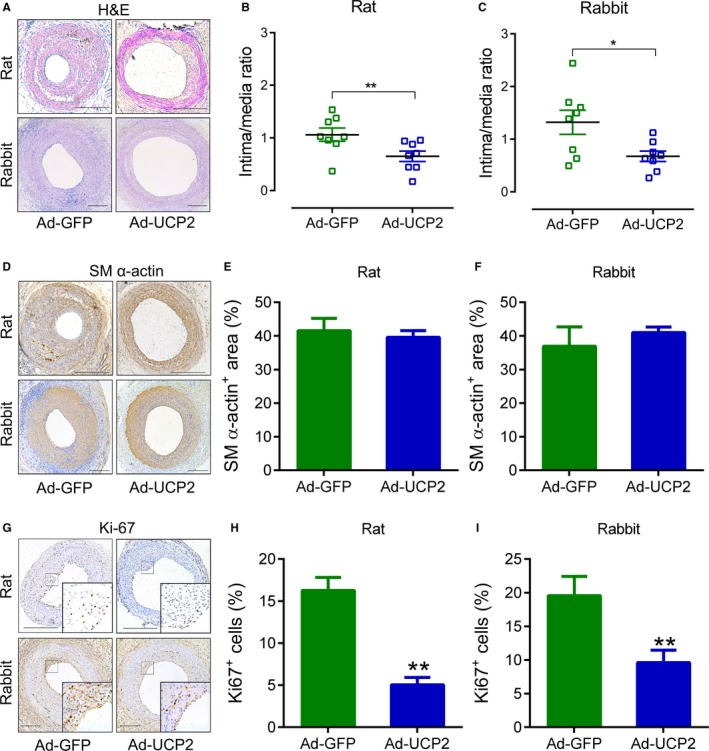
Uncoupling protein 2 (UCP2) ameliorates balloon injury‐induced myointima hyperplasia. Carotid arteries of Sprague‐Dawley rats and New Zealand rabbits were injured with a balloon and locally infected with adenovirus expressing green fluorescent protein (GFP) alone (Ad‐GFP) or both UCP2 and GFP. After 21 days, the injured arteries were harvested for staining. A, Hematoxylin and eosin–stained carotid arterial sections. Bar=200 μm. Intima/media ratio of rat (B) and rabbit (C) injured arteries. n=8 animals in each group. *P<0.05. D, Immunohistochemistry staining of smooth muscle (SM) α‐actin. Bar=200 μm. Percentage of SM α‐actin–positive area to neointima of rat (E) and rabbit (F) injured arteries. n=6. G, Immunohistochemistry staining of Ki67. Bar=200 μm. Percentage of Ki67‐positive cells within neointima of rat (H) and rabbit (I) injured arteries. n=6. **P<0.01 vs Ad‐GFP.
UCP2 Inhibits In‐Stent Stenosis of Porcine Coronary Arteries
To confirm the effects of UCP2 in coronary arteries, we performed the balloon dilation and implanted a bare‐metal stent in minipig coronary artery, followed by local delivery of adenovirus expressing UCP2 or GFP alone to the stented vascular wall with a local drug infusion balloon (Figure S19). Three months after the procedure, follow‐up coronary angiography showed an obvious stenosis in stented segments infected with Ad‐GFP, whereas there is no detectable stenosis in Ad‐UCP2–transfected segments (Figure 6A). Neointima formation in the distal edge of stent was decreased in Ad‐UCP2–treated arteries when compared with Ad‐GFP–treated arteries (Figure 6B). More important, the sections of stented coronary arteries showed a remarkable neointima formation in Ad‐GFP–treated arteries but only minimal neointima in Ad‐UCP2–treated segments (Figure 6C); the in‐stent stenosis was decreased by UCP2 gene transfer (Figure 6D).
Figure 6.
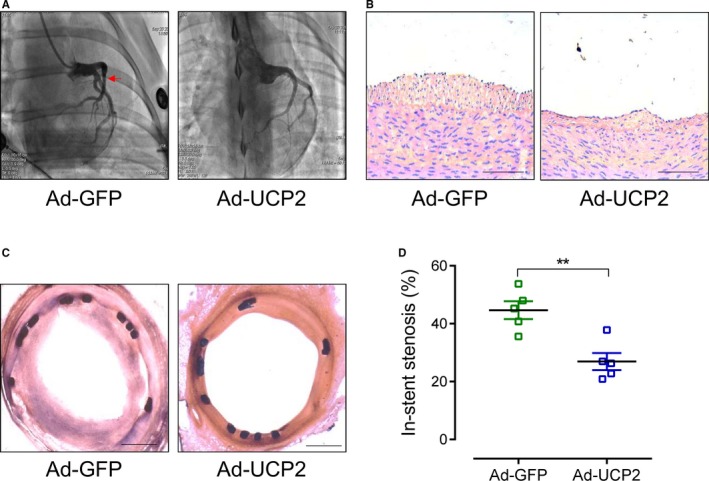
Uncoupling protein 2 (UCP2) inhibits in‐stent stenosis in coronary arteries of minipigs. A, Coronary angiography of Guizhou minipigs 3 months after bare‐metal stent implantation. The stented arterial segments were transfected with adenovirus expressing UCP2 and green fluorescent protein (GFP; Ad‐UCP2) or GFP alone (Ad‐GFP) through a local drug infusion balloon catheter. Red arrow indicates stenosis. B, Hematoxylin and eosin (H&E)–stained sections of porcine coronary arteries distal to the stents. Bar=100 μm. C, H&E‐stained sections of stented coronary arteries. Bar=500 μm. D, In‐stent stenosis of coronary arteries transfected with Ad‐GFP or Ad‐UCP2. n=5. **P<0.01.
