

Abstract
Background
Respiratory muscle weakness contributes to exercise intolerance in patients with heart failure with a preserved ejection fraction (HFpEF)—a condition characterized by multiple comorbidities with few proven treatments. We aimed, therefore, to provide novel insight into the underlying diaphragmatic alterations that occur in HFpEF by using an obese cardiometabolic rat model and further assessed whether exercise training performed only after the development of overt HFpEF could reverse impairments.
Methods and Results
Obese ZSF1 rats (n=12) were compared with their lean controls (n=8) at 20 weeks, with 3 additional groups of obese ZSF1 rats compared at 28 weeks following 8 weeks of either sedentary behavior (n=13), high‐intensity interval training (n=11), or moderate‐continuous training (n=11). Obese rats developed an obvious HFpEF phenotype at 20 and 28 weeks. In the diaphragm at 20 weeks, HFpEF induced a shift towards an oxidative phenotype and a fiber hypertrophy paralleled by a lower protein expression in MuRF1 and MuRF2, yet mitochondrial and contractile functional impairments were observed. At 28 weeks, neither the exercise training regimen of high‐intensity interval training or moderate‐continuous training reversed any of the diaphragm alterations induced by HFpEF.
Conclusions
This study, using a well‐characterized rat model of HFpEF underpinned by multiple comorbidities and exercise intolerance (ie, one that closely resembles the patient phenotype), provides evidence that diaphragm alterations and dysfunction induced in overt HFpEF are not reversed following 8 weeks of aerobic exercise training. As such, whether alternative therapeutic interventions are required to treat respiratory muscle weakness in HFpEF warrants further investigation.
Keywords: diastolic heart failure, metabolism, obesity, respiratory muscle, skeletal muscle exercise, ZSF1
Subject Categories: Animal Models of Human Disease, Contractile function, Physiology, Heart Failure, Exercise
Clinical Perspective
What Is New?
This study, using a rat model of heart failure with a preserved ejection fraction underpinned by multiple comorbidities and exercise intolerance (ie, one that closely resembles the patient phenotype), provides novel and invasive evidence that diaphragm alterations and dysfunction induced by heart failure with a preserved ejection fraction cannot be overcome by the intervention of aerobic exercise training following overt disease development (ie, secondary prevention), which is in contrast to previous studies documenting a benefit of exercise training performed in the period preceding the onset of heart failure with a preserved ejection fraction (ie, primary prevention).
What Are the Clinical Implications?
If these animal findings translate to patients, it would suggest that alternative therapeutic interventions rather than aerobic exercise training may be required to treat respiratory muscle weakness in heart failure with a preserved ejection fraction.
Introduction
Heart failure with a preserved ejection fraction (HFpEF) is now estimated to account for over half of cases of this disease, with its prevalence predicted to increase even further.1 It is disquieting, therefore, that evidence from large clinical drug trials in HFpEF has been largely negative in terms of beneficial outcomes,2, 3 which is in stark contrast to findings in patients with a reduced ejection fraction (HFrEF). This may be related, at least in part, to HFpEF being characterized by multiple comorbidities such as obesity, type II diabetes mellitus, hypertension, and chronic kidney disease, which, in turn, are suggested to induce systemic inflammation and impair multiple extracardiac organs that conspire to drive the HFpEF phenotype.4 Exercise training has been one of the few therapeutic therapies associated with clinical benefits in HFpEF2 and this seems unrelated to cardiac improvements but rather to peripheral adaptations.5 Current evidence, therefore, appears to support the current paradigm that HFpEF and its main symptom exercise intolerance are driven by noncardiac peripheral abnormalities.4
Recent data indicate that respiratory muscle weakness is present in patients with HFpEF. Noninvasive functional measures are reduced compared with age‐matched controls and these are correlated to exercise intolerance, increased sensations of breathless, and an elevated respiratory drive.6, 7 Our laboratory recently confirmed these studies with direct evidence, where a hypertensive rat model of HFpEF demonstrated diaphragm contractile dysfunction that was linked to mitochondrial complex I functional impairments, fiber atrophy, and a fiber type shift from a glycolytic towards an oxidative phenotype.8 Importantly, exercise training performed in the period preceding the onset of HFpEF (ie, primary prevention) was able to abolish diaphragmatic impairments,8 thus providing strong support for exercise training as a key treatment of respiratory muscle weakness in HFpEF. However, the clinical translation of this study was limited by the fact that HFpEF was induced exclusively by hypertension, thereby lacking the multiple comorbidities that typically characterize patients with HFpEF. Furthermore, the exercise training intervention was performed during the development rather than after the onset of HFpEF, with no evidence provided in regards to the optimal training regimen.
The present study, therefore, aimed to first determine the effects of HFpEF on the diaphragm in the more clinically relevant ZSF1 rat model that is known to develop a typical HFpEF phenotype after 20 weeks, which is characterized by multiple comorbidities including obesity, hypertension, type 2 diabetes mellitus, and chronic kidney disease.9, 10, 11, 12 In addition, this study aimed to determine, by assessing 2 different exercise training regimens of high or moderate intensity, the effects of exercise training on the diaphragm after the onset of overt HFpEF, thereby allowing the effects of secondary prevention to be assessed—as is typical in clinical practice. We supposed that answering both of these aims would improve clinical translation and advance our underlying knowledge and treatment of diaphragm dysfunction in HFpEF.
Methods
Study Design
All procedures and experiments were approved by the Norwegian Animal Research Authority in accordance with the national regulations, the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (ETS No.123), and the European Directive 2010/63/EU on the protection of animals used for scientific purposes. Obese diabetic Zucker fatty/spontaneously hypertensive heart failure F1 hybrid (ZSF1) rats (Charles River Laboratories; bought at 8 weeks of age) were used as a model to induce HFpEF, with this strain of rat previously shown to develop typical clinical signs of HFpEF at 20 weeks of age.9, 10, 11, 12 While both lean and obese ZSF1 rats inherit the hypertension gene, only the obese ZSF1 rats inherit a mutation in the leptin receptor gene that drives weight gain and metabolic impairments. A schematic of the study design is presented in Figure 1. Briefly, male obese ZSF1 (ie, HFpEF; n=12) rats were first euthanized at 20 weeks of age and compared with lean ZSF1 rat counterparts (ie, controls; n=8). Furthermore, at 28 weeks of age, 3 additional groups were euthanized, which included sedentary obese rats (n=13) and also exercise trained obese rats that performed either high‐intensity interval training (HFpEF+HIIT; n=11) or moderate continuous training (HFpEF+MCT; n=11). Rats were exposed to identical conditions in a 12‐hour light/dark cycle, fed with standard chow (RM1 chow, SDS) and administered water ad libitum.
Figure 1.
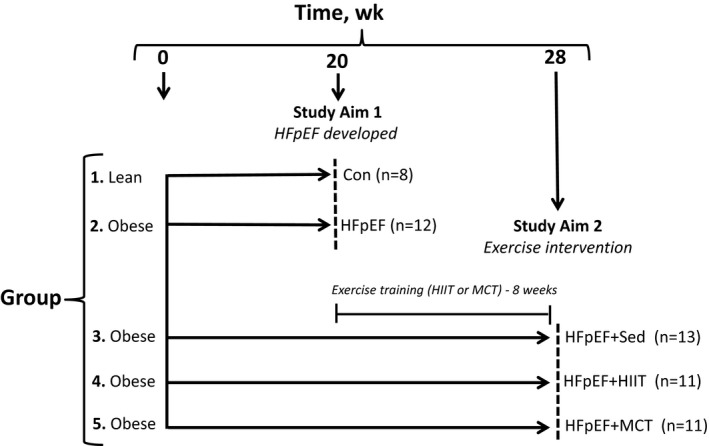
Schematic outline summarizing the present study. Aim 1 of the study evaluated at 20 weeks the effects of an obese cardiometabolic rat model of heart failure with a preserved ejection fraction (HFpEF) on the diaphragm compared with lean controls (Con), while aim 2 of the study evaluated at 28 weeks whether 8 weeks of exercise training (either high‐intensity interval training [HIIT] or moderate‐continuous training [MCT]) could reverse diaphragm alterations as a secondary prevention compared with sedentary (Sed) HFpEF rats. Dashed lines indicate the time point when animals were evaluated (20 or 28 weeks of age), which included assessment of exercise capacity, cardiac function (echocardiography/invasive hemodynamics), muscle function, and subsequent tissue analyses. The sample size (n) for each group are provided in parentheses.
Exercise Training
At 20 and 28 weeks, exercise capacity was measured in all groups as assessed by peak pulmonary oxygen uptake () during a peak treadmill exercise test, as previously described.8 For exercise training, obese ZSF1 rats were randomized at 20 weeks of age to perform either HIIT (3×per week; 4 intervals at 90% for 4 minutes, with 3 minutes of recovery at 60% ) or MCT (5×per week at 60% for 1 hour) for a total of 8 weeks on a treadmill at a gradient of 25°. Exercise was preceded and followed by 10 minutes of running at 40% to 50% .
