

Abstract
Background
Exposure to psychosocial stress is a risk factor for cardiovascular disease, including vascular aging and regeneration. Given that dipeptidyl peptidase‐4 (DPP4) regulates several intracellular signaling pathways associated with the glucagon‐like peptide‐1 (GLP‐1) metabolism, we investigated the role of DPP4/GLP‐1 axis in vascular senescence and ischemia‐induced neovascularization in mice under chronic stress, with a special focus on adiponectin ‐mediated peroxisome proliferator activated receptor‐γ/its co‐activator 1α (PGC‐1α) activation.
Methods and Results
Seven‐week‐old mice subjected to restraint stress for 4 weeks underwent ischemic surgery and were kept under immobilization stress conditions. Mice that underwent ischemic surgery alone served as controls. We demonstrated that stress impaired the recovery of the ischemic/normal blood‐flow ratio throughout the follow‐up period and capillary formation. On postoperative day 4, stressed mice showed the following: increased levels of plasma and ischemic muscle DPP4 and decreased levels of GLP‐1 and adiponectin in plasma and phospho‐AMP‐activated protein kinase α (p‐AMPKα), vascular endothelial growth factor, peroxisome proliferator activated receptor‐γ, PGC‐1α, and Sirt1 proteins and insulin receptor 1 and glucose transporter 4 genes in the ischemic tissues, vessels, and/or adipose tissues and numbers of circulating endothelial CD31+/c‐Kit+ progenitor cells. Chronic stress accelerated aortic senescence and impaired aortic endothelial sprouting. DPP4 inhibition and GLP‐1 receptor activation improved these changes; these benefits were abrogated by adiponectin blocking and genetic depletion.
Conclusions
These results indicate that the DPP4/GLP‐1‐adiponectin axis is a novel therapeutic target for the treatment of vascular aging and cardiovascular disease under chronic stress conditions.
Keywords: aging, stress, vascular biology, vascular disease, vascular endothelium
Subject Categories: Animal Models of Human Disease, Peripheral Vascular Disease
Clinical Perspective
What Is New?
Chronic psychological stress resulted in an unbalance between plasma DPP4 and GLP‐1 levels in animals under ischemic conditions.
Stress decreased GLP‐1‐induced adiponectin production, leading to the reduction of AMP‐activated protein kinase–dependent peroxisome proliferator activated receptor‐γ and PGC‐1α expressions in the vessels and ischemic muscles in mice; and these changes were rectified by genetic and/or pharmacological interventions toward DPP4 or GLP‐1R.
DPP4 inhibition and GLP‐1R activation prevented stress‐related neovascularization impairment associated with endothelial senescence and endothelial progenitor cell mobilization failure and dysfunction.
What Are the Clinical Implications?
Targeting toward the interaction between DPP4‐mediated GLP‐1/GLP‐1R and peroxisome proliferator activated receptor‐γ/PGC‐1α axes can be potential therapeutic avenues of chronic stress‐related cardiovascular disease.
Alterations in plasma DPP4 and GLP‐1 levels are closely linked with the presence of stress in animals.
Introduction
Exposure to chronic psychosocial stress is a risk factor for many diseases, including vascular aging and cardiovascular disease.1, 2, 3 Stress upregulates inflammatory gene expression in leukocytes through a β‐adrenergic induction of myelopoiesis.4 Comprehensive review articles have highlighted the close linking between chronic psychological stress and atherosclerosis‐based cardiovascular disease.5, 6 Patients under moderate or high stress at the time of an acute myocardial infarction exhibited increased 2‐year mortality compared with control subjects under low levels of stress.7 It was also reported that endothelial cells (ECs) and endothelial progenitor cells (EPCs) are sensitive to various pathological stressors.8, 9, 10 However, the changes in vascular aging and regenerative capacity under chronic stress conditions remain largely unknown.
Dipeptidyl peptidase 4 (DPP4) is a complex enzyme that acts as a membrane‐anchored cell surface exopeptidase that transmits intracellular signals through a small intracellular tail.11 DPP4 has gained considerable interest as a therapeutic target, and a variety of DPP4 inhibitors that prolong the insulinotropic effects of glucagon‐like peptide‐1 (GLP‐1) are widely used in clinical settings as a new class of antidiabetic drugs.12, 13, 14 In addition to GLP‐1‐dependent effects on the cardiometabolic risk profile, DPP4 inhibition exhibits vascular protective benefits via the regulation of several substrate factor activities (eg, stromal cell–derived factor‐1α and granulocyte colony‐stimulating factor).15, 16, 17 A recent study reported a close link between increased plasma DPP4 activities and inflammatory and metabolic disorders.18 Moreover, chronic stress increased plasma and tissue DPP4 activities in mice and rats.14, 19
The peroxisome proliferator‐activated receptor‐γ (PPAR‐γ) is a member of the typical nuclear receptor superfamily of ligand‐inducible transcription factors.20 By binding to PPAR‐responsive regulatory elements as obligate heterodimers with retinoid X receptor, PPAR‐γ controls the expression of networks of genes involved in angiogenesis, inflammation, and maintenance of metabolic homeostasis.20 Adiponectin is an important adipocytokine that is downregulated in obesity‐related metabolic and cardiovascular disorders.21 Many studies suggested that adiponectin as well as PPAR‐γ ligands have beneficial effects on cardiac and muscle injuries and angiogenesis.22, 23, 24, 25, 26, 27 In in vitro experiments, the activation of adiponectin‐mediated PPAR‐γ/its coactivator 1α (PGC‐1α) pathway has been implicated in the regulation of a variety of cellular biological events such as cell differentiation, migration, apoptosis, and proliferation.24, 28 PGC‐1α deficiency causes vascular dysfunction by oxidative stress and vascular inflammation.29 Accordingly, we first investigated whether adiponectin‐mediated PPARγ/PGC‐1α signal pathways are involved in the process of ischemia‐induced neovascularization in mice under chronic stress.
Here we examined the hypothesis that increased DPP4 activity may negatively modulate ischemic neovascularization via an adiponectin‐PPARγ/PGC‐1α‐dependent mechanism that is mediated by the reduction of GLP‐1 in mice that have been subjected to chronic stress.
Materials and Methods
Antibodies and Reagents
The following commercially available antibodies were used: Anti‐PECAM‐1 (CD31, sc‐1506), anti‐PPAR‐γ (sc‐7196), and anti‐GAPDH (sc‐20357) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti‐Sirt1 (cat: AP20849c) was from Abgent/Wuxi AppTec (San Diego, CA). Anti‐AMP‐activated protein kinase (AMPK) (#2532), anti‐pAMPKαthr172 (#2535), and anti‐rat IgG antibodies (Alexa Fluor 488 conjugate, 1:100) were from Cell Signaling Technology (Danvers, MA). Anti‐PGC‐1α (ab54481), neutralizing anti‐adiponectin (ab3455) and rabbit control IgG (ab37415) antibodies were purchased from Abcam (Cambridge, MA). Anti‐Mac‐3 (M3/84, 1:50), fluorescein isothiocyanate–conjugated rat anti‐mouse CD31 (MEC13.3, 1:100), and R‐phycoerythrin‐conjugated rat anti‐mouse c‐Kit (CD117, 2B8, 1:100) antibodies were purchased from BD Pharmingen (San Diego, CA). Rabbit anti‐mouse IgG (Alexa Fluor 594 conjugate) antibody was from Molecular Probes (Eugene, OR). Anti‐β‐actin (AC‐15, A1978) and human recombinant DPP4 (1083) was purchased from Sigma‐Aldrich (St. Louis, MO). Anti‐proliferating cell nuclear antigen (clone: PC10, 1:100) was from Merck Millipore (Darmstadt, Germany). Fluorescein Lycopersicon Esculentum (tomato) lectin (cat. FL‐1171) was from Vector Laboratories (Burlingame, CA).
The DPP4‐Glo assay kit and DPP4 inhibitor anagliptin were purchased from Promega (Madison, WI). The ELISA kits for GLP‐1 were from EMD Millipore (EZGLP1T‐36K; Billerica, MA) and adiponectin was from R&D Systems (PMRP300; Minneapolis, MN). Endothelial basal medium‐2 and endothelial growth medium‐2 SingleQuots were purchased from Lonza (Walkersville, MD). Nitrocellulose transfer membrane was from Amersham Bioscience (Piscataway, NJ). RNeasy Micro Kits and SYBR™ Green Master Mix were from Qiagen (Hilden, Germany). The DC™ protein assay kit (cat. 500‐0114) was purchased from Bio‐Rad Laboratories (Hercules, CA). Recombinant human vascular endothelial growth factor (VEGF, 354107) and growth factor‐reduced Matrigel Matrix were from BD Bioscience (Bedford, MA). The SuperScript III First Strand was purchased from Invitrogen (Carlsbad, CA). The Amersham ECL Prime Western Blotting Detection kit was from GE Healthcare (Freiburg, Germany). CD117 MicroBeads and MACS were from Miltenyi Biotec (Bergisch Gladbach, Germany). The In Situ Cell Death Detection Kit (Lot. 10131400), complete and Mini protease inhibitor cocktail, and beta‐galactosidase. staining set (Lot. 10652800) were purchased from Roche Diagnostics (Mannheim, Germany).
Animals
Six‐week‐old C57BL/6J male mice and 5‐week‐old male DPP4+/+ rats (F344/Jcl, Chubu Kagaku Shizai, Nagoya, Japan) and 5‐week‐old male DPP4‐deficient rats (DPP4−/−, F344/DuCr1Cr1j, Charles River Laboratories Japan, Yokohama, Japan) were provided with a standard diet and tap water ad libitum. Adiponectin‐deficient mice (adiponectin−/− 30) on a C57BL/6J background obtained from the same source were used in this experiment. The animal protocols were approved by the Institutional Animal Care and Use Committee of Nagoya University Graduate School of Medicine (Protocol Number 27304) and performed according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Model of Revascularization With or Without Stress
Seven‐week‐old mice (21–24 g) and 6‐week‐old rats (90–105 g) were given immobilized stress in which their movements were restricted by placing them in an appropriately sized animal holder (155‐BSRR for mice; KN‐325‐B Square Rat Holder with 3 sluices for rats; both from Natsume Seisakusho, Tokyo; Figure 1A) for 4 hours per day without food and water intake. Control animals were left undistributed and allowed contact with each other, as described.31 After they underwent the immobilization stress for 4 weeks, the stressed mice and control mice were subjected to unilateral hindlimb ischemic surgery, and we compared the recovery of the ischemic/nonischemic blood‐flow ratio between the stressed and control groups during the follow‐up period.
Figure 1.