UCP2 Inhibits Neointima Development in Human Saphenous Vein
The human great saphenous veins, harvested for coronary artery bypass grafting, were cultured ex vivo to await neointima development. Two weeks later, the Ad‐UCP2–treated vein segments developed fewer neointima when compared with segments infected with Ad‐GFP (Figure 7A and 7B).
Figure 7.
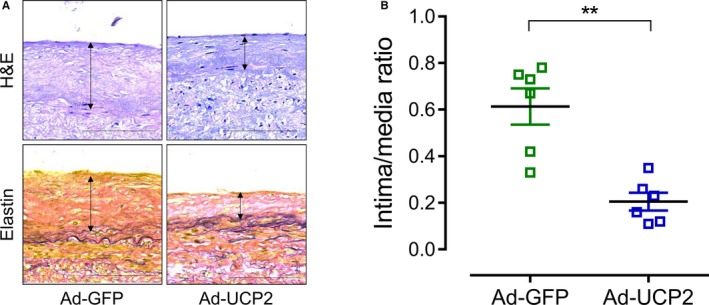
Uncoupling protein 2 (UCP2) inhibits neointima development in human saphenous vein. A, Hematoxylin and eosin–stained (top panel) and Verhoeff–Van Gieson–stained (bottom panel) sections of adenovirus expressing green fluorescent protein (GFP) alone (Ad‐GFP)– and UCP2 and GFP (Ad‐UCP2)–transfected human saphenous veins cultured in RPMI 1640 medium plus platelet‐derived growth factor (PDGF; 10 ng/mL). Bar=100 μm. B, Intima/media ratio of cultured human saphenous veins. n=6. **P<0.01.
Discussion
We have identified the function of UCP2 in suppressing vascular SMC proliferation and myointimal hyperplasia through the reduction of ROS and subsequent inhibition of the NF‐κB pathway. We characterized the dynamic changes of UCP2 expression during myointima development and showed that knockout of UCP2 exacerbated neointima formation in ligated mouse carotid arteries. Overexpression of UCP2 inhibited myointima hyperplasia in rat and rabbit balloon‐injured arteries. We also identified NF‐κB as a critical mediator to promote SMC proliferation after knockdown of UCP2. The finding that UCP2 overexpression inhibits in‐stent stenosis of porcine coronary arteries in vivo and neointimal hyperplasia of human saphenous veins ex vivo suggests that UCP2 is a potential target for the treatment of restenosis. Although the role of UCP2 in cell proliferation was already shown in other cell systems previously,19 this is the first demonstration of its role in neointimal formation with vascular SMCs. A recent study demonstrated that rosiglitazone‐induced inhibition of SMC proliferation is mediated through upregulation of UCP220; however, they did not directly study the function of UCP2 in SMC proliferation and neointima hyperplasia. Mitochondria play an important role in both cell death and proliferation.21 The loss of ΔΨm is a marker of apoptosis,22 whereas the hyperpolarized ΔΨm drives cell proliferation.23 The ΔΨm is tightly regulated by mitochondrial electron transport chain, largely on the basis of metabolic state.24 UCP2, a proton carrier located in the inner membrane of the mitochondria, mildly uncouples oxidative phosphorylation, resulting in a decrease in ΔΨm.25 We found that UCP2 was downregulated during myointima formation in vivo and SMC proliferation in vitro. The transient upregulation of UCP2 may be attributable to the inflammation and oxidative stress early after ligation because treatment with TEMPOL abolished this upregulation, which is consistent with previous studies showing that inflammatory cytokines and ROS increased the expression of UCP2.26, 27 The early increase in UCP2 could also be a compensatory response against vessel inflammation, which warrants further investigation. However, this phenomenon was not present in cultured SMCs because there were no inflammatory cells in the culture dish. The mechanism for the downregulation of UCP2 is still not clear. It might be attributable to phenotypic switch of SMCs and the related metabolic remodeling. The downregulation of UCP2 affected myointimal development, which is supported by the evidence that ablation of UCP2 exacerbated, whereas overexpression of UCP2 ameliorated, myointimal hyperplasia. The beneficial effects of UCP2 on pulmonary artery have been reported previously. Pak et al demonstrated that knockout of UCP2 worsened the pulmonary vascular remodeling by increasing ΔΨm and ROS production.28 Another study showed that UCP2‐deficient mice spontaneously developed pulmonary vascular remodeling because of dysfunction in oxygen sensing.29 Taken together, these studies and the present study are supportive of the beneficial effect of UCP2 in vascular remodeling.
