

Abstract
Background
Cardiomyocytes differentiated from human pluripotent stem cells can serve as an unexhausted source for a cellular cardiac disease model. Although small molecule–mediated cardiomyocyte differentiation methods have been established, the differentiation efficiency is relatively unsatisfactory in multiple lines due to line‐to‐line variation. Additionally, hurdles including line‐specific low expression of endogenous growth factors and the high apoptotic tendency of human pluripotent stem cells also need to be overcome to establish robust and efficient cardiomyocyte differentiation.
Methods and Results
We used the H9–human cardiac troponin T–eGFP reporter cell line to screen for small molecules that promote cardiac differentiation in a monolayer‐based and growth factor–free differentiation model. We found that collaterally treating human pluripotent stem cells with rapamycin and CHIR99021 during the initial stage was essential for efficient and reliable cardiomyocyte differentiation. Moreover, this method maintained consistency in efficiency across different human embryonic stem cell and human induced pluripotent stem cell lines without specifically optimizing multiple parameters (the efficiency in H7, H9, and UQ1 human induced pluripotent stem cells is 98.3%, 93.3%, and 90.6%, respectively). This combination also increased the yield of cardiomyocytes (1:24) and at the same time reduced medium consumption by about 50% when compared with the previous protocols. Further analysis indicated that inhibition of the mammalian target of rapamycin allows efficient cardiomyocyte differentiation through overcoming p53‐dependent apoptosis of human pluripotent stem cells during high‐density monolayer culture via blunting p53 translation and mitochondrial reactive oxygen species production.
Conclusions
We have demonstrated that mammalian target of rapamycin exerts a stage‐specific and multifaceted regulation over cardiac differentiation and provides an optimized approach for generating large numbers of functional cardiomyocytes for disease modeling and in vitro drug screening.
Keywords: apoptosis, cardiac differentiation, human embryonic stem cells, mammalian target of rapamycin
Subject Categories: Stem Cells, Myocardial Biology, Myocardial Regeneration, Cell Signalling/Signal Transduction, Cell Biology/Structural Biology
Clinical Perspective
What Is New?
The present study established a stable and efficient method for reliable cardiomyocyte differentiation and elucidated an undefined mechanism between human pluripotent stem cell apoptosis and efficient cardiac differentiation under the regulation of mTOR signaling pathway.
What Are the Clinical Implications?
This work provided an optimized approach for conceiving and implementing scalable and cost‐effective human pluripotent stem cell–derived cardiomyocyte production systems, which is promising for in vitro disease modeling, high‐throughput drug screening, and prospective human cardiomyocyte‐based myocardial repair.
Introduction
Cardiomyocytes derived from human pluripotent stem cells (hPSCs) have inestimable value for cell‐based therapies, potential drug screening, and cardiac safety pharmacology testing. Over the past decade various cardiac differentiation protocols have been established based on embryonic body (EB) and monolayer methodology.1 Monolayer‐based cardiac differentiation,2, 3 in which hPSCs are cultured to a high density, is believed to be more widely applicable in certain respects because it is easier to manipulate and produces a higher yield than does the EB system. Recently, Palecek's lab reported that sequential activation and suppression of canonical Wnt signaling by small‐molecule inhibitors, including CHIR and IWP2/4, could produce about 85% (human embryonic stem cell [hESC] line H9) purity cardiomyocytes in the growth‐factor‐free monolayer culture.4, 5 However, given the paucity of studies on the monolayer‐based differentiation model, the detailed mechanisms presently remain largely unclear. Previous protocols are not readily transferrable across frequently used hESC lines such as H7. More importantly, the extant methods only focus on the regulatory effect of Wnt signaling. But, whether there is another factor that is essential for this procedure is undefined.
The mammalian target of rapamycin (mTOR), a serine‐threonine protein kinase that was originally identified as a highly specific target of rapamycin, is evolutionarily conserved from yeast to human. mTOR signaling is demonstrated to be the central regulator in a range of vital processes including protein synthesis,6 autophagy,7 cell growth,8 metabolism,9 and survival. During cardiogenesis in vivo and in vitro, mTOR is demonstrated to play indispensable roles in embryonic stem cell maintenance,10 later stages of heart development, and adult heart homeostasis.11 In studies using mouse models, homozygous mutants of mTOR were found to disrupt the proliferation of both inner cell mass and extraembryonic tissues, precipitating embryonic lethality at embryonic day 5.5 (E5.5).12 In addition, cardiac‐specific deletion of mTOR was reported to induce heart failure and massive cardiac dilation as well as to cause early neonatal death.13 Similarly, knockout of mTOR in adult heart leads to a fatal dilated cardiomyopathy.14 However, whether mTOR participates in an early stage of cardiogenesis when cell destiny transforms from embryonic stem cell to cardiac cell is still undetermined.
Previous studies have revealed that hESCs are more sensitive to apoptosis as a result of DNA damage than are differentiated cells due to constitutively active Bax and high mitochondrial priming.15, 16 Recently, Wang et al revealed that apoptosis is important for differentiation initiation through selectively removing stalled murine ESCs via Fas‐activated mitochondrial apoptosis.17 And 2 labs demonstrated that sublethal caspase activation promoted the proliferation and differentiation of cardiac progenitor cells in vivo and in vitro.18, 19 However, a definite reason and effect of the apoptosis of hPSCs during monolayer‐based differentiation, which is essential for hPSC culturing and differentiation, are completely unclear.
In this article we report that mTOR exerts multiple effects on hPSC toward cardiac differentiation. Specifically, gain‐ and loss‐of‐function assays show that inhibiting mTOR signaling is necessary for efficient cardiac induction in the initial stage. Moreover, our results elucidate the detailed mechanisms underlying hPSC apoptosis during the monolayer‐based differentiation process and uncover an undefined link between apoptosis and efficient differentiation. In addition, we reveal the regulatory effects of mTOR on primary cardiogenesis‐related signaling pathways, especially Wnt signaling synergies with CHIR. The functions of mTOR in leading hPSCs toward cardiac differentiation may shed light on a possible mechanism for cardiogenesis in vivo and provide some important theoretical advantages for designing and implementing scalable and cost‐effective hPSC‐derived cardiomyocyte production systems.
Methods
hPSC Culture
Three hESC lines (H9–human cardiac troponin T–eGFP, H9, H7) were obtained from WiCell (Madison, WI) (H9‐hTnnTZ‐pGZ‐D2, WA09, WA07). A human induced PSC (iPSC) line (hiPS‐U‐Q1) was established from human urine cells by a DOX‐induced OKSM expression lentivirus system.20 Human urine cells were obtained from the Department of Cardiology of Changhai Hospital. All human participants in the study had signed informed consent for the use of their urine samples. Isolation and use of human urine cells were approved by the Ethical Committee of Changhai Hospital. All hPSCs were cultured on CF1‐MEF feeders in hESC medium: DMEM/F12 medium supplemented with 20% (vol/vol) KnockOut serum replacer, 10 ng/mL human bFGF, 1 mmol/L l‐glutamine, 0.1 mmol/L NEAA (all from Invitrogen, Carlsbad, CA) and 0.1 mmol/L β‐mercaptoethanol (Sigma‐Aldrich, St. Louis, MO). For a monolayer‐based culture, hPSCs were maintained on human qualified Matrigel (BD Biosciences, San Jose, CA) in mTeSR1 medium (STEMCELL Technologies, Vancouver, Canada).
Screening of Cardiac Differentiation‐Promoting Small Molecules
Based on the monolayer cardiac differentiation protocol found by Murry's laboratory, first we used CHIR99021 (Selleck or STEMCELL Technologies) to substitute activin A, BMP4, and Wnt3a, then used XAV939 (Enzo, Farmingdale, NY) plus KY02111 (Tocris, Bristol, UK) to replace DKK1. Next, according to this modified method, we continued screening using cTnT‐derived eGFP as a reporter:H9‐cTnT‐eGFP cells were cultured in 48‐well plates, and small‐molecule candidates were added during days −3 to 12, at concentrations that were referenced in previous studies; on day 15, the rough ratio of eGFP‐positive cells was adjusted by observation. Small‐molecule candidates included LiCl, HN4Cl, rapamycin, LY294002, Wortmannin, PD98059, PD0325901, SB431542, SB203580, SP600125, retinoic acid, Asiatic acid, Y27632, thiazovivin, z‐VAD‐FMK, VPA, TSA, VO‐OHpic, SF1670, KU‐55933, resveratrol, STR1720, CX‐4945, ABT‐737, nutlin‐3, pifithrin‐α, pifithrin‐μ, GSK1904529A, and FG‐4592.
Human ESC Cardiac Differentiation via Monolayer Model
Human ESC cardiac differentiation was carried out in 24‐well culture plates if there were no special circumstances. At day −5, 2.5×104/cm2 singularized hPSCs were planted on Matrigel in 0.5 mL mTeSR1 for 2 days, followed by another 3 days in the presence of 12 μmol/L CHIR99021 plus 10 nmol/L rapamycin (gene operation). Next, the medium was changed to 0.5 mL RPMI/B27 minus insulin (Invitrogen, Carlsbad, CA) also supplemented with 12 μmol/L CHIR99021 plus 10 nmol/L rapamycin for 1 day, and then cells were treated with 10 μmol/L XAV939 plus 10 μmol/L KY02111 in 0.5 mL RPMI/B27 minus insulin for 4 days without medium replacement. Cells continued to be cultivated in RPMI/B27 minus insulin for 2 days, finally maintained in 1 mL RPMI/B27 with insulin (Invitrogen) until applied in related experiments (changing medium every 3 days).
Human ESC Cardiac Differentiation via EB Model
Prior to formation of EBs, the hESCs were maintained on matrigel for at least 1 generation. To form EBs, hESC colonies were dissociated with dispase (1 mg/mL; Invitrogen) and gently scraped off from the wells. The cell clusters were pipetted to about 20 to 100 cells in size and then cultured in mTeSR1 with 10 μmol/L Y27632 (Selleck Chemicals, Houston, TX). At the second day, the medium was changed to StemPro‐34 (Invitrogen) supplemented with 1 mmol/L ascorbic acid, 2 mmol/L l‐glutamine, and transferrin (Sigma‐Aldrich). Activin A, BMP4 (BD Biosciences), and bFGF were added on this day. After 3 days of culture, human‐VEGF and XAV939 were added for another 4 days. Then the supplemental factors were changed to VEGF (Pepro Tech, Rocky Hill, NJ) and bFGF, and cells were harvested at the 15th or 30th day.
Quantitative Reverse Transcription Polymerase Chain Reaction
Total RNA was prepared with Trizol (Invitrogen) and treated with DNase (Qiagen, Hilden, Germany). RNA (0.5 μg) was reverse transcribed into cDNA via random primer with M‐MLV reverse transcriptase (Promega, Madison, WI). Quantitative real‐time polymerase chain reaction (PCR) was performed using SYBR Green (Toyobo, Osaka, Japan) on a Roche 480 Real‐Time PCR System (Indianapolis, IN). The relative RNA abundance was calculated with 2−ΔΔCt means and normalized with respect to GAPDH expression level. The primer sets are listed in Table S1.
Immunoblot Analysis
Cells with different small‐molecular treatments were harvested at the indicated time points and lysed with Triton buffer (0.5% Triton X‐100 and 20 mmol/L Hepes, pH 7.6)‐containing cocktail. Proteins were separated by 10% or 15% (wt/vol) Tris glycine SDS‐PAGE under denaturing conditions and transferred to a nitrocellulose membrane. After blocking with 5% (wt/vol) milk in Tris‐buffered saline with 0.1% (vol/vol) Tween 20, the samples were incubated with primary antibody overnight at 4°C. The second day, the samples were washed 3 times in Tris‐buffered saline with Tween 20 for 5 minutes and then incubated with an anti‐mouse/rabbit/goat peroxidase‐conjugated secondary antibody at room temperature for 1 hour, finally developed by SuperSignal chemiluminescence (Pierce [Dallas, TX] or Millipore [Billerica, MA]). Each assay was performed at least 3 times independently. Antibodies are listed in Table S2.
Immunostaining
Cells were fixed with 4% (vol/vol) paraformaldehyde for 15 minutes and then permeated with 0.1% (vol/vol) Triton X‐100 for 15 minutes at room temperature. The samples were blocked with a 5% solution of goat serum in PBS and incubated with primary antibody against cTnT (1:250), α‐actinin (1:250), and Brachyury (T) (1:250) overnight at 4°C. Next‐day, samples were incubated with secondary fluoresce‐labeled anti‐mouse/rabbit antibody (1:1000) for 1 hour at room temperature. Nuclei were stained with DAPI (1 μg/mL; Invitrogen) in PBS for 3 minutes. Images were captured under Olympus fluorescent microscopy. Antibodies are listed in Table S2.