Cardiovascular Function
In vivo 2‐dimensional and M‐mode echocardiography, conventional Doppler echocardiography, and tissue Doppler imaging data (Vevo 2100, VisualSonics) were obtained in lightly anesthetized (1.5–2% isoflurane) but spontaneously breathing rats in the supine position using a 24‐MHz transducer to assess cardiac function as previously described,8 which included functional measures of diastole (ie, peak velocity of early mitral flow to tissue velocity [E/E′] and ratio between peak E and A waves of mitral valve velocity [E/A]) and systole (ie, left ventricular [LV] ejection fraction).8 Myocardial velocity (E′) was measured by tissue Doppler imaging at the level of the basal septal segment in the septal wall of the left ventricle in the apical 4‐chamber view, while for early (E) and late (A) filling of the left ventricle the Doppler sample volume was positioned parallel to flow direction and the mitral valve. LV structural parameters were measured from the short‐axis view in M mode to calculate LV ejection fraction. In addition, invasive hemodynamic pressure measurements were measured as the terminal procedure. Under light anesthesia (1.5–2% isoflurane), the right carotid artery was cannulated with a conductance catheter (1.49 F, Transonic Scisense Inc.) and a pressure transducer penetrated the aortic valve into the left ventricle. The LV end‐diastolic pressure, maximum rate of pressure rise (dP/dtmax), maximum rate of pressure fall (dP/dtmin), and time constant (τ) for LV relaxation, after which withdrawal of the catheter into the aorta, and phasic and mean arterial pressures were measured. Data were recorded in LabChart 7 software (AD Instruments). In addition, blood glucose levels were measured using a FreeStyle Mini Meter (nonfasted state at 20 weeks, and a 12‐hour fasted state at 28 weeks). Tissue weights of the left ventricle and the lungs (wet and dry) as well as tibia length were further determined at euthanization.
Diaphragm Analyses
For functional assessment, the left costal diaphragm muscle was prepared in a Krebs‐Hanseleit buffer solution (120.5 NaCl, 4.8 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 20.4 NaHCO3, 1.6 CaCl2, 10 dextrose, and 1 pyruvate [in mmol/L] at a pH of ≈7.40) at room temperature equilibrated with 95% O2 to 5% CO2. Briefly, a muscle bundle connected from the rib to the central tendon was dissected, attached to silk sutures (4‐0) at either end, and mounted vertically in a buffer‐filled organ bath.8 In addition, the right soleus muscle was also dissected and assessed in parallel. The suture connected to the rib was secured to a hook at the bottom of the bath, while the tendon was tied to a force transducer (FORT250, World Precision Instruments Inc.), with the output continuously recorded and digitized (PowerLab 8/30, ADInstruments). In vitro muscle function was assessed by platinum electrodes stimulating the muscle with a supramaximal current (700 mA, 500 ms train duration, 0.25 ms pulse width) from a base stimulator (Grass S88, Grass Technologies) amplified via a high‐power bipolar stimulator (701C, Aurora Scientific Inc.). The muscle bundle was set at an optimal length equivalent to the maximal twitch force produced, after which the bath temperature was increased to 25°C and a 15‐minute thermo‐equilibration period followed. A force‐frequency protocol was then performed at 1, 15, 30, 50, 80, 120, 150, and 300 Hz, respectively, separated with 1‐minute rest intervals. Following a 5‐minute period in which muscle length was measured using digital calipers, the muscle underwent a fatigue protocol over 5 minutes (40 Hz every 2 seconds). Each muscle was subsequently detached, trimmed free from the tendon, blotted dry, and weighed. Muscle force (N) was normalized to muscle cross‐sectional area (cm2) by dividing muscle mass (g) by the product of optimal length (cm) and estimated muscle density (1.06),1 which allowed specific force in N/cm2 to be calculated. Forces generated during the fatigue protocol were normalized to the initial force generated, to provide a relative assessment of fatigability. A further diaphragm bundle was also dissected and permeabilized in saponin, and in situ mitochondrial respiration measurements were performed using high‐resolution respirometry (Oxygraph‐2k, Oroboros Instruments), as described in expansive detail elsewhere.8
In addition, the right costal diaphragm was immediately frozen in liquid N2. Frozen muscle samples were subsequently homogenized in lysis buffer (50 mmol/L Tris, 150 mmol/L sodium chloride, 1 mmol/L EDTA, 1% NP‐40, 0.25% sodium‐deoxycholate, 0.1% SDS, 1% Triton X‐100; pH 7.4) containing a protease inhibitor mix (Inhibitor Mix M, Serva), sonicated, and centrifuged at 16 000g for 5 minutes. The supernatant was isolated and protein content determined (BCA assay, Pierce). Diaphragm homogenates (5–20 μg) mixed with loading buffer (126 mmol/L Tris‐HCl, 20% glycerol, 4% SDS, 1.0% 2‐mercaptoethanol, 0.005% bromophenol blue; pH 6.8) were separated by SDS‐polyacrylamide gel electrophoresis for 1.5 hours at 90 V. Proteins were transferred to a polyvinylidene fluoride membrane and incubated overnight at 4°C with the following primary antibodies: peroxisome proliferator‐activated receptor gamma coactivator 1‐α (PGC1‐α;1/200, Santa Cruz) and MuRF1 and MuRF2 (1/1000, Myomedix Ltd.). Membranes were subsequently incubated with a horseradish peroxidase–conjugated secondary antibody and specific bands visualized by enzymatic chemiluminescence (Super Signal West Pico, Thermo Fisher Scientific Inc.) and densitometry quantified using a 1‐dimensional scan software package (Scanalytics Inc.). Blots were subsequently normalized to the loading control GAPDH (1/30 000, HyTest Ltd). In addition, enzyme activities for citrate synthase and catalase were measured spectrophotometrically, as previously described in detail.13 Protein and enzyme activity data are presented as the fold change relative to the respective control group. The left soleus was also immediately frozen in liquid N2 and underwent the same molecular analyses as described for the diaphragm.
An additional piece of diaphragm was also fixed in 4% PBS‐buffered formalin and embedded in paraffin for subsequent histological analyses of fiber type and cross‐sectional area. Briefly, 3‐μm sections were cut, mounted on glass cover slips, and incubated overnight at 4°C in antibody diluent (Dako) with primary antibody against myosin slow fibers (M8421, 1/400; Sigma). After washing with TBST, sections were first incubated with fluorescently conjugated (Alexa 488) secondary antibodies for 1 hour and then underwent further washing, including once with the dye Hoechst 33342 (1/100, Sigma). Thereafter, sections were visualized under a fluorescent microscope where images were captured with fibers merged with nuclei to establish fiber boundaries (blue), with all stained fibers taken as type I (bright green) and all unstained fibers taken as type II (dark green), allowing fiber type and size to be determined.
Statistical Analyses
Analyses were performed by SPSS version 22 (SPSS Institute Inc). Data are presented as mean±SEM. Between‐group differences were assessed by unpaired Student t test at 20 weeks or by 1‐way ANOVA at 28 weeks. Muscle function (force‐frequency and force‐time relationships) was assessed by 2‐way repeated measures ANOVA (group×frequency or group×time). Post hoc comparisons were calculated using Bonferroni test. Data that failed normality (Kolmogorov and Smirnov) or equal variance tests were compared by Mann‐Whitney (20 weeks) or Kruskal‐Wallis test with Dunn post hoc (28 weeks). Significance was accepted as P<0.05.
Results
Obese ZSF1 Rats Develop HFpEF and an Oxidative Fiber Hypertrophy and Phenotype in the Diaphragm
As expected, at 20 weeks of age, obese rats showed evidence of HFpEF compared with lean controls (Figure 2). Specifically, when compared with controls, obese rats developed typical features associated with the HFpEF syndrome, which included diastolic dysfunction (confirmed by both noninvasive and invasive measures of E/E′ and LV end‐diastolic pressure, respectively; Figure 2A and 2B), a preserved LV ejection fraction (Figure 2C), and LV hypertrophy (Figure 2D), although lung wet to dry ratio was unchanged between the groups (P>0.05). The cardiac alterations occurred in the presence of multiple comorbidities, including hyperglycemia (Figure 2F) and obesity (Figure 2G). Importantly, obese rats also demonstrated a reduced exercise capacity by ≈10% compared with controls (P<0.05) (Figure 2H). In addition and compared with controls, while further echocardiographic measures found that obese animals had an unchanged E/A ratio (1.2±0.1 versus 1.4±0.1; P>0.05), invasive hemodynamics revealed both an increased LV dP/dtmax (7794±469 mm Hg/s versus 9488±382 mm Hg/s; P<0.05) and τ (12.2±0.4 ms versus 13.9±0.4 ms; P<0.01), while the LV dP/dtmin remained unaltered (−8745±721 mm Hg/s versus −8892±296 mm Hg/s; P>0.05). Overall, therefore, these data provide strong evidence that at 20 weeks of age, obese ZSF1 rats developed a similar phenotype to that observed in patients with HFpEF.