The hyperpolarization of ΔΨm links the overproduction of ROS.30 The basic function of UCP2 is mild uncoupling and reducing the ROS level. Although a high level of ROS induces apoptosis and promotes cell death, slightly increased ROS stimulate cell proliferation.8 The TEMPOL, which catalyzes the disproportionation of superoxide, abolished UCP2 ablation‐induced worsening of SMC proliferation and myointimal hyperplasia, suggesting that the antiproliferative effects of UCP2 may be attributable to decreased production of superoxide.
PDGF signal pathway is a major player in SMC proliferation and neointimal hyperplasia after vascular injury.31 Several transcription factors are activated after treatment with PDGF, including NF‐κB, signal transducers and activators of transcription, c‐Jun, c‐Fos, Elk1, and serum response factor.32, 33 After screening the downstream pathway of UCP2 using a protein phosphorylation array, we focused on NF‐κB because the phosphorylated IKKα and IKKβ were most significantly downregulated after overexpression of UCP2. The NF‐κB is a well‐established regulator of cell proliferation and able to be activated by ROS.34, 35 We confirmed that the expression of NF‐κB p65 was increased in both proliferative SMCs in vitro and myointima in vivo, and was decreased by overexpression of UCP2 or treatment with TEMPOL. In addition, the DNA binding activity of p65 to NF‐κB response element was also increased after knockdown of UCP2. Furthermore, NF‐κB inhibitor can abolish UCP2 knockdown‐induced SMC proliferation. Taken together, UCP2 inhibits SMC proliferation and myointimal hyperplasia, probably by decreasing superoxide level and subsequently inhibiting NF‐κB activity.
UCP2 is expressed in both endothelial cells and vascular SMCs. Our previous studies demonstrated that UCP2 protects endothelial function from high‐salt diet– and obesity‐induced damage.17, 36 We measured the endothelial function and recovery because they are also involved in the development of neointima.37 However, there was neither difference in endothelial function between Ucp2 −/− and WT mice after ligation of carotid artery nor reendothelialization between Ad‐GFP– and Ad‐UCP2–infected rat carotid artery. Therefore, the endothelial UCP2 may be less important in the development of neointimal hyperplasia than that in SMCs.
The most exciting part of this study is that UCP2 overexpression by adenovirus‐mediated gene transfer inhibits balloon injury–induced myointimal hyperplasia in rat and rabbit carotid arteries, in‐stent stenosis in swine coronary arteries, and myointimal formation in human saphenous veins. These findings enable our study of translational significance and are complementary to the mouse carotid artery ligation model that does not perfectly mimic the pathophysiological characteristics of restenosis. Especially, the swine model is able to evaluate the effect of UCP2 on the in‐stent stenosis in vivo. The catheter‐based image systems, including quantitative angiography, intravascular ultrasonography, and optical coherence tomography, should be used to quantify the in‐stent stenosis, in addition to histological characteristics. This is a limitation of the current study, which is lack of in vivo quantitative imaging. Adenovirus‐based gene therapy has been used in many clinical trials and will become a novel promising treatment strategy.38, 39 However, the delivery system to achieve the overexpression of UCP2 in a targeted vessel is still a challenge. The lesion‐targeted nanoparticles may be useful in delivering antimicroRNAs to upregulate UCP2.40 The chemical drugs, such as rosiglitazone, that upregulate UCP2 may become candidates for inhibiting restenosis.41 There was a study that demonstrated that oral administration with rosiglitazone significantly reduces in‐stent restenosis in diabetic patients with coronary artery disease.42 However, the antidiabetic effects may also contribute to the decreased restenosis. Therefore, the local delivery of rosiglitazone is worth testing in restenosis.
The role of UCP2 in vascular diseases was broadly studied by our group and other groups. UCP2 protects against endothelial dysfunction, vascular remodeling, and atherosclerosis.17, 18, 36, 43 The present study revealed a novel function of UCP2 in inhibiting myointimal hyperplasia and restenosis after vascular injury. A drug‐eluting stent inhibits restenosis, but it also delays reendothelialization, which causes late and very late in‐stent thrombosis and neoatherosclerosis.44 However, UCP2 inhibits the development of both restenosis and atherosclerosis43 and preserves endothelial function, indicating that UCP2 should become an ideal target for inhibiting myointimal hyperplasia and restenosis after vascular injury.
Sources of Funding
This work was supported by National Natural Science Foundation of China (81470589 and 81100232 to Ma and 81400289 to P. Wang) and Sichuan Youth Science and Technology Foundation (2016JQ0032).