Flow Cytometry
Cultured monolayer hESCs or EB were dissociated by accutase or 0.1% Trypsin into single cells, fixed with 1% (vol/vol) paraformaldehyde for 15 minutes at room temperature, and then stained with primary and secondary antibodies in PBS containing 1% (wt/vol) BSA and 0.1% Triton X‐100. Intracellular eGFP analysis does not need fixation. Data were collected on a Caliber flow cytometer (Beckton Dickinson, Franklin Lakes, NJ) and analyzed by FlowJo (Ashland, OR). Antibodies are listed in Table S2.
RNAi
Human mTOR, TSC1/2, p53, and AMPK1a siRNA sequences were all previously21, 22, 23, 24, 25 described (Table S1) and synthesized by GenePharma Inc (Shanghai, China). The oligos working concentration was 100 nmol/mL, and hESC transfection was exerted by oligofactamine (Invitrogen) 20 hours after hESCs plated as monolayer in 2.5×104/cm2.
Electron Microscopy
The induced‐hPSC‐derived cardiomyocytes were directly scraped off from the dish and then fixed with 2% glutaraldehyde overnight at 4°C.26 These samples were postfixed with 0.25% osmium/0.25% K4Fe(CN)6, 1% tannic acid, followed with 50 mmol/L uranyl acetate. Then specimens were washed 3 times and dehydrated with a series of ethanol. Finally, the cell samples were embedded in araldite 502 resin (Polysciences Inc, Warrington, PA), and polymerization proceeded at 65°C for several days. The ultrathin sections (≈60 nm) obtained by ultramicrotome (Leica EM UC7; Leica, Wetzlar, Germany)) were mounted in EM‐grids, stained with lead citrate, and then observed by FEI Tecnai G2 Spirit TEM (FEI, Hillsboro, OR).
TOPflash Assay
Human iPSCs were cultured in RPMI/B27 differentiation culture, and HEK293 cells cultured in 10% FBS‐DMEM medium (Gibco, Gaithersburg, MD). Cells were transfected with TOPflash plasmid (Addgene, Cambridge, MA) and renila plasmid as internal reference. After 24 hours, CHIR, CHIR plus rapamycin, rapamycin, and mouse Wnt3a (100 ng/mL) were added to the medium. After an additional 24 hours, the luciferase activities were measured by a laser‐scanning Varioskan Flash (Thermo Fisher).
Apoptosis Assay
Annexin V/PI staining was operated with an annexin V/PI kit (Roche) per manufacturer's protocol and analyzed via Caliber flow cytometer (Beckton Dickinson). Intracellular caspase 3/7 activity was measured with Caspase‐Glo 3/7 Assay Systems (Promega) per manufacturer's protocol and analyzed with a laser‐scanning Varioskan Flash (Thermo Fisher). z‐VAD‐FMK (Abcam, Cambridge, UK) working concentration is 50 μmol/L. Cytochrome c released from mitochondria into cytosol during apoptosis was isolated and detected by Cytochrome c Releasing Apoptosis Assay Kit (Abcam).
Intracellular Reactive Oxygen Species Level
Cells cultured in a 96‐well plate were treated with small molecule or special gene RNAi for indicated periods and then incubated in medium containing 25 μmol/L DCFC‐DA (Sigma‐Aldrich, St. Louis, MO) for 30 minutes at 37°C. Stained cells were washed with PBS and analyzed with a laser‐scanning Varioskan Flash (Thermo Fisher).
Mitochondrial Analysis
Mitochondrial transmembrane potential was measured by JC‐1 dye. JC‐1 dye was carried out with JC‐1 kit (QianChen Inc, Shanghai) as per manufacturer's protocol and analyzed via a laser‐scanning Varioskan Flash (Thermo Fisher). Green and red fluorescence were analyzed at 490 and 525 nm, respectively, and the ratio of green/red was considered an indicator of mitochondrial transmembrane potential.
Assessment of Cell Proliferation and Viability
Cell mass was evaluated by determining the cell number in a 24‐well plate for 2 repetition and for 3 parallel samples. Cell viability was measured by Cell Counting Kit‐8 (CCK‐8) (Dojindo Laboratories, Kumamoto).
Colony Growth Assay
H9 cells were dissociated into single cells and seeded in Matrigel‐coated 12‐well plates at a density of 1×105 per well with different treatments of small molecules (0.1% DMSO, 12 μmol/L CHIR, or CHIR plus 10 nmol/L rapamycin). The medium was changed every second day, and colony number was counted at the sixth day.
Cell Cycle Analysis
Cells were incubated with certain small molecules for an indicated time, then harvested, and fixed with 70% ethanol at −20°C overnight. After a washing with PBS, the hESCs were stained with 0.5 mL of 50 μg/mL propidium iodide solution plus 20 U/mL RNase A for 30 minutes at room temperature. Data were collected by Caliber flow cytometer and analyzed with CellQuest Pro software (both from Beckton Dickinson).
BrdU Incorporation Assay for Cell Proliferation
The cells were labeled with 10 μmol/L BrdU for 1 hour and then dissociated by accutase. The hESCs were fixed with 1% paraformaldehyde and washed with PBS; then 1 mL denaturing solution (2 mol/L HCl) was added for 20 minutes. Cells were added with 1 mL wash buffer and then centrifuged for 5 minutes. The cells were resuspended with 0.1 mol/L sodium borate (pH 8.5) and centrifuged for 5 minutes. The hESCs were rinsed again with PBS containing 2% (wt/vol) BSA and 0.1% Triton X‐100 and were then stained with anti‐BrdU antibody for 1 hour at room temperature. After 1 such washing, the hESCs were labeled with the fluorescein isothiocyanate conjugated secondary antibody. Nuclei were stained with 10 μg/mL PI for 10 minutes. Data were collected on a Caliber flow cytometer and analyzed by FlowJo (Ashland, OR).
Glucose Measurement
For glucose measurement, the hESCs were cultured in 12‐well plates with 1 mL of medium containing different inhibitors. At the indicated time points, the medium was collected and pelleted at 1000g for 5 minutes at 4°C. The supernatant was stored at −80°C until the test was performed. Glucose concentration within media was measured using a Glucose Assay Kit (Sigma‐Aldrich, St. Louis, MO).
Oxygen Consumption Rate Measurement
O2 consumption was measured using Seahorse XF24 extracellular Flux analyzer (Agilent, Santa Clara, CA) based on a method described by Zhang and colleagues.27 Briefly, 5×104 hESCs were seeded in each well of a XF24 cell culture plate. The second day, the cells were treated with respective inhibitors for 24 hours. One hour before the analysis, the culture medium was replaced by unbuffered DMEM, and plates were incubated at 37°C for 1 hour to stabilize the pH and temperature; then basal respiration was measured.
Statistical Analysis
Statistical analyses were performed as previously described.28 Briefly, all quantitative data were first evaluated by the Shapiro‐Wilk test to see whether they followed the normal distribution. For the data with normal distribution, a Levene test of homogeneity of variance was further performed. Then the data were analyzed by independent sample t test for comparison between 2 groups. Comparison among multiple groups was carried out by 1‐way ANOVA followed by Tukey's post hoc test unless otherwise specified. Data were presented as means±SEM. For the data that were not suitable for a parametric test, the Mann‐Whitney U test or the Kruskal‐Wallis test followed by a Dunn post hoc comparison was performed. The data were presented as median and range. All P values are 2‐sided, and P<0.05 was considered a statistically significant difference. The significance level is indicated as P<0.05 (*) or P<0.01 (**). Quantitative data plotting was performed in SigmaPlot version 10 (Systat, San Jose, CA), and all statistical calculations were performed using SPSS version 22.0 software (IBM, Armonk, NY).
Results
Rapamycin Potently Improves hPSC Cardiomyogenic Differentiation
We established a screening system using a genetically modified H9 cell line that expresses eGFP driven by human cTnT promoter toward cardiac differentiation in a modified monolayer‐based cardiac differentiation protocol adopted from the work of Murry and colleagues. Driven by the extant findings indicating that Wnt signaling is essential to cardiomyogenesis,29 we first found that addition of Wnt signaling activator CHIR99021 (12 μmol/L) in the first 4 days of culture (3 days in mTeSR1 and 1 day in RPMI B27 medium), and sequential addition of XAV939 (10 μmol/L) and KY02111 (10 μmol/L),30 which are 2 Wnt signaling inhibitors, during the next 4 days without changing the medium to enrich the secretory cytokines, which are believed to be essential for cardiac lineage specification in this stage, could produce ≈10% spontaneously contracting cTnT‐expressing cardiomyocytes without addition of any growth factors. This allowed us to screen a range of small molecules for their cardiomyogenic ability based on this method. Unexpectedly, we found that rapamycin, an mTOR signaling inhibitor, increased the purity of hESC‐derived cardiomyocytes to more than 84% (Figure 1A). Accordingly, treatment with rapamycin prominently enhanced the mRNA expression level of both cardiac‐specific transcription factors and structural proteins, such as NKX2.5, GATA4, MEF2C, MESP1, ISL1, MHY6, cTnT, MLC2a, and MLC2v compared with that in the untreated group at day 5 and day 12, respectively (Figure S1A and S1B). Temporal observations using the H9‐cTnT‐eGFP hESC line showed that GFP‐expressing cells emerged on day 9, whereas day 12 marked the onset of strong, large‐amplitude beating.
Figure 1.
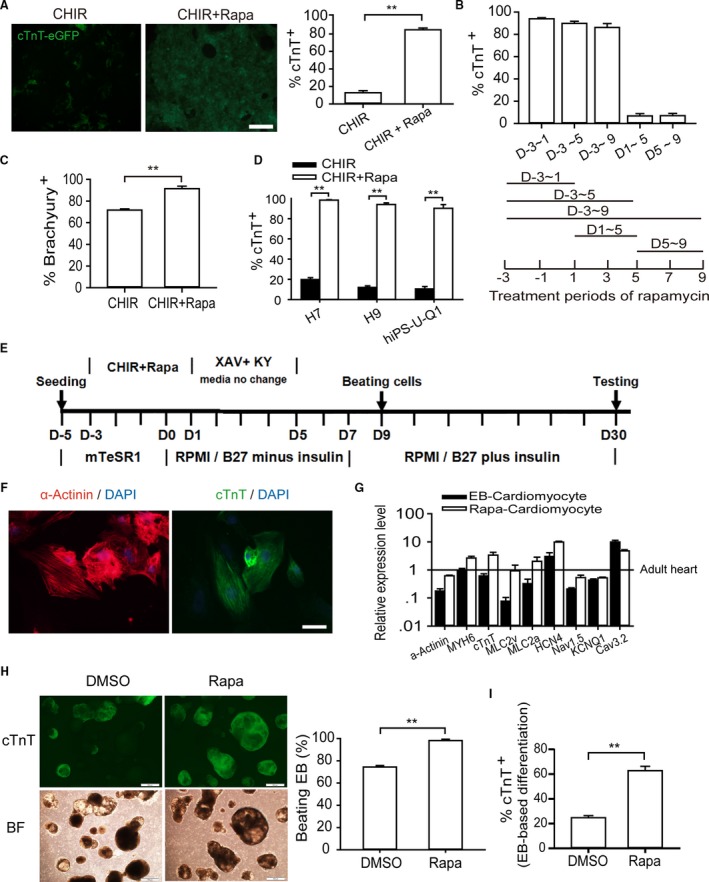
Rapamycin promotes cardiomyocyte differentiation in monolayer‐based growth factor–free system. A, Analyzing the efficiency of cardiomyocyte differentiation in terms of reporter activity. Left and right graphs show the ratio of GFP‐positive cardiomyocytes identified by fluorescence observation and flow cytometry analysis (12 μmol/L CHIR, 10 nmol/L rapamycin). The asterisks indicate statistically significant differences compared with controls (n=5; **P<0.01). Scale bars, 200 μm. B, Effective time window of rapamycin treatment. Addition of rapamycin from day −3 to day 1 is sufficient for highly efficient cardiogenesis based on GFP‐positive cell percentage (n=5). C, Flow cytometry validation of Brachyury‐positive cells at day 4 (n=5; **P<0.01). D, hESC lines H9 and H7 and hiPSC line U‐Q1 were induced into cardiomyocytes with or without rapamycin (n=6; **P<0.01). E, Schematic of protocol for small molecule–mediated cardiomyocyte differentiation from hPSC. F, Immunostaining of cTnT and α‐actinin showed sarcomere organization. Scale bars, 10 μm. G, Gene expression analysis of rapamycin‐induced cardiomyocytes. The expression levels of cardiac marker genes (α‐Actinin,MYH6, cTnT,MLC2v, and MLC2a) and channel genes (HCN4, Nav1.5,KCNQ1, and Cav3.2) were nearly equal to those of EB‐derived cardiomyocyte and adult heart tissue, which was considered to be 100% (n=5). H and I, The effect of rapamycin on EB‐based cardiac differentiation. Rapamycin was added in the first stage (0 to 4 days) of EB differentiation, and cardiac differentiation efficiency was assessed at day 15. The percentage of beating EB (H) and the ratio of GFP‐positive cardiomyocytes (I) were analyzed (n=5; **P<0.01). Scale bars, 500 μm. BF indicates bright field; CHIR, CHIR99021; DMSO, dimethyl sulfoxide; EB, embryoid body; eGFP, enhanced green fluorescent protein; hESC, human embryonic stem cell; KY, KY02111; Rapa, rapamycin; XAV, XAV939.