Figure 2.
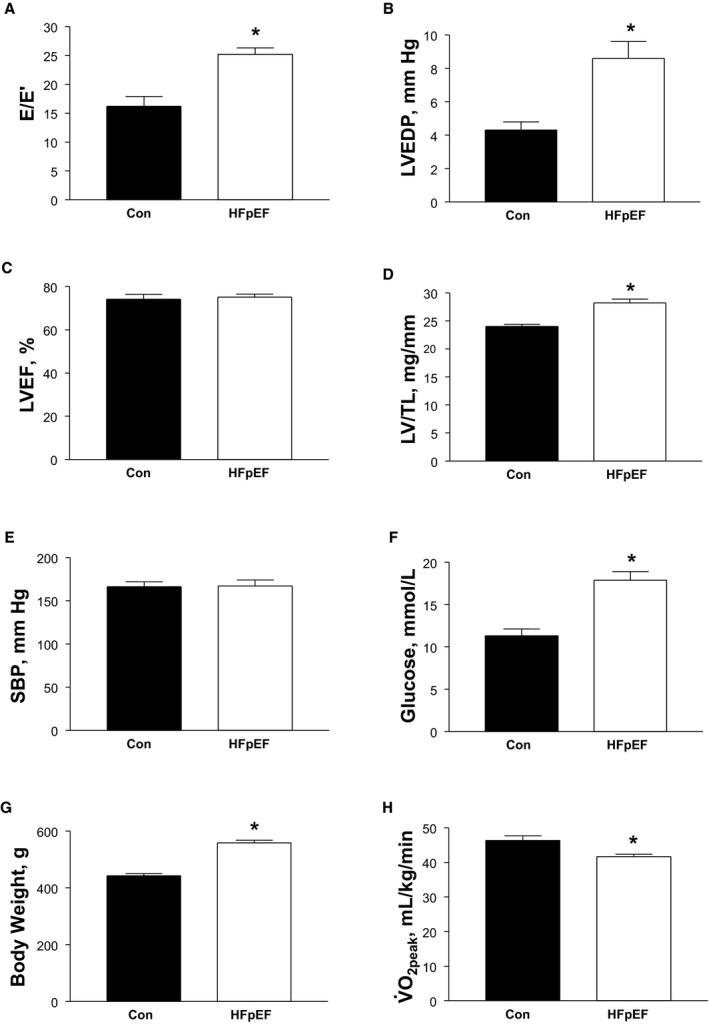
Animal model characteristics at 20 weeks, which demonstrates that a typical heart failure with a preserved ejection fraction (HFpEF) phenotype was developed in obese rats compared with lean controls (Con). HFpEF was confirmed by measures of diastolic dysfunction (A and B) in the presence of normal systolic function (C) in parallel with a cardiac hypertrophy (D). In the absence of an increase in systolic blood pressure (SBP) (E), other comorbidities were developed in HFpEF rats including hyperglycemia (F) and obesity (G), as well as an impaired exercise capacity (H). Data are presented as mean±SEM, with n=8 for Con and n=12 for HFpEF, analyzed with unpaired t test. *P<0.05 vs Con. E/E′ indicates early mitral flow to tissue velocity; LVEF, left ventricular ejection fraction; LVEDP, left ventricular end‐diastolic pressure; LV/TL, left ventricle/tibial length; , peak oxygen uptake.
At 20 weeks, HFpEF also induced numerous alterations in the diaphragm, which included a hypertrophy of type I fibers by 23% (Figure 3C) that corresponded to a decrease of ≈25% in the expression of the atrophy‐related proteins MuRF1 (P=0.065) (Figure 3E) and MuRF2 (P<0.01) (Figure 3F), respectively, with a downregulation of MuRF1 levels inversely correlated to type I fiber size in the HFpEF rats (P=0.01; R 2=0.51). In addition, HFpEF also induced a shift towards an oxidative phenotype, such that the proportion of type I fibers increased by 23% (P<0.05) (Figure 4A and 4B) in parallel to an increase in markers of mitochondrial content and antioxidant capacity in the diaphragm: PGC1‐α protein content and citrate synthase activity were elevated by 51% (P<0.01) (Figure 4C) and 19% (P<0.05) (Figure 4D), respectively, while catalase activity was increased by 68% compared with controls (P<0.01) (Figure 4E). Nevertheless, despite these apparent quantitative improvements, mitochondrial respiration assessed in diaphragm fibers revealed significant qualitative impairments, with HFpEF specifically increasing the leak state by ≈30% (P<0.05; Figure 4F) and reducing the respiratory control ratio by 20%—indicative of an impaired mitochondrial coupling efficiency (P<0.05) (Figure 4G). While no significant differences were discerned between groups in terms of protein expression for contractile proteins (myosin heavy chain and actin), markers of oxidative stress (NOX2), and other atrophy‐related proteins (MAFbx; data not shown), mild impairments to diaphragm contractile function were induced by HFpEF, which included a reduced twitch tension by ≈20% (P<0.05; 4.8±0.3 N/cm2 versus 4.0±0.3 N/cm2, respectively) and trend for maximal force to be reduced by ≈10% (P=0.07; 22±1 N/cm2 versus 19±1 N/cm2, respectively). In contrast, fatigability was not different between groups, with relative force generation (normalized to initial force) unchanged at the end of a 5‐minute protocol (16±1% versus 18±1%; P>0.05).
Figure 3.
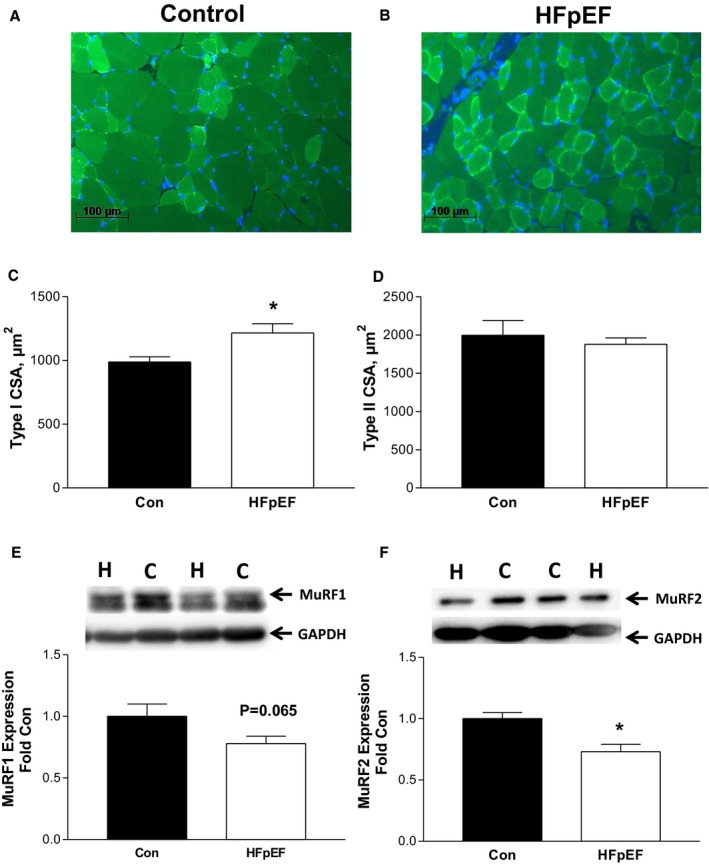
Representative immunofluorescence sections of the diaphragm from control (Con; A) and heart failure with a preserved ejection fraction (HFpEF) (B) rats at 20 weeks, where sections were incubated with an antibody against myosin slow type I fibers to allow detection of type I (bright green) and type II (dark green) fiber proportion and size, with cell nuclei stained (blue) to establish fiber boundaries (scale bars represent 100 μm). The fiber cross‐sectional area (CSA) was increased in type I fibers in the diaphragm from HFpEF rats (C), with no effects observed for the type II isoform (D). Protein expression of MuRF1 (E) and MuRF2 (F) was also lower in HFpEF, with both proteins implicated in cooperating to regulate fiber size. The abbreviations C and H for Western blot lanes represent loaded samples from Con and HFpEF, respectively, with GAPDH used as a loading control. Data are presented as mean±SEM, analyzed with unpaired t test or Mann‐Whitney (type I CSA), with n=8 for Con and n=12 for HFpEF. *P<0.05 vs Con.
