

Abstract
Background
Aberrant Ca2+ handling is a prominent feature of heart failure. Elucidation of the molecular mechanisms responsible for aberrant Ca2+ handling is essential for the development of strategies to blunt pathological changes in calcium dynamics. The peptidyl‐prolyl cis‐trans isomerase peptidyl‐prolyl isomerase 1 (Pin1) is a critical mediator of myocardial hypertrophy development and cardiac progenitor cell cycle. However, the influence of Pin1 on calcium cycling regulation has not been explored. On the basis of these findings, the aim of this study is to define Pin1 as a novel modulator of Ca2+ handling, with implications for improving myocardial contractility and potential for ameliorating development of heart failure.
Methods and Results
Pin1 gene deletion or pharmacological inhibition delays cytosolic Ca2+ decay in isolated cardiomyocytes. Paradoxically, reduced Pin1 activity correlates with increased sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) and Na2+/Ca2+ exchanger 1 protein levels. However, SERCA2a ATPase activity and calcium reuptake were reduced in sarcoplasmic reticulum membranes isolated from Pin1‐deficient hearts, suggesting that Pin1 influences SERCA2a function. SERCA2a and Na2+/Ca2+ exchanger 1 associated with Pin1, as revealed by proximity ligation assay in myocardial tissue sections, indicating that regulation of Ca2+ handling within cardiomyocytes is likely influenced through Pin1 interaction with SERCA2a and Na2+/Ca2+ exchanger 1 proteins.
Conclusions
Pin1 serves as a modulator of SERCA2a and Na2+/Ca2+ exchanger 1 Ca2+ handling proteins, with loss of function resulting in impaired cardiomyocyte relaxation, setting the stage for subsequent investigations to assess Pin1 dysregulation and modulation in the progression of heart failure.
Keywords: cardiomyocyte, Na+/Ca2+ exchange, peptidyl‐prolyl isomerase 1, sarcoplasmic reticulum Ca2+‐ATPase
Subject Categories: Cell Signalling/Signal Transduction, Calcium Cycling/Excitation-Contraction Coupling, Cell Biology/Structural Biology, Myocardial Biology
Clinical Perspective
What Is New?
This study reveals a novel role for peptidyl‐prolyl isomerase 1 in modulating the expression and function of Ca2+‐handling proteins sarco(endo)plasmic reticulum calcium ATPase and Na2+/Ca2+ exchanger 1, with peptidyl‐prolyl isomerase 1 loss of function resulting in impaired cardiomyocyte relaxation.
What Are the Clinical Implications?
Decreased peptidyl‐prolyl isomerase 1 activity could contribute to impairment of Ca2+ cycling in the heart, resulting in diminished contractile function under stress and loss of inotropic responsiveness in the failing heart. Cardiac‐specific peptidyl‐prolyl isomerase 1 overexpression could serve as a molecular interventional strategy to normalize Ca2+ handling within cardiomyocytes laboring under pathological stress.
Introduction
Ca2+ is a ubiquitous intracellular second messenger exerting several fundamental roles in the myocardium, including control of cardiomyocyte mechanical behavior and programmed cell death, as well as regulation of pathological hypertrophic remodeling through Ca2+/calmodulin‐dependent kinase II (CaMKII) signaling.1, 2 Ca2+‐dependent signaling is highly regulated in cardiomyocytes and determines force of cardiac muscle contraction. Ca2+ uptake and release is profoundly altered in failing hearts, resulting in impaired contractility and fatal cardiac arrhythmias.3, 4 Considering the crucial role of Ca2+ in regulating cardiac muscle contraction, much attention has been paid to understanding the role of defects in Ca2+ regulation in heart failure (HF).4
Impaired sarcoplasmic reticulum (SR) Ca2+ uptake causes deterioration to both systolic and diastolic heart function.4, 5 Ca2+ removal from the cytosol occurs by both sarco(endo)plasmic reticulum calcium ATPase (SERCA2a)–mediated uptake and sarcolemmal extrusion, mainly via Na2+/Ca2+ exchanger 1 (NCX‐1). During homeostasis, the amount of Ca2+ recycled by the SR in the relaxation phase equals the amount released, and influx of Ca2+ equals the amount extruded.2 In HF, SERCA2a function is decreased and NCX‐1 activity is enhanced, causing impaired relaxation, which leads to reductions of SR Ca2+ load and cardiomyocyte contractility.2, 4, 6 Cardiac contractility was improved in preclinical and clinical trials by viral overexpression of SERCA2a; it can be accompanied by arrhythmia.4 As with SERCA2a, additional therapeutic targets in key pathways that regulate cardiac Ca2+ dynamics can be revealed by unraveling molecular mechanisms regulating Ca2+ cycling proteins.4
Posttranslational modifications, such as reversible protein phosphorylation, form the basis of several complex signaling cascades governing cellular fate during cardiac pathophysiological conditions.7 However, another layer of regulation beyond binary “on/off” switching mechanisms is added by the peptidyl cis‐to‐trans isomerases (PPIases) that tune intensity and duration of signals by switching the 3‐dimensional conformation of target phosphoproteins.7, 8
Peptidyl‐prolyl isomerase 1 (Pin1) is a unique PPIase that selectively promotes the cis‐to‐trans isomerization on specific recognition of a major regulatory phosphorylation motif (Ser/Thr‐Pro) belonging to the family of Pro‐directed protein kinases that include protein kinase B (Akt), Proto‐oncogene serine/threonine kinase (Pim‐1), cyclin‐dependent protein kinases, and many more canonical cardiac target proteins.7, 9, 10, 11 Pin1 is a small protein (163 amino acids) but highly conserved; it is composed of an N‐terminal WW domain that directs Pin1 to appropriate substrates and a C‐terminal PPIase domain that controls target‐protein conformation switching.9, 10, 11 Previous studies identified Pin1 as an essential regulator of heart hypertrophy, induced by transaortic constriction,12 and of the cardiac progenitor cell cycle.13
PPIase family members other than Pin1 regulate calcium ion channel function, but the influence and role of Pin1 in Ca2+ handling regulation remained unaddressed.14 This study highlights a novel function of Pin1 in the heart as a regulator of Ca2+ cycling, showing that Pin1 controls Ca2+ handling in cardiomyocytes by controlling Ca2+ clearance through modulation of SERCA2a and NCX‐1 protein levels and function. Inclusion of Pin1 as a novel regulator of cardiomyocyte calcium dynamics opens up novel potential avenues for molecular interventional approaches to normalize calcium homeostasis in the pathological myocardium.
Methods
Mice
Pin1+/− and Pin1−/− mice were obtained from Dr Takafumi Uchida (Tohoku University, Miyagi, Japan) and from Dr James Malter (University of Texas Southwestern, Dallas, TX). Nontransgenic (wild‐type [WT]) C57/B6 mice were used as controls. Mice enrolled in the study were ≈6 to 7 months of age, and no differences in the baseline global phenotype were observed in nontransgenic and transgenic mice. An n=5 animals/group was used for the experiment described in Figure 1, whereas an n=3 animals/group was used for all other experiments. All animal protocols and studies were approved by the institutional review board of the Animal Care and Use Committee at San Diego State University (San Diego, CA).
Figure 1.