In order to precisely identify the specific stage in which rapamycin exerted its function, rapamycin was added and withdrawn at the indicated time points. We found that administration of rapamycin in the initial stage of differentiation (days −3 to 1) was sufficient to increase the percentage of cTnT‐positive cardiomyocytes up to 93.5±2.1%. On the other hand, prolonged administration of rapamycin compromised the efficiency of cardiomyocyte induction and significantly reduced the actual yield of cardiac cells (Figure 1B). However, addition of rapamycin during days 1 to 5 or days 5 to 9 resulted in very low cardiac induction and severe cell death. These results indicate that the effect (either positive or negative) of rapamycin on cardiac differentiation is stage‐specific, further suggesting that the pro–cardiac differentiation function of rapamycin is mostly confined to the early stage when hESCs give rise to mesoderm cells, whereas it exhibits a negative effect in subsequent stages. Consistently, we found that the administration of rapamycin increased the mRNA expression level of Brachyury (T), a widely accepted marker of mesoderm, compared with only CHIR treatment, and this promoting effect of rapamycin can be blocked by 3BDO, a novel mTOR activator (Figure S1C and S1D). Accordingly, flow cytometry analysis confirmed that, because of the administration of rapamycin, the Brachyury‐positive cell percentage increased from 75% to over 95% at day 1 (Figure 1C). We then attempted to identify the optimal concentration of rapamycin treatment. The promoting function of rapamycin was significant and stable when its concentrations ranged from 5 to 100 nmol/L, and we chose 10 nmol/L as the working concentration for the following experiments (data not shown).
To evaluate the compatibility of rapamycin across different hPSC lines, we tested the effects of rapamycin within unmodified hESC lines H9, H7, and hiPSC line U‐Q1 (derived from human urine cell).31 In line with the high tendency toward monolayer‐based differentiation of H7, flow cytometry analysis showed that ≈98.3±0.76% of H7‐derived cells and 93.3±2.1% of H9 were positive for the cardiac marker cTnT, respectively. We also obtained more than 90.6±3.6% cTnT‐positive cardiac cells from the U‐Q1 iPSC line, even though hiPSCs are considered much harder to induce than hESCs are (Figure 1D). The optimal induced conditions are illustrated in Figure 1E. (For convenience, the 4 days with rapamycin treatment were marked as days 1 to 4 in the following text and figures.) Treatment combining rapamycin with CHIR in the first 4 days, followed by XAV939 and KY02111 treatment for an additional 4 days without changing media, produced >90% cardiomyocytes that contract spontaneously as coordinated sheets (Videos S1 and S2) in multiple (≥6) independent experiments in 4 hPSC lines. By immunocytological staining of sarcomeric cTnT and α‐actinin, cardiomyocytes derived from hPSCs were shown to possess normal cardiac sarcomere organization (Figure 1F). Electron microscopy also identified the classic cardiomyocyte structural elements, including myofibrils, Z bands, and intercalated disks, within rapamycin‐induced hPSC cardiomyocytes (Figure S1E). Real‐time PCR analysis of rapamycin‐induced cardiomyocytes at day 30 showed that the expression levels of cardiac marker genes (cTnT, MYH6, and HCN4) and important ion channel genes (Nav1.5,Cav3.2, and KCNQ1) examined were similar to those of classic EB‐based differentiation‐derived cardiac cells and adult heart tissue (Figure 1G). After being cultured with 1 mmol/L lactate as the only carbon source for 7 days, the purity of hPSC‐derived cardiac cells was found to increase to 99.9% (Figure S1F).32 We posit that this effect has the potential to minimize the risk of tumorigenesis caused by stem cell contamination in ultimate applications aimed at cell‐based therapies. On average, we obtained 24 cardiomyocytes from 1 original pluripotent cell. Therefore, we have designed and implemented an efficient and high‐yield cardiac differentiation system without the need for additional growth factors.
To investigate whether rapamycin is a general cardiomyocyte differentiation‐promoting agent, we applied rapamycin in classical EB‐based differentiation in line with the approach published by Gordon Keller's lab.33 Interestingly, administration of rapamycin in the early stage (days 0 to 4) increased frequency of beating EB from about 70% to ≈100% in H9‐cTnT‐eGFP cells, while cTnT‐positive cardiomyocyte percentage enhanced from 30% to 60% compared with the solvent‐exposed control (DMSO) group (Figure 1H and 1I). In addition, rapamycin in concentrations below 10 nmol/L had a positive influence on EB growth (Figure S1G). However, addition of CHIR at the same stage led to much larger‐sized EBs with few cTnT‐expressing cells (data not shown). Combined, these results indicate that rapamycin generally promotes cardiac differentiation in different models, and this function is CHIR independent.
Cardiac Differentiation Induced by Rapamycin in hPSCs Is Dependent on mTOR Signaling
The high selectivity of rapamycin in targeting mTOR had been demonstrated in many extant studies, and our results indicate that mTOR activity was dramatically suppressed by rapamycin during 3 days of culturing in mTeSR1, revealed by the decrease of phosphorylation of ribosomal subunit S6 kinase 1 (S6K1) on threnine‐389, a widely used marker for monitoring mTOR activity (Figure 2A). Furthermore, H9‐cTnT‐eGFP hESCs were transfected with siRNA to knock down mTOR expression (Figure 2B). Similar to rapamycin treatment, reducing mTOR expression potently promoted the cardiomyocyte differentiation relative to the negative control. In addition, we used previously described siRNA to knock down TSC1 and TSC2, 2 important negative regulators of the TORC1 pathway,23 resulting in much higher mTOR expression and S6K1 phosphorylation. The overactivated mTOR signaling led to much lower cTnT‐positive cell generation (Figure 2C). In addition, in most cases, all the cells died after day 5. Taken together, these findings indicate that the efficiency of cardiac differentiation from hESC is inversely correlated with the mTOR activity.
Figure 2.
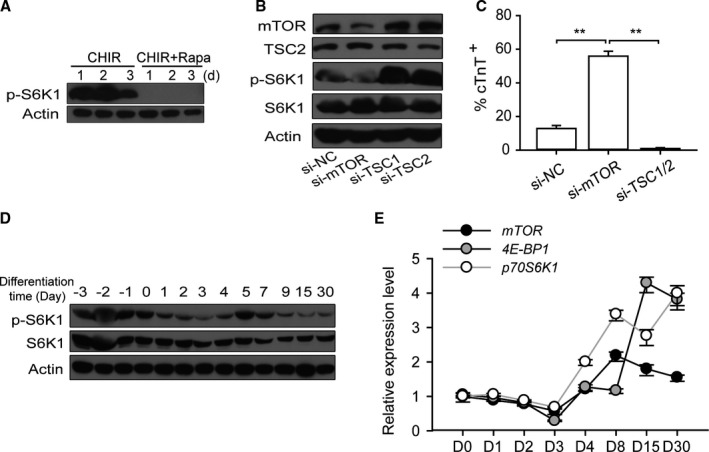
Suppression of mTOR is required for highly efficient cardiac differentiation. A, The activity of mTOR signaling is inhibited by rapamycin administration, shown by the phosphorylation of p‐S6K1 (Thr389). B, The effects of mTOR knockdown and overactivation on cardiomyocyte differentiation. mTOR siRNA significantly decreases mTOR expression and activity. In contrast, TSC1 and TSC2 siRNA increase mTOR expression and activity. C, The percentage of cardiomyocytes following siRNA treatment, as measured by flow cytometry (n=4; **P<0.01). D and E, The dynamic fluctuation of mTOR activity in experiments without rapamycin treatment. mTOR activity was analyzed by Western blot; p70S6K1 (Thr389), S6K1, and β‐actin were used as the loading control (D). The transcription levels of mTOR signal‐associated genes including mTOR, 4E‐BP1, and p70S6K1 as measured by quantitative real‐time PCR (E). CHIR indicates CHIR99021; PCR, polymerase chain reaction; Rapa, rapamycin; si‐NC, negative control siRNA.
Because the dynamic change of mTOR expression during early cardiac development was unclear, we sought to examine the mTOR expression profile during in vitro differentiation. The dynamic fluctuation of mTOR activity in experiments treated with CHIR alone without rapamycin treatment was assessed by analyzing the phosphorylation of S6K1. The results showed that mTOR was persistently activated in the mTeSR1‐cultured stage (days −3 to 0), while dramatically decreasing at the beginning of cardiac induction (days 0 to 3) in the monolayer differentiation model. Thereafter, its activity tended to be restored on the emergence of the cardiac progenitor cells (days 5 to 20) (Figure 2D). Finally, based on our observations, the mRNA levels of mTOR signaling‐associated genes such as mTOR, p70S6K1, and 4E‐BP1 had a similar expression trend and preceded the dynamic fluctuation of mTOR activity (without rapamycin treatment) (Figure 2E).
Combined Treatment With Rapamycin and CHIR Antagonize the Apoptosis of hESCs in a High‐Density Monolayer Culture
Our results showed that rapamycin not only boosted the efficiency of cardiomyocyte differentiation but also increased the total number of cells at the end of differentiation. During the differentiation culture we noted an interesting phenomenon: the rapamycin‐treated hPSCs were morphologically more compact and more resistant to digestion relative to those that had not been treated with rapamycin. We also observed fewer floating dead cells relative to the amount found in the group that had been treated with CHIR only. Interestingly, we found that rapamycin markedly increased the cell count during culturing in mTeSR1. The cell viability measured by Cell Counting Kit‐8 (CCK8) yielded similar results (Figure 3A and 3B). However, if hESCs were seeded at a lower density (0.75×104/cm2), rapamycin did not promote cell viability during the first 2 days. In contrast, on the third day, when cell density increased, the viability of the rapamycin‐treated group exceeded that of the CHIR‐treated group (Figure 3C). In addition, the hESCs colony formation assay revealed that CHIR treatment promoted single‐cell survival and the proliferation to colony compared with the DMSO (vehicle control) and that rapamycin was unable to promote this effect (Figure S2A). Taken together, these results reveal that the growth‐promoting function of rapamycin arose only when hESCs were grown at high density.
Figure 3.
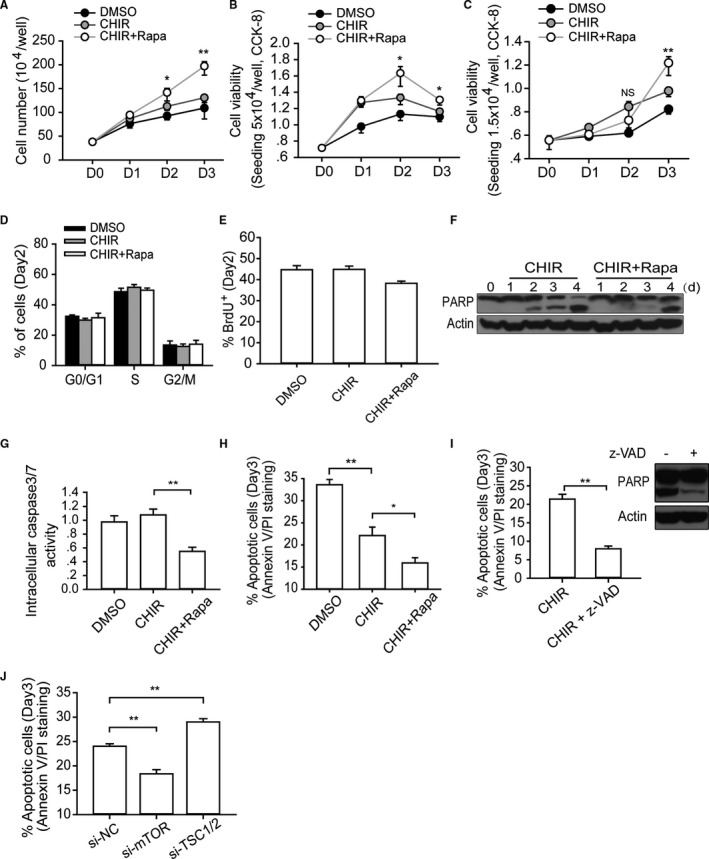
mTOR regulates the apoptosis of hESCs during high‐density monolayer culture. A and B, Cell number calculation and viability measurement. Single H9 cells (5×104/well) were plated in 24‐well plates, cultured with indicated small molecules, and then collected and analyzed at designated time points. One‐way ANOVA with Bonferroni post hoc test was used to calculate statistical significance between CHIR alone and CHIR plus rapamycin–treated cells at each time point (n=4; *P<0.05, **P<0.01). C, Analyzing the dynamic change of cell viability when initial seeding density is much lower. Single H9 cells (1.5×104/well) were plated in 24‐well plates, cultured with indicated small molecules, and then collected and analyzed at designated time points. One‐way ANOVA with Bonferroni post hoc test was used to calculate statistical significance between CHIR alone and CHIR plus rapamycin–treated cells at each time point (n=4; **P<0.01; NS means no significant difference). D and E, Cell cycle and BrdU incorporation analysis of hESCs with different treatments at day 2 (n=4). F through H, The cell death level was analyzed by a different method. Analyzing the cell death levels of hESCs with corresponding treatment by measuring the extent of PARP cleavage (F), intracellular caspase 3/7 activity (n=5) (G), and annexin V/PI staining (n=6) (H) (*P<0.05, **P<0.01). I, Annexin V/PI staining and Western blot analysis of cell death of hESCs with z‐VAD addition (n=4; **P<0.01). J, Annexin V/PI staining analysis of the apoptotic level of hESCs with si‐mTOR or siTSC1/2 treatment at day 3 (n=5; **P<0.01). CHIR indicates CHIR99021; DMSO, dimethyl sulfoxide; hESC, human embryonic stem cell; Rapa, rapamycin; si‐NC, negative control siRNA; z‐VAD, z‐Val‐Ala‐Asp (OMe)‐fluoromethylketone (z‐VAD‐FMK).
