

Abstract
Background
DBZ (Danshensu Bingpian Zhi), a synthetic derivative of a natural compound found in traditional Chinese medicine, has been reported to suppress lipopolysaccharide‐induced macrophage activation and lipid accumulation in vitro. The aim of this study was to assess whether DBZ could attenuate atherosclerosis at early and advanced stages.
Methods and Results
The effects of DBZ on the development of atherosclerosis were studied using apolipoprotein E–deficient (apoE−/−) mice. For early treatment, 5‐week‐old apoE−/− mice were fed a Western diet and treated daily by oral gavage with or without DBZ or atorvastatin for 10 weeks. For advanced treatment, 5‐week‐old apoE−/− mice were fed a Western diet for 10 weeks to induce atherosclerosis, and then they were randomly divided into 4 groups and subjected to the treatment of vehicle, 20 mg/kg per day DBZ, 40 mg/kg per day DBZ, or 10 mg/kg per day atorvastatin for the subsequent 10 weeks. We showed that early treatment of apoE−/− mice with DBZ markedly reduced atherosclerotic lesion formation by inhibiting inflammation and decreasing macrophage infiltration into the vessel wall. Treatment with DBZ also attenuated the progression of preestablished diet‐induced atherosclerotic plaques in apoE−/− mice. In addition, we showed that DBZ may affect LXR (liver X receptor) function and that treatment of macrophages with DBZ suppressed lipopolysaccharide‐stimulated cell migration and oxidized low‐density lipoprotein–induced foam cell formation.
Conclusions
DBZ potentially has antiatherosclerotic effects that involve the inhibition of inflammation, macrophage migration, leukocyte adhesion, and foam cell formation. These results suggest that DBZ may be used as a therapeutic agent for the prevention and treatment of atherosclerosis.
Keywords: atherosclerosis, foam cell, inflammation, LXR
Subject Categories: Atherosclerosis, Peripheral Vascular Disease, Inflammation, Lipids and Cholesterol
Clinical Perspective
What Is New?
DBZ (Danshensu Bingpian Zhi), a synthetic derivative of a natural compound found in traditional Chinese medicine, potentially has antiatherosclerotic effects in apolipoprotein E–deficient mice that involve the inhibition of inflammation, macrophage migration, leukocyte adhesion, and foam cell formation.
What Are the Clinical Implications?
DBZ may be used as a promising therapeutic agent for the prevention and treatment of atherosclerosis.
Introduction
Atherosclerosis is a progressive disease in which lipid metabolism disorder and chronic inflammation coexist.1, 2 It is also the leading cause of mortality and a key risk factor for cardiovascular disease.3, 4 However, the current treatment approach using lipid‐lowering statins prevents only 50% to 60% of all cardiovascular events.5, 6 Monocytes and macrophages are the predominant cellular components of atherosclerotic lesions.2, 3, 7 Inflammation induces the recruitment of monocytes to the subendothelium, and they subsequently transmigrate through the vascular endothelial layer, internalize modified lipoproteins, and then invade the vessel wall to form cholesterol‐rich foam cells; these processes are crucial for the initiation and progression of atherosclerosis.7, 8 NF‐κB (nuclear factor κB) has also been regarded as a proatherogenic factor. Active NF‐κB can induce many inflammatory markers, such as adhesion molecules and chemokines, including endothelial selectin, VCAM‐1 (vascular cell adhesion molecule 1), ICAM‐1 (intracellular adhesion molecule 1), and MCP‐1 (monocyte chemoattractant protein 1).9, 10 Consequently, NF‐κB–mediated vascular inflammation plays a critical role in the initiation and progression of atherosclerosis. Consistent with this notion, inhibition of the NF‐κB signaling pathway in the mouse model significantly reduces the risk of atherosclerosis.11, 12 In addition, multiple studies have demonstrated that inhibition of macrophage foam cell formation by stimulating cholesterol efflux can efficiently protect against atherosclerotic lesion development.13, 14
DBZ is a novel synthetic compound derived from traditional Chinese medicine,15, 16 and the traditional Chinese medicine formula Fufang Danshen is clinically effective for cardiovascular disease.17 We previously showed that DBZ can block lipopolysaccharide‐induced NF‐κB activation and macrophage lipid accumulation. This finding suggested that DBZ modulates key early atherogenic events in vitro15; however, whether DBZ has an antiatherosclerotic effect remains unclear.
In this study, we demonstrated that early DBZ intervention potently reduced atherosclerotic lesion formation by inhibiting inflammatory marker expression and decreasing macrophage infiltration into the vessel walls of mice with apolipoprotein E (apoE) deficiency induced by a Western diet. DBZ treatment also suppressed lipopolysaccharide‐stimulated migration of RAW 264.7 cells and foam cell formation induced by oxidized low‐density lipoprotein (ox‐LDL) in THP‐1 macrophages. Most important, ultrasound and pathological analyses of the aortic tissues showed an obvious protective effect against atherosclerosis development in the advanced DBZ‐treated apoE‐deficient (apoE−/−) mice. Atherosclerotic lesions in DBZ‐treated animals also exhibited increased collagen content and increased of numbers of smooth muscle cells, which are features of plaque stability. Taken together, our results provide evidence that DBZ may contribute to the prevention and treatment of atherosclerosis in apoE−/− mice. This warrants additional research to elucidate the anti‐atherosclerotic role of DBZ, and it is of vital importance for the development of synthetic drugs for atherosclerosis.
Methods and Materials
Reagents
DBZ (purity: 99.6%) was synthesized by Dr Xiaohui Zheng at Northwest University (Xi'an, China). NBD cholesterol (Cayman), apoA1, high‐density lipoprotein (HDL) (ProSpec), atorvastatin (Pfizer Ltd.), lipopolysaccharide, PMA (phorbol 12‐myristate 13‐acetate), T0901317 (hereafter referred to as T1317), and GGPP (geranylgeranyl pyrophosphate) were purchased from Sigma‐Aldrich.
Animals and Experimental Design
Male 5‐week‐old apoE−/− mice and additional age‐matched C57BL/6J mice were obtained from the Department of Laboratory Animal Science at Peking University. All mice (4 to 5 mice per cage) were kept in a temperature‐controlled room on a 12‐hour light/dark cycle with food and water available ad libitum. All mice were fed a high‐cholesterol atherogenic Western diet (21% fat and 1.25% cholesterol). In the prevention experiment, a Western diet and various interventions were started simultaneously in 5‐week‐old mice. In the treatment paradigm, the reagents were administered after 10 weeks of a Western diet (at 15 weeks of age) after significant atherosclerosis had developed. Before each study, the apoE−/− mice were randomly divided into 4 experimental groups (9–12 per group) based on body weight and were administered DBZ (20 or 40 mg/kg per day), Atorvastatin (10 mg/kg per day) or vehicle (equal volume of 50% polyethylene glycol) daily by oral gavage. We used ultrasound analysis to observe aortic lesion progression in mice aged 15 and 25 weeks in vivo. Mice were euthanized by CO2, and tissue and blood were then harvested. The mice were perfused with normal saline through the heart before the tissues were harvested. Finally, the heart with the entire aorta and other tissues were isolated and stored at −80°C for subsequent histological evaluation or gene expression analysis. The animal experiments were performed in strict accordance with the recommendations in the Guide for Institutional Animal Care and Use Committee at the Beijing Normal University, and all experiments conformed to the relevant regulatory standards, which conform to the US National Institutes of Health (NIH) guidelines. The experiments and protocol were approved by the institutional animal care and use committee at Beijing Normal University (approval no. CLS‐EAW‐2013‐011).
In Vivo Ultrasound Analysis
The ultrasound imaging parameters of the ascending aortas were examined with a Vevo 770 Microultrasound system (Visualsonics) equipped with a 30‐MHz transducer. A right parasternal long‐axis view was used to visualize the ascending aorta, aortic arch, and innominate artery in 1 plane.18 Aortic valve peak velocity was measured using Doppler ultrasonography. During the experiments, mice were anesthetized with inhaled 1.5% isoflurane, resulting in a heart rate of ≈300 to 400 beats/min.
Serum Biochemical Analysis
Mice were fasted overnight, and retro‐orbital sinus blood was collected, allowed to coagulate, and centrifuged at 1000g to isolate the serum. The serum was immediately frozen and stored at −80°C. The serum lipid profile, including total cholesterol, triglycerides, LDL cholesterol, and HDL cholesterol, was measured at the Beijing Hospital Clinical Biochemistry Department with an AU 400 fully automated chemistry analyzer (Olympus) using the enzymatic‐colorimetric method. Serum TNF‐α (tumor necrosis factor α), IL‐6 (interleukin 6), and MCP‐1 were evaluated by ELISA, following the manufacturer's instructions.