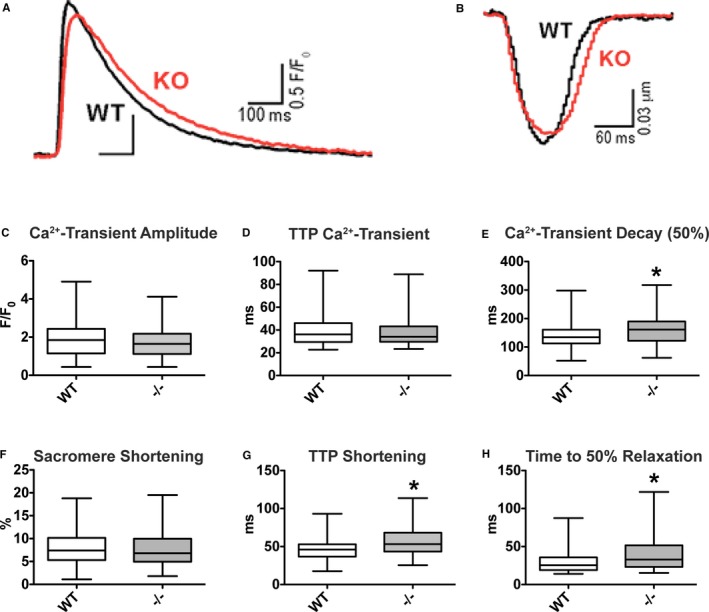
Ca2+ transient decay and time to peak (TTP) shortening and relaxation are delayed in peptidyl‐prolyl isomerase 1 (Pin1)−/− cardiomyocytes. A and B, Pin1−/− cardiac myocytes have delayed Ca2+ transient decay (A) and TTP sarcomere shortening (B), as shown by representative superimposed traces. For each cell, an average trace of >10 beats is reported. C through E, Ca2+ transient amplitude (C) and TTP (D) are unchanged in Pin1−/− myocytes vs wild type (WT), but Ca2+ transient decay (E) is significantly increased by loss of Pin1. WT (n=166 cells from 5 hearts) and Pin1−/− (n=162 cells from 5 hearts) mice are shown. F through H, Sarcomere shortening (F) is unaltered by loss of Pin1, whereas TTP shortening (G) and relaxation (H) are significantly increased in Pin1−/− cardiac myocytes. WT (n=106 cells from 5 hearts) and Pin1−/− (n=108 cells from 5 hearts) mice are shown. Data presented as median and interquartile ranges. *P<0.05 (Student t test).
Ca2+ Cycling and Contractility Assessment
Left ventricular cardiomyocytes, isolated as previously described,15, 16 were placed in a heated bath at 31°C on the stage of inverted microscopes for contractility and Ca2+ transients. Cells were bathed continuously with Tyrode solution containing 1 mmol/L CaCl2, and the myocyte field was stimulated at 1 Hz. Sarcomere shortening was measured with an IonOptix system, whereas intracellular Ca2+ handling was assessed in cells loaded with 0.57 μmol/L Fluo‐4 AM with an epifluorescence system. Setups were coupled with Digidata or DI‐155 interface and pClamp or WinDaq software. Data were analyzed with pClamp or LabChart software. Fluo‐4 signals were expressed as normalized fluorescence (F/F0), where F0 is the diastolic fluorescent level subtracted by the background signal measured in the region adjacent to the cell, as described.16, 17 To assess Ca2+ cycling in response to short‐term Pin1 inhibition, isolated cardiomyocytes were pretreated for 2 hours with either Pin1 inhibitor (1 μmol/L juglone) or water as vehicle control before intracellular Ca2+ handling measurement at room temperature.
Immunoblot Analysis
Whole heart lysates were prepared by homogenizing tissues in lysis buffer containing 1 mol/L Tris‐HCl (pH 7.4), 5 mol/L NaCl, 100 mmol/L EDTA, and 100 mmol/L EGTA, supplemented with protease and phosphatase inhibitor cocktails using a Next Advance bullet blender at 4°C. Tissue homogenates were transferred to clean tubes, Triton X‐100 was added at 1% final concentration, and lysates were incubated on ice for 60 minutes. Homogenates were centrifuged at 16 100g, and supernatants were collected for further experiments. Adult cardiomyocytes were isolated, as previously described.18 Cardiomyocytes were treated with either Pin1 inhibitor (10 nmol/L; or 1 μmol/L juglone) or water as vehicle control for 2 hours. Afterwards, medium was removed and isolation buffer containing 1 mol/L Tris‐HCl (pH 7.4), 5 mol/L NaCl, 100 mmol/L EDTA, 100 mmol/L EGTA, and 1% Triton X‐100, supplemented with protease and phosphatase inhibitor cocktails, was added to the cells for isolating proteins by cell scraping. Tissue and cell lysates were stored at −80°C until further use. Protein concentration was determined by Bradford assay using BSA standards and normalized to the samples having the lowest protein concentration. Proteins lysates were prepared by adding LDS Sample Buffer with 50 mmol/L dithiothreitol. Samples were boiled for 5 minutes at 70°C to denature proteins. Proteins were separated by SDS‐PAGE (15–20 μg in each gel lane) and transferred to Immobilon‐FL polyvinylidene difluoride membranes. Primary antibodies were incubated overnight in TBS Blocking Buffer at 4°C, and secondary antibodies were incubated for 90 minutes at room temperature in blocking buffer. Fluorescent signals were detected using a Scanner Odyssey CLx and quantified using software. Antibodies and dilutions are listed in Table S1.
In Silico Analysis of SERCA2a and NCX‐1 Proteins to Identify Putative Pin1 Binding Sites
SERCA2a and NCX‐1 murine protein sequences were selected from the Protein National Center for Biotechnology Information Database. The accession number for SERCA2a was NP_033852.1; and for NCX‐1, NP_035536.2. The presence of serine next to a proline is highlighted by a black square, whereas the presence of a threonine next to a proline is highlighted by a red square.
Proximity Ligation Assay
Proximity ligation assay was performed as previously described.12, 19, 20, 21, 22 Briefly, hearts fixed in 10% formalin were embedded in paraffin, sectioned at a thickness of 4 µm, and stained for Pin1 and Serca2a/Ncx‐1 primary antibodies. Isolated cardiomyocytes, as described in Immunoblot analysis section, were fixed in 4% paraformaldehyde and permeabilized with 0.3% Triton X‐100 before proceeding with primary antibody staining. Signal was detected using Duolink In Situ Detection Reagent and Probes. Myosin light chain 2 was used to visualize myocardium. Antibody manufacturers and dilutions are listed in Table S1. Duolink In Situ Mounting Medium with 4′,6‐diamidino‐2‐phenylindole was used to counterstain nuclei and coverslip. Scans consisted of Z‐stack random regions for each heart analyzed using an SP8 Confocal Microscope using 40× objective. The number of events was quantified using the ImageJ particles counting tool in each field.
Glutathione S‐Transferase and Glutathione S‐Transferase–Pin1 Recombinant Protein Production
Recombinant glutathione S‐transferase (GST) protein was purchased. The expression plasmid for GST‐Pin1 was also purchased. GST‐Pin1 protein was produced8 with minor adaptation. One Shot Top10 Escherichia coli cells were transformed and amplified in Luria‐Bertani (LB) medium containing ampicillin at 100 μg/mL overnight at 37°C, 67 g overnight. The optical density at 600 optical density was measured by a biophotometer and adjusted with fresh LB medium between 0.4 and 0.6 optical density because less growth or overgrowth will affect future GST‐Pin1 protein expression. LB (400 mL) and 1 mol/L isopropyl β‐d‐1‐thiogalactopyranoside (400 µL) were added to the culture and incubated for another 5 hours at 37°C, 67 g. The bacteria pellets were thereafter obtained by centrifugation at 0°C, 1015 g for 25 minutes. Bacteria pellets were diluted 1:1 in lysis buffer containing 50 mmol/L Tris‐HCl (pH 7.5), 100 mmol/L NaCl, and 5 mmol/L dithiothreitol, supplemented with protease and phosphatase inhibitor cocktails. They were lysed by sonication using 10 short pulses (5–10 seconds), followed by 30‐second pauses to reestablish a low temperature. Cell debris was removed by centrifugation at 0°C, 4872 g for 25 minutes. GST‐Pin1 protein was purified from crude lysates using the Pierce GST Spin Purification Kit, according to the manufacturer's instructions. Protein expression was confirmed by immunoblot for GST, as described in the Immunoblot Analysis subsection of the Methods section.
Pull‐Down Assay
Pull‐down assay using GST‐Pin1 was performed8 with minor adaptation. Briefly, cardiomyocytes were isolated from WT mice18 and incubated in 1 mL of lysis buffer containing 50 mmol/L HEPES (pH 7.4), 150 mmol/L NaCl, 10% glycerol, 1% Triton X‐100, 1.5 mmol/L MgCl2, 1 mmol/L EGTA, 100 mmol/L NaF, 1 mmol/L Na3VO4, and 1 mmol/L dithiothreitol, supplemented with protease and phosphatase inhibitor cocktails for 30 minutes at 4°C on a rotator. Cell lysates were centrifuged at 16 100g for 20 minutes at 4°C to pellet cell debris, and 200 μL of supernatant was transferred to a clean 1.5‐mL tube for each assay. GST or GST‐Pin1 fusion protein at 2 μmol/L was added to supernatant, and samples were rotated at 4°C for 1 hour. GST beads (20 µL) were added to the samples and rotated at 4°C for 1 hour. GST beads were pelleted by centrifugation at 9300g and washed 3 times with 1 mL of lysis buffer. Afterwards, lysis buffer was removed and beads were resuspended in 20 μL of LDS Sample Buffer with 50 mmol/L dithiothreitol. Samples were boiled for 5 minutes at 70°C to denature proteins, GST beads were pelleted by centrifugation at 9300g, and supernatant was applied to the SDS‐PAGE gel. Immunoblot was performed, as described above, using primary antibody dilutions listed in Table S1.