To elucidate how rapamycin increased the cell count, we examined the hESC proliferation and apoptotic levels resulting from different treatments. First, cell proliferation rate was analyzed by cell cycle analysis and BrdU staining at day 2, and there was no significant difference between the CHIR group and the CHIR‐plus‐rapamycin group (Figure 3D and 3E). This indicated that a low concentration (10 nmol/L) of rapamycin did not compromise the proliferation ability of hESCs. Next, we analyzed the extent of cell death induced by different treatments by assessing the cleavage of PARP and the activity of intracellular caspase 3/7. We found that these apoptotic indicators were significantly reduced by rapamycin (Figure 3F and 3G). The annexin V/PI staining assay was subsequently employed to obtain a finer determination. At the end of day 3 the DMSO‐treated group exhibited an average of 33.6% annexin V–positive cells, while CHIR reduced the number of dead cells to 22.1%. Moreover, addition of rapamycin further reduced the proportion of dead cells to 15.9% (Figure 3H). However, it is important to note that hESCs, unlike somatic cells, are more vulnerable to apoptosis following enzymatic dissociation, and the percentage of dissociation‐caused apoptosis is typically about 10%.34
Next, we added 50 μmol/L z‐Val‐Ala‐Asp (OMe)‐fluoromethylketone (z‐VAD‐FMK), a widely‐applied pan caspase inhibitor, to the cell culture, which resulted in no floating cells after 24 hours. Moreover, annexin V/PI staining indicated that more than 90% cells were both annexin V– and PI‐negative (Figure 3I). We speculated that the remaining cell death events were caused by accutase dissociation and other modes of operation damage. However, if the z‐VAD‐FMK treatment lasted for 4 days, all hESCs died when the medium was changed to RPMI/B27, which suggested that an alternative death pathway was activated when apoptosis pathway was blocked. More importantly, we found that elevating mTOR activity by TSC1/2 RNAi obviously aggravated the apoptotic levels of hESC on day 3 (Figure 3J), and similar results were obtained by TSC1/2 RNAi on day 4 (Figure S2B). We also found that the addition of rapamycin effectively hindered the cell death (<40%) during the initial stage of differentiation on day 4 (Figure S2C). However, we posited that the more pronounced cell death noted in this initial differentiation stage resulted from not only the fundamental change in the culture conditions but also greater sensitivity of hESCs to enzymatic dissociation at this stage. Given the complexity of mechanisms leading to cell death following the change to RPMI/B27 medium, here we focus on the cell death that took place in mTeSR1 culture. In sum, rather than suppressing the expansion of hESCs, mTOR inhibition protected hESCs from apoptosis in the high‐density monolayer culture model.
The Apoptosis of hESCs Is p53 Dependent and Can Be Attributed to the Glucose Stress During High‐Density Growth
To identify the cause of apoptosis, we tested the expression of main factors involved in the apoptosis of hESC. First, we found that the cleavage of caspase 8 was not significantly different between groups with and without rapamycin treatment on day 3 (Figure 4A), suggesting that the apoptosis was not triggered by extracellular apoptosis‐inducing ligands. In contrast, we found that p53 progressively increased during DMSO and CHIR treatment; however, these changes were mitigated by rapamycin (Figure 4B). In addition, the phosphorylated p53 (Ser15) was predominantly accumulated in nucleus, and rapamycin prevented this accumulation (Figure 4C). The phosphorylation of p53 on Ser15 was demonstrated to prevent p53 negative regulator MDM2 binding and to result in the accumulation and increased transcriptional activation ability of p53.35 These results indicated that aforementioned apoptotic response was induced by a p53‐dependent endogenous apoptosis pathway. Furthermore, real‐time analysis showed that p53 proapoptotic downstream genes (especially P21) exhibited consistent change patterns on day 2 (Figure 4D). It was noteworthy that the mRNA levels of p53 in the group treated with CHIR only and in that treated with CHIR accompanied by rapamycin were similar, which suggested that rapamycin regulated p53 at the protein level. Next, we used siRNA to knock down p53 protein expression in hESCs. We found that p53‐RNAi hESCs were more resistant to apoptosis compared with the negative control cells (Figure 4E), which demonstrated that the apoptosis of hESCs was mainly mediated by p53 accumulation. More interestingly, p53 knockdown prominently increased the efficiency of cardiac differentiation from 10% to nearly 50% (Figure 4F), which mimics the effect of rapamycin. In sum, these results established a causal link between apoptosis and highly efficient cardiac differentiation.
Figure 4.
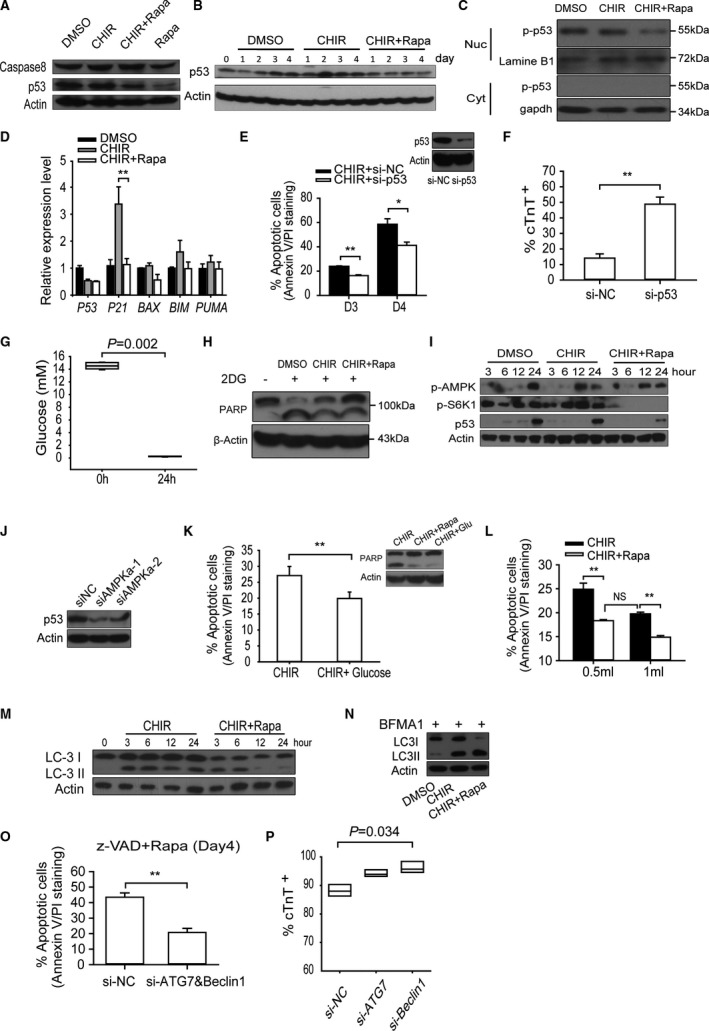
Rapamycin protects the hESCs from environmental stress‐induced apoptosis by reducing p53 accumulation, which is essential for high‐efficient differentiation. A, Western blot analysis of caspase8 and p53 in hESCs between groups with or without rapamycin treatment on day 3; DMSO‐treated cells were used as control. B, Western blots of p53 in cells cultivated in medium with CHIR or CHIR plus rapamycin; DMSO‐treated cells at different time points were used as control. C, Western blot analysis of the distribution of the phosphorylated p53 (Ser15) in cytoplasm and nucleus. D, P53 and its downstream targets (P21,BAX,BIM, and PUMA) expression on day 3 were evaluated by quantitative real‐time PCR (n=5; **P<0.01). E, Knockdown of p53 expression reduced the apoptosis of hESCs on the third and fourth days. The graph on the top right corner shows the effect of p53 siRNA on the expression level of p53 protein (n=5; **P<0.01, *P<0.05). F, Flow cytometry analysis of cTnT‐positive cell percentage in H9‐cTnT‐GFP cells with p53 RNAi and negative control RNAi cells (n=5; **P<0.01). G, The change of medium glucose concentration after 24 hours of culture with CHIR addition (n=6; Mann‐Whitney U test). H, Western blot analysis of cell apoptosis indicated by cleavage of PARP, in groups treated with or without glucose analogue 2DG (2‐deoxy‐d‐glucose), which blocks glycolysis and mimics glucose stress. I, Western blot analysis of the level of p‐AMPK (Thr172), p‐S6K1 (Thr389), and p53 in hESCs cultured with CHIR at indicated time point. J, Knockdown of AMPK by siRNA reduced the accumulation of p53. K, Glucose rescue assay. Supplying the cells with additional 5 mmol/L glucose twice reduced the level of apoptosis revealed by PARP cleavage and annexinV/PI staining (n=6; **P<0.01). L, Analyzing the effect of enlarging culture system, which could reduce environmental stresses on hESC apoptosis. The medium volume was enlarged from 0.5 to 1 mL (n=4; **P<0.01; NS means no significant difference). M and N, Western blot analysis of autophagy level in hESCs with or without rapamycin treatment. LC3II/LC3I expression levels were measured in first 3 days (M), and their accumulation levels were analyzed when hESCs were simultaneously treated with bafilomycin A1 for 6 hours (N). O, Apoptotic levels of hESCs with ATG7 and Beclin1 RNAI, which compare to negative control RNAi in the presence of z‐VAD‐FMK on day 4 (n=5; **P<0.01). P, Knockdown of autophagy essential gene Beclin1 enhanced the efficiency of cardiac differentiation (n=3; Kruskal‐Wallis test followed by Dunn post hoc comparison). BFMA1 indicates bafilomycin A1; CHIR, CHIR99021; Cyt, cytoplasm; DMSO, dimethyl sulfoxide; hESC, human embryonic stem cell; Nuc, nucleus; PCR, polymerase chain reaction; Rapa, rapamycin; si‐NC, negative control siRNA; z‐VAD, z‐Val‐Ala‐Asp (OMe)‐fluoromethylketone (z‐VAD‐FMK).
It is necessary to ascertain which circumstance or molecular pathway is the major trigger of the hESCs apoptosis discussed above. Because the hESCs were in a long‐duration and high‐density culture, nutrients such as glucose and amino acids would be expected to be rapidly consumed due to the significant number of cells and high rate of proliferation. To assess the validity of this premise, we measured the extracellular glucose levels. Glucose concentration declined from the initial 15 to 0.2 mmol/L after 24 hours of culture with the monolayer of cells completely confluent (Figure 4G). Next, we added 1 mmol/L 2‐deoxy‐d‐glucose (2‐DG), an inhibitor of glycolysis, into the culture medium to mimic the conditions of glucose deprivation. As expected, 2‐DG treatment enhanced the cleavage of PARP, whereas rapamycin evidently eliminated it (Figure 4H). Likewise, a decrease in intracellular energy level was also reflected by the enhancement of the phosphorylation of AMP‐dependent kinase (AMPK), an essential sensor for energy limitation, whose activation preceded the accumulation of p53 (Figure 4I). In addition, mTOR remained activated in the CHIR‐treated group, which may be attributed to the obstruction of the critical function of GSK‐3β on TSC1/2 phosphorylation.36 These findings are consistent with those yielded by a previous research, in which the authors reported that glucose deprivation was able to stimulate p53 accumulation and led to apoptosis in TSC2 knockout fibroblast cells through activated AMPK and mTOR signals and rapamycin blocked the mTOR‐dependent p53 translation.37 This was a special case in which rapamycin induced growth rest or apoptosis of cells in common situations. In this scenario, we hypothesized that glucose stress induced p53 accumulation, which, in cooperation with unsuppressed mTOR activity, triggered the hESCs apoptosis. To test this hypothesis, we knocked down AMPK by siRNA and found the p53 accumulation significantly reduced (Figure 4J). Finally, we added 5 mmol/L of glucose into the culture medium twice, at the 6‐ and 12‐hour time points, respectively. Subsequent cleavage of PARP and annexin V/PI staining analysis revealed that the extent of cell apoptosis was partially decreased (Figure 4K). Taken together, these results demonstrate that the shortage of glucose supply was sufficient to trigger p53‐dependent apoptosis in rapidly growing hESCs. We speculated that this shortage of nutrients in limited culture medium can be remedied by an expanded culture system, which would attenuate the apoptosis tendency of hESCs. In order to ascertain whether the fluctuations in the culture conditions are the only reason for hESCs apoptosis in a monolayer‐based culture, we increased the culture medium volume from 0.5 to 1 mL for each well of a 24‐well plate to reduce the extracellular stress. We found that increasing medium volume obviously decreased the apoptotic level, indicating that the energy stress and medium change were the main causes of hESCs cell death. However, we also noted that, in this enlarged culture system, the addition of rapamycin could further prevent cell apoptosis (Figure 4L). These observations indicate that there is another way in which rapamycin could work to reduce hESC apoptosis.