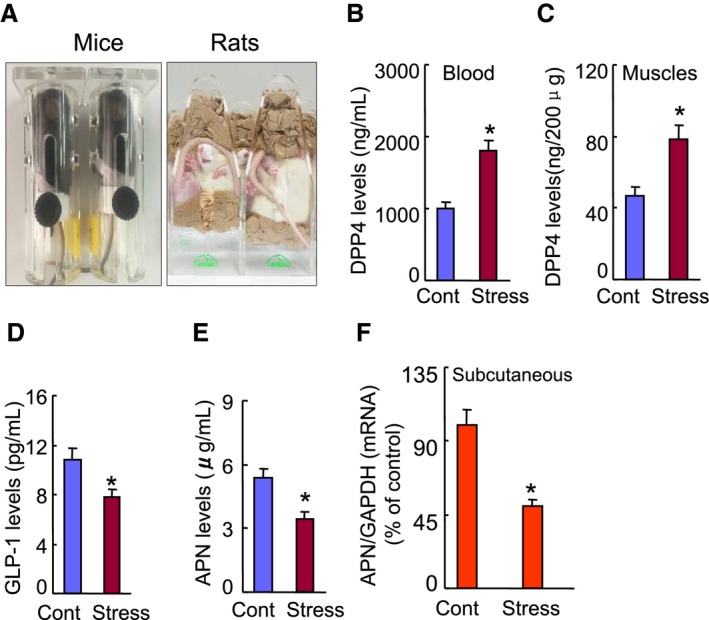
The effects of chronic stress on the levels of DPP4 activities and targeted molecule proteins or genes in the nonstressed and stressed mice. A, The mouse/rat immobilized stress model. B and C, The levels of plasma and muscle DPP4 levels. D and E, ELISA data show the levels of plasma GLP‐1 and adiponectin levels. F, Quantitative real‐time PCR revealed that chronic stress reduced the adiponectin expression in subcutaneous adipose tissue. Values are mean±SE (n=5–7). *P<0.01 vs corresponding controls; NS, not significant by ANOVA and Tukey's post hoc tests. ANOVA indicates analysis of variance; APN adiponectin; DPP4, dipeptidyl peptidase‐4; GAPDH, glyceraldehyde‐3‐phosphate‐dehydrogenase; GLP‐1, glucagon‐like peptide‐1; PCR, polymerase chain reaction.
For the mechanistic studies, 5 independent experiments were performed as follows: (1) In separate DPP4 inhibitor experiments, mice that underwent stress for 4 weeks were assigned to 1 of 3 groups and given (by oral gavage) vehicle (distilled water, Stress), a low dose (30 mg/kg per day, S‐DL) or a high dose of the DPP4 inhibitor anagliptin (60 mg/kg per day, S‐DH, a generous gift from Sanwa Kagaku Pharmaceutical, Mie, Japan) every day for 21 days with continued daily immobilization stress; (2) In specific GLP‐1 receptor agonist experiments, mice that underwent immobilization stress for 4 weeks were assigned to 1 of 2 groups and administered vehicle (saline; stress group) or the GLP‐1R agonist exenatide (5 μg/kg per day, S‐Exe group; AstraZeneca, London, UK) for 14 days; (3) In rat experiments, DPP4+/+ and DPP4−/− rats that were subjected to restraint stress for 4 weeks underwent the hindlimb ischemic surgery, and the stress was continued for 14 days after the surgery; (4) In adiponectin blocking experiments, the mice that were treated with control rabbit IgG (450 μg/kg per day) or rabbit neutralizing adiponectin antibody (N‐adiponectin, 450 μg/kg per day) by subcutaneous injection every week under restraint stress conditions for 2 weeks were subjected to the ischemic surgery and continuously received antibodies and anagliptin (30 mg/kg per day) interventions for 14 days under stress; (5) adiponectin deficiency (adiponectin−/−) mice that underwent immobilization stress for 2 weeks were assigned to 1 of 2 groups and administered vehicle or a dose of oral anagliptin (30 mg/kg per day) for 14 days.
At the end of the experiments, all animals were anesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg; Dainippon Pharmaceutical, Osaka, Japan), and the tissue (muscles, vessels, fats, and femurs) and arterial blood samples were collected for biological analyses (including DPP4 activity, ELISA, gene and protein assays) and histological analyses. All surgical and sampling procedures followed were in accordance with institutional guidelines.
Analysis of Hindlimb Blood Flow
Hindlimb blood flow was detected with laser speckle blood flow imaging (LSBFI, OMEGAZONE, OZ‐1, OMEGAWAVE, Tokyo). LSBFI analyses were performed on the animals' legs and feet before surgery and on postischemic days 0, 4, 7, 14, and/or 21, respectively. Blood flow is shown as changes in the laser frequency with differently colored pixels. The results of the quantitative analysis of blood flow are expressed as the ratio of left (ischemia) to right (nonischemia) LSBFI.8
Measurement of Capillary Density
Ischemic thigh adductor muscles obtained on the indicated days were prepared as 4‐μm‐thick cryosections for the microscopic analysis of capillary density. Endothelial cells were recognized by immunohistochemical staining with anti‐mouse CD31 or fluorescent staining with tomato lectin. The capillary density is expressed as the ratio of the number of capillaries to the number of muscle fibers per high‐power (×200) field measured in 5 randomly chosen microscopic fields from 3 independent cross‐sections in each tissue block, as described.8
Levels of Plasma and Tissue DPP4 Activities
DPP4 levels in plasma and tissues were measured by a DPP4‐Glo Protease Assay with aminoluciferin substrate as previously described.19 Human recombinant DPP4 was used to drive a standard curve. The luminescence intensity was calculated using a luminometer. Based on the standard curve, the plasma and tissue DPP4 levels were derived to represent as ng/mL.19 All measurements were performed in triplicate.
Western Blot Analysis
Rat and mouse tissue samples were obtained on postischemic day 4. The same amounts of protein (40 μg/line) extracted from the ischemic adductor muscle and the descending thoracic aorta vessels were transferred to polyvinylidene difluoride membranes and immunoreacted with targeted primary antibodies. The membranes were then treated with the horseradish peroxidase–conjugated secondary antibody at 1:10 000 to 15 000 dilutions. The Amersham ECL Prime Western Blotting Detection kit was used for the determination of targeted proteins. Protein levels quantitated from Western blots were normalized by loading internal controls.
Gene Expression Assay
Whole RNA was harvested from the tissues with the RNAeasy Mini Kit according to the recommended protocol. The SuperScript III CellsDirect cDNA Synthesis kit was used to generate cDNA. Targeted mRNA levels were evaluated by conducting a TaqMan gene expression assay. Quantitative real‐time polymerase chain reaction was conducted on a 7300 real‐time polymerase chain reaction System (Applied Biosystems, Foster City, CA). All experiments were performed in triplicate. The sequences of primers for targeted genes are shown in Table 1. The targeted gene expression was normalized to the internal housekeeping GAPDH gene.
Table 1.
Primer Sequences for Mice Used for Quantitative Real‐Time PCR
| APN‐F | GATGGCAGAGATGGCACTCC |
| APN‐R | CTTGCCAGTGCTGCCGTCAT |
| GLUT4‐F | TCCCTGTTACCTCCAGGTTG |
| GLUT4‐R | CCTTGCCCTGTCAGGTATGT |
| IRS‐1‐F | CCAGAGTCAAGCCTCACACA |
| IRS‐1‐R | CCCAACTCAACTCCACCACT |
| GAPDH‐F | ATGTGTCCGTCGTGGATCTGA |
| GAPDH‐R | ATGCCTGCTTCACCACCTTCT |
APN indicates adiponectin; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GLUT4, glucose transporter 4; IRS‐1, insulin receptor substrate 1; PCR, polymerase chain reaction.
ELISA and Biochemical Analyses
Blood samples were obtained directly from the left ventricles of mice and rats after a 10‐hour fast and/or combined stress for 2 hours for the ELISA and biochemical analyses. The levels of plasma GLP‐1 and adiponectin were determined using a commercially available ELISA kit according to the manufacturer's instructions. The levels of mouse and rat plasma glucose, corticosterone, triglyceride, total cholesterol, high‐density lipoprotein cholesterol, nonesterified fatty acid, creatinine, and blood urea nitrogen were examined at a commercial laboratory (SRL, Tokyo, Japan).
Gelatin Zymography
On postoperative day 4, the activities of matrix metalloproteinase‐2 (MMP‐2) and MMP‐9 in the muscles were determined by gelatin zymography. First, 20 μg of total tissue protein mixed with a SDS sample buffer without reducing agent were loaded onto a 10% SDS‐polyacrylamide gel containing 1 mg/mL gelatin. After staining with Coomassie blue, the gelatinase activity was quantified by using ImageJ software.8
Aorta‐Ring Culture for Angiogenesis Assay
An ex vivo angiogenesis assay was performed essentially as described.9 Following a careful reversal of the descending thoracic aortas of mice and rats, the aortic rings (1–2 mm) were prepared. The rings were then immediately inserted into layers of growth factor–reduced Matrigel and cultured in endothelial basal medium‐2 containing 50 ng/mL of VEGF for 7 days. Endothelial cells characterized as microvascular sprouting were stained with fluorescein isothiocyanate–labeled mouse anti‐CD31. The sprout length was quantified by using a BZ‐X700 microscope and BZ‐X Analyzer (Keyence, Osaka, Japan). The endothelial cell sprouting density is expressed as the percentage of pixels per image occupied by vessels within a defined area.
Bone‐Marrow (BM)–Derived c‐Kit+ EPC‐Like Cell Isolation
BM‐derived mononuclear cells were obtained from mice and rats (n=5 per group). Following the isolation of lineage− mononuclear cells, BM‐derived c‐Kit+ cells were collected by using MACS and CD117 MicroBeads according to the manufacturer's instructions. c‐Kit+ EPC‐like cells were >90% positive for CD31+ as described.8 After being cultured on fibronectin‐coated dishes (Wako) in endothelial growth medium‐2 and 2% fetal bovine serum for 5 days, the EPC‐like c‐Kit+ cells were isolated and used for the cellular assays.
Endothelial Cell Tube Formation Assay
BM‐derived EPCs (2.5×104 cells/well) were seeded on growth factor–reduced Matrigel in a 24‐well plate and incubated for 24 hours in endothelial basal medium‐2 containing 50 ng/mL of VEGF to induce tube formation. The formation of networks was assessed using a BZ‐X Analyzer to evaluate the number of sprouts in 5 fields (×100) of each well.8
Vascular Senescence Assay
Both the thoracic aorta and aortic arch from mice and rats were used for a vascular cellular senescence assay by β‐galactosidase (β‐Gal) staining set in accordance with the manufacturer's instructions. The degree of vascular cellular senescence for each aorta was quantified with the positive area of activated β‐Gal as previously described.14
Immunohistochemical Staining
On postischemic 4 day, macrophages that had infiltrated the ischemic muscles were detected immunohistochemically in 4‐μm‐thick cryosections with anti‐mouse CD68. Macrophages were counted in 5 random microscopic fields from 3 independent sections, and the infiltration is expressed as the number of CD68+ cells per high‐power field (×200).
Immunocytofluorescent Staining
On postoperative day 4, the vascular progenitor cells isolated from the BM of mice and rats were seeded onto coverslips (2×43 cells/mL) coated with denatured collagen and then cultured in endothelial growth medium‐2 containing 4% fetal bovine serum for 24 hours. Following fixation with 4% paraformaldehyde, the cells were washed 3 times with PBS containing 1% glycerol. The cells were blocked with 0.1% bovine albumin serum/PBS and then treated with the antibodies against ki67 and c‐Kit for the evaluation of c‐Kit+ cell proliferation or with TdT‐mediated dUTP nick‐end labeling staining reagents for their apoptosis assay.