SR‐Membrane Enrichment
SR‐enriched fractions were isolated from WT, Pin1+/−, and Pin1−/− hearts, as previously described,23, 24 with minor adaptations. Briefly, myocardium was homogenized in buffer containing the following: 5 mmol/L HEPES, 250 mmol/L sucrose , 0.2% sodium azide, and a cocktail of phosphatase and protease inhibitors. The homogenate was centrifuged at 5500g for 10 minutes to remove cellular debris. The supernatant was then transferred to clean tubes and centrifuged at 12 500g for 18 minutes. These pellets were discarded, and the spin was repeated. Again, the supernatant was transferred to clean tubes and centrifuged at maximal speed for 1 hour. These pellets were resuspended in homogenizing buffer plus 600 mmol/L KCl and allowed to incubate at 4°C for 30 minutes. This suspension was then centrifuged at maximal speed for 1 hour. The final pellet, enriched in SR membranes, was resuspended in homogenizing buffer and stored at −80°C until assayed.
SERCA2a ATPase Activity
SERCA2a Ca2+‐dependent ATPase activity was measured on 10 μg/mL of SR preparations using the High Throughput Colorimetric ATPase assay kit, according to manufacturer's instructions. This was done in the presence of 2 μmol/L of the calcium ionophore A23187 and of an escalating concentration of free Ca2+ (0–0.5–1–2.5–5–8–10 μmol/L). Reactions were incubated for 30 minutes at room temperature. SERCA‐independent Ca2+‐ATPase activity was measured in the presence of 100 nmol/L of thapsigargin and subtracted from total SR‐ATPase activity to calculate specific SERCA2a‐ATPase hydrolysis activity. The activity of SERCA2a was calculated as hydrolyzed ATP levels (μmol/L) normalized with protein content (mg) and reaction time (minutes) from 3 independent experiments.
SR Uptake and Release
Oxalate‐supported Ca2+‐uptake rates were measured using the Ca2+ fluorescent dye indo‐1, according to published methods,23, 25 with minor adaptation for use on a plate reader (SPECTRAmax Plus). Immediately before collection of emission spectra, 25 μg of isolated SR membranes was added to each well. Baseline reads were collected until fluorescence was not stable; then, CaCl2 was added to each well to produce a consistent starting of [Ca2+]f of 3.5 μmol/L. Shortly after the attainment of a constant [Ca2+]f, 5 mmol/L ATP was added to each well to initiate Ca2+ uptake. Maximum and minimum Ca2+‐dependent fluorescence was determined after the Ca2+ uptake reaction by additions of 250 μmol/L of the Ca2+ chelator EGTA and 1 mmol/L of the CaCl2. 4‐Chloro‐m‐cresol (20 mmol/L) was added to the assay mixture to chemically stimulate Ca2+ release in vitro.
Statistical Analysis
Data in Figure 1 were represented as median and interquartile ranges; in Figures 2, 3, 4 through 5, as mean±SEM; and in Figure 6, as median and interquartile ranges. Statistical analysis was performed using the unpaired Student t test or 1‐way ANOVA, with a Dunnett post‐hoc test to compare each group with a control group using GraphPad Prism version 5.0. When normality or equal variance was not met, nonparametric analysis was performed using a Mann‐Whitney rank sum test.16 P<0.05 was considered statistically significant.
Figure 2.
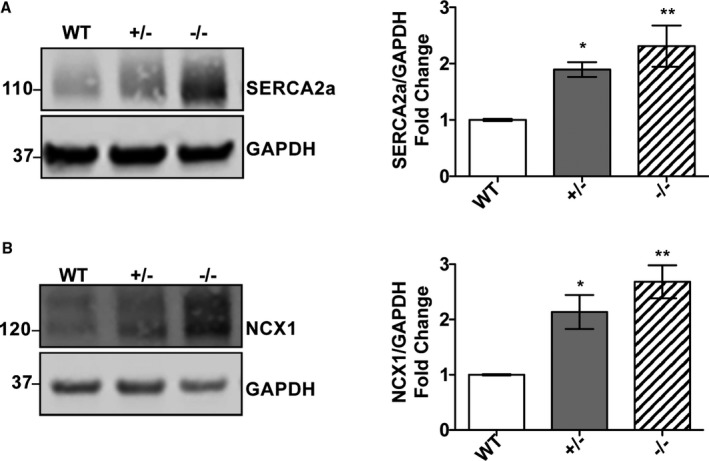
Sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) and Na2+/Ca2+ exchanger 1 (NCX‐1) protein levels are elevated in peptidyl‐prolyl isomerase 1 (Pin1)–deficient hearts. Immunoblot showing SERCA2a (A) and NCX‐1 (B) levels significantly increased in Pin1+/− and Pin1−/− hearts compared with wild type (WT). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a loading control. Quantification of protein expression below each representative blot shown as n‐fold expression/WT. N=3. Data presented as mean±SEM. *P<0.05, † P<0.01 vs WT (1‐way ANOVA, followed by Dunnett post‐hoc analysis).
Figure 3.
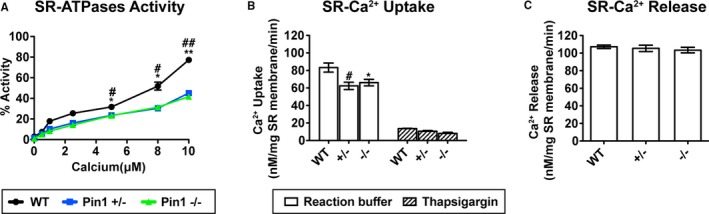
Cardiac sarcoplasmic reticulum (SR) Ca2+‐ATPase activity and uptake are impaired in peptidyl‐prolyl isomerase 1 (Pin1)–deficient hearts. A, Reduced Ca2+‐dependent ATPase activity by purified Pin1+/− and Pin1−/− cardiac SR membranes determined on the basis of release of inorganic phosphate. Specificity of SR Ca2+‐ATPase activity assessed in the presence of 100 nmol/L of thapsigargin. B and C, SR Ca2+ uptake (B) reduced in Pin1+/− and Pin1−/− isolated SR membranes, whereas SR release (C) was not influenced by Pin1 loss, as measured using the Ca2+ fluorescent dye Indo‐1. A through C, N=3±SEM. *P<0.05, † P<0.01 vs Pin1−/−; ‡ P<0.05, § P<0.01 wild type (WT) vs Pin1+/− (1‐way ANOVA, with Dunnett post‐hoc test; A). *P<0.05 WT vs Pin1−/−, † P<0.05 WT vs Pin1+/− (1‐way ANOVA, followed by Dunnett post‐hoc test; B and C).
Figure 4.