Histological Evaluation of Atherosclerotic Lesions
This experiment was performed according to methods reported previously.19, 20 En face pinned aortas were stained with Oil Red O. Images of the aortas were captured with a Nikon digital camera. The proximal aortas attached to the heart were embedded in OCT compound and frozen at −80°C. The aortic root was serially sectioned into 8‐μm sections from the site where the aortic valve appears. A set of 6 consecutive sections were stained with Oil Red O and Masson's Trichrome for morphological analysis of the atherosclerotic plaque. For immunofluorescence, macrophages or the expression of VCAM‐1 in the aortic root were detected by staining sequential sections with rat anti–Monocyte macrophage (MOMA‐2) monoclonal antibody or rabbit polyclonal anti‐VCAM, followed by the corresponding secondary antibodies conjugated with Alexa Fluor 488 or 568 for fluorescence detection. Smooth muscle actin cells were detected with α‐Smooth Muscle–Cy3 (1:400; Sigma‐Aldrich). Images were captured using an Axio Imager M1 (Zeiss). Quantitative analysis of the sections was performed by a technician who was blinded to the treatment group of the mice using Image‐Pro Plus 6.0 software (Media Cybernetics).
Cell Culture
Human embryonic kidney 293 cells were obtained from the American Type Culture Collection and maintained in DMEM containing 10% FBS and 1% penicillin–streptomycin. The THP‐1 and RAW 264.7 cells were maintained in RPMI medium containing 10% heat‐inactivated FBS, antibiotics, and 0.05 mmol/L 2‐mercaptoethanol. Cells were cultured at 5% CO2 and 37°C. To fully differentiate THP‐1 monocytes to macrophages, cells were incubated in growth medium supplemented with 200 nmol/L PMA for 48 hours. The primary peritoneal macrophages were isolated from 25‐week‐old apoE−/− mice fed a Western diet for 20 weeks in each group. Briefly, the mice were euthanized, 5 mL of ice cold 1640 medium (with 3% FBS) was injected into the peritoneal cavity, the peritoneum was gently massaged for 3 minutes, the 1640 medium was collected and centrifuged at 200 g for 5 minutes, the cells were resuspended and cultured at 5% CO2 and 37°C for 2 hours, nonadherent cells were removed, and then cells were cultured for 3 days with 10% FBS/DMEM.
Transwell Migration Assay
The cell migration assay was performed in 24‐well polycarbonate membrane inserts (8.0‐μm pore size). A total of 1×105 RAW 264.7 cells were pretreated with various concentrations of DBZ for 24 hours and then plated onto the upper inserts; the cells were then stimulated with or without lipopolysaccharide (1 μg/mL) for 6 hours. Nonmigrated cells were wiped off with a cotton swab, and migrated cells were fixed with methanol and stained with 0.1% Giemsa. Filters were photographed, and the cells were quantified by counting 5 different microscopic fields using ImageJ (NIH).
Cellular Lipid Analysis
THP‐1 macrophages and the primary peritoneal macrophages were fixed and stained for lipids using Oil Red O staining. To determine intracellular lipid contents, cells were removed from the culture plates and washed twice with PBS. Then, intracellular lipids were extracted using isopropanol. Total and free cholesterol were determined using an enzymatic assay.
Cholesterol Efflux Assay
Cholesterol efflux was measured using the methods modified from Song et al.21 In brief, THP‐1 macrophages were equilibrated with NBD cholesterol (5 μmol/L) for 4 hours. The starved cells were cultured in serum‐free medium with various concentrations of DBZ (5, 10, and 20 μmol/L) for another 16 hours. Cholesterol efflux was induced by the addition of 50 μg/mL recombinant human apoA1 or 50 μg/mL human HDL for an additional 4 hours. The fluorescence‐labeled cholesterol released from the cells into the medium was measured using a Tecan F200 96‐well plate reader. Cholesterol efflux was expressed as a percentage of fluorescence in the medium relative to total fluorescence.
Transient Transfection and Luciferase Reporter Assay
Dual luciferase assays were performed in 24‐well plates with Lipofectamine 2000 transfection reagent, according to the manufacturer's instructions (Invitrogen). The day before the transfection, HEK 293 cells were seeded at a density of 2×105 cells per well such that 70% to 80% confluence was obtained. The cells were transiently transfected with pCMX‐hLXR‐α or pCDG‐mLXR‐β (pCMX‐hLXR‐α or pCDG‐mLXR‐β human LXRα (hLXRα) or mouse LXR‐β(mLXR‐β) protein‐coding region was inserted in pCMX or pCDG1 plasmid) and tk‐(LXRE)×3‐LUC (luciferase reporter vector containing the herpes simplex virus thymidine kinase (HSV‐TK) promoter region and 3 repeats of LXRE (LXR response elements), as well as pRL‐TK (Renilla luciferase reporter vector contains the herpes simplex virus thymidine kinase (HSV‐TK) promoter), which served as an internal control for transfection efficiency. After 24 hours, cells were treated with dimethyl sulfoxide (control), T1317, or the indicated concentration of DBZ. Dual luciferase assays were performed after 24 hours using a Dual‐Luciferase reporter assay kit (Promega).
Molecular Docking
The crystal structure of the human LXR‐α ligand‐binding pocket was prepared (PBD 1UHL). The DBZ structure was built using the Molecule Builder in MOE (Molecular Operating Environment; Chemical Computing Group). The force field, receptor–ligand affinity, and reasonable binding models were analyzed using AutoDock (Scripps Research Institute). Predicted binding conformations were visualized, and ligand–protein interactions were analyzed using PyMOL (Schrödinger), with the binding site defined as a 6‐Å sphere.
Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was isolated from homogenized tissues or cells using TRIzol reagent (Invitrogen). Total RNA (2 μg) was then reverse‐transcribed to cDNA using M‐MLV transcriptase (Promega), according to the manufacturer's instructions. Real‐time polymerase chain reaction was performed on an ABI 7500 System cycler (Applied Biosystems) using SYBRGreen PCR Master Mix (Transgen Biotic), as described previously.22 Gene expression was normalized against the expression of cyclophilin. Data were analyzed using the 2−ΔΔCt method. The primers used are listed in Table S1.
Western Blotting
An equal amount of cell or tissue protein (10 μg) was subjected to SDS‐PAGE, and then Western blotting analysis was performed using the indicated antibodies. Protein detection was performed with rabbit polyclonal antibodies such as anti‐ABCA1 (anti–ATP binding cassette subfamily A member 1), anti‐ABCG1 (anti–ATP binding cassette subfamily G member 1), anti‐MMP9 (anti–matrix metallopeptidase 9), and β‐actin. Immunoreactive proteins were detected with enhanced chemiluminescence detection reagent, as described previously.23 Quantification of the band intensity was performed using ImageJ. Values were normalized to β‐actin.
Statistical Analysis
Statistical analyses were performed with SPSS version 20.0 (IBM Corp). All results are presented as the mean±SEM. Statistical significance was determined by t test or 1‐way ANOVA for data with normal distribution and by Kruskal–Wallis test for nonnormal distribution data or small samples. Statistical significance was set at P<0.05. For the cell experiments, all experiments were performed at least 3 times, and each experiment was performed in triplicate.
Results
Treatment of apoE−/− Mice With DBZ Prevents the Onset of Atherosclerosis
ApoE−/− mice spontaneously develop hyperlipidemia and have been used extensively to study the development of atherosclerosis.24 To determine the effect of DBZ on atherosclerosis, 5‐week‐old apoE−/− mice were fed a Western diet and were treated daily with or without DBZ or atorvastatin (Figure 1A) for 10 weeks by oral gavage. No difference was observed in body weight (Figure S1). En face analysis revealed that DBZ treatment with 40 mg/kg per day significantly reduced the size of Oil Red O–stained atherosclerotic plaques in the aortas by 31.4%, particularly in the aortic arch region, which corresponds to a region that experiences turbulent blood flow. In addition, there was a 38.3% reduction in atherosclerosis, although detectable effects were not observed in the thoracic and abdominal aorta, which correspond to regions of laminar flow (Figure 1B). Atherosclerotic burden was further evaluated in cross‐sections of the aortic root. As expected, treatment with DBZ (20 and 40 mg/kg per day) significantly decreased the lesion area in the aortic root by ≈31.4% and 29.5%, respectively, whereas the positive control (atorvastatin, 10 mg/kg per day) treatment reduced the lesion area by 32.1% (Figure 1C). Histological assessment of atherosclerotic lesions at the aortic root showed considerably reduced macrophage content, as detected by MOMA‐2 staining, in DBZ‐treated apoE−/− mice (Figure 1D), which indicated reduced inflammation. Gene expression analysis also suggested a shift in macrophage phenotype from M1 (proinflammatory) to M2 (anti‐inflammatory) in the aortic arch of DBZ‐treated apoE−/− mice compared with their vehicle‐treated counterparts (Figure 1E).