Because mTOR is the most important regulator of autophagy, we also attempted to establish whether autophagy participated in the antiapoptosis or prodifferentiation functions of rapamycin. In simple terms, autophagy can be understood as a double‐edged sword in cell death. In cell starvation conditions, autophagy is considered to function as a prosurvival mechanism exhibited via a catabolic process that replenishes nutrients for fueling the bioenergetic machinery. In other contexts, excess autophagy can lead to autophagic cell death (Type II cell death) by degrading essential components of cells. First, we examined the level of autophagy among different small molecular–treated groups with or without bafilomycin A1, used here because it disturbs fusion between autophagosomes and lysosomes. The inclusion of bafilomycin A1 revealed that the apparent decrease in LC3‐II in the rapamycin‐treated group was attributed to the lysosomal degradation, which demonstrated that rapamycin promoted autophagic flux in this case (Figure 4M and 4N).
Next, we mitigated the autophagy by ATG7 and Beclin1 RNAi, 2 essential autophagy effectors, in the presence of rapamycin. First, we found that ATG7 or Beclin1 knockdown slightly reduced the cleavage of PARP on day 2 (data not shown). In particular, we observed that on day 4 the extent of cell death was significantly decreased by ATG7 accompanied by Beclin1 knockdown in the presence of z‐VAD (Figure 4O). More importantly, we found that blocking autophagy by Beclin1 RNAi enhanced the efficiency of cardiac differentiation (to 96.2%) within H9‐cTnT‐eGFP cells more than in control groups, which have an efficiency of 88.2% (Figure 4P). Taken together, these results indicate that the increase in autophagy induced by rapamycin has already exceeded the bearing capability of hESCs, which introduced the risk of cell death. This result is reminiscent of the previous conclusion that autophagy‐deficient mice exhibit normal cardiac development.
The Intracellular Reactive Oxygen Species Generation Regulated by mTOR Also Contributes to the hESC Apoptosis
Because it is recognized that DNA damage also stimulates the accumulation of p53, we analyzed the markers of DNA damage, including ATM‐CHK2, ATR‐CHK1, and H2A.X.38 We found that ATM‐CHK2 pathway was activated during cell culture, whereas ATR‐CHK1 activity was underdetected (Figure 5A). We also noted that the phosphorylation of ATM and CHK2 was reduced by rapamycin treatment, indicating that rapamycin reduced DNA damage (Figure 5B). Moreover, the level of histone H2A.X phosphorylation, another marker of DNA damage, was also reduced by rapamycin treatment in a dose‐dependent manner (Figure 5C). ATM kinase is sensed with DNA double‐strand breaks, which arise from various sources such as replication stress, ion radiation, reactive oxygen species (ROS), and others. In several extant studies, hESCs were found to be sensitive to ROS, as they were unable to grow normally without antioxidant ascorbic acid (vitamin C) or 2‐mecaptoethanol (2‐ME). Although basic ROS activity is considered to contribute to hESC differentiation, it will induce caspase‐dependent apoptosis once the oxidative stress exceeds a certain threshold. In our work, we detected the intracellular ROS generation by 2ʹ,7ʹ‐dichlorodihydrofluorescein diacetate staining during hESCs' monolayer‐based culture. Interestingly, we found that, in the CHIR‐treated group, the intracellular ROS activity dramatically increased (by at least 2‐fold) after 16‐hour culture, after which it decreased slightly (Figure 5D). However, rapamycin significantly reduced the intracellular ROS level to less than half of that noted in the CHIR‐treated group (Figure 5E). The dissipation of mitochondrial electrochemical potential gradient (ΔΨ) is known as an original event during apoptosis and contributes to mitochondrial ROS generation. JC‐1 staining showed that the mitochondrial potential gradient of the DMSO and CHIR group gradually deteriorated during a 3‐day culture (mitochondrial depolarization is indicated by an increase in the green/red fluorescence intensity ratio) but that rapamycin treatment effectively alleviated this adverse tendency (Figure 5F). Although the change of ROS and mitochondrial potential emerged at a much earlier stage, before serious apoptosis had occurred, to further confirm that these were the original reasons behind these phenomena rather than byproducts, we examined ROS production and mitochondrial potential after treating the hESCs with p53 and AMPK siRNA. We found that neither blocking the p53 accumulation nor reducing the AMPK activity could reduce the ROS generation or the dissipation of mitochondrial potential (Figure 5G and 5H). Interestingly, we also found that additional glucose or increasing medium volume could stimulate the ROS activity (data not shown). Finally, we found that application of antioxidant ascorbic acid but not N‐acetyl‐l‐cysteine (NAC) mitigated the oxidant‐induced apoptosis (Figure 5I). NAC was reported to have a protective effect against apoptosis. We speculated that the controversial effect of NAC on apoptosis may be due to the pH pressure caused by the high‐concentration solution of NAC (1 mmol/L), which is acidic, because we have found that, apart from nutrient starvation, the pH of the medium also deteriorated during culture, and hESCs underwent apoptosis in relatively low‐pH conditions (data not shown). The great pressure caused by NAC was far beyond the bearing capacity of hESCs in this weak status to overcome and survive. Collectively, these results suggest that, in addition to extracellular stress, intracellular ROS activity and mitochondrial state are also involved in hESC apoptosis in monolayer culture.
Figure 5.
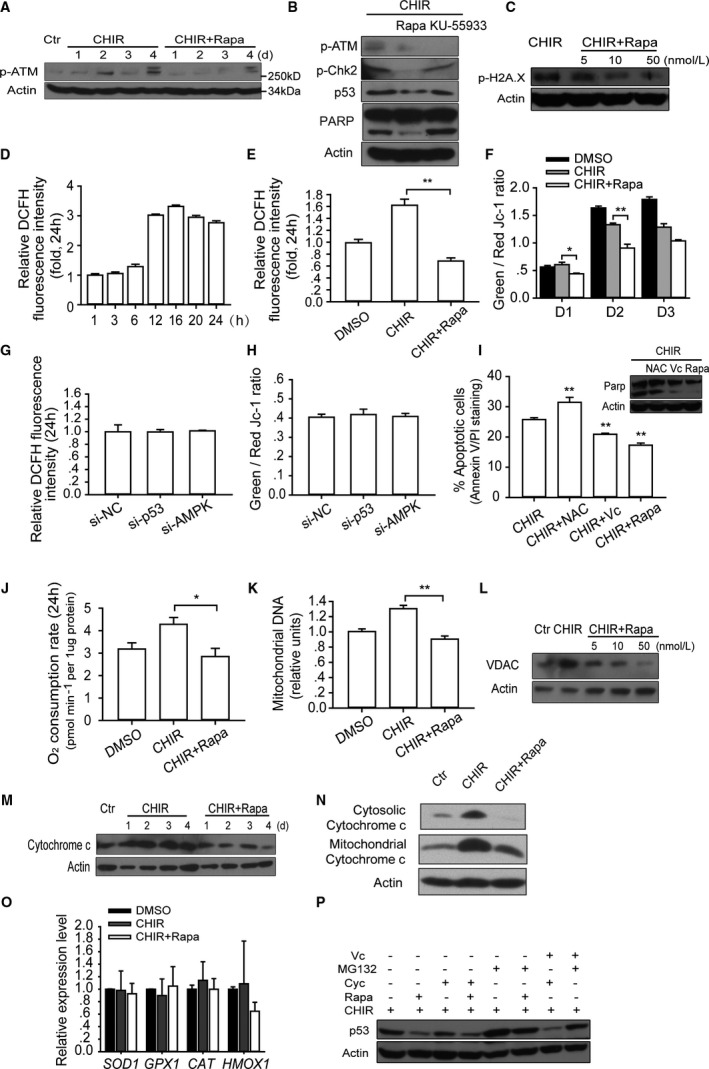
Inhibition of mTOR reduces DNA damage induced apoptosis via decreasing the intracellular ROS generation. A, Western blot analysis of the phosphorylation of ATM (Ser1981), which is a marker of DNA damage, during first 4 days of culture with described treatment. B, Western blot analysis of DNA damage associated with proteins including p‐ATM (Ser1981), p‐Chk2 (Thr68), p53, and PARP in hESCs with described treatment. KU‐55933, an inhibitor of ATM, could block the activation of ATM but could not block the apoptosis as rapamycin did. C, Immunoblot analysis of p‐H2A.X (Ser139) under different rapamycin concentrations. D and E, Intracellular ROS production was analyzed by DCFH (2ʹ,7ʹ‐dichlorodihydrofluorescein) staining at indicated time points and treatment. The dynamic changes of intracellular ROS activity at different time points were analyzed in groups treated with CHIR alone (n=5) (D), and intracellular ROS activity was compared between groups treated with CHIR and CHIR plus rapamycin; DMSO was used as control treatment (n=6; **P<0.01) (E). F, Mitochondrial electrochemical potential gradients (ΔΨ) of hESC were measured by JC‐1 assay within first 3 days. The ratio of green to red fluorescence depends on the mitochondrial membrane potential: green fluorescence indicates low potential, whereas red fluorescence indicates high potential (n=4; *P<0.05, **P<0.01). G and H, Analyzing the effects of p53 and AMPK knockdown on ROS production and mitochondrial electrochemical potential gradients (ΔΨ). The p53 and AMPK protein expression were knocked down by siRNA. Then intracellular ROS activity and ΔΨ were measured after 48 hours in culture with CHIR treatment (n=5). I, Testing the antiapoptosis effect of antioxidant agents (NAC 1 mmol/L and vitamin C 200 μg/mL) in high‐density monolayer growth. The statistical significance of comparisons between testing groups and the CHIR‐alone control group are shown (n=4; **P<0.01). J through N, Rapamycin decrease the oxygen consumption (n=5; *P<0.05) (J), mitochondrial DNA copy number (n=6; **P<0.01) (K), mitochondrial specific protein VDAC (L), cytochrome c expression (M), and the release of cytochrome c from mitochondria into cytosol on day 3 (N). O, Quantitative real‐time PCR analysis of intracellular antioxidant gene expression (n=5). P, Rapamycin affects p53 stability and translation. p53 expression was assessed in a condition in which protein synthesis was blocked by cycloheximide (100 ng/mL) and protein degradation was blocked by MG132 (proteasome inhibitor, 2 μmol/L) and present of antioxidant vitamin C (200 μg/mL). CHIR indicates CHIR99021; Ctr, control; Cyc, cycloheximide; DMSO, dimethyl sulfoxide; hESC, human embryonic stem cell; NAC, N‐acetyl‐l‐cysteine; PCR, polymerase chain reaction; Rapa, rapamycin; ROS, reactive oxygen species; si‐NC, negative control siRNA.
Because the escape of electrons from the aerobic respiratory electron transport chain is 1 of the most critical mechanisms of ROS generation in healthy cells,39 the possibility that rapamycin could influence oxidative function or quantity of mitochondrial in cells was investigated in our system. We discovered that the cell oxygen consumption was decreased by rapamycin after 24‐hour culture (Figure 5J). We further noted that mitochondrial DNA content and protein levels of mitochondrial special genes, such as VDAC1 and cytochrome c, were eliminated after rapamycin treatment (Figure 5K through 5M). Specifically, rapamycin treatment decreased the release of cytochrome c from mitochondria into cytosol, which is an acknowledged assay to assess apoptosis (Figure 5N). In addition, we found that rapamycin treatment had no significant effect on the expression of intracellular antioxidant genes, including SOD1, GPX1, CAT, and HMOX1 (Figure 5O).40 These results indicate that treating hESCs with rapamycin decreased the mitochondrial content and its oxidative function and consequently reduced intracellular ROS generation, which could induce DNA damage and apoptosis.