Disclosures
None.
Supporting information
Data S1. Supplemental Experimental Procedures.Figure S1. Effects of TEMPOL on the expression of Ucp2.Figure S2. Expression of Ucp2 in sham and ligated mouse carotid arteries.Figure S3. Expression of UCP2 in ligated mouse carotid arteries.Figure S4. PDGF decreasesthe expression and function of UCP2.Figure S5. UCP2 ablation exacerbates mouse myointimal hyperplasia.Figure S6. TEMPOL abolishes the UCP2 ablation‐exacerbated myointimal hyperplasia. Area within external elastic lamina (EEL) (A), area within internal elastic lamina (IEL) (B), medial layer area (C), intimal layer area (D), and luminal area (E) were calculated based on the H&E‐stained sections of ligated carotid arteries from Ucp2 −/− mice and wild‐type (WT) littermates treated with or without TEMPOL. n=8 mice in each group. **P<0.01.Figure S7. UCP2 ablation does not affect cell apoptosis. (A) Representative images of TUNNEL assay (green color). White arrows indicate positive cells. (B) The quantification of TUNEL‐positive cells within the intima per section. Scale bar: 50 μm. n=8.Figure S8. Endothelial function of mouse carotid artery. Acetylcholine (ACh)‐induced relaxation of ligated and unligated carotid arteries from wild‐type (WT) and UCP2 knockout (KO) mice onday 3 (A), day7 (B), and day 14 (C) after the procedure. Arterial rings were pre‐contracted with phenylephrine (Phe). Data are mean±SE of 5 animals. **P<0.01 vs WT‐ligated; ## P<0.01 vs KO‐ligated.Figure S9. Knockdown of UCP2 promotessmooth muscle cell (SMC) migration. SMC migration was evaluated by scratch assay. UCP2 specific siRNA‐2 (different from the one used in Figure 3C)‐ and negative siRNA‐transfected SMCs were cultured in monolayer to confluencyand then were scratched witha sterile 200‐μL pipette tip and treated with or without TEMPOL (1 mmol/L). Images taken after 24 hours were used to calculate the recovered area. **P<0.01 vs control cells transfected with the same siRNA; ## P<0.01 vs PDGF‐treated cells transfected with UCP2 specific siRNA; ++ P<0.01 vs PDGF‐treated cells transfected with negative siRNA. n=9 independent experiments.Figure S10. Knockdown of UCP2 promotes smooth muscle cell (SMC) proliferation. SMC proliferation was measured using a colorimetric assay kit (CCK‐8). UCP2 specific siRNA‐2 (different from the one used in Figure 3D)‐ and negative siRNA‐transfected SMCs were treated with or without TEMPOL, incubated with CCK‐8 reagent for 24 hours, and then the absorbance at 450 nm was read using a microplate reader.*P<0.05, **P<0.01 vs control cells transfected with the same siRNA; ## P<0.01 vs PDGF‐treated cells transfected with the same siRNA; ++ P<0.01 vs PDGF‐treated cells transfected with negative siRNA. n=8 independent experiments.Figure S11. Effects of Ucp2 overexpression on the expression of smooth muscle markers. The expression of SM MHC, SM α‐actin, and calponin in primarily cultured smooth muscle cells which were isolated from WT and Ucp2 −/− mice and infected with Ad‐Ucp2 transgene to overexpress Ucp2 (Ucp2 −/−‐tg). Western blotting gel images (A) and quantitation (B). *P<0.05, **P<0.01 vs WT; # P<0.05 vs Ucp2 −/−.Figure S12. Expression of IKK/IκB. Western blots of phospho‐IκB, total IκB, p‐IKK, and IKK in HA‐SMCs transfected with UCP2 specific or negative control siRNA (siR) and treated with or without BAY 11‐7085. *P<0.05, **P<0.01 vs cells transfected with negative siR; ## P<0.01 vs cells transfected with the same siRNA but treated without BAY 11‐7085.
Figure S13. Protein expression of UCP2 in balloon injured rabbit carotid artery. Western blots of UCP2 in lysates from balloon injured and contralateral uninjured rabbit carotid arteries (upper panel) and the quantification of UCP2 level by normalizing to GAPDH. (lower panel). **P<0.01 vs uninjured.
Figure S14. A, Protein expression of GFP in balloon injured ratcarotid artery in week 1, 2, 3, and 4 post infection with Ad‐GFP‐UCP2. B, Protein expression of GFP in balloon injured ratcarotid artery, heart, kidney, lung, and liver in week 1 post infection with Ad‐GFP‐UCP2.
Figure S15. UCP2 ameliorates balloon injury‐induced myointima hyperplasia. Carotid arteries of Sprague–Dawley rats were injured with a balloon and locally infected with adenovirus expressing GFP alone (Ad‐GFP) or both UCP2 and GFP (Ad‐UCP2). Area within external elastic lamina (EEL) (A), area within internal elastic lamina (IEL) (B), medial layer area (C), intimal layer area (D), and luminal area (E) were calculated based on the H&E‐stained sections of injured carotid arteries 21 days after injury and infection. n=8 animals in each group. *P<0.05.