Figure 4.
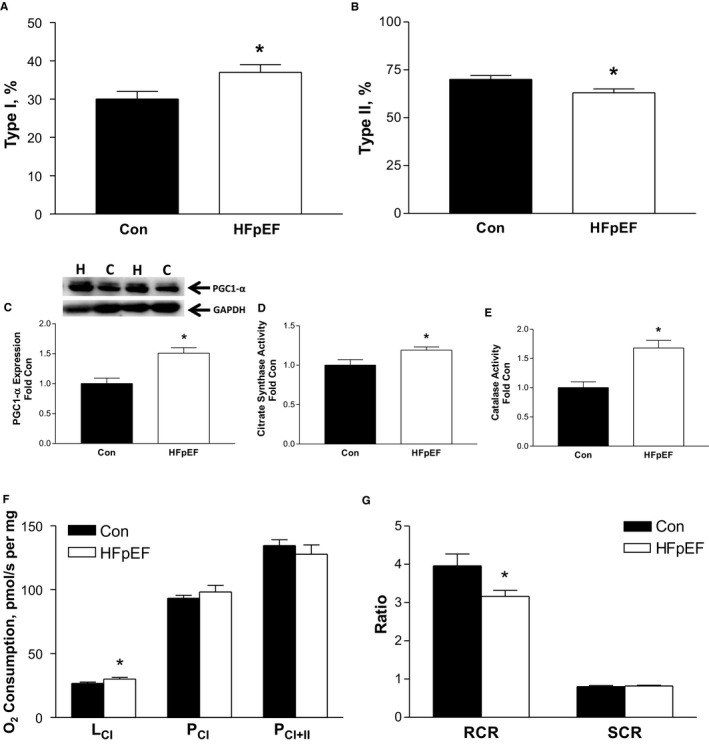
Heart failure with a preserved ejection fraction (HFpEF) induced a shift towards an oxidative phenotype in the diaphragm compared with controls (Con), with a greater proportion of type I (A) and lower proportion of type II fibers (B) at 20 weeks (see Figure 3A and 3B for representative diaphragm sections). This was associated with a greater peroxisome proliferator‐activated receptor gamma coactivator 1‐α (PGC1‐α) protein expression (C) as well as citrate synthase (D) and catalase enzyme activity (E). In contrast, in situ mitochondrial respiration revealed functional deficits in the diaphragm of HFpEF rats, with an increased leak state (LCI) (F) and impaired mitochondrial coupling efficiency (respiratory control ratio [RCR]) (G). Respiration states assessed, in turn, included: LCI, complex I leak respiration following addition of malate and glutamate; PCI, complex I phosphorylated state following addition of ADP; PCI + II, complex II phosphorylated state following addition of succinate. RCR (ie, PCI/LCI) and substrate control ratio (SCR; ie, PCI/PCI + II). The abbreviations C and H for Western blot lanes represent loaded samples from Con and HFpEF, respectively, with GAPDH used as a loading control. Data are presented as mean±SEM, analyzed with unpaired t test, with n=8 for Con and n=12 for HFpEF. *P<0.05 vs Con.
Effects of Exercise Training on the HFpEF Phenotype and on Diaphragm Alterations
Given the molecular and functional alterations we observed in the diaphragm at 20 weeks in rats with HFpEF, we next assessed whether these could be reversed by 8 weeks of exercise training (ie, in the form of secondary prevention by using HIIT or MCT). As anticipated, at 28 weeks, sedentary obese rats still presented with typical HFpEF signs, but these were not influenced by exercise training, which included no change in diastolic function (E/E′ and LV end‐diastolic pressure; Figure 5A and 5B), systolic function (Figure 5C), or LV mass (Figure 5D), alongside no alterations in measures of multiple comorbidities, which included body weight remaining unchanged (Figure 5E through 5G). In addition, further echocardiographic measures found no differences between sedentary, HIIT, or MCT HFpEF rats in terms of the E/A ratio (1.4±0.1, 1.4±0.1, and 1.5±0.1, respectively; P>0.05), while invasive hemodynamics also revealed no differences in terms of the LV dP/dtmax (8841±181, 8146±283, and 8239±306 mm Hg/s, respectively; P>0.05), dP/dtmin (−9005±238, −8267±346, and 8558±427 mm Hg/s, respectively; P>0.05), and τ (14.8±0.4, 15.5±0.7, and 16.0±1.1 ms, respectively; P>0.05). As such, exercise training apparently had limited benefits on hemodynamics, cardiac remodeling, and comorbidities in this rat model of HFpEF. In contrast, however, both HIIT and MCT were able to significantly increase exercise capacity as compared with sedentary HFpEF rats, with increased by ≈15% in both of the exercise trained groups (P<0.05; Figure 5H).
Figure 5.
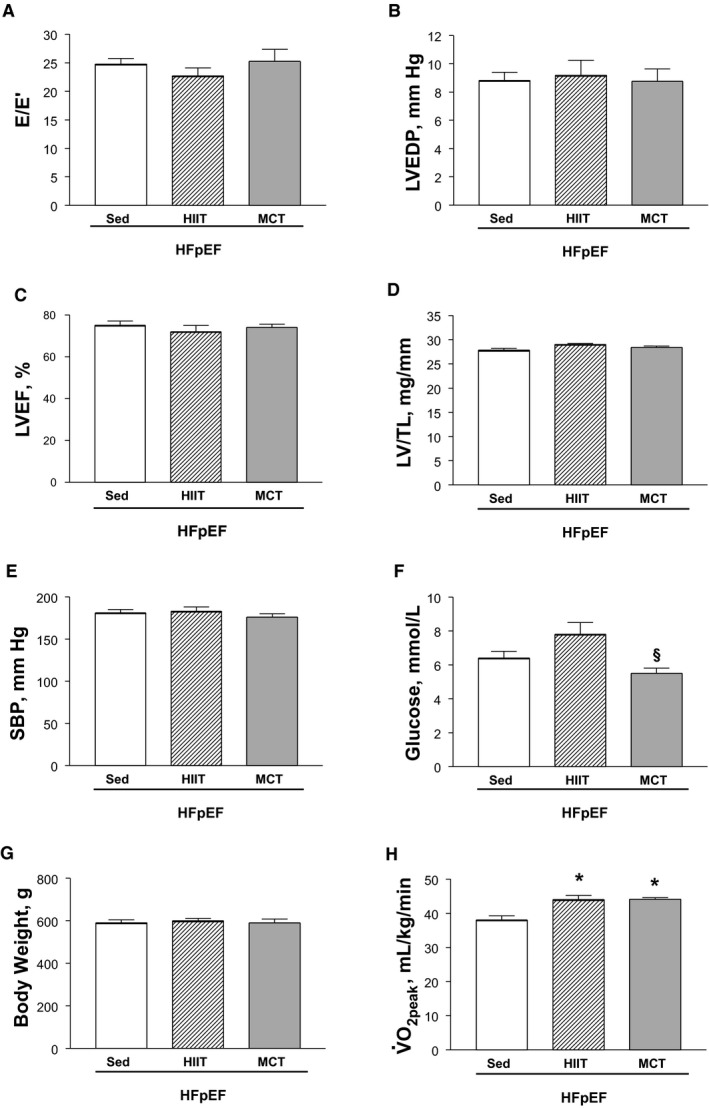
Animal model characteristics at 28 weeks, which demonstrates the heart failure with a preserved ejection fraction (HFpEF) phenotype in sedentary (Sed) obese rats was generally not influenced by 8 weeks of either high‐intensity interval training (HIIT) or moderate‐continuous training (MCT). Compared with sedentary HFpEF rats, exercise training did not influence cardiac measures of diastolic (A and B) or systolic function (C), cardiac hypertrophy (D), or multiple comorbidities such as hypertension (E), hyperglycemia (F), and obesity (G). Importantly, however, both exercise training interventions of HIIT and MCT improved exercise capacity compared with the sedentary HFpEF condition (H). Data are presented as mean±SEM, with n=13 for Sed, n=11 for HIIT, and n=11 for MCT, analyzed with 1‐way ANOVA or Kruskal‐Wallis test with Dunn post hoc (glucose and ). *P<0.05 vs Sed, § P<0.05 vs HIIT. E/E′ indicates early mitral flow to tissue velocity; LVEF, left ventricular ejection fraction; LVEDP, left ventricular end‐diastolic pressure; LV/TL, left ventricle/tibial length; SPB, systolic blood pressure; , peak oxygen uptake.