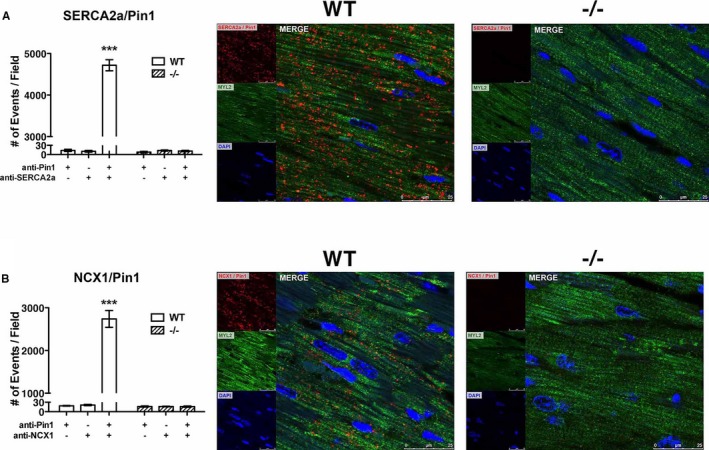
Sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) and Na2+/Ca2+ exchanger 1 (NCX‐1) interact with peptidyl‐prolyl isomerase 1 (Pin1) in vivo. Pin1 interaction with SERCA2a (A) and NCX‐1 (B), as determined by proximity ligation assay (PLA). Number of events detected on tissue sections stained with both primary antibodies and single primary antibodies are shown on the left panels. Representative micrographs of PLA showing direct interaction of Pin1 with NCX‐1 and SERCA2a (red dots) are presented on the right panels. Heart sections were also stained with myosin light chain 2 (MYL2; green) and 4′,6‐diamidino‐2‐phenylindole (DAPI; blue). N=3. Size bar=25 μm. Data are mean±SEM. *P<0.0001 vs wild‐type (WT) NC (1‐way ANOVA, followed by Dunnett post‐hoc test). NC indicate Negative Control.
Figure 5.
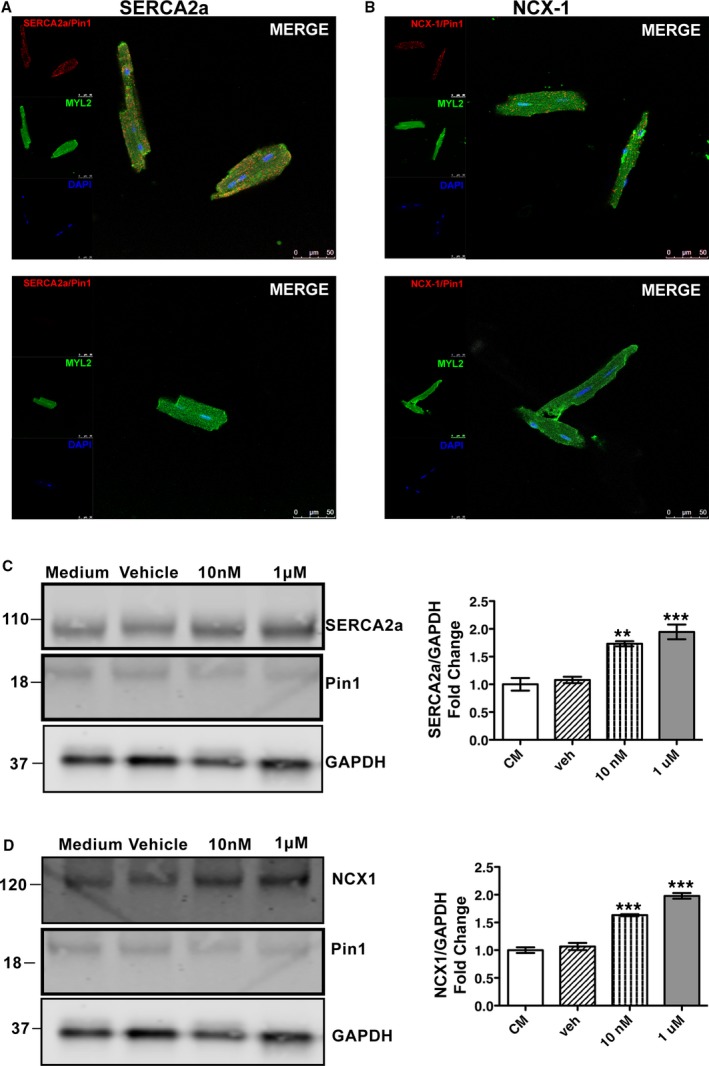
Sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) and Na2+/Ca2+ exchanger 1 (NCX‐1) interaction with peptidyl‐prolyl isomerase 1 (Pin1) is disrupted by Pin1 inhibition. A and B, SERCA2a (A) and NCX‐1 (B) interaction with Pin1 determined by proximity ligation assay on isolated wild‐type (WT) cardiomyocytes treated with either Pin1 inhibitor juglone or vehicle control for 2 hours. N=3. C and D, SERCA2a and NCX‐1 protein expression dose dependently accumulates in cardiomyocytes treated with juglone for 2 hours compared with cardiomyocytes in culture medium (CM). DAPI indicates 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; and MYL2, myosin light chain 2. *P<0.01 vs WT, † P< 0.001 vs WT.
Figure 6.
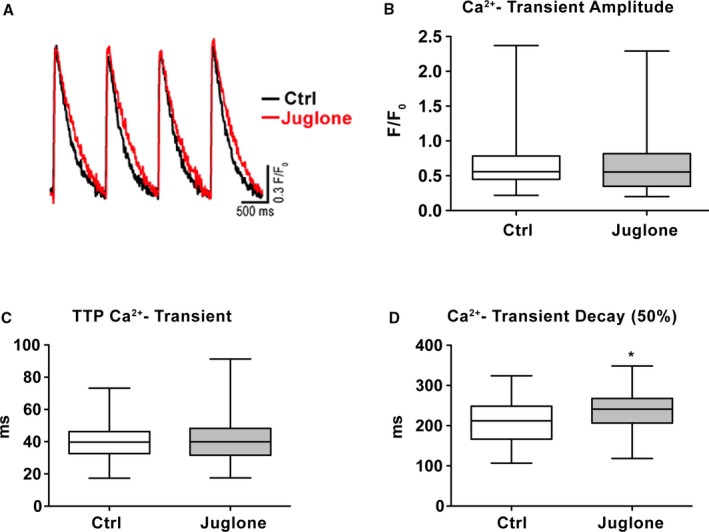
Ca2+ transient decay is delayed by peptidyl‐prolyl isomerase 1 (Pin1) inhibition in vitro. A, Superimposed traces of Ca2+ transient collected in cardiomyocytes treated for 2 hours with Pin1 inhibitor juglone (1 μmol/L) or vehicle control (Ctrl). For each cell, an average trace of >10 beats is reported. B through D, Ca2+ transient amplitude (B) and time to peak (TTP) (C) are unchanged in cardiomyocytes treated with juglone compared with wild type (WT), but Ca2+ transient decay (D) is significantly increased by Pin1 inhibition. Vehicle Ctrl (n=129) and juglone (n=144) from 3 WT hearts are shown. Data are presented as median and interquartile ranges. F/F0, . *P<0.05 (Student t test).
Results
Ca2+ Transient Decay Is Delayed in Pin1−/− Cardiomyocytes
Cardiomyocytes from WT and Pin1−/− mice were isolated to assess the consequences of systemic loss of Pin1 on cardiomyocyte Ca2+ cycling and contractile function. Representative superimposed traces of Ca2+ transients and sarcomere shortening show Pin1−/− cardiomyocytes possess delayed Ca2+ transient decay and slightly longer time to peak compared with WT cardiomyocytes (Figure 1A and 1B). Quantification of Ca2+ transient decay (Figure 1E), time‐to‐peak shortening (Figure 1G), and relaxation (Figure 1H) confirmed a significant delay in Pin1−/− cardiomyocytes compared with WT. Amplitude of Ca2+ transient (Figure 1C), TTP Ca2+‐Transient (Figure 1D), and contractility (Figure 1F) remain unchanged in Pin1−/− cardiomyocytes compared with WT. Collectively, these data indicate loss of Pin1 impairs cardiomyocyte Ca2+ cycling, which may play a critical role in supporting systemic hemodynamic demand in vivo.
SERCA2a and NCX‐1 Protein Levels Are Elevated in Pin1‐Deficient Hearts
Ca2+ influx and efflux from cardiomyocyte SR and cytosol are tightly regulated by expression of several proteins and their phosphorylation status.4 Because Pin1 influences protein expression levels and phosphorylation, the impact of decreased Pin1 expression on SERCA2a, NCX‐1, CaMKII, phospholamban, L‐type Ca2+ channel, and ryanodine receptor 2 protein expression and phosphorylation was assessed using whole heart lysates harvested from WT, Pin1+/−, and Pin1−/− mice. Pin1+/− mice express ≈50% of Pin1 protein compared with WT,26 and have been included in this study to investigate phenotypic consequences of partial Pin1 deletion on calcium handling. SERCA2a protein level was increased ≈1.5‐fold in Pin1+/− and 1.8‐fold in Pin1−/− hearts compared with WT (Figure 2A). NCX‐1 protein level was elevated 1.6‐ and 1.7‐fold in Pin1+/− and Pin1−/−, respectively, compared with WT (Figure 2B). In comparison, expression and phosphorylation of CaMKII, L‐type Ca2+ channels, phospholamban (both pSer16 and pThr17), and ryanodine receptor 2 (both pSer2808 and pSer2814) were unaltered by Pin1 loss (Figure S1A through S1D). These results indicate Pin1 influences SERCA2a and NCX‐1 protein levels as a potential explanation to account for delayed Ca2+ transient decay resolution in isolated cardiomyocytes (Figure 1).