Figure 1.
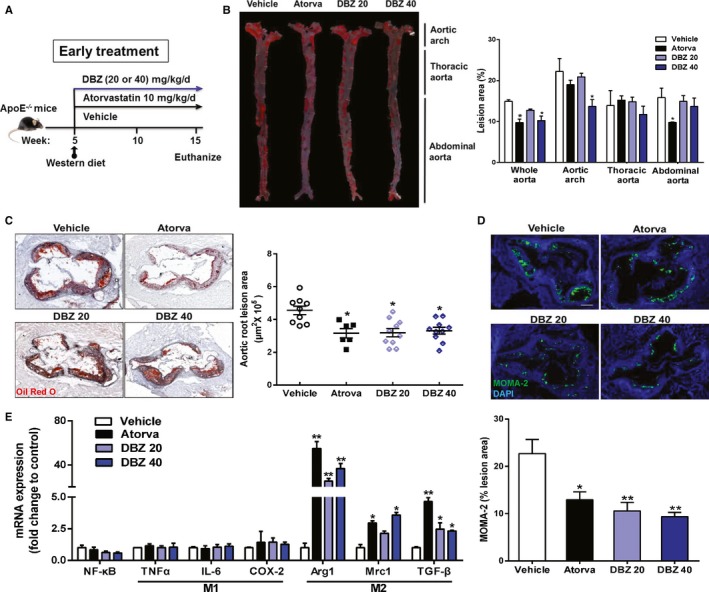
DBZ reduces atherosclerotic plaque formation in apoE−/− mice. Five‐week‐old male apoE−/− mice were fed a Western diet and were administered DBZ (20 mg/kg per day [DBZ 20] or 40 mg/kg per day [DBZ 40]) or atorvastatin (10 mg/kg per day) daily for 10 weeks by gavage. A, Schematic of the experimental procedures. B, Representative images and quantification of Oil Red O–stained en face aortic preparations (n=5 per group). Aortic root section staining with Oil Red O (C) and MOMA‐2 (D) and quantification (scale=200 μm; n=6–10). E, Gene expression analysis of the indicated macrophage phenotypes, M1 (proinflammatory) and M2 (anti‐inflammatory), in the aortic arch with plaques of mice (n=5 per group). Data are presented as the mean±SEM. *P<0.05; **P<0.01. Arg 1 indicates arginase 1; Atorva, atorvastatin; COX‐2, cyclooxygenase 2; DBZ, Danshensu Bingpian Zhi; IL‐6, interleukin 6; MOMA‐2, monocyte macrophages 2; Mrc1, mannose receptor 1; NF‐κB, nuclear factor κB; TNFα, tumor necrosis factor α; TGF‐β, transforming growth factor β.
Treatment of apoE−/− Mice With DBZ Attenuates the Progression of Preestablished Atherosclerotic Plaques
Because atherosclerosis is often treated as an advanced disease in the clinic, we sought to determine whether DBZ treatment affects preestablished atherosclerotic plaques. In this experiment, apoE−/− mice were fed a Western diet for 10 weeks to induce atherosclerosis. The mice were then randomly divided into 4 groups and subjected to treatment with vehicle, DBZ 20 mg/kg per day), DBZ 40 mg/kg per day, or atorvastatin (10 mg/kg per day) for the following 10 weeks while the mice remained on the Western diet (Figure 2A). Aortic valve peak velocity is an important parameter of aortic stenosis, and it correlates with the severity of atherosclerotic plaques.25, 26, 27 We measured aortic valve peak velocity using high‐frequency ultrasound analysis to assess the effects of DBZ treatment (Figure S2). As expected, and as shown in Figure 2B, the vehicle‐treated apoE−/− mice fed with a Western diet for 20 weeks exhibited significantly higher transaortic flow velocity than the age‐matched and chow‐fed C57BL/6J (wild‐type) mice, indicating the success of the model. The Western diet–induced elevation in transaortic flow velocity was substantially attenuated by DBZ and atorvastatin treatment. Moreover, treatment with DBZ at 40 mg/kg restored the transaortic flow velocity to a level similar to that of the chow‐fed wild‐type mice. Results of the echocardiographic analysis showed that DBZ treatment had little effect on cardiac functions (Table S2).
Figure 2.
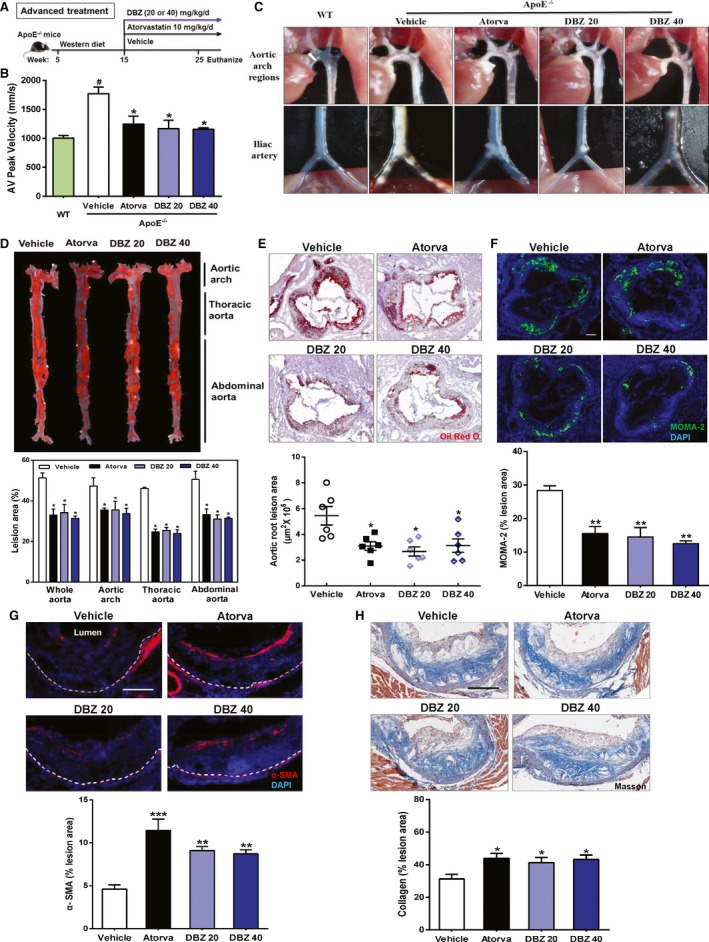
Treatment with DBZ inhibits the development of preestablished atherosclerotic plaques in apolipoprotein E–deficient (apoE−/−) mice. A, Schematic of the experimental procedure for late DBZ intervention. B, Aortic valve (AV) peak velocity was measured using Doppler ultrasonography in mice. # P<0.05 vs WT; *P<0.05 vs vehicle. n=6 per group. C, Representative in situ photographs of the aortic arch and iliac artery regions with white plaques. D, Representative images and quantification of Oil Red O–stained en face aortic preparations (n=5 per group). Representative cross‐sections and quantification of (E) Oil Red O staining (scale bar=200 μm), (F) immunostaining for MOMA‐2 (scale bar=200 μm), (G) α‐SMA staining for smooth muscle actin (scale bar=100 μm), and (H) Masson's Trichrome staining (scale bar=100 μm) in the aortic sinus of the mice. All data are presented as the mean±SEM. *P<0.05; **P<0.01. α‐SMA indicates α‐smooth muscle actin; Atorva, atorvastatin; DBZ, Danshensu Bingpian Zhi; DBZ 20, DBZ 20 mg/kg per day; DBZ 40, DBZ 40 mg/kg per day; MOMA‐2, metallophilic macrophages 2; WT, wild type.
Treatment with DBZ also inhibited the progression of the atherosclerotic plaques. The aortas from Western diet–treated apoE−/− mice exhibited confluent atherosclerotic plaques that were readily visible on exposure of the aortic arch. Of note, the iliac arteries and aortas of these mice were severely occluded. Treatment with DBZ relieved the extent of aortic stenosis in both regions (Figure 2C). As shown in Figure 2D, en face preparations revealed that DBZ at 40 mg/kg ameliorated further plaque development by ≈38.9%, whereas treatment with atorvastatin only ameliorated plaque development by 35.8%. Similar inhibitory effects of DBZ on the progression of atherosclerotic plaques were also observed at the aortic arch, thoracic aorta, and abdominal aorta. Histological evaluation of the aortic root sections showed that the plaques in the DBZ‐treated apoE−/− mice contained fewer lipids, as assessed by Oil Red O staining (Figure 2E), and less macrophage infiltration, as assessed by MOMA‐2 staining (Figure 2F). Although plaque size can indicate the extent of atherosclerotic development, it is believed that plaque stability is a more accurate predictor of clinical outcomes.28, 29 To investigate whether DBZ affects plaque stability, we measured several key parameters that are associated with plaque stability. The number of smooth muscle cells increased in the plaques of DBZ‐treated apoE−/− mice (Figure 2G), and Masson's Trichrome staining revealed that those atherosclerotic plaque lesions contained more collagen (Figure 2H). The reduction in lipid deposition and macrophage infiltration with an increase in smooth muscle cell and collagen content in plaque are features of a more stable plaque phenotype.28 These results indicate that DBZ treatment promotes lesion stability in addition to reducing atherogenesis.