We next assessed the diaphragm from HFpEF rats at 28 weeks of age and found that exercise training had, in general, no significant effect on most measures that were shown to be altered at 20 weeks: fiber size remained unchanged (≈1500 and ≈2000 μm2 for type I and type II fibers across all groups, respectively; P>0.05) (Figure 6D and 6E) while protein expression of MuRF1 and MuRF2 as markers of atrophy were not different after exercise training (P>0.05) (Figure 6F and 6G). Similarly, no fiber type shift was evident following exercise training (all groups demonstrated ≈40% type I and 60% type II fiber proportions; P>0.05) (Figure 7A and 7B), while quantitative markers of mitochondrial density including PGC1‐α protein content (Figure 7C), citrate synthase activity (Figure 7D), and catalase activity (Figure 7E) were not different between the HFpEF groups assigned to either the sedentary, HIIT, or MCT regimens (all P>0.05) (Figure 7A through 7E). While HIIT tended (P=0.06) to improve mitochondrial function by 17% in terms of increasing the respiratory control ratio when compared with sedentary HFpEF rats (only a 5% increase was observed after MCT), no significant differences were otherwise detected (Figure 7F and 7G). Not surprisingly, therefore, exercise training had no benefit on contractile function (P>0.05) (Figure 8A) or on fiber fatigability in HFpEF (P>0.05) (Figure 8B).
Figure 6.
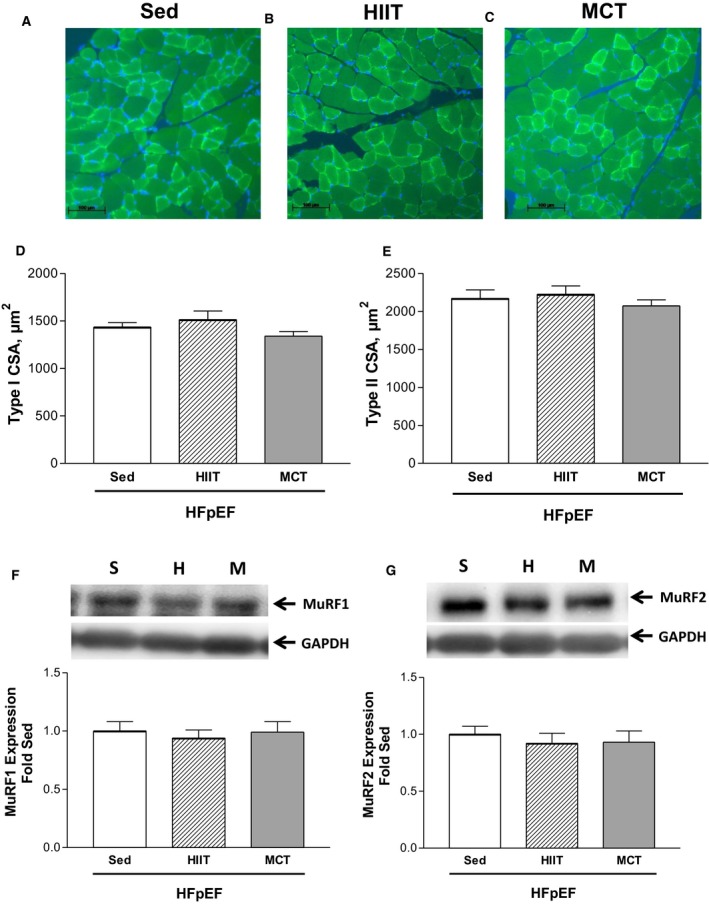
Representative immunofluorescence sections of the diaphragm at 28 weeks from heart failure with a preserved ejection fraction (HFpEF) rats that remained sedentary (Sed; A) or performed 8 weeks of high‐intensity interval training (HIIT; B) or moderate‐continuous training (MCT; C), where sections were incubated with an antibody against myosin slow type I fibers to allow detection of type I (bright green) and type II (dark green) fiber proportion and size, with cell nuclei stained (blue) to establish fiber boundaries (scale bar represents 100 μm). Exercise training in HFpEF rats that performed HIIT or MCT had no effect on fiber cross‐sectional area (CSA) in both type I (D) and II (E) isoforms compared with their sedentary counterparts. Similarly, exercise training had no effect on the protein expression of either MuRF1 (F) or MuRF2 (G). The abbreviations S, H, and M for Western blot lanes represent loaded samples from HFpEF in the sedentary (S), HIIT (H), or MCT (M) groups, respectively, with GAPDH used as a loading control. Data are presented as mean±SEM, analyzed with 1‐way ANOVA, with n=13 for Sed, n=11 for HIIT, and n=11 for MCT.
Figure 7.
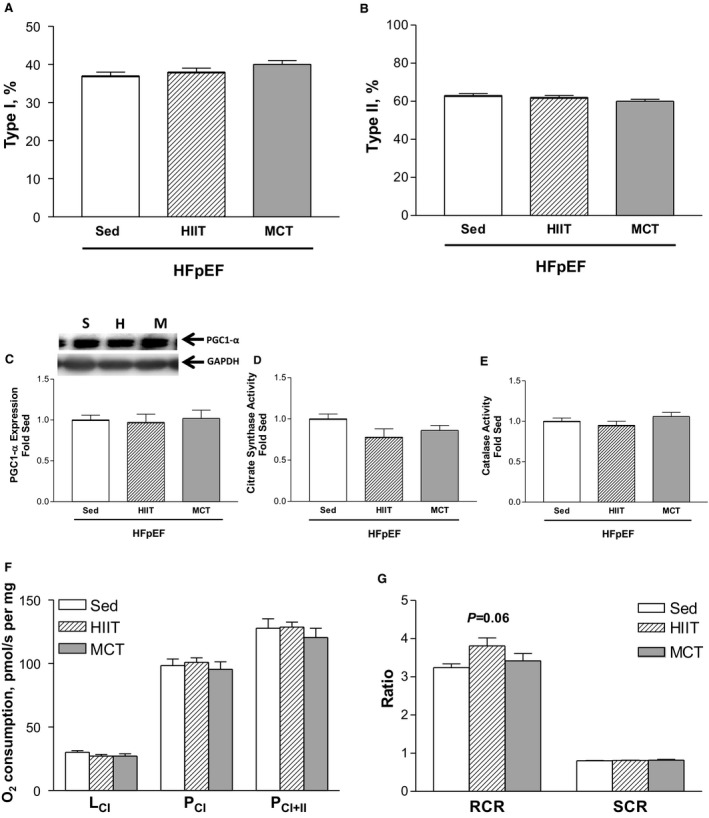
In heart failure with a preserved ejection fraction (HFpEF) rats at 28 weeks, fiber type proportion in the diaphragm for type I (A) and II (B) isoforms was not influenced following either high‐intensity interval training (HIIT) or moderate‐continuous training (MCT) when compared with sedentary (Sed) controls (see Figure 6A through 6C for representative diaphragm sections). Similarly, markers of mitochondrial content remained unchanged, including the protein expression of peroxisome proliferator‐activated receptor gamma coactivator 1‐α (PGC1‐α) (C) and enzyme activities of both citrate synthase (D) and catalase (E), as were functional measures of mitochondrial respiration (F) and the respective control ratios (G). Respiration states assessed, in turn, included: LCI, complex I leak respiration following addition of malate and glutamate; PCI, complex I phosphorylated state following addition of ADP; PCI + II, complex II phosphorylated state following addition of succinate. The abbreviations S, H, and M for Western blot lanes represent loaded samples from HFpEF rats in the sedentary (S), HIIT (H), or MCT (M) groups, respectively, with GAPDH used as a loading control. Data are presented as mean±SEM, analyzed with 1‐way ANOVA, with n=13 for Sed, n=11 for HIIT, and n=11 for MCT. RCR indicates respiratory control ratio; SCR, substrate control ratio.
Figure 8.