Flow Cytometric Quantification of EPCs
On postoperative day 4, peripheral blood and BM were pretreated with mouse anti‐CD16+/CD32+ monoclonal antibody to block the non‐antigen‐specific binding of immunoglobulins to the FcγIII and FcγII receptors of monocytes. EPC‐like mononuclear cells were incubated with an R‐phycoerythrin‐conjugated rat anti‐mouse c‐kit antibody and fluorescein isothiocyanate–conjugated rat anti‐mouse CD31 antibody. Following treatment with flow cytometry lysing solution, the cells were centrifuged and suspended in PBS for the flow cytometry analysis.32
Statistical Analyses
The data are expressed as means±SE. Student t test (for comparisons between 2 groups), and 1‐way ANOVA for comparisons of 3 or more groups followed by Tukey's post hoc tests were used for the statistical analyses. Following test data distribution status, the data were subjected to the statistical analysis. If the homogeneity of variance assumption was violated, the nonparametric Kruskal–Wallis test was used instead. The blood flow data were subjected to a 2‐way repeated‐measures ANOVA and Bonferroni post hoc tests. The comparative incidence of limb amputation was evaluated by the χ2 test. SPSS software ver. 17.0 (SPSS, Chicago, IL) was used for all statistical analyses. P<0.05 was considered significant.
Results
Chronic Stress Impaired Neovascularization in Response to Ischemia
A restricted stress model (Figure 1A) has often been used to investigate psychological stress–related metabolic and inflammatory cardiovascular disorders.31 To test our hypothesis, we created a mouse model of the combination of immobilization and ischemic induction to examine whether chronic stress could influence the levels of DPP4 activity and GLP‐1 protein and angiogenic actions. Compared with the control mice, our ELISA data revealed that stressed mice had dramatically increased DPP4 levels and decreased GLP‐1 levels in plasma and/or ischemic muscles (Figure 1B through 1D). Our serial LSBFI analyses revealed that stressed mice showed decreased recovery of ischemic hindlimb perfusion throughout the follow‐up period, and the ratio of ischemia to nonischemia LSBFI was persistently lower in the stressed mice compared with the nonstressed mice (Figure 2A). Spontaneous amputation (including toenail, foot, and/or below the knee) occurred in 0% of the ischemic limbs of the nonstressed controls, but was markedly increased to 48.5% in the stressed mice. As anticipated, stress significantly reduced the capillary density in the ischemic muscles (Figure 2B), indicating that the chronic stress caused an imbalance between DPP4 activities and GLP‐1 levels and reductions in ischemia‐induced blood recovery and capillary development.
Figure 2.
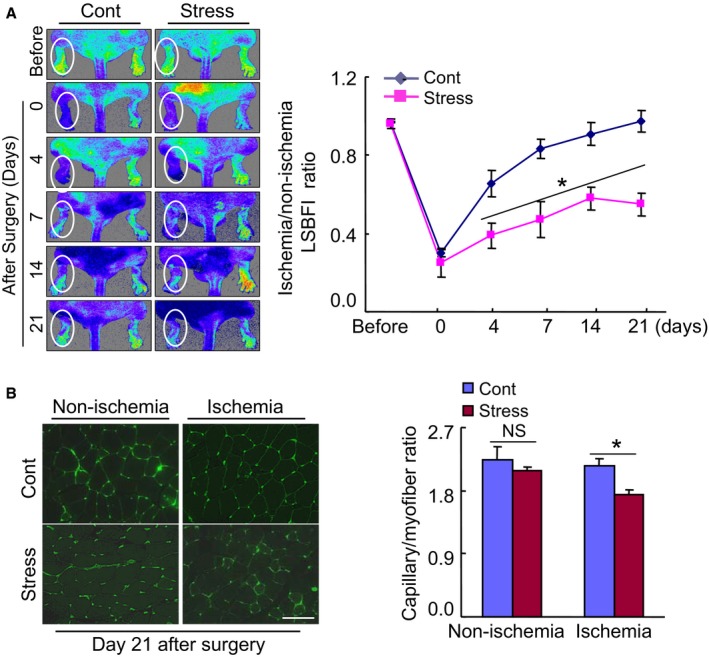
Chronic stress impaired blood flow recovery and capillary formation in ischemic muscles of the mice. A, Representative laser speckle perfusion imaging showed a low perfusion signal (dark blue) in the ischemic hindlimbs of stressed mice and a high signal (red) in control mice. The ratio of ischemic to nonischemic laser speckle blood flow was lower in the stressed mice than in the nonstressed control mice during the follow‐up period. Data are mean±SE (n=6). *P<0.01 by 2‐way repeated‐measures ANOVA and Bonferroni post hoc tests. B, Fluorescent staining was performed using tomato lectin to visualize capillaries in ischemic and nonischemic thigh adductor muscle. Quantitative data showed the reduction of capillary density as expressed by the capillary‐to‐myofiber ratio in ischemic muscle of the stressed mice (n=6). Data are mean±SE. *P<0.05, NS, not significant by Student unpaired t test or ANOVA and Tukey's post hoc tests. Scale bar, 50 μm. LSBFI indicates laser speckle blood flow imaging.
Adiponectin‐mediated PPAR‐γ/PGC‐1α signal pathway inactivation has been implicated in metabolic cardiovascular disorder initiation and progression.22, 25 We thus examined whether these molecules are sensitive to chronic stress. As shown in Figure 1E, a marked reduction of plasma adiponectin levels was observed in the stressed mice on day 4 after ischemic induction. Consistently, the subcutaneous fat of the stressed mice had decreased adiponectin mRNA expression compared with the nonstressed control mice (Figure 1F). We also observed that chronic stress reduced insulin receptor substrate 1 and glucose transporter 4 gene expressions in the inguinal adipose tissues (Table 2). The quantitative analyses of representative immunoblots revealed that the levels of p‐AMPKα, VEGF, PPAR‐γ, and PGC‐1α proteins were lower in the ischemic muscles of the stressed mice compared with those of the control mice (Figure 3A), suggesting a chronic stress‐related inactivation of AdipoR1/AMPKα signaling and the PPAR‐γ/PGC‐1α axis in response to ischemia in the neovascularization process.
Table 2.
Levels of Lipids and Other Parameters in the Control and Stress Groups at 7 Weeks
| Parameter | Control | Stress |
|---|---|---|
| Body weight, g | 23.7±0.4 | 21.5±0.3a |
| Triglyceride, mg/dL | 27.0±5.2 | 18.9±2.0a |
| TCh, mg/dL | 45.0±3.5 | 47.8±4.9 |
| HDL‐C, mg/dL | 30.2±1.7 | 29.1±1.6 |
| NEFA, μEQ/L | 668±50 | 1143±52a |
| Blood urea nitrogen, mg/dL | 29.4±2.1 | 28.6±1.4 |
| Creatinine, mg/dL | 0.2±0.0 | 0.1±0.0 |
| Glucose, mg/dL | 84.3±6.7 | 94.0±7.9 |
| Corticosterone, ng/mL | 170.5±3.2 | 76.3±6.2a |
| Real‐time PCR (adipose) | ||
| IRS‐1, % | 14.0±2.4 | 9.9±1.9a |
| GLUT4, % | 28.4±3.1 | 17.7±1.7a |
Values are mean±SE (n=6–10). GLUT4 indicates glucose transporter 4; HDL‐C, high‐density lipoprotein cholesterol; IRS‐1, insulin receptor substrate 1; NEFA, nonesterified fatty acid; PCR, polymerase chain reaction; TCh, total cholesterol.
P<0.05 by Student t test.
Figure 3.

The levels of targeted proteins in tissues of both experimental groups. A, At d 4 after surgery, equal amounts of total protein extraction were immunoblotted using p‐AMPKα (Thr172), AMPKα, PPAR‐γ, PGC‐1α, VEGF, and β‐actin antibodies. Representative immunoblots and quantitative data show reduced levels of p‐AMPKα, VEGF, PPAR‐γ, and PGC‐1α proteins in ischemic muscles of stressed mice. Data are mean±SE (n=3). *P<0.01, NS, not significant by ANOVA and Tukey's post hoc tests. B, Reversed aorta rings (1–2 mm) (upper left panel) were cultured with EBM‐2 containing 50 ng/mL of VEGF on growth factor‐reduced Matrigel for 7 d, and then microvascular sprouting was characterized by fluorescent staining with FITC‐labeled mouse anti‐CD31 (lower left panel). The aortas (including thoracic aorta and aortic arch) were subjected to vascular cellular senescence by β‐galactosidase (β‐gal) staining. C and D, Quantitative data for endothelial cell sprouting areas and β‐Gal+ areas (×200). The endothelial sprouting areas and β‐Gal+ areas are expressed as the percentage of pixels per image occupied by vessels in the quantitative area. E, Representative Western blots and quantitative data showing the levels of PPAR‐γ and PGC1‐α in the aortas of stressed and control mice. Data are mean±SE (n=4–6). *P<0.01 by Student unpaired t test or ANOVA and Tukey's post hoc tests. Scale bar: 50 μm. ANOVA indicates analysis of variance; EBM, endothelial basal medium; FITC, fluorescein isothiocyanate; p‐AMPKα, phospho‐AMP‐activated protein kinase α; PGC‐1α, PPAR‐γ co‐activator 1α; PPAR‐γ, peroxisome proliferator‐activated receptor‐γ; VEGF, vascular endothelial growth factor.
An ex vivo aortic ring culture assay has been established to investigate in vivo angiogenic states.32, 33 Immunofluorescent images showed endothelial sprouting to tubule formation from the aorta rings cultured at day 7 (Figure 3B). Compared with the controls, the EC‐derived sprouting capacity was reduced in the aortas of the stressed mice (Figure 3B and 3C). We extended our examination to the vascular EC senescence and targeted molecule changes in response to chronic stress. The β‐Gal staining results indicated that stress accelerates vascular senescence (Figure 3B and 3D). Moreover, as anticipated, compared with the control aortas, the stressed aortas exhibited low levels of PPAR‐γ, PGC‐1α, and Sirt1 proteins (Figure 3E). These observations thus suggest that chronic stress caused the decline in PPAR‐γ/PGC‐1α activation and angiogenic action and an acceleration of vascular senescence.
To further clarify the specific effects of chronic stress on vascularization, we sought to compare CD31+/c‐Kit+ EPC‐like cell production and mobilization in both experimental groups after ischemic induction. Flow cytometry analyses revealed that the numbers of CD31+/c‐Kit+ cells were lower in not only BM but also peripheral blood in the stressed mice compared with the nonstressed mice (Figure 4A). Likewise, double immunofluorescence showed that the numbers of BM‐derived CD31+/c‐Kit+ and c‐Kit+/Ki67+ cells were decreased in the stressed mice, whereas c‐Kit+/TdT‐mediated dUTP nick‐end labeling+ cells were increased in the stressed mice (Figure 4B). Therefore, stress appears to reduce vascularization via the reduction of BM EPC production and mobilization.
Figure 4.

Stress decreased CD31+/c‐Kit+ cells in BM and PB. A, Representative dot plots and quantitative data for the numbers of CD31+/c‐Kit+ (per 2×104 cells) in BM and PB (n=6–7, Mann–Whitney U test). B, BM‐derived c‐Kit+ cells were subjected to proliferation and apoptotic assays using Ki67 antibody or TUNEL staining, respectively. Representative images and quantitative data show proliferative and apoptotic cells (n=5–7, Student t test). Data are mean±SE. *P<0.01 vs controls. Scale bar, 50 μm. BM indicates bone marrow; PB, peripheral blood; TUNEL, TdT‐mediated dUTP nick‐end labeling.