The Antiatherosclerotic Activity of DBZ Is Associated With the Inhibition of Inflammation, Macrophage Migration, and Leukocyte Adhesion
Inflammatory processes within the vessel wall mediate the initiation and progression of atherosclerosis. To determine whether the antiatherosclerotic activity of DBZ could be attributed to the anti‐inflammatory actions of DBZ, apoE−/− mice were challenged with the Western diet for 10 weeks; then, serum cytokine concentrations were determined as an index of systemic inflammation. As shown in Figure 3A, treatment with DBZ significantly suppressed serum levels of TNF‐α, IL‐6, and MCP‐1. The anti‐inflammatory activity of DBZ was also demonstrated in vitro. As shown in Figure 3B, treatment of the RAW 264.7 macrophages with DBZ inhibited the lipopolysaccharide‐induced upregulation of TNF‐α, IL‐6, and MCP‐1 in a dose‐dependent manner. Monocyte recruitment and subsequent transendothelial migration are critical steps in early atherosclerosis.4, 10 We also examined the effects of DBZ on lipopolysaccharide‐induced macrophage migration. As expected, exposing RAW 264.7 macrophages to lipopolysaccharide markedly increased cell migration (Figure 3C). Treatment with DBZ dose‐dependently inhibited lipopolysaccharide‐stimulated cell migration. Consistent with the inhibition of cell migration, western blot analysis showed that lipopolysaccharide stimulation induced the expression of MMP9, an important matrix metalloproteinase for migration,30 which was inhibited by DBZ in a dose‐dependent manner in RAW 264.7 macrophages (Figure 3D). Taken together, these results suggest that DBZ suppresses lipopolysaccharide‐induced macrophage cell mobility and may contribute to the antiatherosclerotic effect of DBZ.
Figure 3.
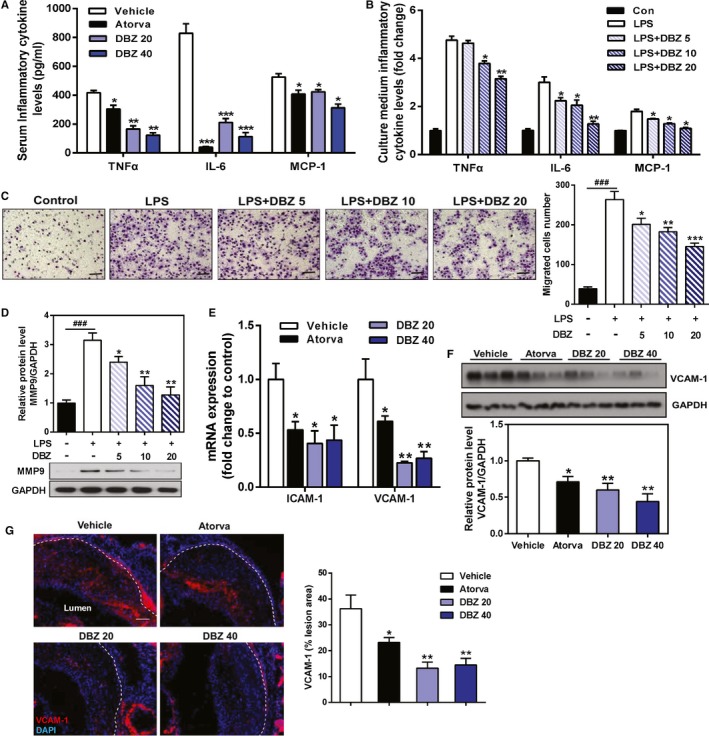
DBZ treatment inhibits systemic inflammation and LPS‐induced cell migration in RAW 264.7 macrophages. A, Quantification of the chemokines TNFα, IL‐6, and MCP‐1 in mouse serum by ELISA, as indicated in Figure 1 (n=6–10). RAW 264.7 cells pretreated with various concentrations of DBZ for 24 h were seeded onto Transwell chambers and stimulated with or without LPS (1 μg/mL) for 6 h in 3 independent experiments. B, Culture medium concentrations of TNFα, IL‐6, and MCP‐1 were assayed by ELISA. C, Representative images and quantification of migrated cells. D, Western blot analysis of MMP9 protein expression in RAW 264.7 macrophages. E, Relative VCAM and ICAM mRNA expression in the aortic arch was measured by quantitative reverse transcriptase polymerase chain reaction and normalized to cyclophilin, as indicated in Figure 1 (n=5 per group). F, VCAM‐1 protein expression in the aorta was assessed by Western blot analysis and normalized to GAPDH (n=3 per group). G, Representative images and quantification of VCAM‐1 staining in the intima layer at the aortic root (scale=100 μm; n=5 per group). Data are presented as the mean±SEM. ### P<0.001 vs control group, *P<0.05; **P<0.01; and ***P<0.001 vs LPS group or vehicle. Atorva indicates atorvastatin; Con, control; DBZ, Danshensu Bingpian Zhi; DBZ 5, DBZ 5 μmol/L; DBZ 10, DBZ 10 μmol/L; DBZ 20, DBZ 20(B,C,D) 20 μmol/L, DBZ 20(A,E,F,G) 20 mg/kg/day; DBZ 40, DBZ 40 mg/kg/day; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; ICAM‐1, intercellular adhesion molecule 1; IL‐6: interleukin 6; LPS, lipopolysaccharide; MCP‐1, monocyte chemoattractant protein 1; MMP9, matrix metallopeptidase 9; TNFα, tumor necrosis factor α; VCAM‐1, vascular cell adhesion molecule 1.
To further clarify the molecular mechanism of DBZ‐induced antiatherogenic effects, the expression of adhesion molecules was analyzed in the aorta of DBZ‐treated apoE−/− mice. The mRNA expression of ICAM‐1 and VCAM‐1, 2 prominent mediators responsible for leukocyte adhesion and recruitment to the vascular wall,31, 32, 33 was significantly decreased in the aortic arch of DBZ‐treated apoE−/− mice compared with their vehicle‐treated counterparts (Figure 3E). At the protein level, treatment with DBZ at 40 mg/kg reduced VCAM‐1 expression by 60%, whereas atorvastatin treatment reduced VCAM‐1 expression by 36% (Figure 3F). Immunostaining of the aortic root sections with the VCAM‐1 antibody confirmed the reduced expression of VCAM‐1 in the atherosclerotic plaques of DBZ‐treated apoE−/− mice (Figure 3G).
DBZ Attenuates ox‐LDL–Induced Foam Cell Formation and Promotes Cholesterol Efflux in Macrophages, Likely Through the Activation of LXR
The transformation of invading macrophages into foam cells by ox‐LDL uptake is another key event of atherogenesis.34, 35 To determine the effects of DBZ on ox‐LDL–induced foam cell formation, THP‐1 monocyte‐derived macrophages were incubated with ox‐LDL (50 μg/mL) for 24 hours. The addition of ox‐LDL to the culture medium induced foam cell formation, as indicated by the cytoplasmic accumulation of lipids. Treatment with DBZ significantly decreased ox‐LDL–induced intracellular lipid accumulation, as revealed by Oil Red O staining (Figure 4A). Cellular lipid content analysis also revealed that 20 μmol/L DBZ decreased cholesteryl ester content by 44% (Figure 4B). These results suggest that DBZ prevents ox‐LDL–induced foam cell formation in THP‐1 macrophages. To further investigate the ability of DBZ inhibiting cholesterol ester accumulation in macrophages, we isolated the primary peritoneal macrophages from 25‐week‐old apoE−/− mice fed a Western diet for 20 weeks in each group. Oil Red O staining and cellular lipid analysis showed that DBZ treatment significantly attenuated intracellular cholesterol ester accumulation in peritoneal macrophages (Figure S3A and S3B).
Figure 4.
