

Abstract
Background
Most novel cancer therapeutics target kinases that are essential to tumor survival. Some of these kinase inhibitors are associated with cardiotoxicity, whereas others appear to be cardiosafe. The basis for this distinction is unclear, as are the molecular effects of kinase inhibitors in the heart.
Methods and Results
We administered clinically relevant doses of sorafenib, sunitinib (cardiotoxic multitargeted kinase inhibitors), or erlotinib (a cardiosafe epidermal growth factor receptor inhibitor) to mice daily for 2 weeks. We then compared the effects of these 3 kinase inhibitors on the cardiac transcriptome using RNAseq and the cardiac kinome using multiplexed inhibitor beads coupled with mass spectrometry. We found unexpectedly broad molecular effects of all 3 kinase inhibitors, suggesting that target kinase selectivity does not define either the molecular response or the potential for cardiotoxicity. Using in vivo drug administration and primary cardiomyocyte culture, we also show that the cardiosafety of erlotinib treatment may result from upregulation of the cardioprotective signal transducer and activator of transcription 3 pathway, as co‐treatment with erlotinib and a signal transducer and activator of transcription inhibitor decreases cardiac contractile function and cardiomyocyte fatty acid oxidation.
Conclusions
Collectively our findings indicate that preclinical kinome and transcriptome profiling may predict the cardiotoxicity of novel kinase inhibitors, and suggest caution for the proposed therapeutic strategy of combined signal transducer and activator of transcription/epidermal growth factor receptor inhibition for cancer treatment.
Keywords: antineoplastic agents, cardiomyopathy, cardiotoxicity, protein kinase inhibitors, proteomics
Subject Categories: Animal Models of Human Disease, Basic Science Research, Mechanisms, Proteomics, Translational Studies
Clinical Perspective
What Is New?
We use advanced proteomics and transcriptome profiling to characterize the molecular response of the heart to multiple kinase inhibitors with unprecedented breadth.
We demonstrate that the molecular effects of both cardiosafe and cardiotoxic kinase inhibitors extend far beyond their target kinases.
We show that upregulation of the signal transducer and activator of transcription 3 pathway protects against myocardial injury and preserves cardiomyocyte fatty acid oxidation in the setting of epidermal growth factor receptor inhibition.
What Are the Clinical Implications?
Our findings suggest that combined epidermal growth factor receptor and signal transducer and activator of transcription inhibition may be cardiotoxic in humans.
Combined kinome and transcriptome profiling may offer a novel platform for predicting the cardiotoxicity of targeted cancer therapies in preclinical studies.
Introduction
Kinase inhibitors (KIs) are the most rapidly growing class of antineoplastic therapeutics and can elicit a highly effective clinical response in many different cancers. Despite putative selectivity for their respective targets, many KIs have unanticipated serious adverse effects. Among the most important of these adverse effects is cardiotoxicity, which can manifest as cardiomyopathy and heart failure.1 KI cardiotoxicity is clinically significant for its inherent morbidity and mortality, and because it can necessitate discontinuation of otherwise optimal cancer therapy. Unfortunately, our current understanding of the mechanisms underlying KI cardiotoxicity is limited and there is no reliable means to predict cardiotoxicity of KIs in development. As the cancer patient population ages, these potentially serious adverse effects will become more prominent, and the need for improved understanding and prediction even more pressing.
KIs vary in their selectivity—some target a single kinase, whereas others have numerous distinct therapeutic targets. It has been proposed that KI cardiotoxicity arises from “on‐target” inhibition of cardioprotective kinases, and that the potential for cardiotoxicity is proportional to the number of kinases targeted.2 However, “off‐target” mechanisms have been proposed as well.3 The true breadth of the molecular effects of therapeutic kinase inhibition on the heart remains unknown. Investigating KI cardiotoxicity in vivo has been difficult using traditional kinase analysis or preclinical models because of limited power to assess the impact of a KI on the kinome en masse. The kinome is a large, highly resilient network, capable of bypassing targeted kinase inhibition through elaborate “reprogramming” in tumors.4 The reprogramming capacity of the cardiac kinome in response to KI treatment is unknown.
We utilized Multiplexed Inhibitor Beads (MIBs)–Sepharose beads with covalently immobilized Type I kinase inhibitors that preferentially bind kinases in their active state.5 MIB capture followed by mass spectrometry (MIB/MS) affords a highly reproducible platform to simultaneously determine changes in the functionality of >70% of the expressed kinome. Particularly when combined with transcriptional profiling, MIB/MS can facilitate a comprehensive high throughput assessment of drug response and may provide a scalable platform to understand cardiotoxicity of approved KIs, predict cardiotoxicity of novel KIs or combination therapies, and develop cardioprotective strategies for patients receiving KIs.
Here we applied MIB/MS and RNAseq to investigate the molecular effects of 3 KIs on the mouse heart. We chose to study sunitinib and sorafenib, as models of multitargeted KIs with known cardiotoxicity,6, 7, 8 as well as erlotinib, a selective epidermal growth factor receptor (EGFR) inhibitor widely considered to be cardiosafe.9, 10 We identified unexpectedly broad kinome reprogramming after treatment with all 3 KIs and potential transcriptional signatures of cardiotoxicity. Paired MIB/MS and RNAseq analysis identified upregulation of the cardioprotective signal transducer and activator of transcription 3 (STAT3) pathway in the hearts of mice treated with erlotinib, analogous to mechanisms of EGFR inhibitor resistance in tumors. Co‐administration of erlotinib and a STAT inhibitor decreased cardiomyocyte fatty acid oxidation in vitro and cardiac contractility in vivo, suggesting that the cardiosafety of erlotinib requires STAT3 upregulation.
Materials and Methods
Animals
Female FVB mice, 14 to 16 weeks old, from Jackson Laboratory were used for all experiments, because female mice are more susceptible to sunitinib‐induced cardiotoxicity.11 Female Sprague‐Dawley rats with newborn litters were from Charles River. Animal care and experimental protocols were approved by the UNC IACUC and complied with Guide for the Care and the Use of Laboratory Animals (National Research Council Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011).
KI Treatment
Mice were gavaged with vehicle (n=4) or erlotinib (Selleck S7786, 50 mg/kg per day, n=5), sorafenib (S7397 30 mg/kg per day, n=5), or sunitinib (S1042, 40 mg/kg per day, n=5) once daily for 14 days in the UNC Lineberger Cancer Center animal core. All mice underwent echocardiography on Days 0, 7, and 14. On Day 14 mice were euthanized by cervical dislocation after an overdose of isoflurane, hearts were removed, weighed, and rapidly transferred to liquid nitrogen. In separate experiments, mice were gavaged with vehicle (n=3) or erlotinib (n=4), and infused with WP‐1066 (Selleck S2796 20 mg/kg per day by osmotic minipump, n=3), or erlotinib+WP 1066 (n=4) for 1 week. These mice underwent conscious echocardiography on Days 0 and 7.
Echocardiography
Conscious transthoracic echocardiography was performed on awake, loosely restrained mice in the McAllister Heart Institute Animal Models Core using a VisualSonics Vevo 2100 ultrasound system (VisualSonics, Inc, Toronto, Ontario, Canada). Two‐dimensional and M‐mode echocardiography were performed in the parasternal long‐axis view at the level of the papillary muscle, and left ventricular systolic function was assessed by fractional shortening (FS, where %FS=[(left ventricular end‐diastolic diameter−left ventricular end‐systolic diameter)/left ventricular end‐diastolic diameter]×100). Reported values represent the average of at least 5 cardiac cycles per mouse. Sonographers and investigators were blinded to mouse treatment condition during image acquisition and analysis.
Lysis and MIB Chromatography
Broad‐spectrum Type I KIs (CTx‐0294885, VI‐16832, PP58, Purvalanol B, UNC‐2147A, and UNC‐8088A), custom‐synthesized with hydrocarbon linkers and terminal amine groups were covalently attached to ECH‐activated Sepharose beads as previously described.12 Mouse left ventricle was rinsed in phosphate‐buffered saline and processed in lysis buffer (50 mmol/L HEPES, 150 mmol/L NaCl, 0.5% Triton X‐100, 1 mmol/L EDTA, 1 mmol/L EGTA, at pH 7.5 containing 10 mmol/L NaF, 2.5 mmol/L NaVO4, complete Protease Inhibitor Cocktail (Roche), and 1% Phosphatase Inhibitor Cocktails 2 and 3 [Sigma]). Four milligram total protein lysate was gravity‐flowed over a mixture of the 6 KI‐linked beads (175 μL total beads), followed by 30 volumes of washes with high salt (1 mol/L NaCl) and low salt (150 mmol/L NaCl) lysis buffer, then 500 μL of low salt lysis buffer containing 0.1% SDS. Bound proteins were eluted by boiling with 0.5% SDS and 1% β‐mercaptoethanol in 100 mmol/L Tris‐HCl, pH 6.8, 2× 15 minutes, treated with DTT (5 mmol/L, 25 minutes at 60°C) and iodoacetamide (20 mmol/L, 30 minutes in the dark at RT), and spin‐concentrated to 100 μL (Amicon Millipore Amicon Ultra‐4, 10K cutoff) before methanol/chloroform precipitation. Proteins were trypsinized overnight at 37°C and labeled with TMT sixplex reagents (Thermo) according to manufacturer instructions, and then dried down in a speed‐vac. Peptides were cleaned with C‐18 spin columns (Pierce).