Figure S16. UCP2 ameliorates balloon injury‐induced myointima hyperplasia. Carotid arteries of New Zealand rabbits were injured with a balloon and locally infected with adenovirus expressing GFP alone (Ad‐GFP) or both UCP2 and GFP (Ad‐UCP2). Area within external elastic lamina (EEL) (A), area within internal elastic lamina (IEL) (B), medial layer area (C), intimal layer area (D), and luminal area (E) were calculated based on the H&E‐stained sections of injured carotid arteries 21 days after injury and infection. n=8 animals in each group. *P<0.05.
Figure S17. Reendothelialization of balloon injured rat carotid artery. A, Representative en face Evans blue staining of rat carotid arteries on day 7 after balloon injury and local Ad‐GFP or Ad‐UCP2 infection. B, Quantification of reendothelialization. n=7 animal in each group. C, Immunohistochemistry staining of CD31 (brown) in rat carotid arteries on day 21 after balloon injury and local Ad‐GFP or Ad‐UCP2 infection. Red arrows indicate positive cells. Scale bar: 200 μm.
Figure S18. Expression of p65 in balloon injured artery. Immunohistochemistry staining of NF‐κB p65 in balloon injured rat (A) and rabbit (B) carotid arteries infected with Ad‐GFP or Ad‐UCP2. DAB (brown in A) and AEC (red in B) substrates were used for rat and rabbit artery, respectively. Scale bar: 200 μm.
Figure S19. Stent implantation and adenovirus delivery of porcine coronary artery. A, Coronary angiography of Guizhou mini‐pig. B, Balloon dilation of left anterior descending coronary artery. C, Bare metal stent deployment. D, Angiography after stent implantation. E and F, ClearWay local drug infusion balloon in stented segment.
Acknowledgments
We thank Weihong Wang, RN, Xiujing Zhi, RN, and Zhen Duan, RN, for assistance during the coronary angiography and intervention of minipigs.
(J Am Heart Assoc. 2017;6:e006593 DOI: 10.1161/JAHA.117.006593.)
Contributor Information
Yongjian Yang, Email: yangyongjian38@sina.com.
Shuangtao Ma, Email: shuangtao.ma@hc.msu.edu.
References
- 1. Unverdorben M, Vallbracht C, Cremers B, Heuer H, Hengstenberg C, Maikowski C, Werner GS, Antoni D, Kleber FX, Bocksch W, Leschke M, Ackermann H, Boxberger M, Speck U, Degenhardt R, Scheller B. Paclitaxel‐coated balloon catheter versus paclitaxel‐coated stent for the treatment of coronary in‐stent restenosis. Circulation. 2009;119:2986–2994. [DOI] [PubMed] [Google Scholar]
- 2. Teirstein PS. Drug‐eluting stent restenosis: an uncommon yet pervasive problem. Circulation. 2010;122:5–7. [DOI] [PubMed] [Google Scholar]
- 3. Bonaa KH, Mannsverk J, Wiseth R, Aaberge L, Myreng Y, Nygard O, Nilsen DW, Klow NE, Uchto M, Trovik T, Bendz B, Stavnes S, Bjornerheim R, Larsen AI, Slette M, Steigen T, Jakobsen OJ, Bleie O, Fossum E, Hanssen TA, Dahl‐Eriksen O, Njolstad I, Rasmussen K, Wilsgaard T, Nordrehaug JE; NORSTENT Investigators . Drug‐eluting or bare‐metal stents for coronary artery disease. N Engl J Med. 2016;375:1242–1252. [DOI] [PubMed] [Google Scholar]
- 4. Dzau VJ, Braun‐Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. [DOI] [PubMed] [Google Scholar]
- 5. Marx SO, Totary‐Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011;4:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ohno T, Gordon D, San H, Pompili VJ, Imperiale MJ, Nabel GJ, Nabel EG. Gene therapy for vascular smooth muscle cell proliferation after arterial injury. Science. 1994;265:781–784. [DOI] [PubMed] [Google Scholar]
- 7. Brahmbhatt A, Remuzzi A, Franzoni M, Misra S. The molecular mechanisms of hemodialysis vascular access failure. Kidney Int. 2016;89:303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal. 2012;16:1150–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rustin P. Mitochondria, from cell death to proliferation. Nat Genet. 2002;30:352–353. [DOI] [PubMed] [Google Scholar]