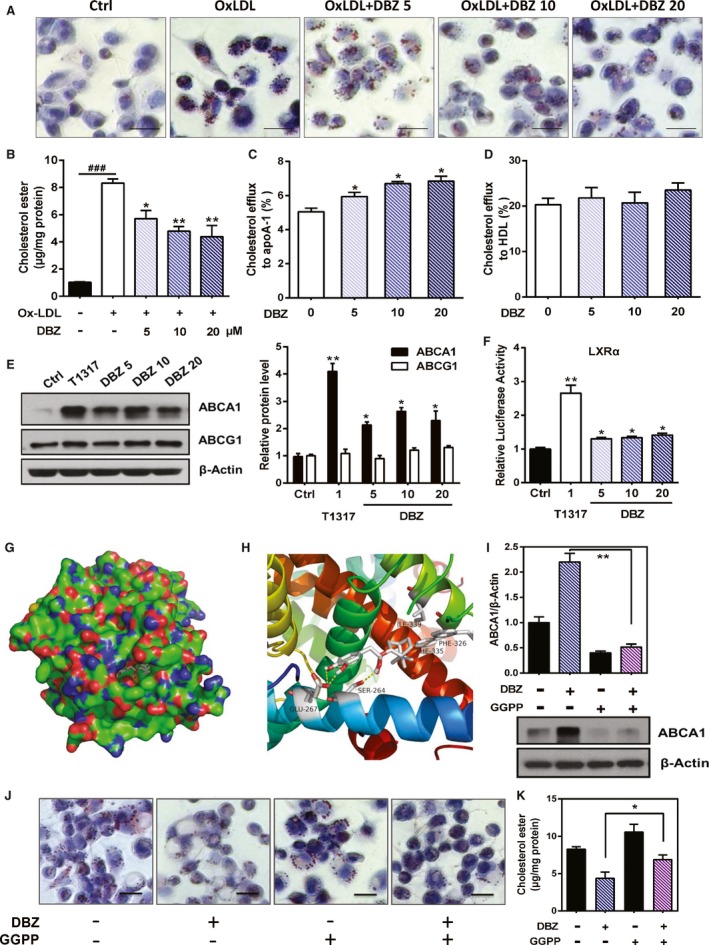
DBZ attenuates ox‐LDL–induced foam cell formation and promotes cholesterol efflux in macrophages. A, THP‐1 macrophages were cotreated with the indicated concentrations of DBZ (5, 10, and 20 μmol/L) and OxLDL (50 μg/mL) for 48 h. Lipid accumulation was observed by Oil Red O staining (scale=20 μm). B, Intracellular cholesteryl ester levels were normalized to cellular protein content in THP‐1 macrophages. Macrophages were treated with DBZ and NBD cholesterol. Cholesterol efflux to apoA‐1 (C) and HDL (D) from THP‐1 macrophages. Cholesterol efflux was expressed as the percentage fluorescence in the medium relative to total fluorescence. E, THP‐1 macrophages were treated with the indicated concentrations of DBZ for 24 h. The relative protein levels of ABCA1 and ABCG1 were determined by Western blot analysis. T1317 (1 μmol/L) was used as a positive control. F, The transcriptional activity of LXRα was assessed by the transactivation reporter assay in 293T cells. G and H, The model structure of the complex formed by the LXRα ligand‐binding pocket and DBZ by molecular docking. I, THP‐1 macrophages were pretreated with GGPP (10 μmol/L) for 2 h and then treated with DBZ (20 μmol/L) for 24 h. The relative protein level of ABCA1 was determined by Western blot analysis. Prior to treatment with DBZ (20 μmol/L), THP‐1 macrophages were treated with GGPP for 2 h and then incubated with 50 μg/mL OxLDL for 48 h. J, Representative Oil Red O staining of THP‐1 macrophage foam cells (scale=20 μm). K, Intracellular cholesteryl ester levels that were normalized to cellular protein content in THP‐1 macrophages. Values are presented as the mean±SEM of at least 3 experiments (### P<0.001, *P<0.05, and **P<0.01). ABCA1 indicates ATP binding cassette subfamily A member 1; ABCG1, ATP binding cassette subfamily G member 1; apoA‐1, apolipoprotein A1; Ctrl, control; DBZ, Danshensu Bingpian Zhi; DBZ 5, DBZ 5 μmol/L; DBZ 10, DBZ 10 μmol/L; DBZ 20, DBZ 20 μmol/L; GGPP, geranylgeranyl pyrophosphate; HDL, high‐density lipoprotein; LXRα, liver X receptor α; OxLDL, oxidized low‐density lipoprotein.
Cholesterol efflux plays a pivotal role in maintaining intracellular cholesterol levels and preventing foam cell formation35, 36; therefore, we evaluated the effect of DBZ on cholesterol efflux from macrophages using an NBD cholesterol efflux assay. As shown in Figure 4C, DBZ dose‐dependently promoted apoA1‐mediated cholesterol efflux from THP‐1 macrophages. Treatment with 20 μmol/L DBZ increased NBD cholesterol efflux to apoA1 by 35.6% (P<0.05), whereas DBZ had little effect on HDL‐induced NBD cholesterol efflux (Figure 4D).
Consistent with the increased cholesterol efflux, the expression of ABCA1 and ABCG1, 2 pivotal factors in cholesterol efflux,37, 38, 39 was upregulated by ≈2‐fold in DBZ‐treated THP‐1 macrophages (Figure 4E). Both cholesterol efflux and the expression of ABCA1 are known to be positively regulated by the nuclear receptor LXR‐α.40 We thus speculated that the effects of DBZ on cholesterol efflux and the expression of ABCA1 may have been mediated by the activation of LXR. Indeed, DBZ treatment of cells cotransfected with the LXR‐α or LXR‐β expression vector and the LXR‐responsive luciferase reporter genes induced luciferase reporter activity, and DBZ showed weaker activation in LXR‐β than LXR‐α (Figure 4F and Figure S4). To determine the interaction of DBZ with LXR‐α, the model structure of the human LXRα ligand‐binding domain and DBZ complex was determined by molecular docking (Figure 4G). In addition, the hydrogen bonds at Glu267 and Ser264, as well as hydrophobic interactions at Ile339, Phe326, and Phe335 (potential), maintain binding stability (Figure 4H). To further determine whether the induction of ABCA1 by DBZ in THP‐1 was an LXR‐α–dependent pathway, THP‐1 macrophages were pretreated with GGPP (10 μmol/L), a pharmacological inhibitor of LXR‐α, which was followed by treatment with DBZ (20 μmol/L). Compared with the THP‐1 macrophages treated with DBZ alone, cotreatment with GGPP decreased ABCA1 protein expression by 80% (Figure 4I). Moreover, cotreatment with GGPP also abrogated the inhibitory effect of DBZ on cholesterol ester accumulation in THP‐1 macrophages (Figure 4J and 4K). Collectively, DBZ, as a putative LXR agonist, attenuates ox‐LDL–induced foam cell formation and promotes cholesterol efflux in macrophages.
DBZ Has Little Effect on Aortic, Hepatic, and Intestinal Reverse Cholesterol Transport
Our lipid profile analysis revealed that treatment with 40 mg/kg DBZ modestly but significantly increased the serum levels of HDL cholesterol but had little effect on the serum levels of triglycerides, total cholesterol, and LDL cholesterol (Table). Treatment with atorvastatin significantly decreased the serum levels of total and LDL cholesterol (Table).
Table 1.
Effects of DBZ on Serum Lipid Profiles in Preestablished Atherosclerotic Plaques of Apolipoprotein E–Deficient Mice
| Vehicle | Atorvastatin | DBZ 20 | DBZ 40 | |
|---|---|---|---|---|
| TG, mg/dL | 123.89±19.46 | 104.61±10.62 | 117.70±34.51 | 100.00±4.43 |
| TC, mg/dL | 1154.51±76.27 | 659.90±43.85** | 1162.20±45.17 | 1028.09±25.82 |
| LDL‐C, mg/dL | 546.65±32.70 | 230.22±26.30** | 527.79±49.33 | 514.55±10.85 |
| HDL‐C, mg/dL | 48.16±5.79 | 43.96±3.95 | 58.95±8.23 | 60.14±2.19* |
All values are expressed as the mean±SEM (n=9 per group). One‐way analysis followed by the Dunnett post hoc test. *P<0.05; **P<0.01 vs vehicle group. DBZ indicates Danshensu Bingpian Zhi; DBZ 20, DBZ 20 mg/kg per day; DBZ 40, DBZ 40 mg/kg per day; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; TC, total cholesterol; TG, triglycerides.
To determine whether DBZ‐induced cholesterol efflux was coupled with increased reverse cholesterol transport in vivo, we measured the expression changes of genes involved in cholesterol metabolism and transport in the aorta, liver, and small intestine. The most significant change in DBZ‐treated apoE−/− mice was an upregulation of the efflux transporters ABCA1 and ABCG1 (≈4‐fold) in the aorta (Figure 5A), as well as ABCG5 (≈8‐fold) and ABCG8 (≈3‐fold) in the small intestine (Figure 5B). In contrast, the mRNA expression of the major cholesterol uptake transporters CD36 and SR‐A (scavenger receptor class A) in the aorta, mRNA expression of CD36 and LDLR (LDL receptor) in the liver, and mRNA expression of NPC1L1 (Niemann‐Pick C1‐like 1) and LDLR in the intestine was unchanged (Figure 5A and Figure S5). The mRNA expression levels of the genes involved in cholesterol biosynthesis, HMG‐CoAR (3‐hydroxy‐3‐methylglutaryl‐coenzyme reductase), and HMG‐CoAS (3‐hydroxy‐3‐methylglutaryl‐coenzyme synthase) were also not affected by DBZ treatment (Figure 5C). The serum lipid and gene expression profiles suggested that the DBZ‐induced cholesterol efflux was not coupled with increased reverse cholesterol transport.