MS and Analysis
Five percent of each sample was first run on a 60‐minute LC gradient and then equalized on total peptide content before combining. Peptides were resuspended in 2% ACN and 0.1% formic acid. Thirty percent of the final peptide suspension was injected onto an Easy nLC‐1000 through a Thermo Easy‐Spray 75 μm×25 cm C‐18 column and separated on a 300‐minute gradient (5%–40% ACN). ESI parameters: 3e6 AGC MS1, 80 ms MS1 max inject time, 1e5 AGC MS2, 100 ms MS2 max inject time, 20 loop count, 1.8 m/z isolation window, 45‐s dynamic exclusion. Spectra were searched against the Uniprot/Swiss‐Prot database with Sequest HT on Proteome Discoverer software. Only peptides with medium or greater confidence (5% FDR) were considered for quantitation, and peptides with >75% co‐isolation interference were omitted. Data for each KI‐treated sample were processed as fold change relative to a pool of 4 vehicle‐treated control samples. After log2, average and SD were calculated to determine consistent changes in kinase MIB‐binding.
RNAseq and Analysis
mRNA‐Seq libraries were constructed using 4 μg total RNA with the Stranded mRNA‐Seq Kit (KAPA Biosystems). Three hearts each were used from each condition (control, erlotinib, sunitinib, sorafenib), multiplexed with Illumina TruSeq adapters, and run on a single 75‐cycle single‐end sequencing run with an Illumina NextSeq‐500. QC‐passed reads were aligned to the mouse reference genome (mm9) using MapSplice.13 The alignment profile was determined by Picard Tools v1.64. Aligned reads were sorted and indexed using SAMtools and translated to transcriptome coordinates and filtered for indels, large inserts, and zero mapping quality using UBU v1.0. Transcript abundance estimates for each sample were performed using an Expectation‐Maximization algorithm.14 Raw RNAseq by Expectation Maximization read counts for all RNAseq samples and raw FASTQ files of RNAseq runs have been uploaded to National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE98973. Reviewers may access these private data at the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=qtsduckchxghladamp;acc=GSE98973.
The DEseq2 algorithm15 was used to determine differential expression analysis of each set of KI‐treated samples versus controls using the expected counts column for each data set. Gene Set Enrichment Analysis (GSEA) was performed on each set of treated versus control data sets using normalized RNAseq by Expectation Maximization read counts. Data were 50‐read filtered such that at least 1 sample for each comparison (3 control versus 3 treated) for each gene must have had a value of at least 50 normalized RNAseq by Expectation Maximization reads. Mouse gene names were converted to their human homolog and GSEA was performed against MSigDB gene sets for Hallmarks, Gene Ontology, KEGG, Reactome, and Oncogenic Signatures. Default parameters were used and only gene sets with nominal P<0.05 and FDR <25% were considered.
Primer Sequences
Nr1d1: ccccaggaagtctacaagtgg; R: agcaccatgccattcagc.
Ednra1 F: tggagtgtttctctgcaag; R: ggaagccactgctctgtacc.
Western Blotting
Proteins were denatured and separated by SDS‐PAGE, transferred to nitrocellulose membranes, and probed with pSTAT3 (Y705 and S727) and STAT3, obtained from Cell Signaling Technology. Secondary horseradish peroxidase–anti‐rabbit was from Jackson Immunoresearch Laboratories. Western blots were visualized by incubation with SuperSignal West Pico or Femto Chemiluminescent Substrate (Thermo Scientific).
Neonatal Rat Ventricular Myocyte Isolation and Culture
Female Sprague‐Dawley rats (n=5) were from Charles River. Neonatal rat ventricular myocytes (NRVMs) were isolated as previously described.16 Briefly, hearts from 1‐to‐2‐day‐old rat pups were minced, digested serially in collagenase (Worthington)‐containing solution, filtered, then preplated to exclude nonmyocytes. NRVMs were then plated on laminin‐coated dishes in DMEM with 5% fetal bovine serum (Sigma F2442) for 24 hours. Experiments were carried out after 36 to 72 hours of serum starvation in the presence of bromodeoxyuridine. Lactate dehydrogenase assays (Sigma MAK066‐1KT) were carried out in 1% fetal bovine serum.
Fatty acid oxidation assays used sodium oleate 500 μmol/L (Sigma O‐7501) with BSA 0.5% (Sigma 05470) and carnitine. 14C‐oleate (Perkin‐Elmer NEC317050UC) was added to the medium and incubated for 2 hours. Medium was transferred to a 48‐well CO2 trapping plate with 200 μL NaOH (1 N). Seventy percent perchloric acid (PCA) was added to each well and CO2 was trapped while agitating on an orbital shaker. One aliquot of medium was transferred to a scintillation vial and labeled CO2 was counted. Cells were scraped on ice, incubated overnight, and then acid‐soluble metabolites were counted in the cell supernatant. Total cellular protein was quantitated using the Bradford reagent. Total oxidation=(disintegrations per minute from CO2 trap−blank)×(200/150)×specific activity+(disintegrations per minute from acid‐soluble metabolites−blank)×(550/300)×specific activity.
Data Analysis and Statistics
Except where otherwise specified, all results are presented as mean±SEM. Two‐group comparisons were made using t test and 3‐group comparisons used ANOVA (1‐way or repeated measures as specified) in GraphPad Prism. Reanalysis with nonparametric tests did not affect the statistical results.
Results
Mouse Body and Heart Weight After KI Treatment
We treated female 14‐to‐16‐week‐old FVB mice daily with vehicle or 1 of 3 kinase inhibitors (erlotinib 50 mg/kg per day, sorafenib 30 mg/kg per day, or sunitinib 40 mg/kg per day) for 14 days in the UNC Lineberger Animal Models core. We chose these doses because they yield plasma drug concentrations similar to the therapeutic range in human studies and have limited systemic toxicities in published mouse experiments.17, 18, 19, 20, 21, 22, 23, 24 On Day 9, 4 mice were euthanized for failure to thrive: 2 from the erlotinib group and 1 each from the sorafenib and sunitinib groups. These mice were included in our analyses. Body weight decreased in sorafenib‐treated mice, but was unchanged in other treatment groups. Heart weight indexed to body weight was higher in erlotinib‐ and sorafenib‐treated mice when compared with vehicle. Treatment with sorafenib and sunitinib, but not erlotinib, led to increased indexed lung weight, consistent with left heart failure (Table 1).
Table 1.
Body and Organ Weights After KI Treatment
| Treatment (n) | Body Weight, Initial (g) | Body Weight, Final (g) | Heart Weight (mg) | Heart/Body Weight (%) | Lung Weight (mg) | Lung Weight/Body Weight (%) |
|---|---|---|---|---|---|---|
| Vehicle (4) | 21.9±0.7 | 21.2±0.4 | 84±4 | 0.40±0.01 | 119±5 | 0.56±0.01 |
| Erlotinib 50 mg/kg per d (5) | 21.1±0.4 | 19.5±0.7 | 87±2 | 0.45±0.02a | 115±3 | 0.59±0.03 |
| Sunitinib 40 mg/kg per d (5) | 20.9±0.5 | 19.9±0.6 | 82±3 | 0.41±0.02 | 128±3a | 0.64±0.03a |
| Sorafenib 30 mg/kg per d (5) | 21.9±0.5 | 20.2±0.2a | 93±3 | 0.46±0.01a | 125±6 | 0.62±0.02a |
KI indicates kinase inhibitor.
All values are mean±SEM, n given in parentheses.
P<0.05 vs vehicle treatment.
Mouse Echocardiography
To assess the effects of KI treatment on cardiac function, we performed conscious echocardiography at baseline and on Days 7 and 14 of treatment. Erlotinib had no significant effect on any echocardiographic parameters. Sunitinib and sorafenib both caused variable decreases in fractional shortening and increases in left ventricular end systolic volume, consistent with impaired contractile function (Figure 1; Table 2, Figure S1). These findings are consistent with previous in vitro studies that showed sorafenib and sunitinib decreased, but erlotinib did not affect, contractile force in engineered rat heart tissue.25
Figure 1.