Cardiac SERCA2a Function Is Impaired in Pin1‐Deficient Hearts
Increased SERCA2a protein level correlates with enhanced Ca2+ uptake, resulting in faster Ca2+ transient decay and relaxation.4 Paradoxically, loss of Pin1 leads to slowing of Ca2+ transient decay (Figure 1) together with elevated SERCA2a protein level (Figure 2). To resolve these superficially discrepant observations, SERCA2a ATPase activity and Ca2+ uptake and release from SR membrane–enriched preparations were measured. Stimulation of Ca2+‐dependent SERCA2a ATPase activity was assessed on the basis of release of inorganic phosphate. SERCA2a‐independent ATPase activity was evaluated in the presence of SERCA2a inhibitor (thapsigargin) and subtracted from total SERCA2a ATPase activity to calculate specific activity. SERCA2a ATPase activity in Pin1+/− and Pin1−/− is reduced compared with WT when [Ca2+] is in the range of 5 to 10 μmol/L (Figure 3A). At a 5 μmol/L Ca2+ concentration, SERCA2a activity was 31.72±1.25% for WT samples compared with 23.57±1.08% for Pin1+/− or 23.33±0.21% for Pin1−/− (P<0.05). At an 8 μmol/L Ca2+ concentration, SERCA2a activity was 51.88±3.83% for WT compared with 30.35±0.19% for Pin1+/− or 31.42±1.25% for Pin1−/− (P<0.05). At a 10 μmol/L Ca2+ concentration, SERCA2a activity was 77.29±1.41% for WT compared with 45.14±2.61% for Pin1+/− or 41.67±1.45% for Pin1−/− (P<0.01). By fluorimetric assay, the SR membrane Ca2+ uptake rate was reduced ≈25% in Pin1+/− and Pin1−/− compared with WT (Figure 3B). Rate of Ca2+ uptake was 83.22±5.3 nmol/L per mg SR membrane/min in WT, but decreased to 62.36±4.22 nmol/L per mg SR membrane/min in Pin1+/− or 66.107±3.717 nmol/L per mg SR membrane/min in Pin1−/− (P<0.05; Figure 3B). SR uptake was comparably inhibited by thapsigargin in all WT, Pin1+/−, and Pin1−/− samples, confirming reduction in SR Ca2+ uptake in Pin1+/− and Pin1−/− membranes with diminished SERCA2a function. In comparison, SR‐mediated Ca2+ release was not affected by Pin1 reduction (Figure 3C). Overall, these results indicate SERCA2a ATPase activity and calcium reuptake are reduced by loss of Pin1.
SERCA2a and NCX‐1 Interact With Pin1 in Vivo
Pin1 selectively recognizes phosphorylated serine or threonine immediately adjacent to a proline.9, 10, 11 Five putative consensus motifs for Pin1 binding were identified on the SERCA2a sequence (Figure S2A). SERCA2a includes Ser661‐Pro662, located in the cytoplasmic domain; and 4 Thr‐Pro dipeptides (3 at 247/8, 499/500, and 537/8, located in cytoplasmic domains, and 1 at 959/60, located in a luminal domain). One putative consensus motif for Pin1 binding instead is present in NCX‐1 at Ser223‐Pro224 (Figure S2B), which is located in the extracellular domain. Thus, interaction of Pin1 with Ca2+ cycling proteins SERCA2a and NCX‐1 protein was evaluated by proximity ligation assay in paraffin tissue sections from WT hearts (Figure 4A and 4B). Proximity between Pin1 and SERCA2a primary antibodies was detected as ≈4700 distinct interactions compared with ≈10 events detected in control sections stained with only 1 antibody (P<0.001; Figure 4A). Furthermore, Pin1 and NCX‐1 interaction events occurred in ≈2700 instances compared with only 18 events in single antibody controls (P<0.001; Figure 4B). Pin1−/− hearts used as a negative control also exhibited a similarly low number of events as single antibody negative control. These findings demonstrate cellular in situ molecular interaction between Pin1 and calcium regulatory proteins SERCA2a and NCX‐1.
Furthering the hypothesis that Pin1 activity influences SERCA2a and NCX‐1 protein expression, isolated cardiomyocytes were pretreated for 2 hours with juglone, a highly selective cell‐permeable, irreversible inhibitor of PPIases or water as vehicle control. SERCA2a or NCX‐1 interaction with Pin1 is disrupted by juglone (Figure 5A and 5B, respectively). Moreover, SERCA2a and NCX‐1 protein levels were dose‐dependently increased by Pin1 inhibition (Figure 5C and 5D). In fact, treatment with only 10 nmol/L juglone elevated SERCA2a levels by 1.8‐fold (P<0.05; Figure 5C) and NCX‐1 levels by 1.5‐fold (P<0.001; Figure 5D) compared with vehicle‐treated controls. Treatment with a higher juglone concentration of 1 μmol/L elevated SERCA2a levels by 2.5‐fold (P<0.001; Figure 5C) and NCX‐1 levels by 1.9‐fold (P<0.001; Figure 5D) compared with vehicle‐treated controls. SERCA2a and NCX‐1 interaction was confirmed by pull‐down assay (Figure S3). GST‐ and GST‐Pin1–specific protein expression was confirmed by immunoblot against GST proteins (Figure S3A). Immunoblot for SERCA2a and NCX‐1 confirmed specific interaction with Pin1, as for other known targets (Figure S3B). Therefore, Pin1 protein interaction and/or activity influences SERCA2a and NCX‐1 protein expression, with loss of Pin1 activity elevating SERCA2a and NCX‐1 protein levels.
Cytosolic Ca2+ Removal Is Delayed by Pin1 Inhibition
Delayed Ca2+ decay correlates with impaired cardiomyocyte relaxation, leading to reduced contractility.2, 4 Interaction between SERCA2a or NCX‐1 and Pin1 to control cardiomyocyte Ca2+ cycling was assessed by observing Ca2+ transients after Pin1 inhibition by juglone. Representative superimposed traces of Ca2+ transients collected from isolated cardiomyocytes treated for 2h with Juglone or water as vehicle control are shown in Figure 6A. Juglone pretreatment of cardiomyocytes for 2 hours prompted significant delay in Ca2+‐transient decay (Figure 6D), whereas Ca2+‐transient amplitude (Figure 6B) and time‐to‐peak Ca2+ transient (Figure 6C) remained unchanged. These results support the regulatory role of Pin1 in controlling cardiomyocyte Ca2+ cycling through modulation of SERCA2a activity and/or expression.
Discussion
Ca2+ signals regulate cardiomyocyte contraction and multiple critical cellular processes, including gene regulation, proliferation, enlargement, and death.2, 4, 27 The exact basis for how a single ion precisely controls such diverse cellular processes remains intriguing.27 However, there is consensus that the magnitude and temporal signature of Ca2+ signal, as well as the cellular localization of these signals, are critical for proper cardiomyocyte function.27 Pin1 exhibits the unique property of acting as a rheostat in the molecular signaling network to regulate intensity and duration of signals.7, 8 Findings in this study define a novel role of Pin1 as a fine‐tuning mechanism for regulation of Ca2+ dynamics through SERCA2a and NCX‐1 proteins.