On the other hand, inflammation has been shown to be involved in the pathogenesis of cardiovascular disease. Here we observed more macrophages in the extracapillary space in the stressed mice on day 4 after ischemic induction compared with the nonstressed control mice (Figure 5A). Compared with control, marked MMP‐2 and MMP‐9 activities were observed in the ischemic muscles of the stressed mice (Figure 5B and 5C), indicating that stress promotes inflammatory action and MMP‐2/‐9 activation in the angiogenesis process. With the exception of body weight and nonesterified fatty acid, triglyceride, and corticosterone, we observed that there were no differences in the levels of total cholesterol, high‐density lipoprotein cholesterol, blood urea nitrogen, glucose, and creatinine between the experimental groups (Table 2).
Figure 5.
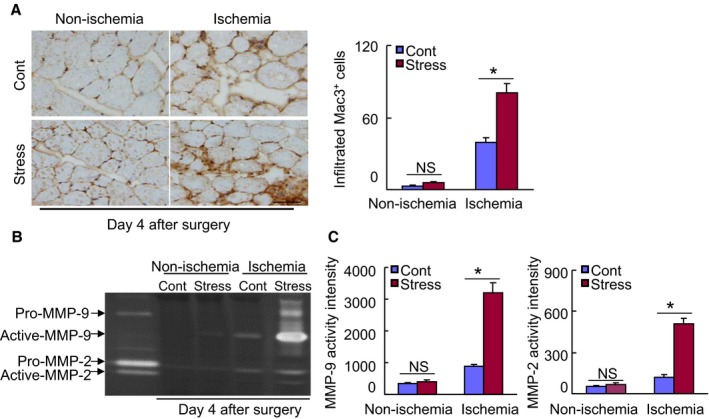
Chronic stress stimulates inflammatory action in ischemic muscles of stressed mice. A, Immunohistochemical staining was performed using antibodies against macrophages (Mac‐3) in ischemic and nonischemic muscles of nonstressed and stressed mice on d 4 after ischemia. Representative images and quantitative data show infiltrated numbers of macrophages (n=6, ANOVA and Tukey's post hoc tests). B and C, Gelatin zymography assay was performed in ischemic and nonischemic muscles of nonstressed and stressed mice on d 4 after ischemia. Representative images and quantitative data show the levels of the MMP‐2 and MMP‐9 gelatinolytic activities (n=3, ANOVA and Tukey's post hoc tests). Values are mean±SE. *P<0.01 vs corresponding controls. Scale bar: 50 μm. MMP‐2 indicates matrix metalloproteinase‐2; NS, not significant.
DPP4 Activity Modulated Neovascularization in Response to Stress
We sought to determine whether pharmacological DPP4 inhibition could rectify an imbalance between DPP4 activities and GLP‐1 protein levels in mice that have been subjected to chronic stress conditions. As anticipated, in comparison with the control mice, DPP4 inhibition by both doses of anagliptin reversed the changes in the DPP4 and GLP‐1 levels in the blood of the stressed mice on postischemic day 4 (Figure 6A and 6B). Likewise, the levels in circulating adiponectin and subcutaneous adipose adiponectin mRNA were increased in the S‐DL and S‐DH mice (Figure 6C and 6D). We have also observed that S‐DL and S‐DH mice had increased levels of inguinal adipose insulin receptor substrate 1 and glucose transporter 4 genes as compared with stress‐alone mice (Table 3). With the exception of nonesterified fatty acid, glucose, and corticosterone, we observed that there were no differences in targeted metabolic parameters among the 3 experimental groups (Table 3).
Figure 6.
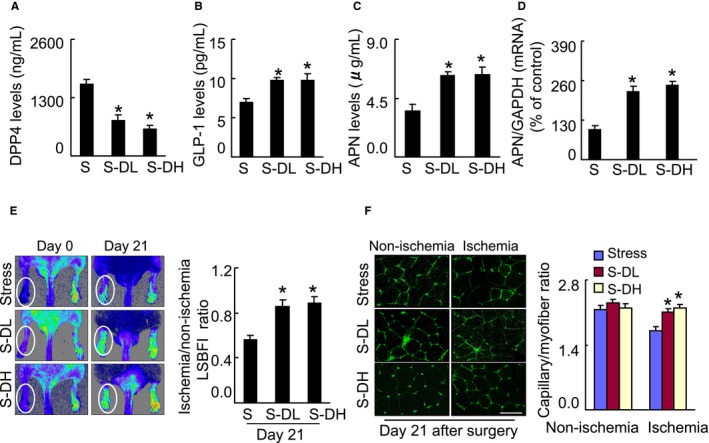
Inhibition of DPP4 reverses GLP‐1 and adiponectin levels in plasma and/or adipose of stressed mice. A through D, Mice were given by oral gavage vehicle (distilled water, Stress), a low dose (30 mg/kg per day, S‐DL) or a high dose of the DPP4 inhibitor anagliptin (60 mg/kg per d, S‐DH) every day from 3 d before undergoing the surgery. At d 4 after surgery, harvested blood and adipose tissues (subcutaneous fat) were analyzed by ELISA or quantitative real‐time PCR, respectively, for the levels of DPP4 activity (A), GLP‐1 protein (B) and adiponectin protein (C), or adiponectin gene (D). Data are mean±SE (n=5–9). *P<0.01 by ANOVA and Tukey's post hoc tests. E, Blood flow was measured by representative laser speckle perfusion imaging, and capillary density was measured by fluorescent staining with tomato lectin. Representative images and quantitative data for blood flow recovery (expressed as the ischemia‐to‐nonischemia LSBFI ratio) (E) and capillary density (expressed as capillary‐to‐myofiber ratio) (F) are shown. Data are mean±SE (n=5–7). *P<0.01 by 1‐way ANOVA and Tukey's post hoc tests. Scale bar: 50 μm. APN indicates adiponectin; DPP4, dipeptidyl peptidase‐4; GAPDH, glyceraldehyde‐3‐phosphate‐dehydrogenase; GLP‐1, glucagon‐like peptide‐1; LSBFI, laser speckle blood flow imaging; PCR, polymerase chain reaction.
Table 3.
Levels of Lipids and Other Parameters in the 3 Experimental Groups at 7 Weeks
| Parameter | Stress | S‐DL | S‐DH |
|---|---|---|---|
| Body weight, g | 21.5±0.3 | 20.5±0.4 | 21.8±0.3 |
| Triglyceride, mg/dL | 17.1 ±1.9 | 16.0±2.4 | 19.2±1.7 |
| TCh, mg/dL | 48.2±4.1 | 47.7±3.9 | 43.9±4.5 |
| HDL‐C, mg/dL | 30.3±1.9 | 32.3±1.3 | 32.1±1.6 |
| NEFA, μEQ/L | 1204±61 | 777±61a | 758±39a |
| Blood urea nitrogen, mg/dL | 29.7± 1.9 | 31.3 ±1.6 | 30.4±1.3 |
| Creatinine, mg/dL | 0.2±0.0 | 0.2±0.0 | 0.2±0.0 |
| Glucose, mg/mL | 101.2±5.6 | 85.5±5.2a | 82.8±4.9a |
| Corticosterone, ng/mL | 80.3±6.1 | 105.6±12.2a | 113.4±10.9a |
| Real‐time PCR (adipose) | |||
| IRS‐1 | 10.4±1.8 | 45.8 ±5.3a | 42.4±3.3a |
| GLUT4 | 16.2±2.7 | 34.5±6.8a | 39.8±4.1a |
Values are mean±SE (n=6–10). GLUT4 indicates glucose transporter 4; HDL‐C, high‐density lipoprotein cholesterol; IRS‐1, insulin receptor substrate 1; NEFA, nonesterified fatty acid; PCR, polymerase chain reaction; S‐DH, stressed mice received high dose of DPP4 inhibitor; S‐DL, stressed mice received low dose of dipeptidyl peptidase 4 inhibitor; TCh, total cholesterol.
P<0.05 vs stress alone mice by analysis of variance and Tukey's post hoc test.
As shown in Figure 6E, marked blood recovery was observed in the S‐DL and S‐DH group mice throughout the follow‐up period. Capillaries were well developed in both treatment groups at day 21 after ischemic induction (Figure 6F). Consistently, DPP4 deletion also mitigated blood recovery and capillary formation in stressed rats (Figure 7A through 7D). Compared with the controls, the rates of amputation were dramatically reduced in the S‐DL and S‐DH groups (Figure 8A). Abundant protein levels of p‐AMPKα, VEGF, PPAR‐γ, and PGC‐1α were observed in the ischemic muscles of S‐DL and S‐DH mice on day 4 after surgery (Figure 8B); these effects were mimicked by DPP4 deletion in the stressed rats (Figure 8C). Thus, the protection of GLP‐1 against degradation by DPP4 inhibition could produce an improvement in the impaired adiponectin/AdipoR1 signaling and PPAR‐γ/PGC‐1α expression and the reduced neovascularization in response to ischemia in mice under chronic stress.
Figure 7.

DPP4 deficiency improved the blood flow recovery and capillary formation in ischemic muscles of stressed rats. A, Representative laser Doppler perfusion images show a low perfusion signal (dark blue) in the ischemic hindlimbs of DPP4+/+ rats and a high signal (red) in DPP4−/− rats. B, The ratio of ischemic to nonischemic LSBFI was higher in the DPP4−/− rats than in the DPP4+/+ rats during the follow‐up period. The data are mean±SE (n=6). *P<0.05 by 2‐way repeated‐measures ANOVA and Bonferroni post hoc tests. C, Immunohistostaining was performed using anti‐CD31 to visualize capillaries in ischemic and nonischemic thigh adductor muscle. D, Quantitative data showing the enhancement of capillary density as expressed by the capillary‐to‐myofiber ratio in ischemic muscle of DPP4−/− rats. Data are mean±SE (n=6). *P<0.01 vs corresponding DPP4+/+ rats, NS, not significant by ANOVA and Tukey's post hoc test. Scale bar: 50 μm. DPP4 indicates dipeptidyl peptidase‐4; LSBFI, laser speckle blood flow imaging.
Figure 8.
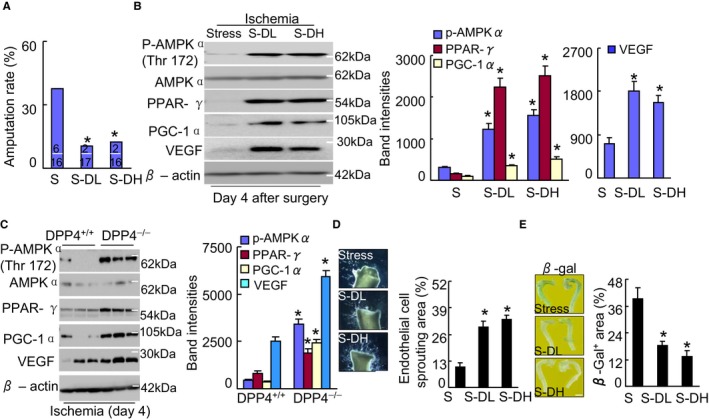
DPP4 inhibition ameliorated amputation and targeted protein levels in stressed mice. A, Quantification of foot amputation (including loss of toenails, foot, and/or below knee) in 3 groups (upper and lower numbers indicate the number of amputations and total animals). *P<0.01 by χ2 test. B and C, Representative immunoblots and quantitative data for the levels of p‐AMPKα, PPAR‐γ, PGC1‐α and VEGF of ischemic muscles of stressed mice (B) and rats (C). D and E, Pharmacological inhibition of DPP4 improved aortic endothelial sprouting and senescence. Quantitative data show the endothelial cell sprouting areas and β‐Gal+ areas (×200). The endothelial sprouting areas and β‐Gal+ areas are expressed as the percentage of pixels per image occupied by vessels in the quantitative area (n=4–5). Data are mean±SE. *P<0.01 by ANOVA and Tukey's post hoc tests. β‐Gal indicates β‐galactosidase; DPP4, dipeptidyl peptidase‐4; p‐AMPKα, phospho‐AMP‐activated protein kinase α; PGC‐1α, PPAR‐γ co‐activator 1α; PPAR‐γ, peroxisome proliferator‐activated receptor‐γ; VEGF, vascular endothelial growth factor.