Figure 5.
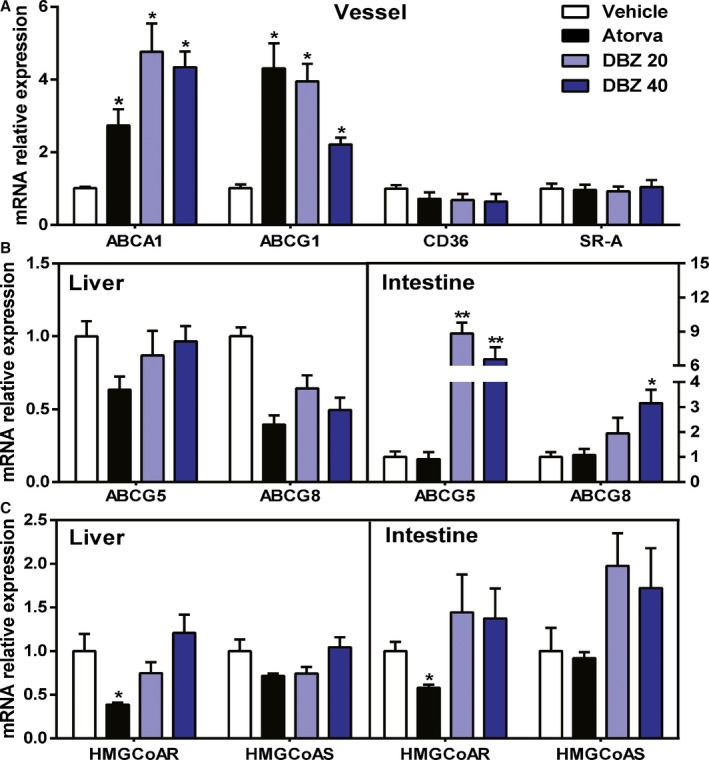
DBZ treatment affects cholesterol efflux gene expression in preestablished atherosclerotic plaque apolipoprotein E–deficient mice. A, Aortic, (B and C) hepatic, and intestinal mRNA levels of genes involved in the metabolism and transport of cholesterol as determined by real‐time polymerase chain reaction and normalized to cyclophilin in the mice, as in Figure 2 (n=6 per group). Data are expressed as the fold change plus or minus SEM (*P<0.05; **P<0.01). ABCA indicates ATP binding cassette subfamily A; ABCG, ATP‐binding cassette subfamily G; Atorva, atorvastatin; CD36, cluster of differentiation 36; DBZ, Danshensu Bingpian Zhi; DBZ 20, DBZ 20 mg/kg per day; DBZ 40, DBZ 40 mg/kg per day; HMGCoAR, 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase; HMGCoAS, 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A synthase; SR‐A, scavenger receptor class A.
Discussion
It is widely recognized that both hyperlipidemia and inflammation are well‐known risk factors in the development of atherosclerosis and have been shown to play causal roles in the progression of atherosclerotic plaques.41, 42 Previous studies have demonstrated that the novel compound DBZ could suppress lipopolysaccharide‐induced monocyte activation and foam cell formation,15 which are the key early events in atherosclerosis. Consequently, we assessed the antiatherosclerotic potential of DBZ in apoE−/− mice, as shown in Figure 6. Our results provided evidence that DBZ significantly reduced atherosclerosis development in vivo, as assessed by the evaluation of en face aortic lesion coverage and aortic root lesions compared with controls (Figure 1B and 1C). Furthermore, the antiatherosclerotic actions of DBZ were attributed to impaired recruitment of macrophages into atherosclerotic lesions (Figure 1D), as well as decreased expression of proinflammatory cytokines and adhesion molecules (Figure 3). Most important, administration of DBZ in apoE−/− mice not only significantly reduced atherosclerotic plaque progression but also promoted plaque regression. In addition to the morphological assessment, the concordant results obtained by noninvasive ultrasound analysis supported the finding that DBZ treatment alleviated aortic stenosis by preventing the elevation of transaortic flow velocity, as shown in the DBZ late‐intervention study (Figure 2).
Figure 6.
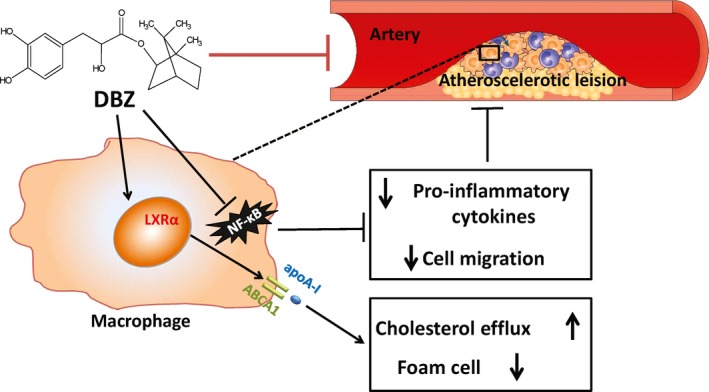
Schematic presentation of the process of atherosclerosis attenuation by DBZ. DBZ suppresses NF‐κB activation and decreases pro‐inflammatory cytokine release and macrophage migration. Meanwhile, DBZ promotes cholesterol efflux and inhibits foam cell formation by activating the LXRα‐ABCA1 signaling pathway. ABCA1 indicates ATP binding cassette subfamily A member 1; apoA‐1, apolipoprotein A‐1; DBZ, Danshensu Bingpian Zhi; LXRα, liver X receptor α; NF‐κB, nuclear factor κB.
To assess additional potential actions of DBZ that are responsible for effects that are protective against atherosclerosis, serum and plaque inflammatory parameters were studied. Treatment with DBZ significantly reduced adhesion molecule expression and the serum concentrations of proinflammatory cytokines that are closely related to the development and progression of atherosclerosis, such as TNF‐α, IL‐6, and MCP‐1, in apoE−/− mice (Figure 3A). This result indicated a potent anti‐inflammatory effect, as reported previously.15 These findings are consistent with previous studies showing that inhibition of the NF‐κB signaling pathway has an atheroprotective effect in apoE−/− mice.11, 12 Further experiments to assess the effects of DBZ on NF‐κB activation in different types of vascular cells, such as endothelial cells, will further demonstrate the anti‐inflammatory activity of DBZ. Meanwhile, we found that DBZ also slightly induced PPAR‐γ (peroxisome proliferator‐activated receptor γ)–responsive luciferase reporter activity and prevented diet‐induced obesity, insulin resistance, hepatic steatosis, systemic low‐grade inflammation, and gut dysbiosis in mice (Y. Zhai, et al, unpublished data, 2017).
In addition to inhibiting lipopolysaccharide‐induced RAW 264.7 cell activation and migration (Figure 3B through 3D), DBZ also promoted cholesterol efflux from THP‐1 macrophages and inhibited ox‐LDL–induced THP‐1–derived foam cell formation (Figure 4A through 4D), which is a hallmark of atherosclerosis progression. As shown in Figure 4B, DBZ markedly inhibited the accumulation of cholesteryl ester, which is the main characteristic of the foam cells that are derived from macrophages.35 The removal of excess cholesterol from macrophages by apoA1 and HDL is thought to play an important role in preventing foam cell formation.39 Our data showed that treatment with DBZ (10 or 20 μmol/L) promoted NBD cholesterol efflux from THP‐1 macrophages to apoA1 (Figure 4C). It is worth noting that ABCA1 mediates cholesterol efflux from cells to apoA1 and is among the earliest identified target genes for LXR‐α, which plays a pivotal role in maintaining macrophage cholesterol homeostasis and reverse cholesterol transport in vivo.43, 44 These results demonstrated that DBZ markedly blocked lipid deposition in THP‐1 macrophages, at least in part, by activating the LXR‐α–ABCA1 signaling pathway.