Decreased Ca2+ reuptake in cardiomyocytes causes impaired myocardial relaxation and slows filling of the ventricle, leading to HF with preserved ejection fraction.4, 27 Results in this study showed that loss of Pin1 activity, by either genetic deletion (Figure 1) or pharmacological inhibition (Figure 6), slows cytosolic Ca2+ clearance in isolated cardiomyocytes. Reduced SERCA2a activity is also a characteristic of HF.4, 5 SR membranes isolated from Pin1‐deficient hearts showed significantly reduced SERCA2a Ca2+‐ATPase activity and uptake (Figure 3). Control of SERCA2a function by Pin1 explains delayed Ca2+ clearance on Pin1 deletion or pharmacological inhibition, substantiating a potential role for Pin1 in contributing to dysregulation of Ca2+ homeostasis associated with HF. Our findings agree with prior studies reporting the pan PPIase inhibitor juglone inhibits SERCA2a Ca2+‐ATPase activity in isolated SR membranes.28 Decreased Pin1 activity concomitant to acute or chronic stress, where faster Ca2+ cycling is required to sustain increased contractile function, could impair cardiac output and contribute to progression of HF because of loss of inotropic responsiveness. SR ATPase activity and SR Ca2+ uptake were similarly impaired in membranes isolated from Pin1+/− and Pin1−/−, indicating that loss of ≈50% Pin protein level is sufficient to impair cardiac SERCA2a function. Results herein reveal another facet of the Pin1 signaling in myocardial biological features associated with regulation of Ca2+ handling within cardiomyocytes. PPIase family members other than Pin1, including cyclophylin D and FK‐506–binding proteins, regulate Ca2+ handling.14 Our findings expand known regulatory roles for the PPIase family by incorporating the parvulin family member Pin1 in regulation of Ca2+ handling within myocardium via influences on SERCA2a and NCX‐1.
This study reveals 2 novel targets regulating Ca2+ homeostasis in the myocardium influenced by Pin1: SERCA2a and NCX‐1 (Figures 4, 5, and S3). Pin1 has been associated with calcium‐dependent signaling through interaction with the Ca2+ regulator protein CaMKII in brain tissue, but whether CaMKII function was affected by Pin1 binding remains unresolved.29 Our hypothesis is that Pin1 binding to SERCA2a and NCX‐1 simultaneously influences both protein function and expression through simultaneous modification of conformation and protein turnover, respectively. Indeed, Pin1 might promote stabilization in the active conformation of SERCA2a and NCX‐1 proteins, as for other targets, like β‐catenin, nuclear factor κB, and p53.9 In this scenario, Pin1 inhibition would impair SERCA2a and NCX‐1 function, which is partially compensated by increased protein production within cardiomyocytes. Furthermore, Pin1 might also be important for SERCA2a and NCX‐1 protein turnover by decreasing stabilization, similar to Pin1‐mediated action on CF‐2, c‐Myc, and cyclin E.9 Consequently, loss of Pin1 might slow SERCA2a and NCX‐1 degradation, resulting in accumulation of protein. The combined impact of SERCA2a‐ and NCX‐1–impaired function, coupled with protein accumulation resulting from diminished Pin1, is consistent with our findings, as summarized in Figure 7. In this model, Pin1 genetic deletion/inhibition reduces overall SERCA2a and NCX‐1 protein functional activity, which is only partially compensated by protein accumulation attributable to impaired SERCA2a and NCX‐1 degradation. Future studies will investigate molecular mechanisms altering SERCA2a and NCX‐1 function on Pin1 loss. SERCA2a was phosphorylated by mass spectrometry in the heart tissue at Ser38 and Ser661.30 Few studies have shown that SERCA2a can be phosphorylated by CaMKII on Ser38, enhancing enzyme activity.31 However, later studies using a polyclonal antibody specific for the phosphorylated Ser38 epitope on the SERCA2a Ca2+ ATPase failed to detect the phosphorylated form in both isolated cardiomyocytes and purified SR vesicles. This suggested that Ser38 phosphorylation on SERCA2a is not a significant regulatory feature of SERCA2a activity.32 No studies have investigated potential consequences of phosphorylation on Ser661 of SERCA2a. NCX‐1 was found, by mass spectrometry in the heart tissue, phosphorylated on Ser389.30 However, no studies have addressed the potential functional consequences of NCX‐1 phosphorylation on these specific sites. Protein kinase C phosphorylates NCX‐1 in vivo, increasing exchange activity, but the precise phosphorylation site has not been identified.33 Previous studies have identified phosphorylation on SERCA2a and NCX‐1 sites that are not in the putative consensus motifs for Pin1 binding.30 However, the tryptic digestion performed before mass spectrometry analysis in this phosphoproteomic study might have led to partial loss of information, leaving phosphorylation sites located within the digested peptide fragments. Further studies would be necessary to characterize if and under which conditions SERCA2a and NCX‐1 are phosphorylated at these motifs to allow Pin1 association.
Figure 7.
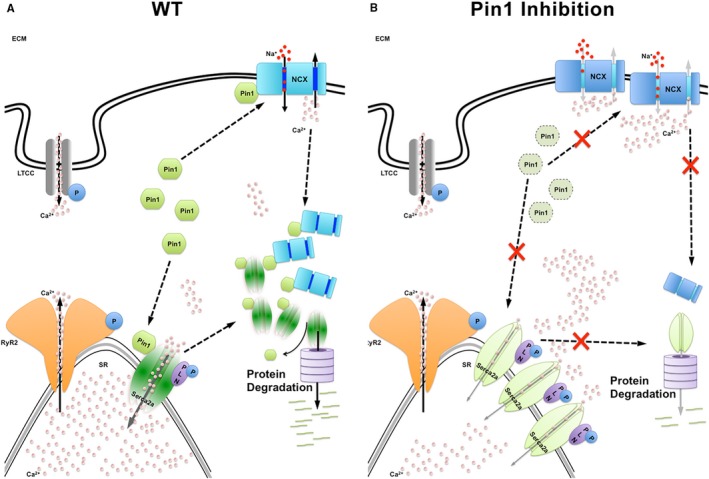
The impact of diminished peptidyl‐prolyl isomerase 1 (Pin1) activity on cardiomyocyte Ca2+ handling. A, Pin1 binding to sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) and Na2+/Ca2+ exchanger 1 (NCX‐1) promotes stabilization of active conformation for SERCA2a and NCX‐1 proteins as well as protein turnover to proteolytic machinery. B, Pin1 gene deletion or pharmacological inhibition impairs SERCA2a and NCX‐1 function, leading to increased compensatory protein production, together with diminished degradation of SERCA2a and NCX‐1 contributing to protein accumulation. ECM indicates extracellular membrane; LTCC, L‐type Ca2+ channel; PLN, phospholamban; RyR2, ryanodine receptor 2; and WT, wild type.
Pin1 dysregulation is implicated in diverse pathological conditions, including aging, immune response, neurodegenerative disease, and multiple cancers.7, 10, 11, 34 Pin1 ubiquitous expression controls homeostasis and growth through regulation of cell proliferation, differentiation, and apoptosis in diverse tissue types.9, 11, 34, 35 Pin1 dual function in regulating both cell signaling and protein folding accounts for why Pin1 expression levels vary widely between tissue types.10 Expression increases proportionally with cellular proliferative capacity, and Pin1 levels are upregulated in most human cancers.9, 10, 11, 34 Pin1 controls cardiac progenitor cell proliferation through influence on cyclin D, cyclin B, p53, and retinoblastoma.13 Pin1 deletion caused cell cycle arrest and senescence, whereas Pin1 overexpression increases differentiation and inhibits senescence of cardiac progenitor cells.13 In stark contrast, Pin1 is highly expressed in neurons from onset of neuronal differentiation and decreases with aging, suggesting Pin1 serves a completely different purpose in this postmitotic cell context.10 A new layer of complexity has been revealed by the overtly oppositional effect of Pin1 on SERCA2a protein expression versus function.