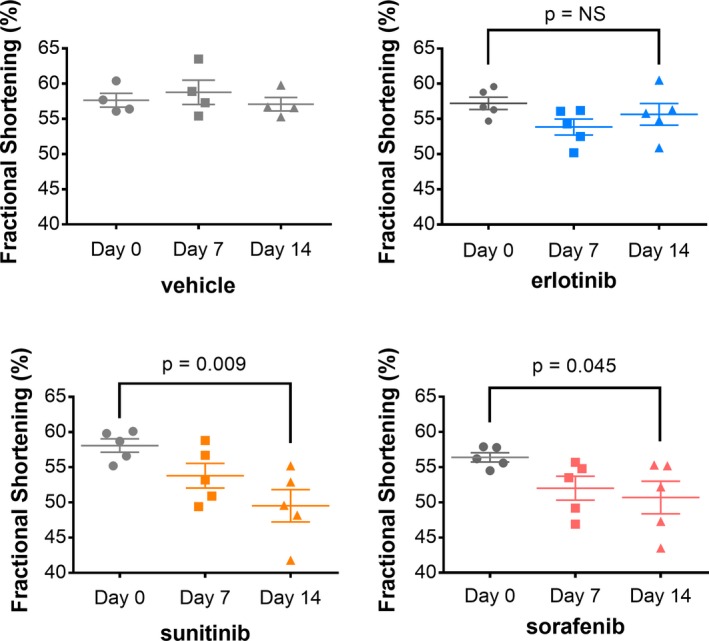
Contractile function is not affected by erlotinib, but is reduced by sunitinib and sorafenib. Female wild‐type FVB mice (n=4 for vehicle, n=5 for KIs) were gavaged with vehicle, erlotinib (50 mg/kg per day), sunitinib (40 mg/kg per day), or sorafenib (30 mg/kg per day) for 14 days. Conscious echocardiograms were performed at baseline, 7 days, and 14 days. Contractile function is reported as fractional shortening. Repeated‐measures ANOVA compared intragroup changes. (*P<0.05). KIs indicates kinase inhibitors; NS, not significant.
Table 2.
Echocardiographic Parameters After KI Treatment
| HR | LVIDd | LVIDs | FS | LVd Vol | LVs Vol | IVSd | PWd | |
|---|---|---|---|---|---|---|---|---|
| Vehicle | ||||||||
| Day 0 | 674±9 | 2.6±0.1 | 1.1±0.0 | 58±1 | 25±2 | 2.8±0.3 | 0.9±0.0 | 1.0±0.0 |
| Day 7 | 681±17 | 2.5±0.1 | 1.0±0.1 | 59±2 | 23±3 | 2.2±0.4 | 1.0±0.0 | 1.0±0.0 |
| Day 14 | 687±14 | 2.7±0.1 | 1.1±0.0 | 57±1 | 28±2 | 3.2±0.3 | 1.0±0.5 | 1.0±0.0 |
| Erlotinib | ||||||||
| Day 0 | 672±23 | 2.7±0.1 | 1.1±0.1 | 57±1 | 26±3 | 3.0±0.5 | 0.9±0.1 | 1.0±0.0 |
| Day 7 | 678±9 | 2.8±0.1 | 1.3±0.1 | 54±1 | 29±3 | 4.1±0.6 | 0.9±0.0 | 1.0±0.1 |
| Day 14 | 668±12 | 2.8±0.1 | 1.3±0.1 | 56±2 | 31±2 | 3.9±0.5 | 1.0±0.0 | 1.0±0.1 |
| Sunitinib | ||||||||
| Day 0 | 684±14 | 2.8±0.1 | 1.2±0.0 | 58±1 | 29±2 | 3.1±0.3 | 0.9±0.0 | 1.0±0.0 |
| Day 7 | 649±28 | 2.7±0.1 | 1.3±0.1 | 54±2 | 28±3 | 3.9±0.5 | 0.9±0.0 | 0.9±0.0 |
| Day 14 | 683±19 | 2.9±0.1 | 1.5±0.1† | 49±3* | 32±2 | 5.7±0.7† | 0.9±0.0 | 1.0±0.0 |
| Sorafenib | ||||||||
| Day 0 | 648±17 | 2.7±0.1 | 1.2±0.0 | 56±1 | 26±2 | 3.1±0.3 | 1.0±0.0 | 0.9±0.0 |
| Day 7 | 661±17 | 2.7±0.1 | 1.3±0.1 | 52±2 | 28±2 | 4.3±0.4 | 0.9±0.0 | 0.9±0.0 |
| Day 14 | 694±15 | 2.7±0.1 | 1.3±0.1 | 52±2* | 28±2 | 4.5±0.8* | 1.0±0.0 | 1.0±0.0 |
| WP1066 | ||||||||
| Day 7 | 683±40 | 3.0±0.1 | 1.4±0.1 | 53±1 | 34±4 | 5.1±0.9 | 0.9±0.0 | 0.9±0.0 |
| Erlotinib+WP1066 | ||||||||
| Day 7 | 653±36 | 2.9±0.1 | 1.5±0.1 | 49±2* | 33±4 | 6.0±0.9* | 0.8±0.0 | 0.8±0.0 |
All values are mean±SEM. FS indicates fractional shortening (%); HR, heart rate (beats per minute); IVSd, interventricular septal thickness, diastole (mm); KI, kinase inhibitor; LVd vol, left ventricular diastolic volume (μL); LVIDd, left ventricular internal diameter, diastole (mm); LVIDs, left ventricular internal diameter, systole (mm); LVs vol, left ventricular systolic volume (μL); PWd, posterior wall, diastole (mm).
*P<0.05, † P<0.01 vs baseline by intragroup repeated‐measures ANOVA (except WP1066 and erlotinib+WP1066, which used 1‐way ANOVA) with Tukey post‐test.
Kinome Profiling Using MIB With MS
We used MIB coupled with MS (MIB/MS) to assess functional cardiac kinome dynamics in response to erlotinib, sunitinib, and sorafenib. All 3 KIs led to unexpectedly broad changes cardiac kinome activity. MIB/MS detected 200 kinases in erlotinib‐treated hearts, 214 kinases in sunitinib‐treated hearts, and 215 kinases in sorafenib‐treated hearts. When compared with vehicle‐treated hearts, 88 kinases were dysregulated by erlotinib, 84 by sunitinib, and 80 by sorafenib (Figures 1 and 2A; Table S1, Figure S2). Of note, more kinases were upregulated than downregulated by each KI, suggesting that the heart, like tumor tissue, undergoes kinome adaptation in response to targeted therapies (Table 3). The 10 most affected kinases for each KI are shown in Figure 2B, with narrow SEs indicating the reproducibility of the findings. Interestingly, metabolic kinases were disproportionately affected by all 3 KIs, relative to other kinase families (Figure 2C). KIs are known to disturb systemic metabolic regulation,26 but their effects on cardiac metabolism have not been described previously.
Figure 2.
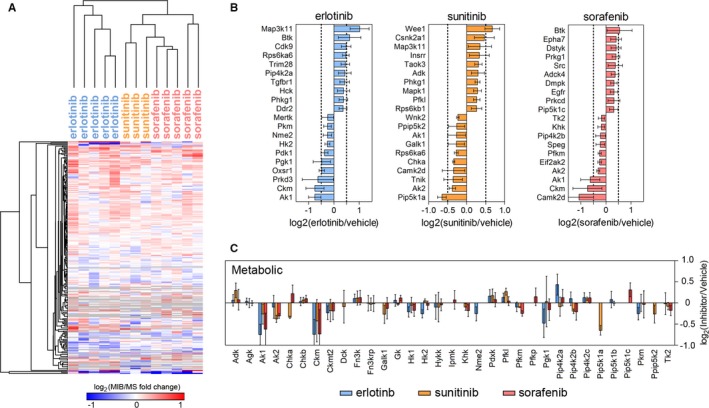
Kinome profiling using multiplexed inhibitor beads with mass spectrometry (MIB/MS) reveals broad changes in the cardiac kinome after kinase inhibitor (KI) treatment. After 14 days of KI treatment, mice were euthanized. The heart was removed and processed for kinome profiling with MIB/MS (n=5 for erlotinib and sorafenib, n=3 for sunitinib). A, Summary heat map of significant changes in kinase activity. B, The 10 kinases most increased and decreased by erlotinib, sunitinib, and sorafenib. C, Kinases involved in regulation of cellular metabolism were disproportionately affected by KI treatment. MIB indicates multiplexed inhibitor beads; MS, mass spectrometry.