- 10. Chiong M, Cartes‐Saavedra B, Norambuena‐Soto I, Mondaca‐Ruff D, Morales PE, Garcia‐Miguel M, Mellado R. Mitochondrial metabolism and the control of vascular smooth muscle cell proliferation. Front Cell Dev Biol. 2014;2:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deuse T, Hua X, Wang D, Maegdefessel L, Heeren J, Scheja L, Bolanos JP, Rakovic A, Spin JM, Stubbendorff M, Ikeno F, Langer F, Zeller T, Schulte‐Uentrop L, Stoehr A, Itagaki R, Haddad F, Eschenhagen T, Blankenberg S, Kiefmann R, Reichenspurner H, Velden J, Klein C, Yeung A, Robbins RC, Tsao PS, Schrepfer S. Dichloroacetate prevents restenosis in preclinical animal models of vessel injury. Nature. 2014;509:641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai Y, Nagel DJ, Zhou Q, Cygnar KD, Zhao H, Li F, Pi X, Knight PA, Yan C. Role of cAMP‐phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ Res. 2015;116:1120–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schulick AH, Newman KD, Virmani R, Dichek DA. In vivo gene transfer into injured carotid arteries: optimization and evaluation of acute toxicity. Circulation. 1995;91:2407–2414. [DOI] [PubMed] [Google Scholar]
- 14. Zoldhelyi P, Chen ZQ, Shelat HS, McNatt JM, Willerson JT. Local gene transfer of tissue factor pathway inhibitor regulates intimal hyperplasia in atherosclerotic arteries. Proc Natl Acad Sci USA. 2001;98:4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang D, Deuse T, Stubbendorff M, Chernogubova E, Erben RG, Eken SM, Jin H, Li Y, Busch A, Heeger CH, Behnisch B, Reichenspurner H, Robbins RC, Spin JM, Tsao PS, Schrepfer S, Maegdefessel L. Local microRNA modulation using a novel anti‐miR‐21‐eluting stent effectively prevents experimental in‐stent restenosis. Arterioscler Thromb Vasc Biol. 2015;35:1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Latif F, Hennebry TA. Successful revascularization of re‐stenosis of lower extremity arteries with localized delivery of paclitaxel. Catheter Cardiovasc Interv. 2008;72:294–298. [DOI] [PubMed] [Google Scholar]
- 17. Ma S, Ma L, Yang D, Luo Z, Hao X, Liu D, Zhu Z. Uncoupling protein 2 ablation exacerbates high‐salt intake‐induced vascular dysfunction. Am J Hypertens. 2010;23:822–828. [DOI] [PubMed] [Google Scholar]
- 18. Ma S, Zhang Y, Wang Q, Yang D, Li D, Tang B, Yang Y. Ablation of uncoupling protein 2 exacerbates salt‐induced cardiovascular and renal remodeling associated with enhanced oxidative stress. Int J Cardiol. 2014;175:206–210. [DOI] [PubMed] [Google Scholar]
- 19. Esteves P, Pecqueur C, Alves‐Guerra MC. UCP2 induces metabolic reprogramming to inhibit proliferation of cancer cells. Mol Cell Oncol. 2015;2:e975024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou Y, Zhang MJ, Li BH, Chen L, Pi Y, Yin YW, Long CY, Wang X, Sun MJ, Chen X, Gao CY, Li JC, Zhang LL. PPARgamma inhibits VSMC proliferation and migration via attenuating oxidative stress through upregulating UCP2. PLoS One. 2016;11:e0154720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Birsoy K, Wang T, Chen WW, Freinkman E, Abu‐Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. [DOI] [PubMed] [Google Scholar]
- 23. Bonnet S, Archer SL, Allalunis‐Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria‐K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. [DOI] [PubMed] [Google Scholar]
- 24. Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, Whitehill GD, Clever D, Eil RL, Palmer DC, Mitra S, Rao M, Keyvanfar K, Schrump DS, Wang E, Marincola FM, Gattinoni L, Leonard WJ, Muranski P, Finkel T, Restifo NP. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23:63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and ucp3. Cell Metab. 2005;2:85–93. [DOI] [PubMed] [Google Scholar]
- 26. Chan SH, Wu CA, Wu KL, Ho YH, Chang AY, Chan JY. Transcriptional upregulation of mitochondrial uncoupling protein 2 protects against oxidative stress‐associated neurogenic hypertension. Circ Res. 2009;105:886–896. [DOI] [PubMed] [Google Scholar]
- 27. Echtay KS, Roussel D, St‐Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. [DOI] [PubMed] [Google Scholar]
- 28. Pak O, Sommer N, Hoeres T, Bakr A, Waisbrod S, Sydykov A, Haag D, Esfandiary A, Kojonazarov B, Veit F, Fuchs B, Weisel FC, Hecker M, Schermuly RT, Grimminger F, Ghofrani HA, Seeger W, Weissmann N. Mitochondrial hyperpolarization in pulmonary vascular remodeling: mitochondrial uncoupling protein deficiency as disease model. Am J Respir Cell Mol Biol. 2013;49:358–367. [DOI] [PubMed] [Google Scholar]
- 29. Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res. 2013;113:126–136. [DOI] [PubMed] [Google Scholar]
- 30. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to pdgf. Science. 1991;253:1129–1132. [DOI] [PubMed] [Google Scholar]
- 32. Millette E, Rauch BH, Kenagy RD, Daum G, Clowes AW. Platelet‐derived growth factor‐bb transactivates the fibroblast growth factor receptor to induce proliferation in human smooth muscle cells. Trends Cardiovasc Med. 2006;16:25–28. [DOI] [PubMed] [Google Scholar]