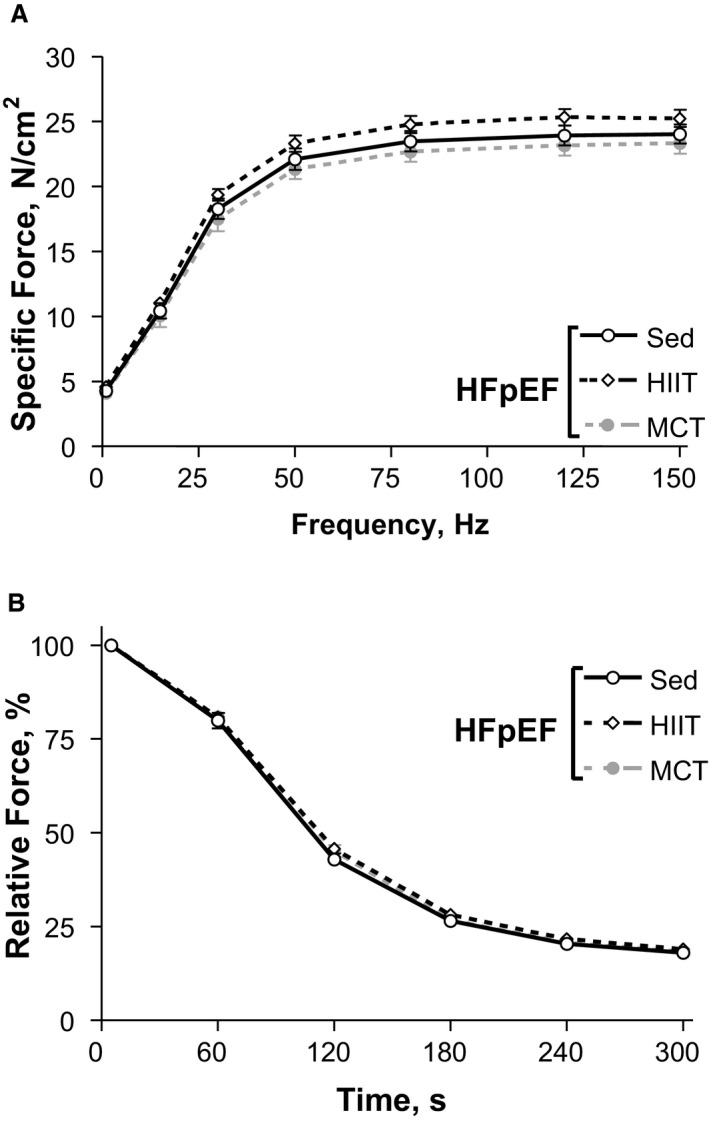
In vitro diaphragm function at 28 weeks in heart failure with a preserved ejection fraction (HFpEF) rats assessed in isolated fiber bundles revealed that compared with sedentary (Sed) rats, the interventions of high‐intensity interval training (HIIT) or moderate‐continuous training (MCT) had no benefits on contractile function (A) or fatigue resistance (B). Data are presented as mean±SEM, analyzed with 2‐way repeated measures ANOVA, with n=13 for Sed, n=11 for HIIT, and n=11 for MCT.
Limb Skeletal Muscle Alterations in HFpEF
In addition, we also assessed alterations in the soleus limb muscle, both at 20 and 28 weeks, in order to provide a comparison to the diaphragm (Figure 9). At 20 weeks, while contractile function and fatigability remained unaltered (P>0.05) (Figure 9A and 9B), muscle atrophy was present in HFpEF rats with muscle mass reduced by ≈15% (P>0.05) (Figure 9C). Further assessment of the molecular alterations revealed that mitochondrial density, as measured by citrate synthase activity, tended (P=0.06) to be increased by 45% in the soleus of HFpEF rats (Figure 9D). Similarly, markers typically related to the atrophy process were also higher in HFpEF rats: protein expression of MuRF1 tended to be increased by ≈20% (P>0.05) (Figure 9F), while MuRF2 increased by ≈25% (P<0.05) (Figure 9G). Interestingly, at 28 weeks, sedentary HFpEF rats tended to generate higher maximal soleus forces (≈10% higher compared with the HIIT HFpEF rats; P<0.05) (Figure 9A), with exercise training also having no influence on soleus fatigability (P>0.05) (Figure 9B). In contrast, however, HIIT was able to attenuate the muscle atrophy induced by HFpEF (P<0.05) (Figure 9C), while molecular measures following exercise training tended to be improved, albeit not significant: citrate synthase activity (P=0.07) (Figure 9D) and PGC1‐α protein content (P>0.05) (Figure 9E) tended to be ≈30% higher in trained animals, while levels of MuRF1 and MuRF2 were generally lower by ≈25% (P>0.05) (Figure 9F and 9G).
Figure 9.
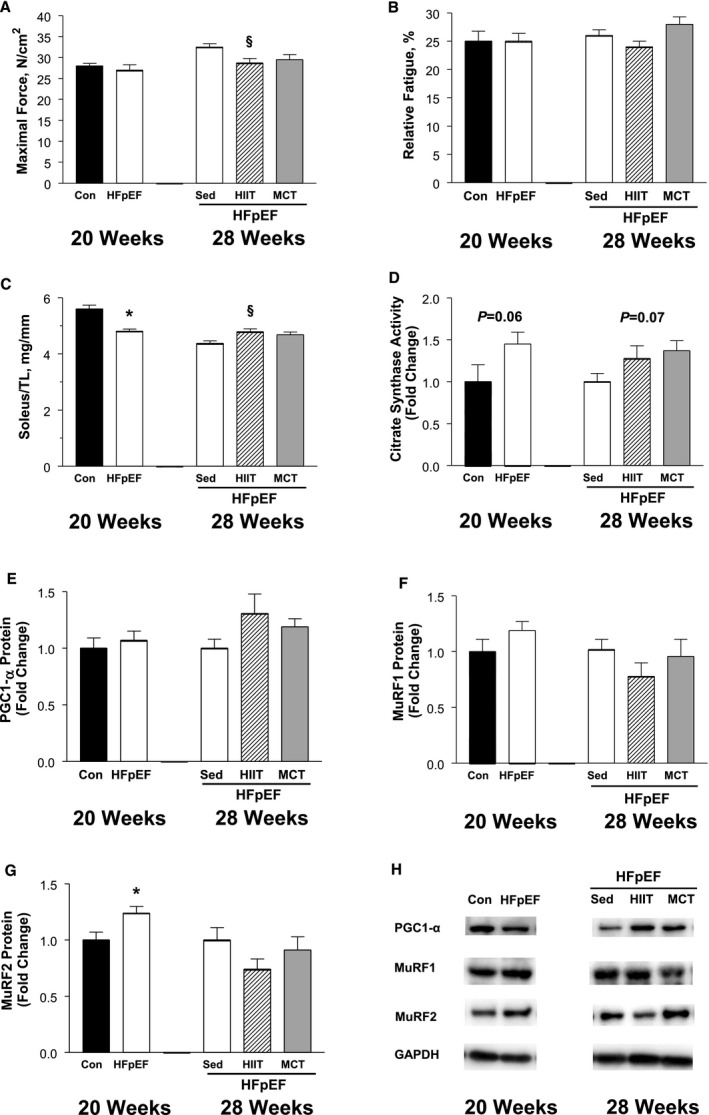
Alterations induced by heart failure with a preserved ejection fraction (HFpEF) in the soleus limb muscle at 20 weeks (control [Con] vs HFpEF) and also following 8 weeks of exercise training at 28 weeks (HFpEF rats that were sedentary [Sed] or performed high‐intensity interval training [HIIT] or moderate‐continuous training [MCT]), where in vitro maximal force generation (A) and fatigability (B) were assessed, followed by tissue analyses including muscle wet‐weight normalized to tibia length (TL) (C), citrate synthase activity (D), and peroxisome proliferator‐activated receptor gamma coactivator 1‐α (PGC1‐α) protein expression (E) as indices of mitochondrial density, and the expression of proteins involved in the muscle remodeling process MuRF1 (F) and MuRF2 (G). Proteins of interest were normalized to loading control GAPDH, with representative Western blot data shown in (H). At 20 weeks, HFpEF induced no functional alterations (A and B) but did reduce muscle mass (C), which was associated with a higher citrate synthase activity (D) and a trend for atrogins MuRF1 and MuRF2 to be increased (F and G). At 28 weeks, exercise training in HFpEF tended to increase muscle mass (C) and mitochondrial markers (D and E) and reduce the expression of atrogins (F and G). Data are presented as mean±SEM, analyzed with unpaired t test (or Mann‐Whitney for soleus force and weight measures) at 20 weeks or 1‐way ANOVA (Bonferroni post hoc) at 28 weeks, with n=8 for Con and n=12 for HFpEF at 20 weeks, while n=13 for Sed, n=11 for HIIT, and n=11 for MCT in HFpEF rats at 28 weeks. *P<0.05 vs Con. § P<0.05 vs Sed.
Discussion
This study, using a well‐characterized rat model of HFpEF underpinned by multiple comorbidities and exercise intolerance (ie, one that closely resembles the patient phenotype),9, 10, 11, 12 provides the first evidence in rats that diaphragm alterations and dysfunction induced in overt HFpEF were not reversed following 8 weeks of aerobic exercise training (ie, secondary prevention). This was observed for both the HIIT and MCT exercise training regimens, which indicates an apparent metabolic inflexibility of the diaphragm in HFpEF—a finding that opposes previous data where exercise training as primary prevention was beneficial in the period preceding the development of HFpEF.8 As such, these data may have important clinical implications for treating respiratory muscle weakness in patients with HFpEF,6, 7 suggesting that alternative therapeutic interventions may provide greater benefits compared with aerobic exercise training. Nevertheless, the finding that both training regimens still improved exercise capacity in HFpEF supports the current notion that exercise training is an effective treatment strategy for the HFpEF syndrome.2
Does HFpEF Induce a Myopathy in the Diaphragm?