Table 3.
Summary of KI Regulation of the Cardiac Kinome and Transcriptome
| Treatment | MIB/MS Kinases Assayed | MIB/MS Significant Upregulation | MIB/MS Significant Downregulation | RNAseq Upregulation | RNAseq Downregulation |
|---|---|---|---|---|---|
| Erlotinib 50 mg/kg per d | 200 | 70 | 18 | 484 (4%) | 1333 (10%) |
| Sunitinib 40 mg/kg per d | 214 | 54 | 30 | 627 (5%) | 424 (3%) |
| Sorafenib 30 mg/kg per d | 215 | 62 | 18 | 516 (4%) | 726 (5%) |
KI indicates kinase inhibitor; MIB/MS, multiplexed inhibitor beads/mass spectrometry.
We analyzed MIB/MS data to identify a potential kinome signature of KI cardiotoxicity, comparing cardiosafe erlotinib with cardiotoxic sorafenib and sunitinib. MIB/MS kinome response signatures accurately clustered hearts by treatment and separated erlotinib from sunitinib and sorafenib (Figure 3A). Erlotinib induced a series of kinases including Map3k11, Pip4k2a, Pip4K2b, Ddr2, Cdk9, Bmx, and Hck. Ingenuity Pathway Analysis (IPA, Qiagen) performed on the top‐20 most‐induced kinases for each of the treatments (within SD) indicated that erlotinib uniquely induces STAT3 (z‐score 2.8, P=5.0×10−9), nuclear factor kappa B (NFκB) (z‐score 2.2, P=2.2×10−7), and Tec kinase (z‐score 1.9, P=4.0×10−6) pathway activity in the mouse heart (Figure 3B). Protein interactions for the erlotinib‐activated kinases were identified using the STRING database and shown in Figure 3C, illustrating multiple connections to STAT3 and NFκB.
Figure 3.

MIB/MS comparison of cardiosafe erlotinib with cardiotoxic sunitinib and sorafenib indicates differential regulation of multiple signaling pathways, including STAT3. To identify a kinome signature of cardiotoxicity, MIB/MS results unique to erlotinib were compared with results for sunitinib and sorafenib. A, Summary heatmap displaying kinases with significantly altered activity in erlotinib‐treated hearts compared with hearts treated with sunitinib or sorafenib. B, Ingenuity Pathway Analysis (Qiagen) compared the kinome profile of erlotinib with sorafenib and sunitinib. Differentially regulated canonical pathways are displayed. C, STRING database interaction of erlotinib‐induced kinases. D, Immunoblot of heart lysates from mice treated for 14 days with kinase inhibitors. MIB/MS indicates multiplexed inhibitor beads/mass spectrometry; NFκB, nuclear factor kappa B; STAT3, signal transducer and activator of transcription 3.
To confirm the IPA analysis, we immunoblotted 14‐day KI‐treated heart lysates for pSTAT3 and STAT3. We found that erlotinib increased STAT3 phosphorylation at Y705 by 2.2‐fold when compared with vehicle (n=8 per group); STAT3 S727 phosphorylation was unaffected by erlotinib. Sunitinib and sorafenib administration either did not change or decreased STAT3 activation (Figure 3D). All 3 KIs inhibited EGFR (data not shown). STAT3 and NFκB both are known to play context‐dependent cardioprotective roles in the heart,27, 28 though the function of Tec kinase is largely unknown.
In summary, we applied MIB/MS to KI‐treated mouse hearts and identified unexpectedly broad changes in kinome regulation, suggesting that kinome reprogramming occurs in the heart. The extent of these changes was similar in a highly selective and cardiosafe KI (erlotinib) and multitargeted cardiotoxic KIs (sorafenib and sunitinib).
Transcriptome Profiling Using RNAseq
We then performed deep mRNA sequencing (RNAseq) on 3 of the same hearts that underwent MIB/MS analysis for each treatment group. We used the differential expression algorithm DEseq2 15 to determine significant changes compared with vehicle‐treated mice for each of the KIs. Erlotinib treatment significantly affected expression (adjusted P<0.05) of 535 genes (4.4% of the measured transcriptome), with sorafenib affecting 227 genes (1.9%) and sunitinib only 98 genes (0.8%) (Figure 4A; Table 3, Table S2). Several transcription factors were among the most differentially expressed transcripts in each of the conditions, including an upregulation of Junb hearts from erlotinib‐treated mice and Foxo3 in sunitinib‐treated hearts. JunB is induced in cardiomyocyte hypertrophy,29 whereas FOXO3 is a central regulator of cardiac autophagy and atrophy.30
Figure 4.
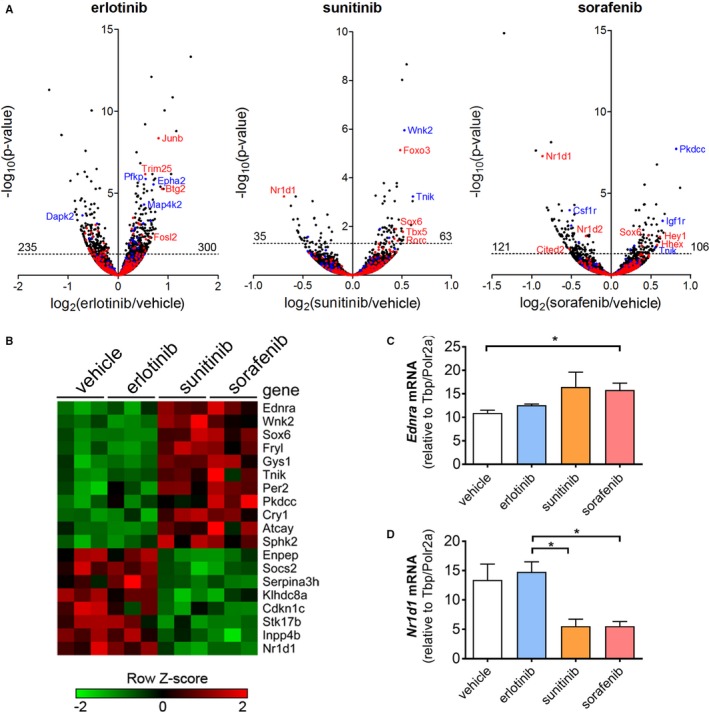
RNAseq demonstrates transcriptional changes in the mouse heart after treatment with kinase inhibitors (KIs). To assess the effect of KI treatment on the cardiac kinome, heart tissue from female FVB mice (n=3 per group) treated for 14 days was processed for RNAseq. A, Volcano plots of statistically significant (DEseq2) transcriptional responses to KI treatment, with kinases in blue and transcription factors in red. B, Heat map displaying genes that distinguish cardiosafe treatment with vehicle or erlotinib from cardiotoxic treatment with sunitinib or sorafenib. C and D, Confirmatory qRT‐PCR of selected differentially regulated genes Ednra and Nr1d1. (*P<0.05 by 1‐way ANOVA). qRT‐PCR indicates quantitative real‐time polymerase chain reaction; TFs, transcription factors.
Next we compared the control and erlotinib‐treated groups with the sunitinib and sorafenib‐treated hearts to identify a putative transcriptional signature of cardiotoxicity. Interestingly, 19 genes were commonly regulated by both sunitinib and sorafenib, but unaffected by erlotinib (Figure 4B). Transcription factors (Figure 4A in red) were disproportionately represented among the most discrepantly regulated genes. This pattern of gene expression also includes sunitinib‐ and sorafenib‐induced upregulation of endothelin receptor A (Ednra), the receptor for endothelin‐1 and a well‐recognized mediator of cardiac hypertrophy and heart failure (reviewed in31). Sunitinib is known to upregulate the endothelin system in the vasculature,32 but its effects in the heart have not been studied. Interestingly, endothelin receptor A upregulation also is implicated in tumor chemoresistance, and endothelin receptor antagonists have been proposed as novel cancer therapies.33, 34 We identified common upregulation of Sox6 in the sunitinib and sorafenib groups (Figure 4A and 4B). Overexpression of Sox6 causes cardiomyopathy and heart failure in mice, with induction of pathological isoforms of sarcomeric proteins.35 Genes downregulated in this cardiotoxic signature include Nr1d1 (RevErbα), a transcription factor involved in circadian rhythm and metabolic regulation,36 as well as mitochondrial biogenesis, in skeletal muscle.37 Quantitative reverse transcriptase PCR confirmed the RNAseq findings for both Ednra and Nr1d1 (Figure 4C and 4D).