Next, we explored the mechanisms through which rapamycin disturbed p53 accumulation. Because rapamycin was previously found capable of reducing p53 accumulation by blocking its translation,37 cycloheximide was applied to block the translation in hESC. We found that cycloheximide markedly reduced the difference in p53 accumulation that resulted from treatment with and without rapamycin. On the other hand, when MG132 was used to block the protein degradation, which eliminated the ROS‐induced resistance to degradation, the difference in p53 accumulation narrowed further. Finally, we found that, by adding 200 μg/mL vitamin C together with cycloheximide into the CHIR group, p53 accumulation was completely eliminated (Figure 5P). These results suggest that the influence of rapamycin on p53 accumulation is based on both translation and degradation pathways. Conclusively, the antiapoptotic properties of rapamycin stem from its 2 coordinated effects on multiple extracellular stress‐induced p53 accumulation and intracellular ROS generation.
Rapamycin Promotes Multiple Signaling Pathways Essential for Mesoderm Induction
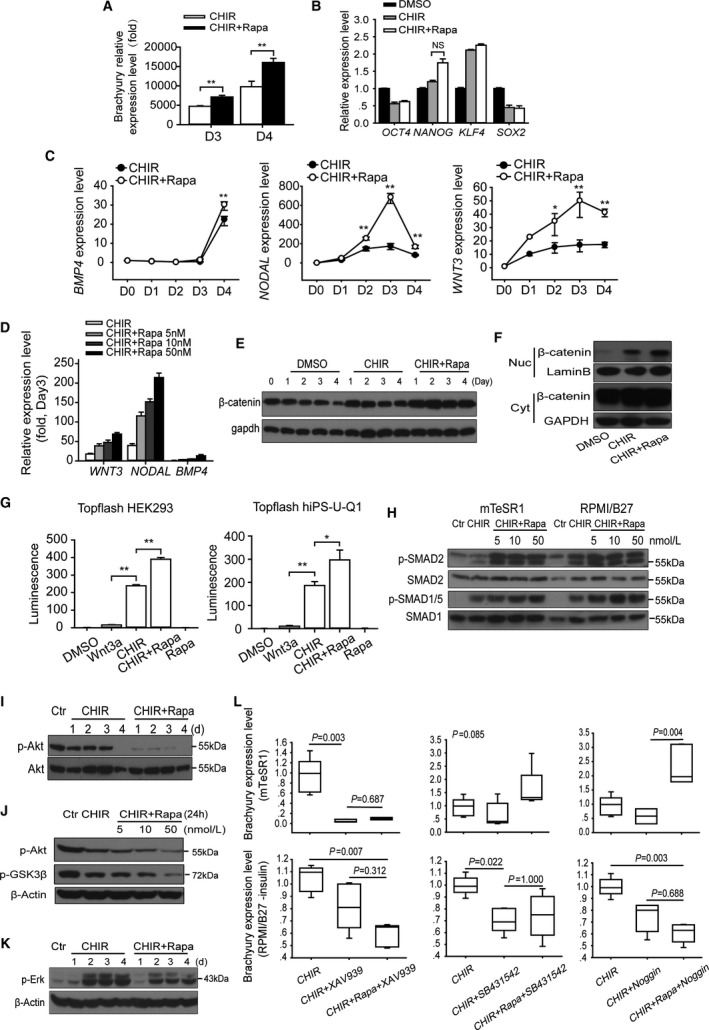
We have proved that rapamycin can significantly promote mesoderm differentiation, as shown by a common feature of mesoderm, Brachyury (T). Specifically, we found that the level of Brachyury expression was upregulated by additional rapamycin treatment compared with the CHIR treatment not only in the mTeSR1 medium but also in the RPMI B27 medium (Figure 6A). On the other hand, the difference in the effects of treatments with or without rapamycin on the mRNA expression of hESC self‐renewal core factors, including OCT4, NANOG, KLF4, and SOX2, was not statistically significant (Figure 6B). Collectively, these results confirmed the ability of rapamycin to promote mesoderm commitment from hPSC. To further analyze the mechanism by which rapamycin promoted mesoderm derivation and eventually accelerated the cardiac differentiation efficiency, we checked the mesoderm‐associated signaling pathways. It is widely accepted that mesoderm formation is a collaborative process induced by Wnt, TGF‐β, FGF, and AKT signaling. Extant results have shown that Brachyury is one of the most important downstream factors of Wnt and TGF‐β signaling pathways during cardiac genesis and differentiation in vivo and in vitro. Thus, we postulated that the Wnt or TGF‐β signaling cascade may be implicated in the prodifferentiation function of rapamycin. We detected multiple mesoderm commitment‐associated genes via quantitative real time PCR test and found that BMP4, NODAL, and WNT3 were all upregulated by 4 days of treatment with rapamycin, and this effect is dose dependent (Figure 6C and 6D). Particularly, we found that the endogenous BMP4 expression was not sensitive to CHIR or rapamycin treatment in the first 3 days. In contrast, the endogenous NODAL expression was upregulated sharply at day 3 by rapamycin. Although the upregulated level of NODAL is much higher than that of WNT3, unlike the persistent promoting effect of rapamycin on WNT3 expression, after the medium changed to RPMI B27 in the first day of differentiation, the level of NODAL went down similar to the control group. Because Murry's group have shown that the endogenous Wnt level is essential for the difference among different hESC lines toward cardiomyocyte differentiation, we take special notice of Wnt signaling. To further confirm the regulatory function of rapamycin on the Wnt signaling pathway, we detected the influence of rapamycin treatment on its downstream process. We found that rapamycin increased the protein expression of β‐catenin, a key mediator of Wnt signaling, on the basis of CHIR induction (Figure 6E). In addition, rapamycin further stimulated the translocation of β‐catenin into the nucleus after a relatively short treatment (12 hours), implying that rapamycin has a direct effect on Wnt signaling transduction (Figure 6F). Finally, to obtain a more precise measurement of the effect of rapamycin on the Wnt signaling pathway, the TOPflash assay, a generally accepted reporter system used for canonical Wnt signaling activities measurement, was employed using U‐Q1‐hiPSCs and HEK293 cells. As expected, addition of 10 nmol/L rapamycin further elevated the luciferase activity to about 1.6‐fold higher levels compared with those noted in the CHIR group (Figure 6G). Taken together, these results substantiate the assertion that rapamycin is an activator of Wnt signaling in both hPSCs and differentiated cells. CHIR is the most effective Wnt signaling activator presently known, and the directed regulation of mTOR on Wnt signaling is unclear. So our finding that rapamycin further stimulates Wnt signaling when CHIR concentration is already high (12 μmol/L) is not only useful for practically inducing Wnt signaling but also indicates that the regulatory modes of rapamycin and CHIR on Wnt signaling are independent.
Figure 6.
Rapamycin promotes multiple signaling pathways essential for mesoderm induction. A, The transcriptional level of Brachyury with or without rapamycin treatment in mTeSR1 (D3) and RPMI B27 (D4) medium (n=5; **P<0.01). B, The transcriptional level of hESC self‐renewal core factors, including OCT4,NANOG,KLF4, and SOX2 with or without rapamycin treatment (n=5; NS means no significant difference). C, Daily transcriptional levels of endogenous growth factors including BMP4,NODAL, and WNT3 in H9 hESCs induced by CHIR with or without rapamycin (10 nmol/L). Treated cells were collected and analyzed at indicated time points, and 1‐way ANOVA followed by post hoc Bonferroni test was used to calculate significant difference between groups at each time point (n=4; *P<0.05, **P<0.01). D, Quantitative real‐time PCR analysis of WNT3,NODAL, and BMP4 expression with different concentrations of rapamycin at day 3 (n=5). E, The expression of β‐catenin with or without rapamycin treatment. F, Nucleocytoplasmic separation experiment showed the distribution of β‐catenin, which reflects the activity of Wnt signaling. G, Topflash reporter assay performed using hiPS‐U‐Q1 and HEK293T cells. The effect of CHIR, rapamycin, or DMSO was examined, and Wnt3a (100 ng/mL) was added as a positive control (n=5; *P<0.05, **P<0.01). H, Expression of phosphorylation of SMAD2 (Ser467), SMAD1/5 (Ser463/465), and total SMAD2 and SMAD1/5 were analyzed via Western blots in H9 cells treated with 12 μmol/L CHIR or CHIR plus 10 nmol/L rapamycin for 6 hours. I, The activity of PI3K/AKT signaling with or without rapamycin treatment was tested by the phosphorylation of AKT at Ser473 via Western blot with total AKT expression as reference. J, Western blot analysis shows that the suppression effect of rapamycin on the Akt pathway was concentration dependent, and the level of GSK‐3β (Ser9) phosphorylation, which is an index of Akt activity, had a similar change. K, The activity of FGF‐Erk signaling pathway was shown by the Erk (Ser44/42) phosphorylation with or without rapamycin treatment at different time points. L, Analyzing the influences of inhibition of Wnt, NODAL, and BMP4 signaling pathways on rapamycin‐induced Brachyury expression in mTeSR1 and RPMI/B27 conditions (n=5; Kruskal‐Wallis test followed by Dunn post hoc comparison). CHIR indicates CHIR99021; Ctr, control; Cyt, cytoplasm; DMSO, dimethyl sulfoxide; hESC, human embryonic stem cell; Nuc, nucleus; PCR, polymerase chain reaction; Rapa, rapamycin.
To determine whether TGF‐β signaling pathway was required in the monolayer‐based differentiation performed in this work, we quantified cardiomyocytes derived from H9‐cTnT‐GFP cells when NODAL or BMP4 signaling antagonists were added at day 4 for 24 hours. Both SB431542 and Noggin completely eliminated cardiomyocyte specification at concentrations of 2 μmol/L and 100 ng/mL, respectively (data not shown). Moreover, addition of activin A or BMP4, together with CHIR, was able to further increase Brachyury transcription in both mTeSR1 and RPMI/B27 culture conditions (Figure S3A). These results revealed that the activation of the TGF‐β superfamily was a prerequisite for cardiac differentiation, although the exogenous activin A and BMP4 were not required. Next, we examined endogenous mRNA expression of NODAL, activin A (INHB1), BMP2, BMP4, and BMP7 after 3 days of CHIR treatment with or without rapamycin (Figure S3B). The transcription of NODAL and BMP4 but not activin A (INHB1) immediately increased following the CHIR treatment, whereas the increase of BMP4 was milder than that noted in NODAL. Importantly, the addition of rapamycin further boosted their expression. Moreover, all primary downstream targets—including ID1, SMAD6, SMAD7, LEFTY1, and LEFTY2—were unregulated by rapamycin (Figure S3C). In addition, rapamycin markedly stimulated the phosphorylation of Smad2 and Smad1/5, which are transcriptional effectors of NODAL and BMP signaling, respectively. These observations are based on the CHIR treatment and were noted in both mTeSR1 and RPMI/B27 conditions after 6 hours of treatment. Moreover, these promotion effects were dose dependent (Figure 6H). These results confirm our assumption that both NODAL and BMP signaling pathways are activated by rapamycin treatment.
Earlier findings indicate that the suppression of PI3K‐Akt signaling pathway is a prerequisite for mesendoderm induction.41 In our study, we found that rapamycin decreased Akt phosphorylation (Ser473) during hESC differentiation, and this effect was dependent on the rapamycin concentration. We also noted that the level of GSK‐3β (Ser9) phosphorylation, which is an index of Akt activity, exhibited the same change (Figure 6I and 6J). Although previous studies have demonstrated that rapamycin could suppress Akt activity mediated by mTORC2 in some cell types under sustained treatment, this important function has not been confirmed in hESC. On the other hand, in the present study, rapamycin did not stimulate FGF/Erk signaling pathway activity, which is another important cardiac differentiation‐related signaling mode (Figure 6K).
To further elucidate which signal primarily contributed to the prodifferentiation function of rapamycin, we assessed the expression‐promoting effects of rapamycin and CHIR on Brachyury in the presence of correlated inhibitors, including XAV939, SB431542, and Noggin. We found that Wnt inhibitor XAV939 was able to totally block the increase of Brachyury in mTeSR1 condition, irrespective of whether rapamycin was included in the treatment. On the other hand, in the RPMI/B27 condition, suppressing any 1 of Wnt, NODAL, and BMP4 pathways could partially eliminate the promoting functions of CHIR and rapamycin (Figure 6L). These results suggest that, in the predifferentiation stage (mTeSR1 culturing), rapamycin and CHIR effects can be primarily attributed to Wnt signaling. When the differentiation formally starts, these 3 signaling pathways contribute to the formation of mesoderm independently.