To determine the consequences of DPP4 inhibition, the aortas of both experimental groups were applied to vascular senescence and tubulogenesis assays for the evaluation of angiogenesis. The results indicated that the genetic and pharmacological inhibition of DPP4 mitigated not only stress‐related aorta‐derived EC sprouting but also vascular senescence (Figures 8D, 8E, 9A, and 9B). Abundant protein levels of PPAR‐γ, PGC‐1α, and Sirt1 proteins were observed in the aortas of the S‐DL and S‐DH mice (Figure 9C), and these effects were mimicked by DPP4 deletion (Figure 9D), indicating that DPP4 could modulate stress‐related vascular endothelial senescence and angiogenesis failure, which are accompanied by PPAR‐γ/PGC‐1α inactivation. As shown in Figure 10A, we observed that the numbers of CD31+/c‐Kit+ cells in the BM and peripheral blood were higher in both the S‐DL and S‐DH mice compared with the control mice (Figure 10A). DPP4 inhibition also ameliorated the BM c‐Kit+ cell proliferation, apoptosis, and tubulogenic action (Figure 10B through 10D). Taken together, these results indicate that DPP4 may modulate angiogenesis and vasculogenesis in response to ischemia in mice under stress.
Figure 9.
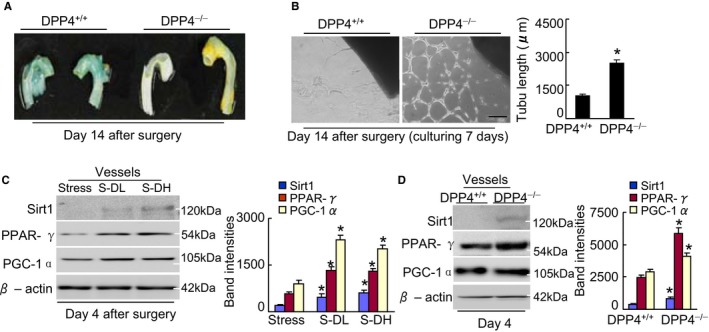
DPP4 deletion ameliorates aortic senescence and endothelial sprouting. A, On postoperative d 14, the aortas were subjected to β‐gal staining. Representative β‐gal staining images show that DPP4 deficiency ameliorated aortic senescence. B, On operative d 14, the aortas from rats of both genotypes were cultured in matrix gel for 7 d. Representative microscopy images and quantitative data for aortic endothelial sprouting length (n=5, Student t test). C and D, Representative immunoblots and quantitative data show the levels of targeted proteins in the aortas of mice (C) and rats (D). Data are mean±SE (n=3). *P<0.01 by ANOVA and Tukey's post hoc tests. Scale bar, 50 μm. β‐Gal indicates β‐galactosidase; DPP4, dipeptidyl peptidase‐4; PGC‐1α, PPAR‐γ co‐activator 1α; PPAR‐γ, peroxisome proliferator‐activated receptor‐γ.
Figure 10.
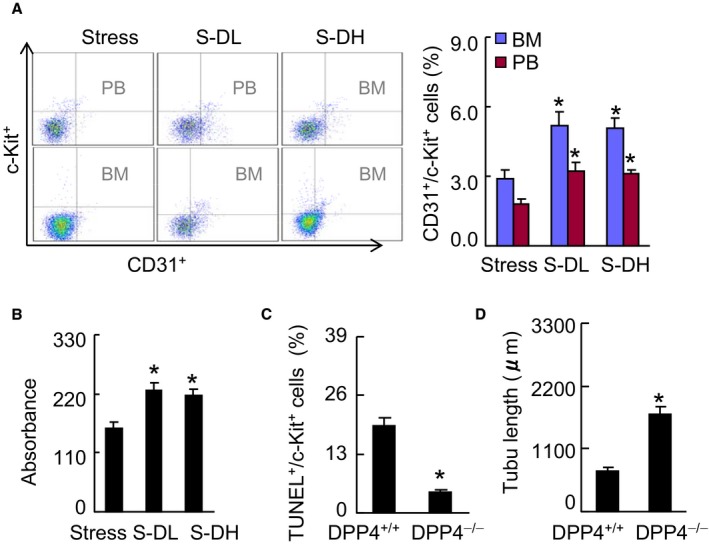
DPP4 inhibition ameliorates EPC production and mobilization in stressed mice. A, Representative dot plots and quantitative data for the numbers of CD31+/c‐Kit+ (per 2×104 cells) in BM and PB (n=5–6, Mann–Whitney U test). B through D, BM‐derived c‐Kit+ cells were subjected to proliferation (B), apoptosis (C), and tubule formation (D) assays (Student t test). Data are mean±SE (n=5–7). *P<0.01 by ANOVA and Tukey's post hoc tests or Student t test. BM indicates bone marrow; DPP4, dipeptidyl peptidase‐4; EPC, endothelial progenitor cell; PB, peripheral blood; TUNEL, TdT‐mediated dUTP nick‐end labeling.
Quantitative gelatin zymography analysis showed that DPP4 inhibition suppressed the MMP‐2 and MMP‐9 activities in the ischemic muscles of the stressed mice (Figure 11A). Interestingly, DPP4 deficiency mitigated the MMP‐related proteolysis and microphage infiltration (Figure 11B and 11C). We also observed that a GLP‐1 agonist improved inflammatory action in the stressed mice (Figure 12). These findings thus suggest that a DPP4 inhibition‐ and GLP‐1R activation‐mediated anti‐inflammatory effect may contribute to neovascularization in mice under chronic stress. In addition, with the exception of nonesterified fatty acid and glucose, we observed that there were no significant differences in targeted metabolic parameters between the DPP4+/+ and DPP4−/− experimental groups (Table 4).
Figure 11.
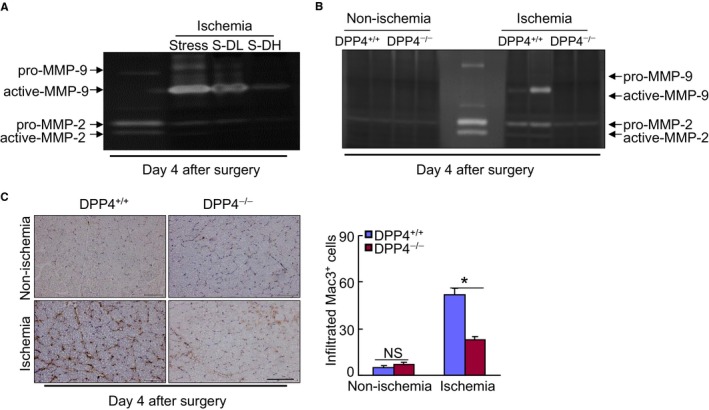
DPP4 inhibition decreased inflammation and MMP activity. A and B, Representative gelatin zymographic images showing the levels of MMP‐2 and MMP‐9 gelatinolytic activities in the nonischemic and ischemic muscles of stressed mice (A) and rats (B) at the noted days after ischemic surgery. C, Representative microscopy images and quantitative data for the macrophage infiltration. Data are mean±SE (n=6‐7). *P<0.01 vs DPP4+/+ by ANOVA and Tukey's post hoc test. Scale bar, 50 μm. DPP4 indicates dipeptidyl peptidase‐4; MMP‐2, matrix metalloproteinase‐2; NS, not significant.
Figure 12.
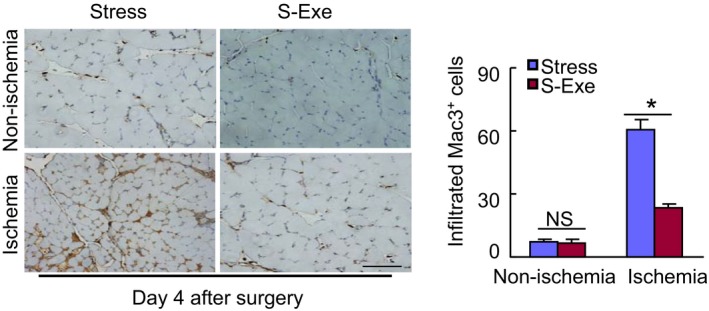
GLP‐1R activation suppressed inflammatory action in the ischemic muscles of stressed mice. Representative images and quantitative analysis of infiltrated macrophages in muscles of both experimental groups on postoperative d 4. Data are mean±SE (n=5). *P<0.01 by ANOVA and Tukey's post hoc test. Scale bar, 50 μm. GLP‐1R indicates glucagon‐like peptide‐1 receptor; NS, not significant.
Table 4.
Levels of Lipids and Other Parameters in 2 Experimental Groups at 4 Weeks
| Parameter | DPP4+/+ | DPP4−/− |
|---|---|---|
| Body weight, g | 203.8±6.9 | 228.3±3.7 |
| Triglyceride, mg/dL | 37.7±2.4 | 30.6±2.3 |
| TCh, mg/dL | 4.5±0.3 | 4.3±0.2 |
| HDL‐C, mg/dL | 20.0±1.0 | 20.7±0.8 |
| NEFA, μEQ/L | 663.1±57.2 | 459.4±37.2a |
| Blood urea nitrogen, mg/dL | 18.3±1.8 | 21.0±0.7 |
| Creatinine, mg/dL | 0.2±0.0 | 0.2±0.0 |
| Glucose, mg/dL | 103.5±6.0 | 85.8±5.9a |
Values are mean±SE (n=6–8). DPP4 indicates dipeptidyl peptidase‐4; DPP4−/−, DPP4 knockout; DPP4+/+, wild‐type; HDL‐C, high‐density lipoprotein cholesterol; NEFA, nonesterified fatty acid.; TCh, total cholesterol.
P<0.05 vs DPP4+/+ by Student t tests.
A GLP‐1 Analogue Ameliorated Neovascularization in Response to Stress
We examined whether GLP‐1 supplementation could ameliorate ischemia‐induced neovascularization under chronic stress. As shown in Figure 13A and 13B, marked improvements of blood recovery and capillary density were observed in the S‐Exe group mice compared with the stress alone mice. Likewise, the GLP‐1R agonist exenatide also attenuated the amputation events of ischemic hindlimbs in mice (Figure 13C). We observed abundant levels of plasma adiponectin protein and adipose adiponectin mRNA as well as targeted p‐AMPKα, PPAR‐γ, PGC‐1α, and VEGF proteins in S‐Exe mice compared with the control mice (Figures 13D, 13E, and 14A). Moreover, exenatide treatment enhanced the levels of plasma corticosterone (143.4±18.9 versus 89.1±5.6 ng/mL) and adipose insulin receptor substrate 1 (56.3±5.2 versus 12.5±2.1) and glucose transporter 4 (29.9±3.1 versus 10.1±1.4; P<0.01 for each) mRNAs in the stressed mice. Thus, these findings indicating that GLP‐1 supplementation can produce a beneficial effect on impaired angiogenic action in response to ischemia in mice that received stress.