GSEA was performed on these RNAseq data to further define functional gene expression patterns induced by the KIs. Gene sets significantly induced or repressed by each treatment are shown in Table S3. When compared with vehicle treatment, sunitinib and sorafenib both generated a loss of established gene set signatures, with 204 (sunitinib) and 175 (sorafenib) gene sets enriched in control (lost in treatment). Gene sets commonly repressed by sunitinib and sorafenib primarily involved extracellular matrix assembly and the ribosome/translation (Table S3). In contrast, 43 gene sets were induced significantly by erlotinib treatment. Figure 5A displays gene sets that are both significantly induced by erlotinib and lost upon treatment with sorafenib, sunitinib, or both cardiotoxic agents. In concordance with MIB/MS kinome response data, this GSEA analysis identified erlotinib‐induced upregulation of transcripts related to NFκB (HALLMARK TNFA SIGNALING VIA NFKB) and STAT3 (HALLMARK IL6 JAK STAT3 SIGNALING). GSEA plots are shown in Figure 5B and complementary heat maps are shown in Figure S3.
Figure 5.
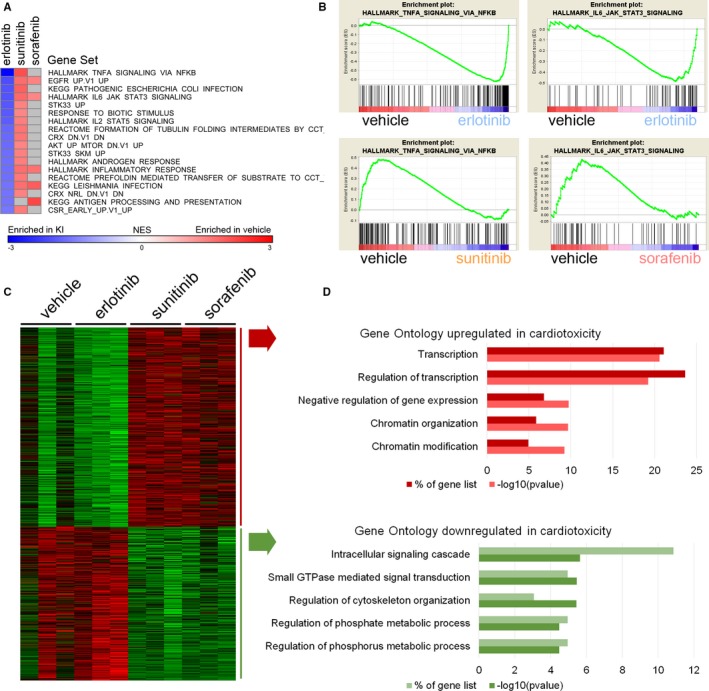
Gene Set Enrichment Analysis of RNAseq data suggests that regulation of STAT3 and NFκB pathways and regulation of transcription distinguish erlotinib from sorafenib and sunitinib. We analyzed RNAseq data to identify transcriptomic signatures of cardiotoxic kinase inhibitors. A, Gene Set Enrichment Analysis of RNAseq data for kinase inhibitors sorted for fold change in erlotinib. B, Enrichment plots for Hallmark TNFA signaling via NFκB and Hallmark IL6 JAK STAT3 signaling. C, Heat map and D summary figure for Gene Ontology terms that distinguish transcriptomic changes in cardiosafe (vehicle and erlotinib) from cardiotoxic (sunitinib and sorafenib) treatments. EGFR indicates epidermal growth factor receptor; IL6, interleukin 6; JAK, Janus kinase; NES, Normalized Enrichment Score; NFκB, nuclear factor kappa B; STAT3, signal transducer and activator of transcription 3; TNFA, tumor necrosis factor α.
To profile the transcriptomic impact of cardiotoxic KIs with greater statistical power, we performed DEseq2 combining control and erlotinib against combined sorafenib and sunitinib. This yielded 555 genes upregulated and 428 genes downregulated (P<0.05) by sorafenib and sunitinib compared with control‐ and erlotinib‐treated hearts (Figure S4). Gene Ontology enrichment analysis of the cardiotoxic KIs revealed highly significant upregulation of genes involved in transcription and chromatin modification (Figure 5C and 5D).
In summary, RNAseq revealed focused transcriptional changes after KI treatment. Pathway analysis of the common transcriptional effects of sunitinib and sorafenib indicated that the cardiotoxic KIs affected transcriptional regulation and chromatin modification. Erlotinib uniquely induced upregulation of NFκB and STAT3 pathways, recapitulating the MIB/MS findings.
Fatty Acid Oxidation Assays in NRVMs
In our study, both MIB‐MS and RNAseq analysis suggested that KIs affect cardiac metabolism, consistent with previous studies indicating that sunitinib and sorafenib cause ATP depletion and mitochondrial dysfunction.38 To evaluate further the functional consequences of our MIB‐MS and RNAseq findings, we treated NRVMs with vehicle or KIs in vitro for 24 hours. Given that fatty acids are the primary energy substrate in cardiomyocytes, we assayed fatty acid oxidation in NRVMs after treatment with KIs, using the carnitine palmitoyl transferase inhibitor, etomoxir, as a control. Erlotinib caused a dose‐dependent increase in fatty acid oxidation, whereas sunitinib and sorafenib decreased fatty acid oxidation (Figure 6A). None of the KIs significantly increased cell death at the chosen concentrations, as measured by lactate dehydrogenase release (Figure 6B).
Figure 6.
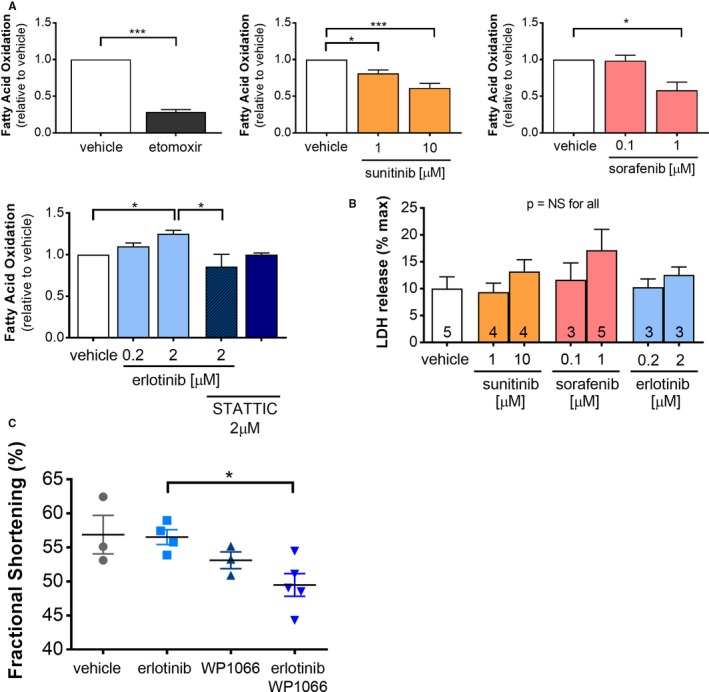
Co‐administration of a STAT inhibitor abrogates erlotinib‐induced increase in cardiomyocyte fatty acid oxidation in vitro and impairs contractile function in vivo. A, Neonatal rat ventricular myocytes (n=4 independent experiments) were treated with vehicle, the carnitine phosphatoyl transferase inhibitor, etomoxir, or kinase inhibitors (including the STAT inhibitor, STATTIC) for 24 h, then fatty acid oxidation was assayed using 14C‐oleate. B, Cell death in NRVMs was assayed by LDH release (n=3–5 independent experiments, as indicated). C, Female FVB mice were treated with vehicle, erlotinib, and/or the STAT inhibitor WP1066 for 7 days and contractile function was assessed using conscious echocardiography. Groups for all experiments were compared using 1‐way ANOVA, *P<0.05, **P<0.01. LDH indicates lactate dehydrogenase; NRVMs, neonatal rat ventricular myocytes; NS, not significant; STAT, signal transducer and activator of transcription.
Erlotinib‐induced enhancement of fatty acid oxidation was unexpected, as EGFR has no recognized role in regulating fatty acid oxidation. MIB‐MS and RNAseq both indicated that cardiac STAT3 activity was upregulated by erlotinib treatment, and 1 previous study suggested that STAT3 stimulates cardiac fatty acid oxidation in response to leptin.39 To test whether the effect of erlotinib on fatty acid oxidation required STAT3 activation, we co‐treated NRVMs with erlotinib and the selective STAT3 inhibitor, STATTIC. We found that STATTIC abrogated erlotinib‐mediated increase in fatty acid oxidation, suggesting that STAT3 upregulation may contribute to this energetically favorable metabolic adaptation (Figure 6A).