In line with the in vitro analysis, significant upregulation of aortic and intestinal ABC transporters (ABCA1, ABCG1, and ABCG5/8), which are also known LXR‐α target genes, was observed in DBZ‐treated apoE−/− mice (Figure 5). Our findings are in agreement with reports that LXR activation stimulates cholesterol efflux and delays the progression of atherosclerosis in hyperlipidemic mice.44, 45 Although DBZ induces LXR‐α target gene expression, it is not a direct agonist of LXR‐α. We hypothesize that DBZ triggers the recruitment of the coactivators of LXR‐α, thus activating its target gene expression. Choi et al reported that PPAR‐γ ligands, such as rosiglitazone and MRL24 (selective PPAR? modulators 24), activate PPAR‐γ by blocking Cdk5‐mediated phosphorylation and are completely independent of classical receptor transcriptional agonism.46 Consequently, the effects and regulatory mechanisms of LXR by DBZ remain to be elucidated.
Atorvastatin, a widely used lipid‐lowering drug to treat dyslipidemia and prevent cardiovascular disease, works by inhibiting HMG‐CoAR, an enzyme found in the liver that plays a key role in the production of cholesterol in vivo.47, 48 Our results showed that DBZ 40 mg/kg exhibits a protective effect against atherosclerosis that is similar to that of atorvastatin (Figure 1). In addition, the atheroprotective role of DBZ was also observed in the DBZ late‐intervention group and was comparable to that of atorvastatin (Figure 2). As shown in Figure 5, real‐time polymerase chain reaction results indicate that DBZ‐treated apoE−/− mice display increased cholesterol efflux gene expression in the aorta and small intestine compared with controls, which may also contribute to the antiatherogenic effect of DBZ beyond inhibiting the inflammatory response. Accordingly, it can be inferred that combination therapy with DBZ and atorvastatin may be more effective for prevention and treatment of atherosclerosis.
Compared with plaque size, plaque stability is a more accurate predictor of plaque rupture in clinical events, and the stabilization of plaques is becoming more important for atherosclerosis treatment.28, 29 We found that DBZ‐treated apoE−/− mice exhibited the features of more stable atherosclerotic plaques, including less lipid content, less infiltration of macrophages, more α–smooth muscle actin, and more collagen content. Taken together, the present data suggest that DBZ not only reduces atherosclerotic lesion formation but also has an impact on plaque stability.
In conclusion, our results demonstrate that DBZ potently attenuates atherosclerosis development in apoE−/− mice. The potential antiatherosclerotic actions of DBZ that are responsible for the protection against and treatment of atherosclerosis involve inflammation suppression and the promotion of reverse cholesterol transport. These data indicate that DBZ may represent a promising therapeutic drug for the prevention or treatment of atherosclerosis.
Sources of Funding
This work was supported by National Natural Science Foundation of China (nos. 31571164 and 31271207), Program for Innovative Research Team in the Ministry of Education of China (IRT‐15R55) and the Inner Mongolia Science Foundation (no. 2017BS0801).
Disclosures
None.
Supporting information
Table S1. Sequences of Real‐Time Polymerase Chain Reaction Primers
Table S2. The Basic Echocardiography Data After DBZ (Danshensu Bingpian Zhi) Treatment for 10 Weeks in Each Group at 25 Weeks in Apolipoprotein E–Deficient Mice
Figure S1. Comparisons of body weight resulting from different treatments in apolipoprotein E–deficient mice fed a Western diet for 10 weeks. The results are presented as the mean±SEM (n=9 per group). Atorva indicates atorvastatin.
Figure S2. Representative in vivo ultrasound images show the long‐axis view of the mouse ascending aorta and innominate artery (IA) in various groups. Peak diastolic flow velocity at the aortic valve (AV; indicated by arrows) was measured and quantified after outlining the Doppler signal. Atorva indicates atorvastatin.
Figure S3. The primary peritoneal macrophages from 25‐week‐old apolipoprotein E–deficient mice fed a Western diet for 20 weeks in each group. A, Cells were cultured for 72 h in complete medium and subsequently stained with Oil Red O (scale=50 μm). B, Relative intracellular cholesteryl ester levels were normalized to cellular protein content in peritoneal macrophages. Values are presented as the mean±SEM (n=3 per group. *P<0.05. Atorva indicates atorvastatin.
Figure S4. The transcriptional activity of LXRβ was assessed by the transactivation reporter assay in 293T cells. GW3965 was induced as a positive control. Values are presented as the mean±SEM of at least 3 experiments (*P<0.05 and **P<0.01). LXRβ indicates liver X receptor β.
Figure S5. Hepatic and intestinal mRNA levels of genes involved in the transporter of cholesterol as determined by real‐time polymerase chain reaction in the mice and normalized to cyclophilin in the mice, as indicated in Figure 2 (n=6 per group). Atorva indicates atorvastatin; CD36, cluster of differentiation 36; LDLR, low‐density lipoprotein receptor; NCP1L1, Niemann‐Pick C1‐like 1.
Acknowledgments
We thank Prof. Wen Xie from Center for Pharmacogenetics at the University of Pittsburgh for supplying plasmids pCMX‐hLXR‐α, pCDG‐mLXR‐β, and tk‐(LXRE)×3‐LUC and helping us improve the English writing and Dr Jingwei Xu for the molecular docking assay.
(J Am Heart Assoc. 2017;6:e006297 DOI: 10.1161/JAHA.117.006297.)
Contributor Information
Xiaohui Zheng, Email: Zhengxh@nwu.edu.cn.
Yonggong Zhai, Email: ygzhai@bnu.edu.cn.
References
- 1. Back M, Hansson GK. Anti‐inflammatory therapies for atherosclerosis. Nat Rev Cardiol. 2015;12:199–211. [DOI] [PubMed] [Google Scholar]
- 2. Nahrendorf M, Swirski FK. Immunology neutrophil‐macrophage communication in inflammation and atherosclerosis. Science. 2015;349:237–238. [DOI] [PubMed] [Google Scholar]
- 3. Del Toro R, Chevre R, Rodriguez C, Ordonez A, Martinez‐Gonzalez J, Andres V, Mendez‐Ferrer S. Nestin(+) cells direct inflammatory cell migration in atherosclerosis. Nat Commun. 2016;7:12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong ND. Epidemiological studies of CHD and the evolution of preventive cardiology. Nat Rev Cardiol. 2014;11:276–289. [DOI] [PubMed] [Google Scholar]
- 5. Khan R, Spagnoli V, Tardif JC, L'Allier PL. Novel anti‐inflammatory therapies for the treatment of atherosclerosis. Atherosclerosis. 2015;240:497–509. [DOI] [PubMed] [Google Scholar]
- 6. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. [DOI] [PubMed] [Google Scholar]
- 7. Tabas I, Garcia‐Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baker RG, Hayden MS, Ghosh S. Nf‐kappa B, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cesar L, Suarez SV, Adi J, Adi N, Vazquez‐Padron R, Yu H, Ma Q, Goldschmidt‐Clermont PJ, Agatston A, Kurlansky P, Webster KA. An essential role for diet in exercise‐mediated protection against dyslipidemia, inflammation and atherosclerosis in Apoe−/− mice. PLoS One. 2011;6:e17263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanters E, Gijbels MJJ, van der Made I, Vergouwe MN, Heeringa P, Kraal G, Hofker MH, de Winther MPJ. Hematopoietic NF‐kappa B1 deficiency results in small atherosclerotic lesions with an inflammatory phenotype. Blood. 2004;103:934–940. [DOI] [PubMed] [Google Scholar]
- 12. Sun XH, He SL, Wara AKM, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li DZ, Blackwell TS, Sukhova GK, Croce K, Feinberg MW. Systemic delivery of microRNA‐181b inhibits nuclear factor‐kappa B activation, vascular inflammation, and atherosclerosis in apolipoprotein E‐deficient mice. Circ Res. 2014;114:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taqueti VR, Jaffer FA. High‐resolution molecular imaging via intravital microscopy: illuminating vascular biology in vivo. Integr Biol (Camb). 2013;5:278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Julve J, Llaverias G, Blanco‐Vaca F, Escola‐Gil JC. Seeking novel targets for improving in vivo macrophage‐specific reverse cholesterol transport: translating basic science into new therapies for the prevention and treatment of atherosclerosis. Curr Vasc Pharmacol. 2011;9:220–237. [DOI] [PubMed] [Google Scholar]
- 15. Xie XN, Wang SX, Xiao L, Zhang J, Wang J, Liu J, Shen XJ, He DC, Zheng XH, Zhai YG. DBZ blocks LPS‐induced monocyte activation and foam cell formation via inhibiting nuclear factor‐kappa B. Cell Physiol Biochem. 2011;28:649–662. [DOI] [PubMed] [Google Scholar]