Figure 13.
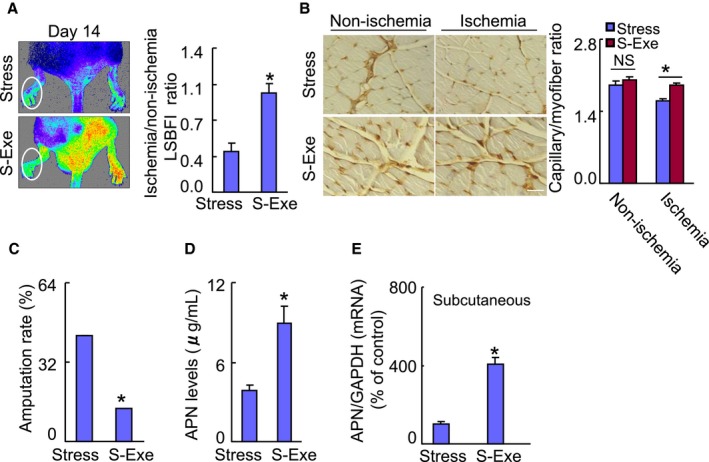
A GLP‐1 agonist ameliorated the blood flow recovery and capillary formation in stressed mice. A, Representative laser speckle perfusion imaging showing a low perfusion signal (dark blue) in the ischemic hindlimbs of stressed mice and a high signal (red) in S‐Exe mice. The ratio of ischemic‐to‐nonischemic laser speckle blood flow was higher in the S‐Exe mice than in the nonstressed control mice during the follow‐up periods. B, Immunostaining was performed using CD31 antibody to visualize capillaries in ischemic and nonischemic thigh adductor muscles. Quantitative data show an enhancement of capillary density as expressed by the capillary‐to‐myofiber ratio in ischemic muscle of the S‐Exe mice. C, Quantification of foot amputation (including loss of toenails, foot, and/or below knee) in 2 groups. D and E, At d 4 after surgery, harvested blood and tissues were analyzed by ELISA and quantitative real‐time PCR for plasma APN protein and its gene of the subcutaneous fat. Data are mean±SE (n=5–6). *P<0.01, NS, not significant by Student unpaired t test or ANOVA and Tukey's post hoc tests. Scale bar: 50 μm. APN indicates adiponectin; GAPDH, glyceraldehyde‐3‐phosphate‐dehydrogenase; GLP‐1, glucagon‐like peptide‐1; LSBFI, laser speckle blood flow imaging; PCR, polymerase chain reaction; S‐Exe, stressed group mice treated with exenatide.
Figure 14.
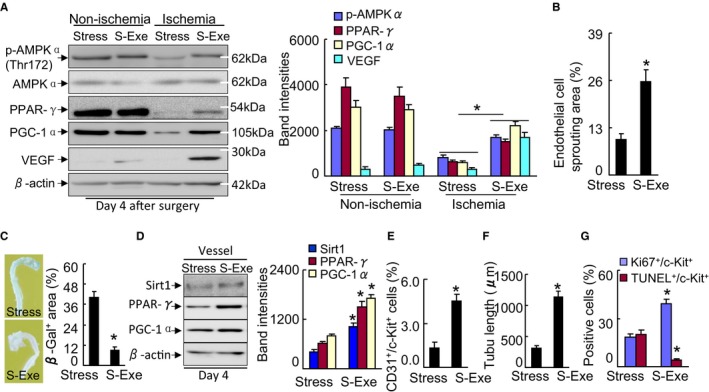
GLP‐1R activation ameliorated targeted protein levels in stressed mice. A, Representative immunoblots and quantitative data show the levels of p‐AMPKα, PPAR‐γ, PGC‐1α, and VEGF of the muscles of stressed mice. B, Quantification of aortic endothelial sprouting area in both experimental groups. C, Representative images and quantitative data show that exenatide improved aortic endothelial senescence (n=4). D, Representative immunoblots and quantitative data show the levels of PPAR‐γ and PGC‐1α of the vessels of stressed mice (n=3). E, Quantification of the numbers of peripheral blood CD31+/c‐Kit+ EPC numbers (per 2×104 cells). F, BM‐derived c‐Kit+ cells (5×103 cells) were cultured on growth factor–reduced Matrigel for 24 h and then the tubule lengths were calculated. G, BM‐derived c‐Kit+ cells (5×103 cells) were seeded on a 4‐chamber culture slide and cultured for 72 h. The cells were subjected to proliferation and apoptosis induced by H2O2 (250 μmol/L) assays using Ki67 antibody or a TUNEL staining kit, respectively (n=5–6). Data are mean±SE. *P<0.01 by Student unpaired t test or ANOVA and Tukey's post hoc tests. BM, indicates bone marrow; EPC, endothelial progenitor cell; GLP‐1R, glucagon‐like peptide‐1 receptor; p‐AMPKα, phospho‐AMP‐activated protein kinase α; PGC‐1α, PPAR‐γ co‐activator 1α; PPAR‐γ, peroxisome proliferator‐activated receptor‐γ; TUNEL, TdT‐mediated dUTP nick‐end labeling; VEGF, vascular endothelial growth factor.
In addition, the aorta‐ring culture and β‐Gal staining assays showed that exenatide prevented aortic endothelial cell sprouting and senescence (Figure 14B and 14C). As anticipated, representative immunoblots showed that the levels of PPAR‐γ, PGC‐1α, and Sirt1 proteins were higher in the aortas of the S‐Exe mice compared with those of the control mice (Figure 14D). Moreover, exenatide mitigated the CD31+/c‐Kit+ EPC‐like cell mobilization in the stressed mice (Figure 14E). GLP‐1R activation also improved the BM‐derived c‐Kit+ cell tubule formation, proliferation, and apoptosis (Figure 14F and 14G). Collectively, GLP‐1R activation appears to have promoted angiogenic and vasculogenic actions via the activation of adiponectin/AdopR1 signaling and PPAR‐γ/PGC‐1α expression in the stressed mice.
Adiponectin Deficiency Diminished DPP4 Inhibition‐Mediated Vasculoprotective Actions in Mice Under Chronic Stress
As shown in Figure 15A and 15B, adiponectin depletion with its neutralizing antibody resulted in decreased blood flow recovery and capillary formation in stressed mice treated with anagliptin. The fluorescence activated cell sorter analysis revealed that the circulating levels of CD31+/c‐Kit+ were lower in the S‐Ana mice treated with N‐adiponectin than in the S‐Ana mice treated rabbit control IgG (Figure 15C). Likewise, adiponectin deficiency also abrogated DPP4 inhibition‐related vasculoprotection (Figure 15D). Together with the Western blotting analysis showing that DPP4 inhibition had no effect on PPAR‐γ and PGC‐1α protein expressions (Figure 15E), the present findings suggest that an adiponectin/AdopR1 signal may regulate ischemia‐induced neovascularization through PPAR‐γ/PGC‐1α activation under our experimental conditions. In addition, anagliptin exhibited no effect on the numbers of blood and BM CD31+/c‐Kit+ cells in adiponectin−/− mice (Figure 16).
Figure 15.

Adiponectin depletion abrogated DPP4 inhibition‐related vascular benefits in stressed mice. A and B, During the stress protocol, anagliptin (30 mg/kg per d)‐loaded stress mice were injected subcutaneously with control IgG (N‐APN[−]) or neutralizing antibody against APN (N‐APN [+]) every 5 d. Representative blood perfusion images and/or quantitative data show the blood flow (A) and capillary density (B) (n=5). C, Quantification of circulating CD31+/c‐Kit+ in both experimental groups (n=5). D and E, APN−/− mice pretreated with vehicle or anagliptin (30 mg/kg per d) for 3 d underwent ischemic surgery and were subjected to the LSBFI and immunoblotting analyses. Representative images and quantitative data show blood flow recovery and the levels of PPAR‐γ and PGC‐1α proteins in muscles of APN−/− mice (n=3–5). Data are mean±SE. *P<0.01 vs corresponding controls; NS, not significant by Student unpaired t test or ANOVA and Tukey's post hoc tests. APN indicates adiponectin; DPP4, dipeptidyl peptidase‐4; LSBFI, laser speckle blood flow imaging; PGC‐1α, PPAR‐γ co‐activator 1α; PPAR‐γ, peroxisome proliferator‐activated receptor‐γ.
Figure 16.
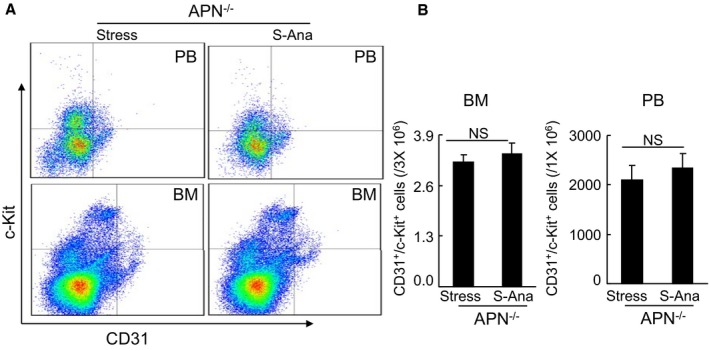
Anagliptin had no effect on CD31+/c‐Kit+ cells in BM and PB in adiponectin−/− mice at d 14 after surgery. A and B, Representative dot plots and quantitative data for the numbers of CD31+/c‐Kit+ in BM and PB (n=6, Mann–Whitney U test). Data are mean±SE. APN indicates adiponectin; BM, bone marrow; NS, no significant; PB, peripheral blood; S‐Ana, stressed mice treated with anagliptin.
Discussion
This study revealed the novel finding that chronic stress accelerates DPP4‐mediated GLP‐1 degradation and alters plasma adiponectin. The decrease in the adiponectin/AdipoR1 signal lowered the expression of regulatory angiogenic genes PPAR‐γ and PGC‐1α in the vessels and the ischemic muscles, thereby accelerating insulin resistance and vascular senescence and ischemia‐induced angiogenic action failure in stressed mice. These changes also resulted in BM‐derived EPC mobilization failure and dysfunction, reducing the vascular regeneration capacity. DPP4 inhibition and GLP‐1R activation ameliorated ischemia‐induced neovascularization under our experimental conditions, and this vascular benefit was diminished by the genetic or pharmacological depletions of adiponectin.