Combined Treatment With Erlotinib and STAT‐Inhibitor In Vivo
Profiling both the cardiac kinome and transcriptome indicated that erlotinib treatment is associated with upregulation of the STAT3 pathway in the heart, a particularly interesting finding given that STAT3 activation is a primary mechanism of tumor resistance to EGFR inhibitor treatment in multiple types of cancer.40, 41 We sought to determine whether upregulation of cardioprotective STAT3 might account for the lack of cardiotoxicity of erlotinib by treating female FVB mice for 7 days with erlotinib (50 mg/kg per day) and/or WP1066 (20 mg/kg per day by osmotic minipump), a STAT inhibitor with demonstrated in vivo activity.42, 43
Treatment with erlotinib did not affect contractile function. WP1066 was associated with a nonsignificant trend towards decreased contractile function (fractional shortening 53±2% on Day 7), but co‐administration of erlotinib and WP1066 significantly decreased fractional shortening to 49±2% (Table 2; Figure 6C). Collectively, these findings suggest that activation of STAT3 contributes to the heart's biological response to erlotinib by increasing fatty acid oxidation and preserving contractile function.
Discussion
We utilized MIB‐MS and RNAseq to provide a comprehensive characterization of the molecular effects of KIs in the heart. This combined approach revealed unexpectedly broad effects on both the expressed kinome and transcriptome, underscoring the complexity of KI cardiotoxicity. We found that erlotinib unexpectedly upregulated STAT3 activity in the mouse heart and that co‐administration of erlotinib and a STAT inhibitor decreased cardiac contractility and cardiomyocyte fatty acid oxidation. Collectively, these findings position combined MIB‐MS and RNAseq as a platform for preclinical prediction of KI cardiotoxicity in vivo.
MIB‐MS is a recently developed technique that quantifies changes in the functional kinome rather than merely kinase abundance as provided by standard proteomics approaches. Kinase capture occurs reproducibly as a function of kinase abundance, the affinity of kinases for the inhibitor beads, and the activation state of the kinase. As such, our MIB‐MS data extend the published explication of the cardiac kinome.44 This platform has been applied to tumor tissue,5, 12, 45 but has not been used previously in normal tissue. Somewhat surprisingly, given that myocardium consists largely of somatic, nondividing issue, we find robust reprogramming of the cardiac kinome after KI treatment, similar in scope to the changes observed in malignant tissue. Many of the observed alterations in kinase activity are not clearly predicted by the molecular targeting of the drugs, suggesting that the cardiac response to KI treatment represents a complex interaction of both “on‐target” and “off‐target” signaling.
MIB/MS and RNAseq yielded distinct but complementary data sets. The majority of MIB/MS‐detected changes in kinases were not predicted by transcript abundance, suggesting that KIs remodel the cardiac kinome largely through post‐translational signaling. Predictably, the transcriptional changes detected by RNAseq data offer a more extensive characterization of KI‐induced molecular effects. The most salient signal from pathway analysis of the RNAseq data is the suggestion that the cardiotoxic KIs, sunitinib and sorafenib, affect transcriptional regulation and chromatin remodeling, likely yielding a net repression of transcription that blunts adaptive responses.
We chose to study erlotinib as an exemplar of a highly selective KI without known cardiotoxicity; sunitinib and sorafenib were chosen for their recognized cardiotoxicity. It has been suggested that KIs such as sunitinib and sorafenib are inherently more likely to be cardiotoxic as they are multitargeted,2, 46 in contrast with highly selective KIs like erlotinib. However, our findings suggest that KI selectivity likely does not predict the molecular response in the heart or the propensity for adverse cardiac effects: erlotinib treatment led to more changes in the kinome and transcriptome than either sunitinib or sorafenib.
The most consistent signal from combined kinome and transcriptome analysis was the upregulation of STAT3 and NFκB pathways after erlotinib treatment. These pathways were downregulated by sunitinib and sorafenib, consistent with previous studies in tumor tissue.40, 41 Upregulation of both NFκB47 and STAT3 activity has been implicated in the development of resistance to EGFR inhibitors (such as erlotinib) in numerous types of cancer including non–small cell lung cancer,48, 49 and pancreatic cancer.50 Preclinical studies have shown that the addition of a STAT3 inhibitor to an EGFR inhibitor can overcome resistance in ovarian cancer,51 soft tissue sarcoma,52 head and neck cancer, and bladder cancer.53
The ErbB family of receptors, including EGFR, mediates numerous essential roles in cardiomyocytes54; hence it is not immediately evident why erlotinib is not cardiotoxic. By contrast, trastuzumab, an ErbB2 antagonist used extensively in the treatment of breast cancer, has widely recognized cardiotoxicity (reviewed in55). Our analysis of the kinome and transcriptome led us to consider whether STAT3 activation might confer protection against otherwise putatively cardiotoxic effects of erlotinib, given that STAT3 activation through Y705 phosphorylation can be cardioprotective in the setting of numerous insults (56, 57 reviewed in 27). Of note, STAT3 exerts its adaptive effects both through signaling and as a transcription factor,58 potentially accounting for the detection of its upregulation in both MIB‐MS and RNAseq. We found that co‐treatment of cardiomyocytes with erlotinib and the STAT3 inhibitor, STATTIC, abrogated the beneficial effect of erlotinib on cardiomyocyte fatty acid oxidation. We also found that co‐treatment of mice with erlotinib and the STAT inhibitor, WP1066, decreased contractile function in vivo, whereas neither treatment affected fractional shortening when administered alone. Collectively, these findings suggest that cardiac STAT3 upregulation after EGFR inhibition represents a prosurvival response analogous to the development of treatment resistance in tumors.
These findings may provide some caution regarding proposed combination therapy with EGFR and STAT3 inhibitors, such as the Phase 1 trial that is already under way for the treatment of EGFR‐mutated lung cancer with combined inhibition of EGFR and the JAK/STAT pathway (clinicaltrials.gov: NCT02145637). Such potential interactions would not be evident with standard focused analysis of kinase activation, and highlight the potential for combined MIB/MS and RNAseq in the preclinical prediction of cardiotoxicity in combination KI treatment. This platform may be particularly useful in assessing the potential toxicity of combination therapies, given the identified similarities between tumor and cardiac kinome reprogramming in the setting of KI administration.
Our study has several limitations. We assessed the cardiac response to a single dose of each KI. Though we chose KI doses that are well established in the literature, we cannot exclude the possibility that our findings are dose dependent. We also studied female mice from a single strain and acknowledge that there may be strain‐related differences in drug response. Interestingly, in contrast to most types of cardiac insult, female mice are more prone to sunitinib‐induced cardiotoxicity than male mice.11 It is possible that the inclusion of 4 mice that were euthanized early because of excessive weight loss could have affected our data analysis; however, none of those mice showed evidence of heart failure (mean fractional shortening 56% at Day 7). Lastly, we did not pursue the possibility that upregulation of cardioprotective NFκB signaling,59 in addition to STAT3 activation, might buffer the cardiac effects of EGFR inhibition with erlotinib. We will pursue that avenue of investigation in future studies.
Collectively, our findings suggest that combined kinome and transcriptome profiling could generate a molecular signature of KI cardiotoxicity that could be useful in predicting potential adverse cardiac effects of novel KIs in the preclinical setting. That signature will be enriched by application of this platform to other extant KIs, as well as proposed combination therapies. Though routine sampling of human myocardium poses unacceptable risk, understanding the effects of KIs on the mouse cardiac kinome and transcriptome may facilitate identification of circulating markers of pathway‐specific cardiac injury that could mitigate the risk of cardiac toxicity during early phase clinical trials of KIs in development.
Sources of Funding
This work was supported by the National Institutes of Health (UL1TR001111 to Jensen and Johnson through UNC NC TraCS) and the McAllister Research Foundation (to Jensen).
Disclosures
None.
Supporting information
Table S1. MIB‐MS Summary Data
Table S2. RNAseq by Expectation Maximization (RSEM) Data for RNAseq
Table S3. Gene Set Enrichment Analysis (GSEA) Results
Figure S1. Conscious echocardiography compared structural and functional cardiac parameters. Female wild‐type FVB mice (n=4 for vehicle, n=5 for KIs) were gavaged with vehicle, erlotinib (50 mg/kg per d), sunitinib (40 mg/kg per d), or sorafenib (30 mg/kg per d) for 14 days. Conscious echocardiograms were performed at baseline, 7 days, and 14 days. Repeated‐measures ANOVA compared intragroup changes. (*P<0.05). LVID indicates left ventricular internal diameter.
Figure S2. Graphical summary of Multiplex Inhibitor Beads–Mass Spectrometry (MIB‐MS) kinome profiling of kinase inhibitor‐treated mouse hearts. Kinase activity depicted with reference to vehicle‐treated hearts.