In the myocardium, Pin1 serves a central modulator role for pathological hypertrophy and fibrosis.12, 36, 37 In the myocardium, Pin1 controls hypertrophic growth through regulation of Akt and Mitogen‐activated protein kinase (MEK).12 Either Pin1 genetic deletion or cardiac‐specific overexpression blunted hypertrophic responses induced by transaortic constriction through modulation of different pathways.12 Loss of Pin1 diminished hypertrophic signaling of Akt and MEK, whereas overexpression of Pin1 increases Rapidly Accelerated Fibrosarcoma serine/threonine‐specific protein kinases (Raf‐1) phosphorylation on the autoinhibitory site Ser259, leading to reduced MEK activation.12 In addition, Pin1 inhibition by juglone attenuates cardiac extracellular matrix deposition in a diabetic mouse model by regulating the phosphorylation of Akt, transforming growth factor‐β1, and matrix metalloproteinases.36 Furthermore, juglone treatment decreased transforming growth factor‐β1 activity in the promyelocytic leukemia nuclear bodies and hypertrophic response in cardiac fibroblasts challenged with arsenic trioxide and angiotensin II,36 corroborating a role for Pin1 in regulating myocardial hypertrophy. Delayed cytosolic Ca2+ reuptake, caused by Pin1 inhibition, could be at a subpathologic level of disturbance. It could contribute to a prohypertrophic phenotype, with activation of cytosolic Ca2+ pathways through the calcineurin‐Nuclear Factor of Activated T cell protein (NFAT) pathway and Ca2+‐calmodulin–dependent kinase Mitogen activated protein kinase Kinase (MEF2).1, 38 In the latter case, overt lack of hypertrophic phenotype in Pin1 knockout mice might be maintained by the collective influence of Pin1 on multiple regulatory cascade pathways in parallel that collectively influence cell biological features. Pin1 mediation of hypertrophic responsiveness is complex, because cardiac‐specific Pin1 overexpression blunted hypertrophic response induced by transaortic constriction by influencing pathways different from those affected by Pin1 deletion.12 Preservation of homeostasis is consistent with Pin1 acting as a rheostat on multiple concurrent pathways to maintain optimal function. Cardiac‐specific Pin1 overexpression could be explored as a molecular interventional strategy to enhance SERCA2a activity and normalize Ca2+ homeostasis within myocardial cells.
Sources of Funding
Sacchi was supported by Swiss National Found Fellowship (P2BSP3_155252). Wang is supported by the Rees Stealy Research Foundation Fellowship. Kubli is supported by NIH grant F32 HL136064‐01. Jin was supported by the American Heart Association Postdoctoral Fellowship 16POST27510010. Malter is supported by NIH grants P01HL088594. Glembotski is supported by NIH grants R01 HL75573, R01 HL104535, R01 HL127439, and P01 HL085577. Sussman is supported by NIH grants R01HL067245, R37HL091102, R01HL105759, R01HL113647, R01HL117163, P01HL085577, and R01HL122525, as well as an award from the Fondation Leducq.
Disclosures
Sussman is founder and co‐owner of CardioCreate Inc. The other authors report no conflicts.
Supporting information
Table S1. Antibody List
Figure S1. pCamKII/CamKII (A), pLTCC/LTCC(B), pPLN (Ser16 and Thr 17)/PLN (C) and pRyR2 (Ser2808 and Ser2814)/RyR2 (D) expression and phosphorylation levels are unaltered in Pin1 deficient hearts. Phosphorylated Ca2+/Calmodulin‐dependent kinase II (pCamKII)/CamKII (A), phosphorylated L‐type Ca2+ channel (LTCC)/LTCC(B), phosphorylated Phospholamban (PLN) (Ser16 and Thr 17)/PLN (C) and pRyR2 (Ser2808 and Ser2814)/RyR2 expression and phosphorylation levels are unaltered in peptidyl prolyl isomerase (Pin1)+/− and Pin1−/− hearts compared to wild‐type (WT) by immunoblot analysis. GAPDH used as loading control. Quantitation of protein expression shown below each representative blot as n‐fold expression/WT. Data presented as mean±SEM, N=4.
Figure S2. Putative consensus motifs for Pin1 binding on SERCA2a and NCX‐1 protein sequences identified by in silico analysis. A, Sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) sequence accession number NP_033852.1 (murine; NCBI protein database) or (B) Na2+/Ca2+ exchanger 1 (NCX‐1) sequence accession number NP_035536.2 (murine; NCBI protein database) analyzed for serine next to a proline residue (black squares) or threonine next to a proline (red squares).
Figure S3. SERCA2a and NCX‐1 physically interact with GST‐Pin1. A, Immunoblot for Glutathione S‐Transferase (GST) (left) and GST‐Pin1 (right) showing GST and GST‐Pin1 protein expression at 25 and 45 kDa respectively. B, Na2+/Ca2+ exchanger 1 (NCX‐1) and sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) interacted with GSTPin1 but not with GST upon pull‐down as shown by immunoblot. β‐catenin and AKT were used as positive control for the pull‐down since are known targets of Pin1. N=3.
Acknowledgments
We thank Mansa Melvin and Julie Hu (University of Texas Southwestern) for providing assistance with Pin1−/− and Pin1+/− mice handling and transfer; Debora Eaton, Remus Berretta (Temple University), and Dr Dong Sun (New York Medical College) for technical support with isolated cardiomyocyte measurements; and all the members of the Sussman laboratory for their critical feedback.
(J Am Heart Assoc. 2017;6:e006837 DOI: 10.1161/JAHA.117.006837.)
References
- 1. van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. [DOI] [PubMed] [Google Scholar]
- 3. Gorski PA, Ceholski DK, Hajjar RJ. Altered myocardial calcium cycling and energetics in heart failure: a rational approach for disease treatment. Cell Metab. 2015;21:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. 2013;123:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stammers AN, Susser SE, Hamm NC, Hlynsky MW, Kimber DE, Kehler DS, Duhamel TA. The regulation of sarco(endo)plasmic reticulum calcium‐ATPases (SERCA). Can J Physiol Pharmacol. 2015;93:1–12. [DOI] [PubMed] [Google Scholar]
- 6. Hobai IA, O'Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation‐contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. [DOI] [PubMed] [Google Scholar]
- 7. Hariharan N, Sussman MA. Pin1: a molecular orchestrator in the heart. Trends Cardiovasc Med. 2014;24:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yaffe MB. Sequence‐specific and phosphorylation‐dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. [DOI] [PubMed] [Google Scholar]
- 9. Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. [DOI] [PubMed] [Google Scholar]
- 10. Driver JA, Zhou XZ, Lu KP. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer's disease. Biochim Biophys Acta. 2015;1850:2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liou Y‐C, Zhou XZ, Lu KP. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci. 2011;36:501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toko H, Konstandin MH, Doroudgar S, Ormachea L, Joyo E, Joyo AY, Din S, Gude NA, Collins B, Völkers M, Thuerauf DJ, Glembotski CC, Chen CH, Lu KP, Müller OJ, Uchida T, Sussman MA. Regulation of cardiac hypertrophic signaling by prolyl isomerase Pin1. Circ Res. 2013;112:1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Toko H, Hariharan N, Konstandin MH, Ormachea L, McGregor M, Gude NA, Sundararaman B, Joyo E, Joyo AY, Collins B, Din S, Mohsin S, Uchida T, Sussman MA. Differential regulation of cellular senescence and differentiation by prolyl isomerase Pin1 in cardiac progenitor cells. J Biol Chem. 2014;289:5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perrucci GL, Gowran A, Zanobini M, Capogrossi MC, Pompilio G, Nigro P. Peptidyl‐prolyl isomerases: a full cast of critical actors in cardiovascular diseases. Cardiovasc Res. 2015;106:353–364. [DOI] [PubMed] [Google Scholar]