In contrast to HFrEF,14 the effects of HFpEF on the diaphragm remain relatively unknown attributable, in part, to this patient cohort only more recently becoming apparent and the lack of appropriate animal models. Nevertheless, current evidence suggests that diaphragm weakness is an important driving factor that exacerbates the main symptom of exercise intolerance in patients with HFpEF.6, 7 While the underlying intracellular mechanisms responsible for diaphragm dysfunction in HFpEF remain poorly explored, our laboratory recently provided initial evidence using a hypertensive rat model of HFpEF.8, 15 Indeed, and consistent with the present study, rats with HFpEF also had a shift towards a type I fiber proportion, an increase in mitochondrial quantitative markers despite impaired qualitative function, and a reduction in maximal contractile function.8, 15
However, our previous animal model used Dahl salt‐sensitive rats—a model where HFpEF was driven exclusively by hypertension in the absence of the main symptom exercise intolerance.8, 15 In the present study, ZSF1 rats were used where multiple comorbidities are developed (eg, obesity, diabetes mellitus, hypertension, chronic kidney disease) as well as exercise intolerance,9, 10, 11, 12 thus more closely reflecting the HFpEF patient phenotype. This may explain, at least in part, why the current obese HFpEF animals demonstrated an ≈20% type I fiber hypertrophy and no impairment to fatiguing contractions, which is in contrast to our hypertensive HFpEF model where a fiber atrophy of ≈30% was observed with greater fatigability.8 These opposing findings are likely related to the obesity developed in the present animal model, which is prevalent in >80% of patients with HFpEF.4 Obesity is known to elevate the mechanical work of breathing that is suggested to drive a fiber hypertrophy and increase mitochondrial oxidative capacity, thereby allowing the muscle to become more resistant to fatigue.16, 17 Indeed, a greater proportion of type I fibers are assumed not only to increase the efficiency of ATP hydrolysis,18 which may, in turn, delay fatigue, but also reduce isometric force generation caused by type I compared with type II fibers generating lower specific forces.19 The latter may provide a partial explanation for the lower contractile forces we observed in the HFpEF rats compared with controls at 20 weeks, although single fiber analyses are required to answer this suggestion indefinitely. In addition, the type I fiber hypertrophy we observed in the diaphragm of our obese HFpEF rats was also associated with a reduction in the atrophy‐related proteins MuRF1 and MuRF2, which may suggest a role for the MuRF family of proteins in mediating this effect. That is, a lower protein expression of both MuRF1 and MuRF2 in the diaphragm would be predicted to downregulate pathways related to protein degradation and upregulate pathways related to protein synthesis. Indeed, previous evidence has shown that genetic deletion of both MuRF1 and MuRF2 cooperatively induce a fiber hypertrophy,20 while the MuRF proteins have also been implicated as regulators of protein synthesis and energy metabolism.20, 21 As such, the increased work of breathing consequent to obesity in HFpEF may provide some partial protection against fiber atrophy in the diaphragm that acts to maintain muscle function.
Despite the shift towards an oxidative phenotype, we still observed functional mitochondrial impairments, which included a higher leak state and an apparent reduction in mitochondrial coupling efficiency (ie, a lower respiratory control ratio). Previous data from rat skeletal muscle have shown that when mitochondrial proton flow is uncoupled from oxidative phosphorylation using 2, 4‐dinitrophenol, a higher leak state and reduced mitochondrial efficiency is observed and corresponds to an increase in markers of mitochondrial content and a shift towards an oxidative fiber proportion.22 As such, whether a greater work of breathing or rather an increase in mitochondrial uncoupling mediated the shift towards an oxidative phenotype in the diaphragm of our HFpEF rats remains a matter of debate, but further research is clearly warranted. Despite the increase in mitochondrial quantitative measures, the functional mitochondrial deficits in combination with the fiber contractile impairments in HFpEF rats support the assumption that an underlying myopathy may occur in HFpEF—a suggestion reinforced by our findings that exercise training was unable to provide any beneficial effects in the diaphragm. Clearly, however, more evidence is still required from animal models and patient biopsies to better understand the intracellular perturbations associated with diaphragm dysfunction in HFpEF.
Does Exercise Training Improve Diaphragm Function in HFpEF?
At present, unlike many drug‐based treatments, aerobic exercise training seems to be one of the few effective therapies available for treating patients with HFpEF.2 In the present study, our main focus was to closely mirror the clinical situation, where patients commonly begin exercise training once HFpEF has been diagnosed (ie, secondary prevention). As such, we initially allowed our rats to develop the HFpEF syndrome over 20 weeks, which is consistent with earlier studies.9, 10, 11, 12 Based on our previous study where exercise training (ie, HIIT) performed in the period preceding HFpEF development was able to attenuate diaphragm dysfunction and normalize mitochondrial function,8 we also expected to see a similar response in the current study. To our surprise, however, exercise training had no apparent benefits on the diaphragm, at least in relation to the measures we performed, and thus was unable to reverse the functional impairments we also observed at 20 weeks. Furthermore, these findings were independent of the training regimen as both HIIT and MCT were ineffective. The intensity of the exercise was apparently adequate, as previous evidence has shown this intensity of the HIIT protocol is able to induce positive alterations in the diaphragm,8 while was improved in both trained groups compared with sedentary HFpEF rats. Overall, therefore, these data seem to suggest that in HFpEF, a certain degree of metabolic inflexibility may exist that prevents the diaphragm from responding to the stress of exercise training.
Whether these findings also translate to patients remain to be determined, but they suggest that following the development of overt HFpEF, aerobic exercise training may be ineffective in reversing diaphragm weakness. Intriguingly, exercise capacity still increased in HFpEF rats that performed the HIIT or MCT compared with their sedentary counterparts, which suggests that improvements in vascular and/or limb muscle function were likely responsible (discussed below in more detail). Indeed, as exercise training in our HFpEF rats also had no benefits on cardiac parameters supports previous patient data,5 where cardiac function was also not influenced. As such, the present study seems to support a role that improvements in limb skeletal muscle and/or vascular/endothelial function may be the primary mechanism that allows exercise capacity to be elevated following exercise training in HFpEF.
Limb Muscle Alterations in HFpEF
In order to provide a comparison to the diaphragm, we also analyzed a limb muscle—the soleus. It has been suggested that in HFrEF, the diaphragm may be more sensitive to alterations compared with limb muscle23; however, with regards to HFpEF, initial evidence indicates that the limb muscles may be similarly affected.8 While we found no obvious functional deficits in the soleus muscle from the current group of obese HFpEF rats (ie, contractile function and fatigue indices remained unchanged), we did reveal numerous tissue alterations such that at 20 weeks an ≈15% muscle atrophy was present and this was associated with an upregulation in atrogins (ie, the MuRF1 and MuRF2), while mitochondrial capacity (as inferred from citrate synthase activity) tended to be higher in HFpEF rats compared with controls. The latter may suggest a potential compensatory response to the obese phenotype attributable to a reduction in blood flow or consequent to an increase in weight‐bearing load. Indeed, this suggestion is supported by previous data whereby obesity was shown to increase measures of oxidative capacity in numerous limb muscles.24 Yet, and in contrast, biopsies collected from limb muscle in patients with HFpEF recently reported that mitochondrial content and oxidative capacity were reduced.25 However, as this patient cohort were widely heterogeneous, older, and not primarily characterized by obesity,25 whether our current findings translate to patients remains to be determined.
In addition, and similar to the diaphragm, the present study found that exercise training had limited benefits on most soleus tissue indices in HFpEF rats, although the HIIT intervention was clearly able to attenuate the muscle atrophy observed in sedentary HFpEF rats. This was associated with a trend for levels of atrogins (MuRF1 and MuRF2) to be reduced and mitochondrial markers (PGC1‐α and citrate synthase) to be increased. Collectively, therefore, these data confirm that in an obese model of HFpEF, numerous alterations in limb skeletal muscle are induced, with evidence demonstrating that HIIT may provide an intervention to reverse potential underlying impairments, which, in turn, may contribute to improving exercise capacity following training. Alternatively, the primary mechanism that may have underpinned the increase in exercise capacity in HFpEF following the HIIT or MCT regimens may be directly related to improvements in endothelial function. It is well established that HFpEF can induce endothelial dysfunction, with available evidence showing that this can be reversed by exercise training.26 While inclusion of endothelial measurements was beyond the scope of the present study, where the main interest was to investigate the diaphragm, the potential for exercise training to improve peripheral endothelial function remains likely. The latter would be predicted to elevate O2 diffusion from capillary to myocyte, thereby driving improvements in subsequent exercise capacity27—a finding that was observed in the present study following 8 weeks of exercise training.