Figure S3. Heat maps for Gene Set Enrichment Analysis of RNAseq data. Upregulation of transcripts in NFκB and STAT3 pathways distinguishes erlotinib from sunitinib and sorafenib. IL6 indicates interleukin 6; NFκB, nuclear factor kappa B; STAT3, signal transducer and activator of transcription 3; TNFα, tumor necrosis factor α.
Figure S4. Volcano plots comparing statistically significant (DEseq2) transcriptional responses to cardiosafe (control and erlotinib) or cardiotoxic (sorafenib and sunitinib) treatments. Kinases are in blue and transcription factors are in red. TF indicates transcription factor.
Acknowledgments
The authors would like to thank Naim Rashid (UNC Lineberger Cancer Center) for his statistical expertise.
(J Am Heart Assoc. 2017;6:e006635 DOI: 10.1161/JAHA.117.006635.)
References
- 1. Chen ZI, Ai DI. Cardiotoxicity associated with targeted cancer therapies. Mol Clin Oncol. 2016;4:675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hasinoff BB, Patel D. The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro. Toxicol Appl Pharmacol. 2010;249:132–139. [DOI] [PubMed] [Google Scholar]
- 3. Kerkela R, Woulfe KC, Durand JB, Vagnozzi R, Kramer D, Chu TF, Beahm C, Chen MH, Force T. Sunitinib‐induced cardiotoxicity is mediated by off‐target inhibition of AMP‐activated protein kinase. Clin Transl Sci. 2009;2:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Graves LM, Duncan JS, Whittle MC, Johnson GL. The dynamic nature of the kinome. Biochem J. 2013;450:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, Usary J, Kuan PF, Smalley DM, Major B, He X, Hoadley KA, Zhou B, Sharpless NE, Perou CM, Kim WY, Gomez SM, Chen X, Jin J, Frye SV, Earp HS, Graves LM, Johnson GL. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple‐negative breast cancer. Cell. 2012;149:307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Di Lorenzo G, Autorino R, Bruni G, Carteni G, Ricevuto E, Tudini M, Ficorella C, Romano C, Aieta M, Giordano A, Giuliano M, Gonnella A, De Nunzio C, Rizzo M, Montesarchio V, Ewer M, De Placido S. Cardiovascular toxicity following sunitinib therapy in metastatic renal cell carcinoma: a multicenter analysis. Ann Oncol. 2009;20:1535–1542. [DOI] [PubMed] [Google Scholar]
- 7. Schmidinger M, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C, Ruhsam M, Hejna M, Schmidinger H. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008;26:5204–5212. [DOI] [PubMed] [Google Scholar]
- 8. Narayan V, Keefe SM, Haas N, Wang L, Puzanov I, Putt ME, Catino A, Fang J, Agarwal N, Hyman D, Smith AM, Finkelman BS, Narayan HK, Ewer S, ElAmm C, Lenihan DJ, Ky B. Prospective evaluation of sunitinib‐induced cardiotoxicity in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2017;23:3601–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doherty KR, Wappel RL, Talbert DR, Trusk PB, Moran DM, Kramer JW, Brown AM, Shell SA, Bacus S. Multi‐parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol Appl Pharmacol. 2013;272:245–255. [DOI] [PubMed] [Google Scholar]
- 10. Hervent AS, De Keulenaer GW. Molecular mechanisms of cardiotoxicity induced by ErbB receptor inhibitor cancer therapeutics. Int J Mol Sci. 2012;13:12268–12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harvey PA, Leinwand LA. Oestrogen enhances cardiotoxicity induced by sunitinib by regulation of drug transport and metabolism. Cardiovasc Res. 2015;107:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KA, Granger DA, Reuther RA, Graves LM, Gomez SM, Kuan PF, Parker JS, Chen X, Sciaky N, Carey LA, Earp HS, Jin J, Johnson GL. Inhibition of lapatinib‐induced kinome reprogramming in ERBB2‐positive breast cancer by targeting BET family bromodomains. Cell Rep. 2015;11:390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, MacLeod JN, Chiang DY, Prins JF, Liu J. MapSplice: accurate mapping of RNA‐seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simpson P, Savion S. Differentiation of rat myocytes in single cell cultures with and without proliferating nonmyocardial cells. Cross‐striations, ultrastructure, and chronotropic response to isoproterenol. Circ Res. 1982;50:101–116. [DOI] [PubMed] [Google Scholar]
- 17. Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. [DOI] [PubMed] [Google Scholar]
- 18. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–3140. [DOI] [PubMed] [Google Scholar]
- 19. Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, Bortolon E, Ichetovkin M, Chen C, McNabola A, Wilkie D, Carter CA, Taylor IC, Lynch M, Wilhelm S. Sorafenib (BAY 43‐9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59:561–574. [DOI] [PubMed] [Google Scholar]
- 20. Hashita T, Katsuyama Y, Nakamura K, Momose Y, Komatsu D, Koide N, Miyagawa S, Nakamura T, Yamamoto K, Ohmori S. Treatment of a GIST patient with modified dose of sunitinib by measurement of plasma drug concentrations. Oncol Lett. 2012;4:501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- 22. Schem C, Bauerschlag D, Bender S, Lorenzen AC, Loermann D, Hamann S, Rosel F, Kalthoff H, Gluer CC, Jonat W, Tiwari S. Preclinical evaluation of sunitinib as a single agent in the prophylactic setting in a mouse model of bone metastases. BMC Cancer. 2013;13:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frohna P, Lu J, Eppler S, Hamilton M, Wolf J, Rakhit A, Ling J, Kenkare‐Mitra SR, Lum BL. Evaluation of the absolute oral bioavailability and bioequivalence of erlotinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in a randomized, crossover study in healthy subjects. J Clin Pharmacol. 2006;46:282–290. [DOI] [PubMed] [Google Scholar]
- 24. Deeken JF, Beumer JH, Anders NM, Wanjiku T, Rusnak M, Rudek MA. Preclinical assessment of the interactions between the antiretroviral drugs, ritonavir and efavirenz, and the tyrosine kinase inhibitor erlotinib. Cancer Chemother Pharmacol. 2015;76:813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacob F, Yonis AY, Cuello F, Luther P, Schulze T, Eder A, Streichert T, Mannhardt I, Hirt MN, Schaaf S, Stenzig J, Force T, Eschenhagen T, Hansen A. Analysis of tyrosine kinase inhibitor‐mediated decline in contractile force in rat engineered heart tissue. PLoS One. 2016;11:e0145937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li W, Croce K, Steensma DP, McDermott DF, Ben‐Yehuda O, Moslehi J. Vascular and metabolic implications of novel targeted cancer therapies: focus on kinase inhibitors. J Am Coll Cardiol. 2015;66:1160–1178. [DOI] [PubMed] [Google Scholar]
- 27. Fischer P, Hilfiker‐Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130‐STAT3 axis. Basic Res Cardiol. 2007;102:279–297. [DOI] [PubMed] [Google Scholar]
- 28. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF‐kappaB in the heart: to be or not to NF‐kappaB. Circ Res. 2011;108:1122–1132. [DOI] [PubMed] [Google Scholar]
- 29. Zheng JS, Boluyt MO, Long X, O'Neill L, Lakatta EG, Crow MT. Extracellular ATP inhibits adrenergic agonist‐induced hypertrophy of neonatal cardiac myocytes. Circ Res. 1996;78:525–535. [DOI] [PubMed] [Google Scholar]
- 30. Schips TG, Wietelmann A, Hohn K, Schimanski S, Walther P, Braun T, Wirth T, Maier HJ. FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc Res. 2011;91:587–597. [DOI] [PubMed] [Google Scholar]
- 31. Giannessi D, Del Ry S, Vitale RL. The role of endothelins and their receptors in heart failure. Pharmacol Res. 2001;43:111–126. [DOI] [PubMed] [Google Scholar]
- 32. Kappers MH, Smedts FM, Horn T, van Esch JH, Sleijfer S, Leijten F, Wesseling S, Strevens H, Jan Danser AH, van den Meiracker AH. The vascular endothelial growth factor receptor inhibitor sunitinib causes a preeclampsia‐like syndrome with activation of the endothelin system. Hypertension. 2011;58:295–302. [DOI] [PubMed] [Google Scholar]
- 33. Rosano L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Caprara V, Semprucci E, Ferrandina G, Natali PG, Bagnato A. Endothelin A receptor/beta‐arrestin signaling to the WNT pathway renders ovarian cancer cells resistant to chemotherapy. Cancer Res. 2014;74:7453–7464. [DOI] [PubMed] [Google Scholar]