- 16. Zhao X, Zheng X, Fan TP, Li Z, Zhang Y, Zheng J. A novel drug discovery strategy inspired by traditional medicine philosophies. Science. 2015;347:S38–S40. [Google Scholar]
- 17. Cheng TO. Cardiovascular effects of Danshen. Int J Cardiol. 2007;121:9–22. [DOI] [PubMed] [Google Scholar]
- 18. Wang YH, Yang Y, Xie JJ, Li ZA, Zhang XS, Li RJ. Noninvasive assessment of atherosclerosis in apolipoprotein‐e knockout mice by ultrasound biomicroscopy. Ultrasound Med Biol. 2011;37:892–899. [DOI] [PubMed] [Google Scholar]
- 19. Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. [DOI] [PubMed] [Google Scholar]
- 20. Kako Y, Takuma S, Goldberg IJ. Quantitative assessment of atherosclerotic lesions with echocardiography in transgenic mice. Circulation. 1998;98:799. [Google Scholar]
- 21. Song W, Wang W, Wang Y, Dou L, Chen L, Yan X. Characterization of fluorescent NBD‐cholesterol efflux in THP‐1‐derived macrophages. Mol Med Rep. 2015;12:5989–5996. [DOI] [PubMed] [Google Scholar]
- 22. Xu P, Hong F, Wang J, Cong Y, Dai S, Wang S, Wang J, Jin X, Wang F, Liu J, Zhai Y. Microbiome remodeling via the montmorillonite adsorption‐excretion axis prevents obesity‐related metabolic disorders. EBioMedicine. 2017;16:251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu P, Wang J, Hong F, Wang S, Jin X, Xue T, Jia L, Zhai Y. Melatonin prevents obesity through modulation of gut microbiota in mice. J Pineal Res. 2017;62:e12399. [DOI] [PubMed] [Google Scholar]
- 24. Matsushima Y, Hayashi S, Tachibana H. Spontaneously hyperlipidemic (SHL) mice: Japanese wild mice with apolipoprotein E deficiency. Mamm Genome. 1999;10:352–357. [DOI] [PubMed] [Google Scholar]
- 25. Rajani R, Hancock J, Chambers JB. The art of assessing aortic stenosis. Heart. 2012;98:Iv14–Iv22. [DOI] [PubMed] [Google Scholar]
- 26. Yeang C, Cotter B, Tsimikas S. Experimental animal models evaluating the causal role of lipoprotein(a) in atherosclerosis and aortic stenosis. Cardiovasc Drugs Ther. 2016;30:75–85. [DOI] [PubMed] [Google Scholar]
- 27. Verweij SL, van der Valk FM, Stroes ESG. Novel directions in inflammation as a therapeutic target in atherosclerosis. Curr Opin Lipidol. 2015;26:580–585. [DOI] [PubMed] [Google Scholar]
- 28. Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282–1292. [DOI] [PubMed] [Google Scholar]
- 29. Koenig W, Khuseyinova N. Biomarkers of atherosclerotic plaque instability and rupture. Arterioscler Thromb Vasc Biol. 2007;27:15–26. [DOI] [PubMed] [Google Scholar]
- 30. Hanania R, Sun HS, Xu KW, Pustylnik S, Jeganathan S, Harrison RE. Classically activated macrophages use stable microtubules for matrix metalloproteinase‐9 (MMP‐9) secretion. J Biol Chem. 2012;287:8468–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulation of VCAM‐1 and ICAM‐1 at atherosclerosis‐prone sites on the endothelium in the APoE‐deficient mouse. Arterioscler Thromb Vasc Biol. 1998;18:842–851. [DOI] [PubMed] [Google Scholar]
- 32. Linton MF, Fazio S. Macrophages, inflammation, and atherosclerosis. Int J Obesity. 2003;27:S35–S40. [DOI] [PubMed] [Google Scholar]
- 33. Park JG, Ryu SY, Jung IH, Lee YH, Kang KJ, Lee MR, Lee MN, Sonn SK, Lee JH, Lee H, Oh GT, Moon K, Shim H. Evaluation of VCAM‐1 antibodies as therapeutic agent for atherosclerosis in apolipoprotein E‐deficient mice. Atherosclerosis. 2013;226:356–363. [DOI] [PubMed] [Google Scholar]
- 34. Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–1242. [DOI] [PubMed] [Google Scholar]
- 35. Ghosh S, Zhao B, Bie JH, Song JM. Macrophage cholesteryl ester mobilization and atherosclerosis. Vascul Pharmacol. 2010;52:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ouimet M, Marcel YL. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol. 2012;32:575–581. [DOI] [PubMed] [Google Scholar]
- 37. Ji AL, Wroblewski JM, Cai L, de Beer MC, Webb NR, van der Westhuyzen DR. Nascent HDL formation in hepatocytes and role of ABCA1, ABCG1, and SR‐BI. J Lipid Res. 2012;53:446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Westerterp M, Murphy AJ, Wang M, Pagler TA, Vengrenyuk Y, Kappus MS, Gorman DJ, Nagareddy PR, Zhu XW, Abramowicz S, Parks JS, Welch C, Fisher EA, Wang N, Yvan‐Charvet L, Tall AR. Deficiency of ATP‐binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res. 2013;112:1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yvan‐Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol. 2010;30:139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Larrede S, Quinn CM, Jessup W, Frisdal E, Olivier M, Hsieh V, Kim MJ, Van Eck M, Couvert P, Carrie A, Giral P, Chapman MJ, Guerin M, Le Goff W. Stimulation of cholesterol efflux by LXR agonists in cholesterol‐loaded human macrophages is ABCA1‐dependent but ABCG1‐independent. Arterioscler Throm Vasc Biol. 2009;29:1930–1936. [DOI] [PubMed] [Google Scholar]
- 41. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roth Flach RJ, Skoura A, Matevossian A, Danai LV, Zheng W, Cortes C, Bhattacharya SK, Aouadi M, Hagan N, Yawe JC, Vangala P, Menendez LG, Cooper MP, Fitzgibbons TP, Buckbinder L, Czech MP. Endothelial protein kinase MAP4K4 promotes vascular inflammation and atherosclerosis. Nat Commun. 2015;6:8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oosterveer MH, Grefhorst A, Groen AK, Kuipers F. The liver X receptor: control of cellular lipid homeostasis and beyond implications for drug design. Prog Lipid Res. 2010;49:343–352. [DOI] [PubMed] [Google Scholar]
- 44. Lo Sasso G, Murzilli S, Salvatore L, D'Ernco I, Petruzzelli M, Conca P, Jiang ZY, Calabresi L, Parini P, Moschetta A. Intestinal specific LXR activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab. 2010;12:187–193. [DOI] [PubMed] [Google Scholar]
- 45. Joseph SB, McKilligin E, Pei LM, Watson MA, Collins AR, Laffitte BA, Chen MY, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang XP, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA. 2002;99:7604–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti‐diabetic drugs inhibit obesity‐linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Komatsu T, Ayaori M, Uto‐kondo H, Hayashi K, Tamura K, Sasaki M, Yogo M, Ogura M, Takiguchi S, Yakushiji E, Nakaya K, Ikewaki K. Atorvastatin reduces atherosclerotic plaque inflammation and serum high‐sensitivity CRP levels in patients with carotid atherosclerosis. Circulation. 2013;128:S11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, Zamvil SS. The HMG‐CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences of Real‐Time Polymerase Chain Reaction Primers
Table S2. The Basic Echocardiography Data After DBZ (Danshensu Bingpian Zhi) Treatment for 10 Weeks in Each Group at 25 Weeks in Apolipoprotein E–Deficient Mice
Figure S1. Comparisons of body weight resulting from different treatments in apolipoprotein E–deficient mice fed a Western diet for 10 weeks. The results are presented as the mean±SEM (n=9 per group). Atorva indicates atorvastatin.
Figure S2. Representative in vivo ultrasound images show the long‐axis view of the mouse ascending aorta and innominate artery (IA) in various groups. Peak diastolic flow velocity at the aortic valve (AV; indicated by arrows) was measured and quantified after outlining the Doppler signal. Atorva indicates atorvastatin.
Figure S3. The primary peritoneal macrophages from 25‐week‐old apolipoprotein E–deficient mice fed a Western diet for 20 weeks in each group. A, Cells were cultured for 72 h in complete medium and subsequently stained with Oil Red O (scale=50 μm). B, Relative intracellular cholesteryl ester levels were normalized to cellular protein content in peritoneal macrophages. Values are presented as the mean±SEM (n=3 per group. *P<0.05. Atorva indicates atorvastatin.
Figure S4. The transcriptional activity of LXRβ was assessed by the transactivation reporter assay in 293T cells. GW3965 was induced as a positive control. Values are presented as the mean±SEM of at least 3 experiments (*P<0.05 and **P<0.01). LXRβ indicates liver X receptor β.
Figure S5. Hepatic and intestinal mRNA levels of genes involved in the transporter of cholesterol as determined by real‐time polymerase chain reaction in the mice and normalized to cyclophilin in the mice, as indicated in Figure 2 (n=6 per group). Atorva indicates atorvastatin; CD36, cluster of differentiation 36; LDLR, low‐density lipoprotein receptor; NCP1L1, Niemann‐Pick C1‐like 1.