Our view of the cross‐talk between GLP‐1/GLP‐1R and adiponectin‐AdipoR1 axes as a potential regulator of several intracellular signals has key clinical implications. The ability of chronic stress to exert deleterious changes of both axes is likely to have contributed to the decline in the vascular senescence and regeneration capacity. Our animal studies demonstrated that the administration of the DPP4 inhibitor anagliptin will restore impaired ischemia‐induced blood flow recovery and capillary formation in stressed mice. Our observations show that stress resulted in decreased levels of plasma GLP‐1 and adiponectin proteins and adipose adiponectin gene, accompanied by increased plasma DPP4 activity; these changes were reversed by the pharmacological and genetic disruption of DPP4 gene. Exenatide also exhibited a positive effect on plasma and tissue adiponectin levels in the stressed mice. Thus, the reduction of adiponectin by an acceleration of DPP4‐mediated GLP‐1 degradation appears to contribute to the decline in ischemia‐induced neovascularization in mice subjected to chronic stress. In the data presented here, genetic or pharmacological intervention toward DPP4 and GLP‐1R restored the vascular regenerative capacity. Western blotting assays demonstrated that the levels of p‐AMPKα, PPAR‐γ, PGC‐1α, and VEGF proteins were ameliorated in ischemic muscles of stressed mice that underwent both interventions. Moreover, the genetic and pharmacological depletion of adiponectin diminished the DPP4 inhibition‐related vascular protection. Adiponectin protected ischemic stress‐related cardiovascular injury.22, 24 Nuclear transcription factor PPAR‐γ/PGC‐1α signaling has been implicated in metabolic cardiovascular disease initiation and progression.20 Based on the currently available information, a comprehensive review article noted that PPAR‐γ activation can represent a common mechanism in the protection of cardiovascular tissues against various types of stress.20 Taken together, these findings indicate that increased DPP4 activity may have negatively modulated vascular regeneration actions via the GLP‐1‐mediated adiponectin/AdipoR1 and PPARγ/PGC‐1α interaction in the mice subjected to our experimental conditions.
It is well established that there is a close link between vascular senescence and dysfunction and cardiovascular disorders. We have previously shown that old human umbilical vein endothelial cells exhibit lower proliferative and tubulogenic abilities compared with young cells.8 The aortic rings from aged mice had impaired EC sprouting.9 In the present study, chronic stress accelerated vascular senescence in the stressed mice compared with the nonstressed controls. Likewise, the aorta‐ring culture assay showed a reduction in the microvessel sprouting of stressed aortas, suggesting that impaired angiogenesis was associated with the acceleration of vascular endothelial senescence under stress. Here we have demonstrated that anagliptin and exenatide improved the tubulogenic capacity of stressed aortas. Both therapies also resulted in increased levels of not only plasma adiponectin and but also the levels of aortic PPAR‐γ, PGC‐1α, and Sirt1 protein in the stressed mice. It was reported that adiponectin improved endothelial dysfunction under pathophysiological conditions.23, 24 A recent study showed that a variety of endothelial cellular biological events including cell migration and proliferation were modified by the activation of several PPAR‐γ‐dependent signaling pathways.32 The upregulation of adiponectin by DPP4 inhibition and GLP‐1R activation could therefore represent a common mechanism in the protection of vascular tissues against chronic stress.
It has become clear that the function and numbers of EPCs are modified by pathological conditions such as aging and metabolic disorder.9, 34 In the present study, the BM‐derived EPC angiogenic actions were lower and apoptosis was higher in the stressed mice than in the nonstressed mice; these changes were ameliorated by DPP4 inhibition and GLP‐1R activation. It was reported that DPP4 depletion increased the homing of circulating C‐X‐C chemokine receptor type 4+ (CXCR4+) stem cells into ischemic myocardial vasculature.15 Fadini and colleagues documented that diabetes mellitus differentially influenced DPP4 activity in peripheral blood and impaired stem/progenitor cell mobilization after ischemia and granulocyte–colony‐stimulating factor administration.35 Our present findings show that the numbers of blood CD31+/c‐Kit+ cells were lower in the stressed mice than in the control mice; this alteration was restored by both a DPP4 inhibitor and a GLP‐1R agonist. Conversely, we observed that adiponectin blocking reduced BM EPC mobilization in S‐Ana mice. We recently demonstrated that adiponectin/AdipoR1 activation produces BM‐derived stem cell mobilization and regenerative capacity in a senescence‐accelerated mouse model.36 Adiponectin protects EPCs against apoptosis and therefore could modulate the ability of EPCs to induce the repair of vascular damage.37 Another study demonstrated that adiponectin depletion resulted in decreased BM‐derived EPC cell survival and differentiation ability in mice.38 Thus, the ability of DPP4 inhibition and GLP‐1R activation to improve EPC function and recruitment exerts salutary effects on ischemic vasculature by enhancing adiponectin production, thereby promoting revascularization under stress conditions.
Inflammatory over‐reaction has been shown to reduce the neovascularization of ischemic limbs in aged mice.8, 39 The exercise‐mediated prevention of inflammatory responses to ischemia has been shown to ameliorate revascularization in aged animals.8 It was reported that inflammatory responses (ie, inflammatory cytokine expression and related cell infiltration) are accelerated by stress.31 Power and colleagues reported that social stress upregulated inflammatory gene expression in the leukocyte transcriptome.40 Here we observed that stressed ischemic muscles had dramatically increased numbers of infiltrated macrophages and decreased blood flow recovery and capillary density compared with nonstressed ischemic muscles, suggesting that stress accelerates inflammatory over‐reaction to the contribution to impaired vascular regeneration in response to ischemia. This concept was further supported by our observation that the levels of infiltrated macrophage‐derived MMP‐9 and MMP‐2 gelatinolytic activities were higher in ischemic muscles of the stressed mice compared with those of the control mice. Macrophage‐specific PPAR‐γ signaling controls macrophage activation in insulin resistance status.41 It was reported that a PPAR‐γ agonist protected kidney from injury via the attenuation of inflammatory actions.42 Accumulating evidence indicates that adiponectin facilitates macrophage polarization toward an anti‐inflammatory phenotype.43, 44, 45 On the other hand, GLP‐1/GLP‐1R signal also induces M2 polarization of human macrophages.46 In the data presented here, stress had decreased in plasma GLP‐1 and adiponectin as well as tissue PPAR‐γ/PGC‐1α, which was accompanied by increased plasma DPP4 activities. We observed that DPP4 deficiency and anagliptin ameliorated these stress‐related changes and inflammatory action. Moreover, exenatide exerts an anti‐inflammatory effect in stressed mice. Thus, these findings suggest that the DPP4 inhibition‐ and GLP‐1R activation‐mediated improvement of vascular regeneration capacity may be partially attributable to the reduction of inflammation through adiponectin/AdipoR1‐dependent PPAR‐γ/PGC‐1α signaling activation under our experimental conditions.
It is well established that psychological stress can accelerate cardiovascular disorder initiation and progression via either depression or activation of the hypothalamic–pituitary–adrenal feedback mechanisms.6 Previous studies reported that GLP‐1R agonists have a sustained stimulatory effect on neurohormone (ie, corticosterone) secretion in animals.47, 48 We have shown that chronic stress markedly suppressed the plasma corticosterone level; this effect was restored by the treatment with anagliptin and exenatide. Recently, we have demonstrated that chronic stress also increased plasma adrenaline and noradrenaline levels, and these changes were reversed by both drugs.14, 19 In addition, our observations here show that pharmacological interventions targeted toward DPP4 activity and GLP‐1R activation ameliorated the levels of plasma glucose and adipose insulin receptor substrate 1 and glucose transporter 4 genes in the stressed mice. Adiponectin has been shown to improve glucose metabolism and insulin resistance in metabolic disorder.26, 27 Thus, both pharmacological therapies may rectify the alterations in the neurohormone levels, resulting in adiponectin‐mediated beneficial effects on glucose metabolism and insulin resistance, contributing to the mitigation of vasculoprotection in mice under our experimental conditions.
Conclusions
Taken together, the previous and present findings show a critical role of the interaction between DPP4‐related GLP‐1/GLP‐1R and adiponectin/AdiopR1 signaling pathways in ischemic vascular regeneration under chronic stress conditions. This novel biological function of the cross‐talk may be exploited for the therapeutic management of chronic psychosocial stress‐related vascular aging and cardiovascular disease.
Sources of Funding
This work was supported in part by the National Natural Science Foundation of China (nos. 81560240, 81560149, 81460082); by the Ministry of Education, Culture, Sports, Science and Technology of Japan Society for the Promotion of Science (JAPAN) (nos. 15H04801, 15H04802); and by the Novartis Aging and The Japan Geriatrics Society (JAPAN) (no. 27‐007756).
Disclosures
None.
Acknowledgments
The authors gratefully acknowledge K. Suzuki (The Department of Community Healthcare and Geriatrics) for her contributions (mice handling [ie, mice case cleaning] and experimental buffer preparations [eg, lysis buffer, gelatin zymography reaction buffer, etc]) and M. Tanaka and I. Mizukuchi (The Division for Medical Research Engineering of Nagoya University Graduate School of Medicine) for their excellent technical supports (instruction on how to use flow cytometry and confocal microscopy).
(J Am Heart Assoc. 2017;6:e006421 DOI: 10.1161/JAHA.117.006421.)