Discussion
We revealed that mTOR possesses a wide variety of effects on cardiogenesis from hPSCs (Figure 7). Specifically, mTOR functioned as a “master switch” of multiple differentiation‐promoting signals, including Wnt, NODAL, and BMP4. Our findings also indicated that multiple stresses, including glucose starvation and increasing ROS levels, when introduced along the monolayer‐based hPSC culture, resulted in a dramatic augmentation of p53 activity, eventually resulting in cell apoptosis. However, mTOR inhibition by rapamycin effectively mitigated all these stresses and reduced hPSC apoptosis, which enhanced the stability of cardiac differentiation. Because it takes advantage of the reasonable regulation of mTOR and Wnt signaling pathways, our cardiomyocyte differentiation strategy is the most efficient method among the existing protocols, and it also produces the highest yield. Moreover, our method provides some other potential benefits during application. First, in the initial stage from day −3 to day 1, the combined treatment of CHIR and rapamycin in our protocol was performed in 50% usage of the medium in previous work,5 and in the later stage, a reduction of medium happened from day 1 to day 5. The addition of rapamycin reduces medium consumption by about 50%, thereby generating significant savings in the cost of cardiomyocyte generation, which can be particularly important in a large‐scale production. Second, we have noticed that the cardiac differentiation efficiency and stability of CHIR alone are low in our experimental system. These results are similar to a recent report that the outcomes of cardiac differentiation efficiency with CHIR‐alone treatment are variable from lot to lot and even from experiment to experiment.42 And we have also verified other protocols with CHIR‐alone treatment: the efficiency of cardiac differentiation is still variable among experimental repeats with a range of 5.6% to 80% (data not shown). Additional rapamycin treatment in these conditions can always boost the efficiency to no more than 90% and improve the variability of CHIR efficacy (data not shown). However, the application of rapamycin in our optimized protocol overcame the variability in cardiac differentiation among different hESC lines and experimental repeats and gained a higher efficiency up to 87% to 99.06%. Third, our method reduces the number of times and operation complexity of culturing medium replacement, which is now much easier to manipulate compared with the existing protocols. Taken together, our findings confirm that the addition of rapamycin not only strengthens the function of CHIR, but also remedies its defects. Our data further suggest that rapamycin, an existing and clinically approved drug, would be widely applied in large‐scale hPSC culture and hPSC‐derived terminal cell production. These assertions are supported by those reported in a previous study,43 where the authors reported that hESCs were unable to survive during large‐scale suspension culture without rapamycin, which was posited to prevent the dissociation of hESC aggregates by avoiding the fibroblast differentiation.
Figure 7.
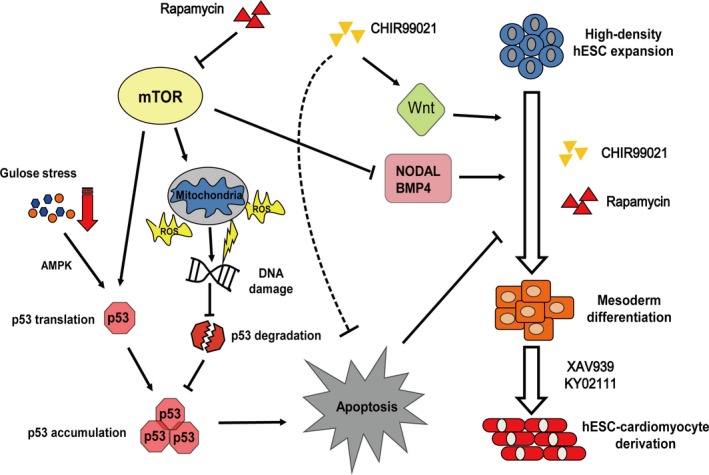
The mTOR signal has multiple effects on cardiac differentiation. Schematic model for the role of mTOR in regulating differentiation and apoptosis of hESCs and cardiomyocytes (dotted line indicates unidentified mechanisms). AMPK indicates AMP‐dependent kinase; hESC, human embryonic stem cell; mTOR, mammalian target of rapamycin; ROS, reactive oxygen species.
We have also revealed detailed mechanisms along the monolayer‐based differentiation path. The findings reported here suggest that, in the prime stage, endogenous NODAL expression first responded to the inducible factors (CHIR and rapamycin), which was followed by endogenous WNT3 and BMP4 expression, both of which exhibited modest change compared with NODAL. However, all these signaling pathways were indispensable for cardiac induction, and their inhibition could completely block this process. Given that the influence of mTOR on these pathways was immediate, we posited that mTOR physically stimulated SMAD and β‐catenin, both of which were implicated in recent studies.44, 45, 46 Nonetheless, the exact mechanism underpinning this process remains unclear, and further studies are required in order to elucidate how mTOR regulates Wnt signaling.
Dramatic cell death is a common observation in hPSC monolayer‐based differentiation. However, it is presently unknown whether it is necessary for normal differentiation because cell death could not be fully eliminated in previous studies. We have demonstrated that this cell death process is in fact p53‐dependent apoptosis and is partially autophagy dependent. Moreover, its extent is controlled by mTOR activity. It is well known that mTOR is the most important regulator of autophagy. Our studies have also revealed that autophagy is activated during the initial stage of differentiation when rapamycin is added. And we found that knockdown of autophagy‐associated genes ATG7 and Beclin1 in the presence of rapamycin further decreased apoptosis and increased the efficiency of cardiac differentiation. We speculate that, in the process of cell fate transition from stem cells to mesoderm lineages, autophagy can also play a role as a remodeling mechanism to erase excess organelles and to rebuild essential organelles such as mitochondria for cardiomyocyte survival. Thus, moderate autophagy is appropriate and essential for successful cell fate remodeling, but excess autophagy can result in apoptosis due to the dramatic loss of organelles. It is possible when considering the shortage of amino acids in our differentiation system that the fundamental autophagy level in this process can be high and rapamycin‐induced autophagy is incompatible with cell survival. So excess autophagy induces autophagic cell death by degrading essential components of cells and further blunts the differentiation, and our results suggest that appropriate intensity of autophagy is essential for cardiac differentiation. Our unpublished data showed that increasing extracellular glucose concentration or amplifying medium volume significantly increased ROS generation, which compromises their antiapoptosis effects. In addition, our unpublished data also suggest that TGF‐β and CHIR could elevate the hESCs' apoptosis threshold through increasing the expression of antiapoptosis gene Mcl‐1, which is consistent with a previous study.47 We established that reducing the extent of hPSC apoptosis by p53 knockdown or rapamycin treatment significantly increased the efficiency of cardiac induction. Intuitively, this effect resulted from the higher density of robust hESCs that could secrete and accumulate much greater quantity of growth factors, such as WNT3, NODAL, and BMP4. However, further research is required in order to establish whether there is another underlying mechanism contributing to these phenomena. Finally, our results suggest that p53‐dependent apoptosis constitutes a main barrier to differentiation, which may prevent suboptimal parental cells (such as the cells carrying persistent DNA damage) from surviving and transferring their defect to abnormal or cancer cells. However, rapamycin not only protects the cells from apoptosis but also improves the state of hESCs fundamentally, which is essential for highly efficient and robust differentiation.
Our findings provide an important contribution to the current understanding of the exact functions of mTOR in the initial stages of cardiogenesis during embryo development. However, the precise manner in which the mTOR activity is regulated in hESC in vitro and in vivo remains to be established. Our findings imply that mTOR activity could be gradually suppressed by nutrients and hypoxia, all of which naturally emerge during cardiogenesis in a monolayer mode or immediately after embryo implantation. This explains why a certain proportion of cardiomyocytes are generated without rapamycin treatment and why this important relationship between mTOR and cardiogenesis had not been defined until now. More importantly, this result implies that nutrient availability and environmental conditions could regulate the fate of embryonic stem cells, which is consistent with the previously reported findings indicating that inhibition of mTOR by amino acid deprivation impaired blastocyst implantation and outgrowth in vivo.48 It is, however, important to note that other regulatory mechanisms may be employed. One extant study demonstrated that BMP4 can stimulate the expression of miR‐23a in hESC.49 We also found presence of a predicted miR‐23a banding site on raptor 3′‐UTR, which may contribute to the downregulation of mTORC1 during cardiac induction. Combined with previous studies, our findings confirm that hESCs are highly sensitive to the status of mTOR. Hence, the precise regulatory mechanisms by which hESCs dominate the mTOR status should be the focus of future studies in this field.
Sources of Funding
This work was supported in part by the National Natural Science Foundation of China (91339205, 81130005, 31229002, 81470407, and 81570208), the National Key Research and Development Program of China (2017YFA0103700), and the Chinese Academy of Sciences (XDA01040306).
Disclosures
None.
Supporting information
Table S1. Primer Sequences Used for qPCR and siRNA Sequences Used for RNAi
Table S2. Antibodies Used for Western Blot, FACS Analysis, and Immunofluorescence Analysis
Figure S1. Rapamycin promotes cardiomyocyte differentiation. A and B, Quantitative real‐time PCR analysis of the expression of cardiac‐specific transcription factors and structural proteins such as NKX2.5, GATA4, RALDH2, MEF2C, MESP1, ISL1, cTnT, HCN4, MHY6, MLC2a, and MLC2v with or without rapamycin treatment at day 5 and day 12, respectively (n=6). C, The effect of different concentration of rapamycin on T (Brachyury) mRNA expression at day 3 (n=5). D, The induction of mesoderm marker gene Brachyury was blocked by 3BDO, a novel mTOR activator, and this effect was reduced by rapamycin addition (n=5). E, Transmission electron microscopic images of H9‐derived cardiomyocytes produced by the method described in Figure 1E. The white arrow indicates mature mitochondria, myofibrils with Z‐band, and intercalated disks. F, Representative flow cytometry data of GFP‐positive cardiomyocytes after lactate‐culturing purification. G, The concentration of rapamycin treatment influences the growth of EBs. Comparison of cell number (total) within indicated treatment. During days 0 to 3, 5 nmol/L rapamycin treatment increased the total cell number of EBs compared with the DMSO‐treated group. But 20 nmol/L rapamycin treatment inhibited the growth of EBs. The cell number was counted at day 10 (n=5). **P<0.01, *P<0.05; Mann‐Whitney U test was applied in B. 3BDO indicates 3‐benzyl‐5‐((2‐nitrophenoxy) methyl)‐dihydrofuran‐2(3H)‐one; CHIR, CHIR99021; DMSO, dimethyl sulfoxide; ID, intercalated disks; MF, myofibrils; Mt, mitochondria; PCR, polymerase chain reaction; Rapa, rapamycin.
Figure S2. The antiapoptosis effect of rapamycin on hESCs in high‐density monolayer culture. A, Rapamycin was unable to promote the colony formation of hESCs. H9 cells were dissociated into single cells and seeded in Matrigel‐coated 12‐well plates at a density of 1×105 per well with different treatment with small molecules: 0.1% DMSO, 12 μmol/L CHIR or CHIR plus 10 nmol/L rapamycin (n=5; Kruskal‐Wallis test followed by Dunn post hoc comparison). B and C, The activity of mTOR is essential for the apoptosis of hESCs at day 4. Annexin V/PI staining analysis of the apoptotic level of hESCs with si‐mTOR or si‐TSC1/2 treatment at day 4 (n=6; **P<0.01, one way ANOVA) (B). Annexin V/PI staining analysis of the apoptotic level of hESCs with or without rapamycin treatment at day 4 (n=6; Mann‐Whitney U test) (C). CHIR indicates CHIR99021; DMSO, dimethyl sulfoxide; hESC, human embryonic stem cell; Rapa, rapamycin; si‐NC, negative control siRNA.
Figure S3. TGF‐β signaling pathway is involved in mesoderm induction of rapamycin. A, The effects of activin A and BMP4 on Brachyury transcription level in mTeSR1 and RPMI/B27 culture conditions, respectively. Level of mRNA expression was normalized to DMSO group (n=5). B and C, Quantitative real‐time PCR analysis of associated TGF‐β superfamily members and their downstream genes (n=6). CHIR indicates CHIR99021; DMSO, dimethyl sulfoxide; PCR, polymerase chain reaction; Rapa, rapamycin; TGF‐β, transforming growth factor β.
Video S1. Day 15 cardiomyocytes induced from H9‐cTnT‐eGFP cells (×10).
Video S2. Day 15 cardiomyocytes induced from H7 cells (×20).
Acknowledgments
We thank Professors Lei Xiao and Guo‐Liang Xu for kindly providing DOX‐induced OKSM expression lentivirus plasmids; Professor Jun‐Ying Miao for providing small‐molecule 3BDO; Professors Jian Lu and Jia‐Qi Zhu for comments on statistical analysis; and staff in the core facility (Institute of Health Sciences) for technical assistance. We thank all Jing laboratory members for comments.
(J Am Heart Assoc. 2017;6:e005295 DOI: 10.1161/JAHA.116.005295.)