Study Limitations
While the present study assessed the effects of HFpEF in ZSF1 rats at one of the most advanced time points yet studied (ie, 28 weeks of age), whether these findings translate to the typically elderly HFpEF population remains unclear. In addition, whether our findings can be generalized to the more general female HFpEF population is unknown, with only male rats used in the present study. We also only assessed the effects of aerobic exercise training and thus it remains unclear whether other training modalities such as respiratory muscle training may have greater benefits on diaphragm function. Further, we also did not systemically evaluate food intake in the current study, which may have provided useful insight into the caloric response to exercise training in HFpEF.
Conclusions
Diaphragm alterations induced by an obesity‐driven rat model of HFpEF were not reversed following 8 weeks of aerobic exercise training, which suggests alternative therapeutic interventions may be required to treat respiratory muscle weakness in patient resembling this phenotype.
Sources of Funding
This study was funded by the European Commission: The Seventh Framework Programme for Research (FP7‐Health/602405).
Disclosures
None.
(J Am Heart Assoc. 2017;6:e006416 DOI: 10.1161/JAHA.117.006416.)
References
- 1. Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB, Shah SJ, Ahmed A, Bonow RO, Cleland JG, Cody RJ, Chioncel O, Collins SP, Dunnmon P, Filippatos G, Lefkowitz MP, Marti CN, McMurray JJ, Misselwitz F, Nodari S, O'Connor C, Pfeffer MA, Pieske B, Pitt B, Rosano G, Sabbah HN, Senni M, Solomon SD, Stockbridge N, Teerlink JR, Georgiopoulou VV, Gheorghiade M. Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Fail. 2014;2:97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fukuta H, Goto T, Wakami K, Ohte N. Effects of drug and exercise intervention on functional capacity and quality of life in heart failure with preserved ejection fraction: a meta‐analysis of randomized controlled trials. Eur J Prev Cardiol. 2016;23:78–85. [DOI] [PubMed] [Google Scholar]
- 3. Sharma K, Kass DA. Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ Res. 2014;115:79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype‐specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation. 2016;134:73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pandey A, Parashar A, Kumbhani DJ, Agarwal S, Garg J, Kitzman D, Levine BD, Drazner M, Berry JD. Exercise training in patients with heart failure and preserved ejection fraction: meta‐analysis of randomized control trials. Circ Heart Fail. 2015;8:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lavietes MH, Gerula CM, Fless KG, Cherniack NS, Arora RR. Inspiratory muscle weakness in diastolic dysfunction. Chest. 2004;126:838–844. [DOI] [PubMed] [Google Scholar]
- 7. Yamada K, Kinugasa Y, Sota T, Miyagi M, Sugihara S, Kato M, Yamamoto K. Inspiratory muscle weakness is associated with exercise intolerance in patients with heart failure with preserved ejection fraction: a preliminary study. J Cardiac Fail. 2016;22:38–47. [DOI] [PubMed] [Google Scholar]
- 8. Bowen TS, Rolim NP, Fischer T, Baekkerud FH, Medeiros A, Werner S, Bronstad E, Rognmo O, Mangner N, Linke A, Schuler G, Silva GJ, Wisloff U, Adams V; Optimex Study G . Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur J Heart Fail. 2015;17:263–272. [DOI] [PubMed] [Google Scholar]
- 9. Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite‐Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N . Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016;4:312–324. [DOI] [PubMed] [Google Scholar]
- 10. Hamdani N, Franssen C, Lourenco A, Falcao‐Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, Gonzalez A, Tschope C, Diez J, Linke WA, Leite‐Moreira AF, Paulus WJ. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239–1249. [DOI] [PubMed] [Google Scholar]
- 11. Leite S, Oliveira‐Pinto J, Tavares‐Silva M, Abdellatif M, Fontoura D, Falcao‐Pires I, Leite‐Moreira AF, Lourenco AP. Echocardiography and invasive hemodynamics during stress testing for diagnosis of heart failure with preserved ejection fraction: an experimental study. Am J Physiol Heart Circ Physiol. 2015;308:H1556–H1563. [DOI] [PubMed] [Google Scholar]
- 12. van Dijk CG, Oosterhuis NR, Xu YJ, Brandt M, Paulus WJ, van Heerebeek L, Duncker DJ, Verhaar MC, Fontoura D, Lourenco AP, Leite‐Moreira AF, Falcao‐Pires I, Joles JA, Cheng C. Distinct endothelial cell responses in the heart and kidney microvasculature characterize the progression of heart failure with preserved ejection fraction in the obese ZSF1 rat with cardiorenal metabolic syndrome. Circ Heart Fail. 2016;9:e002760. [DOI] [PubMed] [Google Scholar]
- 13. Bowen TS, Eisenkolb S, Drobner J, Fischer T, Werner S, Linke A, Mangner N, Schuler G, Adams V. High‐intensity interval training prevents oxidant‐mediated diaphragm muscle weakness in hypertensive mice. FASEB J. 2017;31:60–71. [DOI] [PubMed] [Google Scholar]
- 14. Kelley RC, Ferreira LF. Diaphragm abnormalities in heart failure and aging: mechanisms and integration of cardiovascular and respiratory pathophysiology. Heart Fail Rev. 2017;22:191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seiler M, Bowen TS, Rolim N, Dieterlen MT, Werner S, Hoshi T, Fischer T, Mangner N, Linke A, Schuler G, Halle M, Wisloff U, Adams V. Skeletal muscle alterations are exacerbated in heart failure with reduced compared with preserved ejection fraction: mediated by circulating cytokines? Circ Heart Fail. 2016;9:(9). [DOI] [PubMed] [Google Scholar]
- 16. Farkas GA, Gosselin LE, Zhan WZ, Schlenker EH, Sieck GC. Histochemical and mechanical properties of diaphragm muscle in morbidly obese Zucker rats. J Appl Physiol (1985). 1994;77:2250–2259. [DOI] [PubMed] [Google Scholar]
- 17. Powers SK, Farkas GA, Demirel H, Coombes J, Fletcher L, Hughes MG, Hodge K, Dodd SL, Schlenker EH. Effects of aging and obesity on respiratory muscle phenotype in Zucker rats. J Appl Physiol (1985). 1996;81:1347–1354. [DOI] [PubMed] [Google Scholar]
- 18. Crow MT, Kushmerick MJ. Chemical energetics of slow‐ and fast‐twitch muscles of the mouse. J Gen Physiol. 1982;79:147–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geiger PC, Cody MJ, Macken RL, Sieck GC. Maximum specific force depends on myosin heavy chain content in rat diaphragm muscle fibers. J Appl Physiol (1985). 2000;89:695–703. [DOI] [PubMed] [Google Scholar]
- 20. Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008;27:350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, Chiba T, Doi N, Kitamura F, Tanaka K, Abe K, Witt SH, Rybin V, Gasch A, Franz T, Labeit S, Sorimachi H. Muscle RING‐finger protein‐1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J Mol Biol. 2008;376:1224–1236. [DOI] [PubMed] [Google Scholar]
- 22. Schlagowski AI, Singh F, Charles AL, Gali Ramamoorthy T, Favret F, Piquard F, Geny B, Zoll J. Mitochondrial uncoupling reduces exercise capacity despite several skeletal muscle metabolic adaptations. J Appl Physiol (1985). 2014;116:364–375. [DOI] [PubMed] [Google Scholar]
- 23. van Hees HW, Ottenheijm CA, Granzier HL, Dekhuijzen PN, Heunks LM. Heart failure decreases passive tension generation of rat diaphragm fibers. Int J Cardiol. 2010;141:275–283. [DOI] [PubMed] [Google Scholar]
- 24. Torgan CE, Brozinick JT Jr, Kastello GM, Ivy JL. Muscle morphological and biochemical adaptations to training in obese Zucker rats. J Appl Physiol. 1989;67:1807–1813. [DOI] [PubMed] [Google Scholar]
- 25. Molina AJ, Bharadwaj MS, Van Horn C, Nicklas BJ, Lyles MF, Eggebeen J, Haykowsky MJ, Brubaker PH, Kitzman DW. Skeletal muscle mitochondrial content, oxidative capacity, and Mfn2 expression are reduced in older patients with heart failure and preserved ejection fraction and are related to exercise intolerance. JACC Heart Fail. 2016;4:636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adams V, Alves M, Fischer T, Rolim N, Werner S, Schütt N, Bowen TS, Linke A, Schuler G, Wisloff U. High‐intensity interval training attenuates endothelial dysfunction in a Dahl salt‐sensitive rat model of heart failure with preserved ejection fraction. J Appl Physiol. 2015;119:745–752. [DOI] [PubMed] [Google Scholar]
- 27. Hirai DM, Musch TI, Poole DC. Exercise training in chronic heart failure: improving skeletal muscle O2 transport and utilization. Am J Physiol Heart Circ Physiol. 2015;309:H1419–H1439. [DOI] [PMC free article] [PubMed] [Google Scholar]