- 33. Karin M, Hunter T. Transcriptional control by protein phosphorylation: signal transmission from the cell surface to the nucleus. Curr Biol. 1995;5:747–757. [DOI] [PubMed] [Google Scholar]
- 34. Mehrhof FB, Schmidt‐Ullrich R, Dietz R, Scheidereit C. Regulation of vascular smooth muscle cell proliferation: role of NF‐kappaB revisited. Circ Res. 2005;96:958–964. [DOI] [PubMed] [Google Scholar]
- 35. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF‐kappaB signaling. Cell Res. 2011;21:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tian XY, Wong WT, Xu A, Lu Y, Zhang Y, Wang L, Cheang WS, Wang Y, Yao X, Huang Y. Uncoupling protein‐2 protects endothelial function in diet‐induced obese mice. Circ Res. 2012;110:1211–1216. [DOI] [PubMed] [Google Scholar]
- 37. Kipshidze N, Dangas G, Tsapenko M, Moses J, Leon MB, Kutryk M, Serruys P. Role of the endothelium in modulating neointimal formation: vasculoprotective approaches to attenuate restenosis after percutaneous coronary interventions. J Am Coll Cardiol. 2004;44:733–739. [DOI] [PubMed] [Google Scholar]
- 38. Berger JS, Hiatt WR. Medical therapy in peripheral artery disease. Circulation. 2012;126:491–500. [DOI] [PubMed] [Google Scholar]
- 39. Wolfram JA, Donahue JK. Gene therapy to treat cardiovascular disease. J Am Heart Assoc. 2013;2:e000119 DOI: 10.1161/JAHA.113.000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma S, Tian XY, Zhang Y, Mu C, Shen H, Bismuth J, Pownall HJ, Huang Y, Wong WT. E‐selectin‐targeting delivery of micrornas by microparticles ameliorates endothelial inflammation and atherosclerosis. Sci Rep. 2016;6:22910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Villarroya F, Iglesias R, Giralt M. PPARs in the control of uncoupling proteins gene expression. PPAR Res. 2007;2007:74364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choi D, Kim SK, Choi SH, Ko YG, Ahn CW, Jang Y, Lim SK, Lee HC, Cha BS. Preventative effects of rosiglitazone on restenosis after coronary stent implantation in patients with type 2 diabetes. Diabetes Care. 2004;27:2654–2660. [DOI] [PubMed] [Google Scholar]
- 43. Blanc J, Alves‐Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, Mallat Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation. 2003;107:388–390. [DOI] [PubMed] [Google Scholar]
- 44. Finn AV, Otsuka F. Neoatherosclerosis: a culprit in very late stent thrombosis. Circ Cardiovasc Interv. 2012;5:6–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Experimental Procedures.Figure S1. Effects of TEMPOL on the expression of Ucp2.Figure S2. Expression of Ucp2 in sham and ligated mouse carotid arteries.Figure S3. Expression of UCP2 in ligated mouse carotid arteries.Figure S4. PDGF decreasesthe expression and function of UCP2.Figure S5. UCP2 ablation exacerbates mouse myointimal hyperplasia.Figure S6. TEMPOL abolishes the UCP2 ablation‐exacerbated myointimal hyperplasia. Area within external elastic lamina (EEL) (A), area within internal elastic lamina (IEL) (B), medial layer area (C), intimal layer area (D), and luminal area (E) were calculated based on the H&E‐stained sections of ligated carotid arteries from Ucp2 −/− mice and wild‐type (WT) littermates treated with or without TEMPOL. n=8 mice in each group. **P<0.01.Figure S7. UCP2 ablation does not affect cell apoptosis. (A) Representative images of TUNNEL assay (green color). White arrows indicate positive cells. (B) The quantification of TUNEL‐positive cells within the intima per section. Scale bar: 50 μm. n=8.Figure S8. Endothelial function of mouse carotid artery. Acetylcholine (ACh)‐induced relaxation of ligated and unligated carotid arteries from wild‐type (WT) and UCP2 knockout (KO) mice onday 3 (A), day7 (B), and day 14 (C) after the procedure. Arterial rings were pre‐contracted with phenylephrine (Phe). Data are mean±SE of 5 animals. **P<0.01 vs WT‐ligated; ## P<0.01 vs KO‐ligated.Figure S9. Knockdown of UCP2 promotessmooth muscle cell (SMC) migration. SMC migration was evaluated by scratch assay. UCP2 specific siRNA‐2 (different from the one used in Figure 3C)‐ and negative siRNA‐transfected SMCs were cultured in monolayer to confluencyand then were scratched witha sterile 200‐μL pipette tip and treated with or without TEMPOL (1 mmol/L). Images taken after 24 hours were used to calculate the recovered area. **P<0.01 vs control cells transfected with the same siRNA; ## P<0.01 vs PDGF‐treated cells transfected with UCP2 specific siRNA; ++ P<0.01 vs PDGF‐treated cells transfected with negative siRNA. n=9 independent experiments.Figure S10. Knockdown of UCP2 promotes smooth muscle cell (SMC) proliferation. SMC proliferation was measured using a colorimetric assay kit (CCK‐8). UCP2 specific siRNA‐2 (different from the one used in Figure 3D)‐ and negative siRNA‐transfected SMCs were treated with or without TEMPOL, incubated with CCK‐8 reagent for 24 hours, and then the absorbance at 450 nm was read using a microplate reader.