References
- 1. Burridge PW, Keller G, Gold JD, Wu JC. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012;10:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, O'Sullivan C, Collins L, Chen Y, Minami E, Gill EA, Ueno S, Yuan C, Gold J, Murry CE. Cardiomyocytes derived from human embryonic stem cells in pro‐survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. [DOI] [PubMed] [Google Scholar]
- 3. Paige SL, Osugi T, Afanasiev OK, Pabon L, Reinecke H, Murry CE. Endogenous Wnt/β‐catenin signaling is required for cardiac differentiation in human embryonic stem cells. PLoS One. 2010;5:e11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lian XJ, Hsiao C, Wilson G, Zhu KX, Hazeltine LB, Azarin SM, Raval KK, Zhang JH, Kamp TJ, Palecek SP. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA. 2012;109:E1848–E1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lian XJ, Zhang JH, Azarin SM, Zhu KX, Hazeltine LB, Bao XP, Hsiao C, Kamp TJ, Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β‐catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raught B, Gingras AC, Sonenberg N. The target of rapamycin (TOR) proteins. Proc Natl Acad Sci USA. 2001;98:7037–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Noda T, Ohsumi Y. TOR, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. [DOI] [PubMed] [Google Scholar]
- 8. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. [DOI] [PubMed] [Google Scholar]
- 9. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. [DOI] [PubMed] [Google Scholar]
- 10. Zhou J, Su P, Wang L, Chen J, Zimmermann M, Genbacev O, Afonja O, Horne MC, Tanaka T, Duan E, Fisher SJ, Liao J, Chen J, Wang F. mTOR supports long‐term self‐renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc Natl Acad Sci USA. 2009;106:7840–7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, Kiyama H, Yonezawa K, Yamanaka S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24:6710–6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu Y, Pires KM, Whitehead KJ, Olsen CD, Wayment B, Zhang YC, Bugger H, Ilkun O, Litwin SE, Thomas G, Kozma SC, Abel ED. Mechanistic target of rapamycin (mTOR) is essential for murine embryonic heart development and growth. PLoS One. 2013;8:e54221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, Krishnan J, Lerch R, Hall MN, Ruegg MA, Pedrazzini T, Brink M. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. [DOI] [PubMed] [Google Scholar]
- 15. Liu JC, Guan X, Ryan JA, Rivera AG, Mock C, Agrawal V, Letai A, Lerou PH, Lahav G. High mitochondrial priming sensitizes hESCs to DNA‐damage‐induced apoptosis. Cell Stem Cell. 2013;13:483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dumitru R, Gama V, Fagan BM, Bower JJ, Swahari V, Pevny LH, Deshmukh M. Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol Cell. 2012;46:573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang ES, Reyes NA, Melton C, Huskey NE, Momcilovic O, Goga A, Blelloch R, Oakes SA. Fas‐activated mitochondrial apoptosis culls stalled embryonic stem cells to promote differentiation. Curr Biol. 2015;25:3110–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abdul‐Ghani M, Dufort D, Stiles R, De Repentigny Y, Kothary R, Megeney LA. Wnt11 promotes cardiomyocyte development by caspase‐mediated suppression of canonical Wnt signals. Mol Cell Biol. 2011;31:163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bulatovic I, Ibarra C, Osterholm C, Wang H, Beltran‐Rodriguez A, Varas‐Godoy M, Mansson‐Broberg A, Uhlen P, Simon A, Grinnemo KH. Sublethal caspase activation promotes generation of cardiomyocytes from embryonic stem cells. PLoS One. 2015;10:e0120176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H, Jaenisch R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vander Haar E, Lee S‐I, Bandhakavi S, Griffin TJ, Kim D‐H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. [DOI] [PubMed] [Google Scholar]
- 22. Ma H, Pederson T. Depletion of the nucleolar protein nucleostemin causes G1 cell cycle arrest via the p53 pathway. Mol Biol Cell. 2007;18:2630–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. [DOI] [PubMed] [Google Scholar]
- 24. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1‐binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. [DOI] [PubMed] [Google Scholar]
- 25. Ryu JH, Cho YS, Chun YS, Park JW. Myocardial SSAT induction via AMPK signaling and its implication for ischemic injury. Biochem Biophys Res Commun. 2008;366:438–444. [DOI] [PubMed] [Google Scholar]
- 26. Snir M, Kehat I, Gepstein A, Coleman R, Itskovitz‐Eldor J, Livne E, Gepstein L. Assessment of the ultrastructural and proliferative properties of human embryonic stem cell‐derived cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;285:H2355–H2363. [DOI] [PubMed] [Google Scholar]
- 27. Zhang J, Nuebel E, Wisidagama DR, Setoguchi K, Hong JS, Van Horn CM, Imam SS, Vergnes L, Malone CS, Koehler CM. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat Protoc. 2012;7:1068–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tian FJ, An LN, Wang GK, Zhu JQ, Li Q, Zhang YY, Zeng A, Zou J, Zhu RF, Han XS, Shen N, Yang HT, Zhao XX, Huang S, Qin YW, Jing Q. Elevated microRNA‐155 promotes foam cell formation by targeting HBP1 in atherogenesis. Cardiovasc Res. 2014;103:100–110. [DOI] [PubMed] [Google Scholar]
- 29. Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE. Biphasic role for Wnt/β‐catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci USA. 2007;104:9685–9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Minami I, Yamada K, Otsuji TG, Yamamoto T, Shen Y, Otsuka S, Kadota S, Morone N, Barve M, Asai Y, Tenkova‐Heuser T, Heuser JE, Uesugi M, Aiba K, Nakatsuji N. A small molecule that promotes cardiac differentiation of human pluripotent stem cells under defined, cytokine‐ and xeno‐free conditions. Cell Rep. 2012;2:1448–1460. [DOI] [PubMed] [Google Scholar]
- 31. Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, Wang Y, Zhang Y, Zhuang Q, Li Y, Bao X, Tse HF, Grillari J, Grillari‐Voglauer R, Pei D, Esteban MA. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7:2080–2089. [DOI] [PubMed] [Google Scholar]
- 32. Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K. Distinct metabolic flow enables large‐scale purification of mouse and human pluripotent stem cell‐derived cardiomyocytes. Cell Stem Cell. 2013;12:127–137. [DOI] [PubMed] [Google Scholar]
- 33. Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G. Stage‐specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–240. [DOI] [PubMed] [Google Scholar]
- 34. Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J, Li J, Song Z, Qu X, Zhou P, Wu J, Ding M, Deng H. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem. 2007;282:5842–5852. [DOI] [PubMed] [Google Scholar]
- 35. Shieh S‐Y, Ikeda M, Taya Y, Prives C. DNA damage‐induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. [DOI] [PubMed] [Google Scholar]
- 36. Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. [DOI] [PubMed] [Google Scholar]
- 37. Lee CH, Inoki K, Karbowniczek M, Petroulakis E, Sonenberg N, Henske EP, Guan KL. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 2007;26:4812–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hirao A, Kong Y‐Y, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage‐induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. [DOI] [PubMed] [Google Scholar]
- 39. Brookes PS. Mitochondrial H+ leak and ROS generation: an odd couple. Free Radic Biol Med. 2005;38:12–23. [DOI] [PubMed] [Google Scholar]
- 40. Chaudhari P, Ye Z, Jang Y‐Y. Roles of reactive oxygen species in the fate of stem cells. Antioxid Redox Signal. 2014;20:1881–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McLean AB, D'Amour KA, Jones KL, Krishnamoorthy M, Kulik MJ, Reynolds DM, Sheppard AM, Liu H, Xu Y, Baetge EE, Dalton S. Activin A efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3‐kinase signaling is suppressed. Stem Cells. 2007;25:29–38. [DOI] [PubMed] [Google Scholar]
- 42. Kim MS, Horst A, Blinka S, Stamm K, Mahnke D, Schuman J, Gundry R, Tomita‐Mitchell A, Lough J. Activin‐A and BMP4 levels modulate cell type specification during CHIR‐induced cardiomyogenesis. PLoS One. 2015;10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hunt MM, Meng G, Rancourt DE, Gates ID, Kallos MS. Factorial experimental design for the culture of human embryonic stem cells as aggregates in stirred suspension bioreactors reveals the potential for interaction effects between bioprocess parameters. Tissue Eng Part C Methods. 2014;20:76–89. [DOI] [PubMed] [Google Scholar]
- 44. Deng Z, Lei X, Zhang X, Zhang H, Liu S, Chen Q, Hu H, Wang X, Ning L, Cao Y, Zhao T, Zhou J, Chen T, Duan E. mTOR signaling promotes stem cell activation via counterbalancing BMP‐mediated suppression during hair regeneration. J Mol Cell Biol. 2015;7:62–72. [DOI] [PubMed] [Google Scholar]
- 45. Lee K‐W, Yook J‐Y, Son M‐Y, Kim M‐J, Koo D‐B, Han Y‐M, Cho YS. Rapamycin promotes the osteoblastic differentiation of human embryonic stem cells by blocking the mTOR pathway and stimulating the BMP/Smad pathway. Stem Cells Dev. 2010;19:557–568. [DOI] [PubMed] [Google Scholar]
- 46. Wahdan‐Alaswad RS, Bane KL, Song K, Shola DTN, Garcia JA, Danielpour D. Inhibition of mTORC1 kinase activates Smads 1 and 5 but not Smad8 in human prostate cancer cells, mediating cytostatic response to rapamycin. Mol Cancer Res. 2012;10:821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase‐3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL‐1. Mol Cell. 2006;21:749–760. [DOI] [PubMed] [Google Scholar]
- 48. Martin PM, Sutherland AE, Van Winkle LJ. Amino acid transport regulates blastocyst implantation. Biol Reprod. 2003;69:1101–1108. [DOI] [PubMed] [Google Scholar]
- 49. Musto A, Navarra A, Vocca A, Gargiulo A, Minopoli G, Romano S, Romano MF, Russo T, Parisi S. miR‐23a, miR‐24 and miR‐27a protect differentiating ESCs from BMP4‐induced apoptosis. Cell Death Differ. 2015;22:1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer Sequences Used for qPCR and siRNA Sequences Used for RNAi
Table S2. Antibodies Used for Western Blot, FACS Analysis, and Immunofluorescence Analysis
Figure S1. Rapamycin promotes cardiomyocyte differentiation. A and B, Quantitative real‐time PCR analysis of the expression of cardiac‐specific transcription factors and structural proteins such as NKX2.5, GATA4, RALDH2, MEF2C, MESP1, ISL1, cTnT, HCN4, MHY6, MLC2a, and MLC2v with or without rapamycin treatment at day 5 and day 12, respectively (n=6). C, The effect of different concentration of rapamycin on T (Brachyury) mRNA expression at day 3 (n=5). D, The induction of mesoderm marker gene Brachyury was blocked by 3BDO, a novel mTOR activator, and this effect was reduced by rapamycin addition (n=5). E, Transmission electron microscopic images of H9‐derived cardiomyocytes produced by the method described in Figure 1E. The white arrow indicates mature mitochondria, myofibrils with Z‐band, and intercalated disks. F, Representative flow cytometry data of GFP‐positive cardiomyocytes after lactate‐culturing purification. G, The concentration of rapamycin treatment influences the growth of EBs. Comparison of cell number (total) within indicated treatment. During days 0 to 3, 5 nmol/L rapamycin treatment increased the total cell number of EBs compared with the DMSO‐treated group. But 20 nmol/L rapamycin treatment inhibited the growth of EBs. The cell number was counted at day 10 (n=5). **P<0.01, *P<0.05; Mann‐Whitney U test was applied in B. 3BDO indicates 3‐benzyl‐5‐((2‐nitrophenoxy) methyl)‐dihydrofuran‐2(3H)‐one; CHIR, CHIR99021; DMSO, dimethyl sulfoxide; ID, intercalated disks; MF, myofibrils; Mt, mitochondria; PCR, polymerase chain reaction; Rapa, rapamycin.
Figure S2. The antiapoptosis effect of rapamycin on hESCs in high‐density monolayer culture. A, Rapamycin was unable to promote the colony formation of hESCs. H9 cells were dissociated into single cells and seeded in Matrigel‐coated 12‐well plates at a density of 1×105 per well with different treatment with small molecules: 0.1% DMSO, 12 μmol/L CHIR or CHIR plus 10 nmol/L rapamycin (n=5; Kruskal‐Wallis test followed by Dunn post hoc comparison). B and C, The activity of mTOR is essential for the apoptosis of hESCs at day 4. Annexin V/PI staining analysis of the apoptotic level of hESCs with si‐mTOR or si‐TSC1/2 treatment at day 4 (n=6; **P<0.01, one way ANOVA) (B). Annexin V/PI staining analysis of the apoptotic level of hESCs with or without rapamycin treatment at day 4 (n=6; Mann‐Whitney U test) (C). CHIR indicates CHIR99021; DMSO, dimethyl sulfoxide; hESC, human embryonic stem cell; Rapa, rapamycin; si‐NC, negative control siRNA.
Figure S3. TGF‐β signaling pathway is involved in mesoderm induction of rapamycin. A, The effects of activin A and BMP4 on Brachyury transcription level in mTeSR1 and RPMI/B27 culture conditions, respectively. Level of mRNA expression was normalized to DMSO group (n=5). B and C, Quantitative real‐time PCR analysis of associated TGF‐β superfamily members and their downstream genes (n=6). CHIR indicates CHIR99021; DMSO, dimethyl sulfoxide; PCR, polymerase chain reaction; Rapa, rapamycin; TGF‐β, transforming growth factor β.
Video S1. Day 15 cardiomyocytes induced from H9‐cTnT‐eGFP cells (×10).
Video S2. Day 15 cardiomyocytes induced from H7 cells (×20).