- 15. Jaleel N, Nakayama H, Chen X, Kubo H, MacDonnell S, Zhang H, Berretta R, Robbins J, Cribbs L, Molkentin JD, Houser SR. Ca2+ influx through T‐ and L‐type Ca2+ channels have different effects on myocyte contractility and induce unique cardiac phenotypes. Circ Res. 2008;103:1109–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Signore S, Sorrentino A, Ferreira‐Martins J, Kannappan R, Shafaie M, Del Ben F, Isobe K, Arranto C, Wybieralska E, Webster A, Sanada F, Ogórek B, Zheng H, Liu X, Del Monte F, D'Alessandro DA, Wunimenghe O, Michler RE, Hosoda T, Goichberg P, Leri A, Kajstura J, Anversa P, Rota M. Inositol 1, 4, 5‐trisphosphate receptors and human left ventricular myocytes. Circulation. 2013;128:1286–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Signore S, Sorrentino A, Borghetti G, Cannata A, Meo M, Zhou Y, Kannappan R, Pasqualini F, O'Malley H, Sundman M, Tsigkas N, Zhang E, Arranto C, Mangiaracina C, Isobe K, Sena BF, Kim J, Goichberg P, Nahrendorf M, Isom LL, Leri A, Anversa P, Rota M. Late Na(+) current and protracted electrical recovery are critical determinants of the aging myopathy. Nat Commun. 2015;6:8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kubli DA, Cortez MQ, Moyzis AG, Najor RH, Lee Y, Gustafsson ÅB. PINK1 is dispensable for mitochondrial recruitment of parkin and activation of mitophagy in cardiac myocytes. PLoS One. 2015;10:e0130707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius K‐J, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson L‐G, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. [DOI] [PubMed] [Google Scholar]
- 20. Zieba A, Wählby C, Hjelm F, Jordan L, Berg J, Landegren U, Pardali K. Bright‐field microscopy visualization of proteins and protein complexes by in situ proximity ligation with peroxidase detection. Clin Chem. 2010;56:99–110. [DOI] [PubMed] [Google Scholar]
- 21. Din S, Mason M, Völkers M, Johnson B, Cottage CT, Wang Z, Joyo AY, Quijada P, Erhardt P, Magnuson NS, Konstandin MH, Sussman MA. Pim‐1 preserves mitochondrial morphology by inhibiting dynamin‐related protein 1 translocation. Proc Natl Acad Sci USA. 2013;110:5969–5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Volkers M, Toko H, Doroudgar S, Din S, Quijada P, Joyo AY, Ornelas L, Joyo E, Thuerauf DJ, Konstandin MH, Gude N, Glembotski CC, Sussman MA. Pathological hypertrophy amelioration by PRAS40‐mediated inhibition of mTORC1. Proc Natl Acad Sci USA. 2013;110:12661–12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Brien PJ. Calcium sequestration by isolated sarcoplasmic reticulum: real‐time monitoring using ratiometric dual‐emission spectrofluorometry and the fluorescent calcium‐binding dye indo‐1. Mol Cell Biochem. 1990;94:113–119. [DOI] [PubMed] [Google Scholar]
- 24. Tupling R, Green H, Senisterra G, Lepock J, McKee N. Effects of 4‐h ischemia and 1‐h reperfusion on rat muscle sarcoplasmic reticulum function. Am J Physiol Endocrinol Metab. 2001;281:867–877. [DOI] [PubMed] [Google Scholar]
- 25. Duhamel TA, Green HJ, Stewart RD, Foley KP, Smith IC, Ouyang J. Muscle metabolic, SR Ca(2+) ‐cycling responses to prolonged cycling, with and without glucose supplementation. J Appl Physiol. 2007;103:1986–1998. [DOI] [PubMed] [Google Scholar]
- 26. Baik SH, Fane M, Park JH, Cheng YL, Fann DYW, Yun UJ, Choi Y, Park JS, Chai BH, Park JS, Back SH, Jeong JI, Jang YJ, Bahn G, Lee JY, Li YI, Sobey CG, Uchida T, Park JH, Kim HT, Tang SC, Arumugam TV, Jo DG. Pin1 promotes neuronal death in stroke by stabilizing notch intracellular domain. Ann Neurol. 2015;77:504–516. [DOI] [PubMed] [Google Scholar]
- 27. Aronsen JM, Louch WE, Sjaastad I. Cardiomyocyte Ca2+ dynamics: clinical perspectives. Scand Cardiovasc J. 2016;50:65–77. [DOI] [PubMed] [Google Scholar]
- 28. Floreani M, Forlin A, Bellin S, Carpenedo F. Structure‐activity relationship for the inhibition of cardiac sarcoplasmic reticulum Ca2+ ATPase activity by naphthoquinones. Biochem Mol Biol Int. 1995;37:757–763. [PubMed] [Google Scholar]
- 29. Tatara Y, Terakawa T, Uchida T. Identification of Pin1‐binding phosphorylated proteins in the mouse brain. Biosci Biotechnol Biochem. 2010;74:2480–2483. [DOI] [PubMed] [Google Scholar]
- 30. Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. A tissue‐specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Toyofuku T, Curotto Kurzydlowski K, Narayanan N, MacLennan DH. Identification of Ser38 as the site in cardiac sar‐ coplasmic reticulum Ca2+‐ATPase that is phosphorylated by Ca2+/calmodulin‐dependent protein kinase. J Biol Chem. 1994;269:26492–26496. [PubMed] [Google Scholar]
- 32. Rodriguez P, Jackson WA, Colyer J. Critical evaluation of cardiac Ca2+‐ATPase phosphorylation on serine 38 using a phosphorylation site‐specific antibody. J Biol Chem. 2004;279:17111–17119. [DOI] [PubMed] [Google Scholar]
- 33. Iwamoto T, Pan Y, Wakabayashi S, Imagawa T, Yamanaka H, Shigekawa M. Phosphorylation‐dependent regulation of cardiac Na+/Ca2+ exchanger via protein kinase C. J Biol Chem. 1996;271:13609–13615. [DOI] [PubMed] [Google Scholar]
- 34. Lin C‐H, Li H‐Y, Lee Y‐C, Calkins MJ, Lee K‐H, Yang C‐N, Lu P‐J. Landscape of Pin1 in the cell cycle. Exp Biol Med. 2015;240:403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sorrentino G, Comel A, Mantovani F, Del Sal G. Regulation of mitochondrial apoptosis by Pin1 in cancer and neurodegeneration. Mitochondrion. 2014;19:88–96. [DOI] [PubMed] [Google Scholar]
- 36. Liu YY‐X, Zhao D, Qiu F, Zhang L‐L, Liu S‐K, Li Y‐Y, Liu M‐T, Wu D, Wang J‐X, Ding X‐Q, Liu YY‐X, Dong C‐J, Shao X‐Q, Yang B‐F, Chu W‐F. Manipulating PML SUMOylation via silencing UBC9 and RNF4 regulates cardiac fibrosis. Mol Ther. 2017;25:666–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu X, Liang E, Song X, Du Z, Zhang Y, Zhao Y. Inhibition of Pin1 alleviates myocardial fibrosis and dysfunction in STZ‐induced diabetic mice. Biochem Biophys Res Commun. 2016;479:109–115. [DOI] [PubMed] [Google Scholar]
- 38. Zarain‐Herzberg A, Fragoso‐Medina J, Estrada‐Avilés R. Calcium‐regulated transcriptional pathways in the normal and pathologic heart. IUBMB Life. 2011;63:847–855. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibody List
Figure S1. pCamKII/CamKII (A), pLTCC/LTCC(B), pPLN (Ser16 and Thr 17)/PLN (C) and pRyR2 (Ser2808 and Ser2814)/RyR2 (D) expression and phosphorylation levels are unaltered in Pin1 deficient hearts. Phosphorylated Ca2+/Calmodulin‐dependent kinase II (pCamKII)/CamKII (A), phosphorylated L‐type Ca2+ channel (LTCC)/LTCC(B), phosphorylated Phospholamban (PLN) (Ser16 and Thr 17)/PLN (C) and pRyR2 (Ser2808 and Ser2814)/RyR2 expression and phosphorylation levels are unaltered in peptidyl prolyl isomerase (Pin1)+/− and Pin1−/− hearts compared to wild‐type (WT) by immunoblot analysis. GAPDH used as loading control. Quantitation of protein expression shown below each representative blot as n‐fold expression/WT. Data presented as mean±SEM, N=4.
Figure S2. Putative consensus motifs for Pin1 binding on SERCA2a and NCX‐1 protein sequences identified by in silico analysis. A, Sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) sequence accession number NP_033852.1 (murine; NCBI protein database) or (B) Na2+/Ca2+ exchanger 1 (NCX‐1) sequence accession number NP_035536.2 (murine; NCBI protein database) analyzed for serine next to a proline residue (black squares) or threonine next to a proline (red squares).
Figure S3. SERCA2a and NCX‐1 physically interact with GST‐Pin1. A, Immunoblot for Glutathione S‐Transferase (GST) (left) and GST‐Pin1 (right) showing GST and GST‐Pin1 protein expression at 25 and 45 kDa respectively. B, Na2+/Ca2+ exchanger 1 (NCX‐1) and sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) interacted with GSTPin1 but not with GST upon pull‐down as shown by immunoblot. β‐catenin and AKT were used as positive control for the pull‐down since are known targets of Pin1. N=3.