References
- 1. Rosengren A, Hawken S, Ounpuu S, Sliwa K, Zubaid M, Almahmeed WA, Blackett KN, Sitthi‐amorn C, Sato H, Yusuf S. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case‐control study. Lancet. 2004;364:953–962. [DOI] [PubMed] [Google Scholar]
- 2. Roepke SK, Allison M, Von Kanel R, Mausbach BT, Chattillion EA, Harmell AL, Patterson TL, Dimsdale JE, Mills PJ, Ziegler MG, Ancoli‐Israel S, Grant I. Relationship between chronic stress and carotid intima‐media thickness (IMT) in elderly Alzheimer's disease caregivers. Stress. 2012;15:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chandola T, Brunner E, Marmot M. Chronic stress at work and the metabolic syndrome: prospective study. BMJ. 2006;332:521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, Kobor MS, Reader BF, Sheridan JF, Cole SW. Social stress up‐regulates inflammatory gene expression in the leukocyte transcriptome via beta‐adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA. 2013;110:16574–16579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steptoe A, Kivimaki M. Stress and cardiovascular disease. Nat Rev Cardiol. 2012;9:360–370. [DOI] [PubMed] [Google Scholar]
- 6. Gu HF, Tang CK, Yang YZ. Psychological stress, immune response, and atherosclerosis. Atherosclerosis. 2012;223:69–77. [DOI] [PubMed] [Google Scholar]
- 7. Arnold SV, Smolderen KG, Buchanan DM, Li Y, Spertus JA. Perceived stress in myocardial infarction: long‐term mortality and health status outcomes. J Am Coll Cardiol. 2012;60:1756–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng XW, Kuzuya M, Kim W, Song H, Hu L, Inoue A, Nakamura K, Di Q, Sasaki T, Tsuzuki M, Shi GP, Okumura K, Murohara T. Exercise training stimulates ischemia‐induced neovascularization via phosphatidylinositol 3‐kinase/Akt‐dependent hypoxia‐induced factor‐1 alpha reactivation in mice of advanced age. Circulation. 2010;122:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng XW, Kuzuya M, Nakamura K, Maeda K, Tsuzuki M, Kim W, Sasaki T, Liu Z, Inoue N, Kondo T, Jin H, Numaguchi Y, Okumura K, Yokota M, Iguchi A, Murohara T. Mechanisms underlying the impairment of ischemia‐induced neovascularization in matrix metalloproteinase 2‐deficient mice. Circ Res. 2007;100:904–913. [DOI] [PubMed] [Google Scholar]
- 10. Dong Y, Sun Q, Liu T, Wang H, Jiao K, Xu J, Liu X, Liu H, Wang W. Nitrative stress participates in endothelial progenitor cell injury in hyperhomocysteinemia. PLoS One. 2016;11:e0158672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lei Y, Hu L, Yang G, Piao L, Jin M, Cheng X. Dipeptidyl peptidase‐IV inhibition for the treatment of cardiovascular disease—recent insights focusing on angiogenesis and neovascularization. Circ J. 2017;81:770–776. [DOI] [PubMed] [Google Scholar]
- 12. Shigeta T, Aoyama M, Bando YK, Monji A, Mitsui T, Takatsu M, Cheng XW, Okumura T, Hirashiki A, Nagata K, Murohara T. Dipeptidyl peptidase‐4 modulates left ventricular dysfunction in chronic heart failure via angiogenesis‐dependent and ‐independent actions. Circulation. 2012;126:1838–1851. [DOI] [PubMed] [Google Scholar]
- 13. Zhong J, Maiseyeu A, Davis SN, Rajagopalan S. DPP4 in cardiometabolic disease: recent insights from the laboratory and clinical trials of DPP4 inhibition. Circ Res. 2015;116:1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lei Y, Yang G, Hu L, Piao L, Inoue A, Jiang H, Sasaki T, Zhao G, Yisireyili M, Yu C, Xu W, Takeshita K, Okumura K, Kuzuya M, Cheng XW. Increased dipeptidyl peptidase‐4 accelerates diet‐related vascular aging and atherosclerosis in ApoE‐deficient mice under chronic stress. Int J Cardiol. 2017;243:413–420. [DOI] [PubMed] [Google Scholar]
- 15. Zaruba MM, Theiss HD, Vallaster M, Mehl U, Brunner S, David R, Fischer R, Krieg L, Hirsch E, Huber B, Nathan P, Israel L, Imhof A, Herbach N, Assmann G, Wanke R, Mueller‐Hoecker J, Steinbeck G, Franz WM. Synergy between CD26/DPP‐IV inhibition and G‐CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell. 2009;4:313–323. [DOI] [PubMed] [Google Scholar]
- 16. Huber BC, Brunner S, Segeth A, Nathan P, Fischer R, Zaruba MM, Vallaster M, Theiss HD, David R, Gerbitz A, Franz WM. Parathyroid hormone is a DPP‐IV inhibitor and increases SDF‐1‐driven homing of CXCR4(+) stem cells into the ischaemic heart. Cardiovasc Res. 2011;90:529–537. [DOI] [PubMed] [Google Scholar]
- 17. Teraa M, Sprengers RW, Westerweel PE, Gremmels H, Goumans MJ, Teerlink T, Moll FL, Verhaar MC. Bone marrow alterations and lower endothelial progenitor cell numbers in critical limb ischemia patients. PLoS One. 2013;8:e55592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang G, Li Y, Cui L, Jiang H, Li X, Jin C, Jin D, Zhao G, Jin J, Sun R, Piao L, Xu W, Fang C, Lei Y, Yuan K, Xuan C, Ding D, Cheng X. Increased plasma dipeptidyl peptidase‐4 activities in patients with coronary artery disease. PLoS One. 2016;11:e0163027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu E, Hu L, Wu H, Piao L, Zhao G, Inoue A, Kim W, Yu C, Xu W, Bando YK, Li X, Lei Y, Hao CN, Takeshita K, Kim WS, Okumura K, Murohara T, Kuzuya M, Cheng XW. Dipeptidyl peptidase‐4 regulates hematopoietic stem cell activation in response to chronic stress. J Am Heart Assoc. 2017;6:e006394 DOI: 10.1161/JAHA.117.006394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shibata R, Ouchi N, Murohara T. Adiponectin and cardiovascular disease. Circ J. 2009;73:608–614. [DOI] [PubMed] [Google Scholar]
- 22. Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115:1408–1416. [DOI] [PubMed] [Google Scholar]
- 23. Cao Y, Tao L, Yuan Y, Jiao X, Lau WB, Wang Y, Christopher T, Lopez B, Chan L, Goldstein B, Ma XL. Endothelial dysfunction in adiponectin deficiency and its mechanisms involved. J Mol Cell Cardiol. 2009;46:413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li R, Wang WQ, Zhang H, Yang X, Fan Q, Christopher TA, Lopez BL, Tao L, Goldstein BJ, Gao F, Ma XL. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. Am J Physiol Endocrinol Metab. 2007;293:E1703–E1708. [DOI] [PubMed] [Google Scholar]
- 25. Lee WS, Kim J. Peroxisome proliferator‐activated receptors and the heart: lessons from the past and future directions. PPAR Res. 2015;2015:271983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iwabu M, Yamauchi T, Okada‐Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. Adiponectin and AdipoR1 regulate PGC‐1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464:1313–1319. [DOI] [PubMed] [Google Scholar]
- 27. Okada‐Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura‐Someya T, Shirouzu M, Ogata H, Tokuyama K, Ueki K, Nagano T, Tanaka A, Yokoyama S, Kadowaki T. A small‐molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2015;503:493–499. [DOI] [PubMed] [Google Scholar]
- 28. Yamamoto K, Ohishi M, Ho C, Kurtz TW, Rakugi H. Telmisartan‐induced inhibition of vascular cell proliferation beyond angiotensin receptor blockade and peroxisome proliferator‐activated receptor‐gamma activation. Hypertension. 2009;54:1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kroller‐Schon S, Jansen T, Schuler A, Oelze M, Wenzel P, Hausding M, Kerahrodi JG, Beisele M, Lackner KJ, Daiber A, Munzel T, Schulz E. Peroxisome proliferator‐activated receptor gamma, coactivator 1alpha deletion induces angiotensin II‐associated vascular dysfunction by increasing mitochondrial oxidative stress and vascular inflammation. Arterioscler Thromb Vasc Biol. 2013;33:1928–1935. [DOI] [PubMed] [Google Scholar]
- 30. Kondo M, Shibata R, Miura R, Shimano M, Kondo K, Li P, Ohashi T, Kihara S, Maeda N, Walsh K, Ouchi N, Murohara T. Caloric restriction stimulates revascularization in response to ischemia via adiponectin‐mediated activation of endothelial nitric‐oxide synthase. J Biol Chem. 2009;284:1718–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uchida Y, Takeshita K, Yamamoto K, Kikuchi R, Nakayama T, Nomura M, Cheng XW, Egashira K, Matsushita T, Nakamura H, Murohara T. Stress augments insulin resistance and prothrombotic state: role of visceral adipose‐derived monocyte chemoattractant protein‐1. Diabetes. 2012;61:1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li X, Cheng XW, Hu L, Wu H, Guo P, Hao CN, Jiang H, Zhu E, Huang Z, Inoue A, Sasaki T, Du Q, Takeshita K, Okumura K, Murohara T, Kuzuya M. Cathepsin S activity controls ischemia‐induced neovascularization in mice. Int J Cardiol. 2015;183:198–208. [DOI] [PubMed] [Google Scholar]
- 33. Jiang H, Cheng XW, Shi GP, Hu L, Inoue A, Yamamura Y, Wu H, Takeshita K, Li X, Huang Z, Song H, Asai M, Hao CN, Unno K, Koike T, Oshida Y, Okumura K, Murohara T, Kuzuya M. Cathepsin K‐mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat Commun. 2014;5:3838. [DOI] [PubMed] [Google Scholar]
- 34. Di Q, Cheng Z, Kim W, Liu Z, Song H, Li X, Nan Y, Wang C, Cheng X. Impaired cross‐activation of beta3 integrin and VEGFR‐2 on endothelial progenitor cells with aging decreases angiogenesis in response to hypoxia. Int J Cardiol. 2013;168:2167–2176. [DOI] [PubMed] [Google Scholar]
- 35. Fadini GP, Albiero M, Seeger F, Poncina N, Menegazzo L, Angelini A, Castellani C, Thiene G, Agostini C, Cappellari R, Boscaro E, Zeiher A, Dimmeler S, Avogaro A. Stem cell compartmentalization in diabetes and high cardiovascular risk reveals the role of DPP‐4 in diabetic stem cell mobilopathy. Basic Res Cardiol. 2013;108:313. [DOI] [PubMed] [Google Scholar]
- 36. Inoue A, Cheng XW, Huang Z, Hu L, Kikuchi R, Jiang H, Piao L, Sasaki T, Itakura K, Wu H, Zhao G, Lei Y, Yang G, Zhu E, Li X, Sato K, Koike T, Kuzuya M. Exercise restores muscle stem cell mobilization, regenerative capacity and muscle metabolic alterations via adiponectin/AdipoR1 activation in SAMP10 mice. J Cachexia Sarcopenia Muscle. 2017;8:370–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lavoie V, Kernaleguen AE, Charron G, Farhat N, Cossette M, Mamarbachi AM, Allen BG, Rheaume E, Tardif JC. Functional effects of adiponectin on endothelial progenitor cells. Obesity. 2010;19:722–728. [DOI] [PubMed] [Google Scholar]
- 38. Eren P, Camus S, Matrone G, Ebrahimian TG, Francois D, Tedgui A, Sebastien Silvestre J, Blanc‐Brude OP. Adiponectinemia controls pro‐angiogenic cell therapy. Stem Cells. 2009;27:2712–2721. [DOI] [PubMed] [Google Scholar]
- 39. Cheng XW, Kuzuya M, Sasaki T, Inoue A, Hu L, Song H, Huang Z, Li P, Takeshita K, Hirashiki A, Sato K, Shi GP, Okumura K, Murohara T. Inhibition of mineralocorticoid receptor is a renoprotective effect of the 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitor pitavastatin. J Hypertens. 2011;29:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur Muhlen C, Bode C, Fricchione GL, Denninger J, Lin CP, Vinegoni C, Libby P, Swirski FK, Weissleder R, Nahrendorf M. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20:754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Odegaard JI, Ricardo‐Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage‐specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, Kohno N, Yorioka N. PPAR‐gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF‐beta. Lab Invest. 2009;89:47–58. [DOI] [PubMed] [Google Scholar]
- 43. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, Pedersen AA, Kalthoff C, Tullin S, Sams A, Summer R, Walsh K. Adiponectin promotes macrophage polarization toward an anti‐inflammatory phenotype. J Biol Chem. 2010;285:6153–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin‐dependent M2 macrophage polarization: link between the metabolic and innate immune activity of full‐length adiponectin. J Biol Chem. 2011;286:13460–13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van Stijn CM, Kim J, Lusis AJ, Barish GD, Tangirala RK. Macrophage polarization phenotype regulates adiponectin receptor expression and adiponectin anti‐inflammatory response. FASEB J. 2015;29:636–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shiraishi D, Fujiwara Y, Komohara Y, Mizuta H, Takeya M. Glucagon‐like peptide‐1 (GLP‐1) induces M2 polarization of human macrophages via STAT3 activation. Biochem Biophys Res Commun. 2012;425:304–308. [DOI] [PubMed] [Google Scholar]
- 47. Krass M, Volke A, Runkorg K, Wegener G, Lund S, Abildgaard A, Vasar E, Volke V. GLP‐1 receptor agonists have a sustained stimulatory effect on corticosterone release after chronic treatment. Acta Neuropsychiatr. 2015;27:25–32. [DOI] [PubMed] [Google Scholar]
- 48. Jessen L, Smith EP, Ulrich‐Lai Y, Herman JP, Seeley RJ, Sandoval D, D'Alessio D. Central nervous system GLP‐1 receptors regulate islet hormone secretion and glucose homeostasis in male rats. Endocrinology. 2017;158:2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]