- 34. Kim SW, Choi HJ, Lee HJ, He J, Wu Q, Langley RR, Fidler IJ, Kim SJ. Role of the endothelin axis in astrocyte‐ and endothelial cell‐mediated chemoprotection of cancer cells. Neuro Oncol. 2014;16:1585–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ding J, Chen J, Wang Y, Kataoka M, Ma L, Zhou P, Hu X, Lin Z, Nie M, Deng ZL, Pu WT, Wang DZ. Trbp regulates heart function through microRNA‐mediated Sox6 repression. Nat Genet. 2015;47:776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y, Fang B, Emmett MJ, Damle M, Sun Z, Feng D, Armour SM, Remsberg JR, Jager J, Soccio RE, Steger DJ, Lazar MA. GENE REGULATION. Discrete functions of nuclear receptor Rev‐erbalpha couple metabolism to the clock. Science. 2015;348:1488–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Woldt E, Sebti Y, Solt LA, Duhem C, Lancel S, Eeckhoute J, Hesselink MK, Paquet C, Delhaye S, Shin Y, Kamenecka TM, Schaart G, Lefebvre P, Neviere R, Burris TP, Schrauwen P, Staels B, Duez H. Rev‐erb‐alpha modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat Med. 2013;19:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. French KJ, Coatney RW, Renninger JP, Hu CX, Gales TL, Zhao S, Storck LM, Davis CB, McSurdy‐Freed J, Chen E, Frazier KS. Differences in effects on myocardium and mitochondria by angiogenic inhibitors suggest separate mechanisms of cardiotoxicity. Toxicol Pathol. 2010;38:691–702. [DOI] [PubMed] [Google Scholar]
- 39. Sharma V, Mustafa S, Patel N, Wambolt R, Allard MF, McNeill JH. Stimulation of cardiac fatty acid oxidation by leptin is mediated by a nitric oxide‐p38 MAPK‐dependent mechanism. Eur J Pharmacol. 2009;617:113–117. [DOI] [PubMed] [Google Scholar]
- 40. Hung MH, Tai WT, Shiau CW, Chen KF. Downregulation of signal transducer and activator of transcription 3 by sorafenib: a novel mechanism for hepatocellular carcinoma therapy. World J Gastroenterol. 2014;20:15269–15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsu FT, Liu YC, Chiang IT, Liu RS, Wang HE, Lin WJ, Hwang JJ. Sorafenib increases efficacy of vorinostat against human hepatocellular carcinoma through transduction inhibition of vorinostat‐induced ERK/NF‐kappaB signaling. Int J Oncol. 2014;45:177–188. [DOI] [PubMed] [Google Scholar]
- 42. Judd LM, Menheniott TR, Ling H, Jackson CB, Howlett M, Kalantzis A, Priebe W, Giraud AS. Inhibition of the JAK2/STAT3 pathway reduces gastric cancer growth in vitro and in vivo. PLoS One. 2014;9:e95993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qu X, Zhuang G, Yu L, Meng G, Ferrara N. Induction of Bv8 expression by granulocyte colony‐stimulating factor in CD11b+Gr1+ cells: key role of Stat3 signaling. J Biol Chem. 2012;287:19574–19584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fuller SJ, Osborne SA, Leonard SJ, Hardyman MA, Vaniotis G, Allen BG, Sugden PH, Clerk A. Cardiac protein kinases: the cardiomyocyte kinome and differential kinase expression in human failing hearts. Cardiovasc Res. 2015;108:87–98. [DOI] [PubMed] [Google Scholar]
- 45. Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares‐Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU, Chen X, Duncan JS, Whittle MC, Angus SP, Velarde SH, Golitz BT, He X, Santos C, Darr DB, Gallagher K, Graves LM, Perou CM, Carey LA, Earp HS, Johnson GL. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P‐TEFb complex. Cancer Discov. 2017;7:302–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7:332–344. [DOI] [PubMed] [Google Scholar]
- 47. Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, Moonsamy P, Dahlman K, Miller VA, Costa C, Hannon G, Sawyers CL. FAS and NF‐kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tang J, Guo F, Du Y, Liu X, Qin Q, Liu X, Yin T, Jiang L, Wang Y. Continuous exposure of non‐small cell lung cancer cells with wild‐type EGFR to an inhibitor of EGFR tyrosine kinase induces chemoresistance by activating STAT3. Int J Oncol. 2015;46:2083–2095. [DOI] [PubMed] [Google Scholar]
- 49. Harada D, Takigawa N, Ochi N, Ninomiya T, Yasugi M, Kubo T, Takeda H, Ichihara E, Ohashi K, Takata S, Tanimoto M, Kiura K. JAK2‐related pathway induces acquired erlotinib resistance in lung cancer cells harboring an epidermal growth factor receptor‐activating mutation. Cancer Sci. 2012;103:1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ioannou N, Seddon AM, Dalgleish A, Mackintosh D, Solca F, Modjtahedi H. Acquired resistance of pancreatic cancer cells to treatment with gemcitabine and HER‐inhibitors is accompanied by increased sensitivity to STAT3 inhibition. Int J Oncol. 2016;48:908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wen W, Wu J, Liu L, Tian Y, Buettner R, Hsieh MY, Horne D, Dellinger TH, Han ES, Jove R, Yim JH. Synergistic anti‐tumor effect of combined inhibition of EGFR and JAK/STAT3 pathways in human ovarian cancer. Mol Cancer. 2015;14:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang X, Goldstein D, Crowe PJ, Yang M, Garrett K, Zeps N, Yang JL. Overcoming resistance of targeted EGFR monotherapy by inhibition of STAT3 escape pathway in soft tissue sarcoma. Oncotarget. 2016;7:21496–21509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, Maxwell J, Wang L, Gooding WE, Johnson DE, Grandis JR. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin Cancer Res. 2012;18:4986–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fuller SJ, Sivarajah K, Sugden PH. ErbB receptors, their ligands, and the consequences of their activation and inhibition in the myocardium. J Mol Cell Cardiol. 2008;44:831–854. [DOI] [PubMed] [Google Scholar]
- 55. Nemeth BT, Varga ZV, Wu WJ, Pacher P. Trastuzumab cardiotoxicity: from clinical trials to experimental studies. Br J Pharmacol. 2016. Available at: http://onlinelibrary.wiley.com/wol1/doi/10.1111/bph.13643/full. Accessed October 11, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bolli R, Stein AB, Guo Y, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu Y, Xuan YT. A murine model of inducible, cardiac‐specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol. 2011;50:589–597. [DOI] [PubMed] [Google Scholar]
- 57. Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res. 2011;109:1302–1308. [DOI] [PubMed] [Google Scholar]
- 58. Zouein FA, Kurdi M, Booz GW. Dancing rhinos in stilettos: the amazing saga of the genomic and nongenomic actions of STAT3 in the heart. JAKSTAT. 2013;2:e24352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zelarayan L, Renger A, Noack C, Zafiriou MP, Gehrke C, van der Nagel R, Dietz R, de Windt L, Bergmann MW. NF‐kappaB activation is required for adaptive cardiac hypertrophy. Cardiovasc Res. 2009;84:416–424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. MIB‐MS Summary Data
Table S2. RNAseq by Expectation Maximization (RSEM) Data for RNAseq
Table S3. Gene Set Enrichment Analysis (GSEA) Results
Figure S1. Conscious echocardiography compared structural and functional cardiac parameters. Female wild‐type FVB mice (n=4 for vehicle, n=5 for KIs) were gavaged with vehicle, erlotinib (50 mg/kg per d), sunitinib (40 mg/kg per d), or sorafenib (30 mg/kg per d) for 14 days. Conscious echocardiograms were performed at baseline, 7 days, and 14 days. Repeated‐measures ANOVA compared intragroup changes. (*P<0.05). LVID indicates left ventricular internal diameter.
Figure S2. Graphical summary of Multiplex Inhibitor Beads–Mass Spectrometry (MIB‐MS) kinome profiling of kinase inhibitor‐treated mouse hearts. Kinase activity depicted with reference to vehicle‐treated hearts.
Figure S3. Heat maps for Gene Set Enrichment Analysis of RNAseq data. Upregulation of transcripts in NFκB and STAT3 pathways distinguishes erlotinib from sunitinib and sorafenib. IL6 indicates interleukin 6; NFκB, nuclear factor kappa B; STAT3, signal transducer and activator of transcription 3; TNFα, tumor necrosis factor α.
Figure S4. Volcano plots comparing statistically significant (DEseq2) transcriptional responses to cardiosafe (control and erlotinib) or cardiotoxic (sorafenib and sunitinib) treatments. Kinases are in blue and transcription factors are in red. TF indicates transcription factor.