*P<0.05, **P<0.01 vs control cells transfected with the same siRNA; ## P<0.01 vs PDGF‐treated cells transfected with the same siRNA; ++ P<0.01 vs PDGF‐treated cells transfected with negative siRNA. n=8 independent experiments.Figure S11. Effects of Ucp2 overexpression on the expression of smooth muscle markers. The expression of SM MHC, SM α‐actin, and calponin in primarily cultured smooth muscle cells which were isolated from WT and Ucp2 −/− mice and infected with Ad‐Ucp2 transgene to overexpress Ucp2 (Ucp2 −/−‐tg). Western blotting gel images (A) and quantitation (B). *P<0.05, **P<0.01 vs WT; # P<0.05 vs Ucp2 −/−.Figure S12. Expression of IKK/IκB. Western blots of phospho‐IκB, total IκB, p‐IKK, and IKK in HA‐SMCs transfected with UCP2 specific or negative control siRNA (siR) and treated with or without BAY 11‐7085. *P<0.05, **P<0.01 vs cells transfected with negative siR; ## P<0.01 vs cells transfected with the same siRNA but treated without BAY 11‐7085.
Figure S13. Protein expression of UCP2 in balloon injured rabbit carotid artery. Western blots of UCP2 in lysates from balloon injured and contralateral uninjured rabbit carotid arteries (upper panel) and the quantification of UCP2 level by normalizing to GAPDH. (lower panel). **P<0.01 vs uninjured.
Figure S14. A, Protein expression of GFP in balloon injured ratcarotid artery in week 1, 2, 3, and 4 post infection with Ad‐GFP‐UCP2. B, Protein expression of GFP in balloon injured ratcarotid artery, heart, kidney, lung, and liver in week 1 post infection with Ad‐GFP‐UCP2.
Figure S15. UCP2 ameliorates balloon injury‐induced myointima hyperplasia. Carotid arteries of Sprague–Dawley rats were injured with a balloon and locally infected with adenovirus expressing GFP alone (Ad‐GFP) or both UCP2 and GFP (Ad‐UCP2). Area within external elastic lamina (EEL) (A), area within internal elastic lamina (IEL) (B), medial layer area (C), intimal layer area (D), and luminal area (E) were calculated based on the H&E‐stained sections of injured carotid arteries 21 days after injury and infection. n=8 animals in each group. *P<0.05.
Figure S16. UCP2 ameliorates balloon injury‐induced myointima hyperplasia. Carotid arteries of New Zealand rabbits were injured with a balloon and locally infected with adenovirus expressing GFP alone (Ad‐GFP) or both UCP2 and GFP (Ad‐UCP2). Area within external elastic lamina (EEL) (A), area within internal elastic lamina (IEL) (B), medial layer area (C), intimal layer area (D), and luminal area (E) were calculated based on the H&E‐stained sections of injured carotid arteries 21 days after injury and infection. n=8 animals in each group. *P<0.05.
Figure S17. Reendothelialization of balloon injured rat carotid artery. A, Representative en face Evans blue staining of rat carotid arteries on day 7 after balloon injury and local Ad‐GFP or Ad‐UCP2 infection. B, Quantification of reendothelialization. n=7 animal in each group. C, Immunohistochemistry staining of CD31 (brown) in rat carotid arteries on day 21 after balloon injury and local Ad‐GFP or Ad‐UCP2 infection. Red arrows indicate positive cells. Scale bar: 200 μm.
Figure S18. Expression of p65 in balloon injured artery. Immunohistochemistry staining of NF‐κB p65 in balloon injured rat (A) and rabbit (B) carotid arteries infected with Ad‐GFP or Ad‐UCP2. DAB (brown in A) and AEC (red in B) substrates were used for rat and rabbit artery, respectively. Scale bar: 200 μm.
Figure S19. Stent implantation and adenovirus delivery of porcine coronary artery. A, Coronary angiography of Guizhou mini‐pig. B, Balloon dilation of left anterior descending coronary artery. C, Bare metal stent deployment. D, Angiography after stent implantation. E and F, ClearWay local drug infusion balloon in stented segment.