

Abstract
Neuroinflammation is a prominent component of several neurodegenerative diseases, including multiple sclerosis, Alzheimer’s disease, Parkinson’s disease, tauopathies, amyotrophic lateral sclerosis and prion diseases. In such conditions, the ability to decrease neuroinflammation by drug therapy may influence disease progression. Statins have been used to treat hyperlipidemia as well as reduce neuroinflammation and oxidative stress in various tissues. In previous studies, treatment of scrapie-infected mice with the type 1 statins, simvastatin or pravastatin, showed a small beneficial effect on survival time. In the current study, to increase the effectiveness of statin therapy, we treated infected mice with atorvastatin, a type 2 statin that has improved pharmacokinetics over many type 1 statins. Treatments with either simvastatin or pravastatin were tested for comparison. We evaluated scrapie-infected mice for protease-resistant PrP (PrPres) accumulation, gliosis, neuroinflammation and time until advanced clinical disease requiring euthanasia. All three statin treatments reduced total serum cholesterol ≥40 % in mice. However, gliosis and PrPres deposition were similar in statin-treated and untreated infected mice. Time to euthanasia due to advanced clinical signs was not changed in statin-treated mice relative to untreated mice, a finding at odds with previous reports. Expression of 84 inflammatory genes involved in neuroinflammation was also quantitated. Seven genes were reduced by pravastatin, and one gene was reduced by atorvastatin. In contrast, simvastatin therapy did not reduce any of the tested genes, but did slightly increase the expression of Ccl2 and Cxcl13. Our studies indicate that none of the three statins tested were effective in reducing scrapie-induced neuroinflammation or neuropathogenesis.
Keywords: statin, prion, scrapie, neuroinflammation, gliosis, chemokine
Abbreviations
AD, Alzheimer’s disease; ANOVA, analysis of variance; CNS, central nervous system; DAB, 3, 3′-diaminobenzidine; EAE, experimental autoimmune encephalomyelitis; GFAP, glial fibrillary acidic protein; HMG-CoA, 3-hydroxy-3-methyl-glutaryl-coenzyme A; IBA-1, ionized calcium-binding adapter-1; i.c., intracerebrally; p.i., post-inoculation; PrPres, protease-resistant PrP; PrPSc , scrapie-associated PrP ; PrP, prion protein.
Introduction
Prion diseases are transmissible fatal brain disorders that are characterized by the accumulation of a misfolded host protein (PrP) in the CNS, accompanied by grey matter spongiosis and neuroinflammation consisting of astro- and micro-gliosis [1, 2]. Although prion pathogenesis is not completely understood, damage and/or loss of neurons during disease is likely a major contributing factor. Neuronal damage after prion infection may occur through multiple mechanisms, including excitotoxicity [3, 4], inflammatory cytokine exposure [5–7], mitochondrial dysfunction [8–12], or targeted cell death through direct interaction with the prion protein [13–15].
Neurodegeneration and neuroinflammation are also common in multiple sclerosis (MS), and prion-like diseases, such as Alzheimer’s disease (AD), Parkinson’s disease and tauopathies [5, 6, 16–21]. In these diseases, as well as in prion diseases, neuroinflammation might contribute to the pathogenic process. Therefore, treatment to reduce neuroinflammation may be useful. One class of drugs shown to lessen inflammation in various models of neurodegenerative disease are statins [22, 23].
Statins have been utilized to treat atherosclerosis, where the primary mechanism is reduction of hyperlipidemia. However, statins also have anti-inflammatory effects that increase their effectiveness against atherosclerosis. These effects include modulation of endothelial function and disruption of signalling pathways, resulting in reduction of inflammation [24]. Atorvastatin and simvastatin affect neuroinflammation in mouse models of Parkinson’s disease by reducing proinflammatory cytokines in the brain [25–28]. Furthermore, in rodent models of AD, atorvastatin reduces the production of pro-inflammatory cytokines and decreases the number of microglia in the hippocampus [29, 30]. Similarly, in studies using the experimental autoimmune encephalomyelitis (EAE) rodent model for MS, statins reduced proinflammatory cytokines, increased anti-inflammatory responses, decreased infiltration of monocytes into the central nervous system (CNS) and decreased adhesion molecule expression on immune cells [31–34]. Therefore, statin treatment has shown promise in reducing the neuroinflammation associated with several neurodegenerative disease models.
In the present study of scrapie-infected mice, we tested atorvastatin, a commonly prescribed type 2 lipophilic statin, which is structurally different from and pharmacokinetically superior to many of the type 1 statins [35], and has not been studied in prion diseases. For comparison, we performed similar treatment experiments using simvastatin and pravastatin (type 1 statins) that had been tested previously by others and showed modest statistically significant improvements for survival times [36–39]. We hypothesized that treatment of scrapie-infected mice with atorvastatin would be an improvement on treatment with simvastatin or pravastatin. Various aspects of scrapie pathogenesis were analysed using biochemical, histological and immunological means to determine whether statin treatment could reduce chronic neuroinflammation and alter survival following scrapie infection.
Results
Evaluation of serum cholesterol in mice with statin treatment
Serum cholesterol levels were measured in uninfected C57BL/10 mice that were either untreated or treated with statins for approximately 150 days (Fig. 1). Untreated mice had a mean serum cholesterol of 91 mg dl−1. Oral treatment with pravastatin, atorvastatin, or simvastatin similarly reduced serum cholesterol in mice by ≥40 %. These results indicated the statin doses in our mice were sufficient to affect the target enzyme HMG-CoA reductase and significantly reduced serum cholesterol in treated mice after 150 days of therapy. Thus, the food pellets formulated with drug used in this study appeared to be an effective method to administer statins to mice.
Fig. 1.
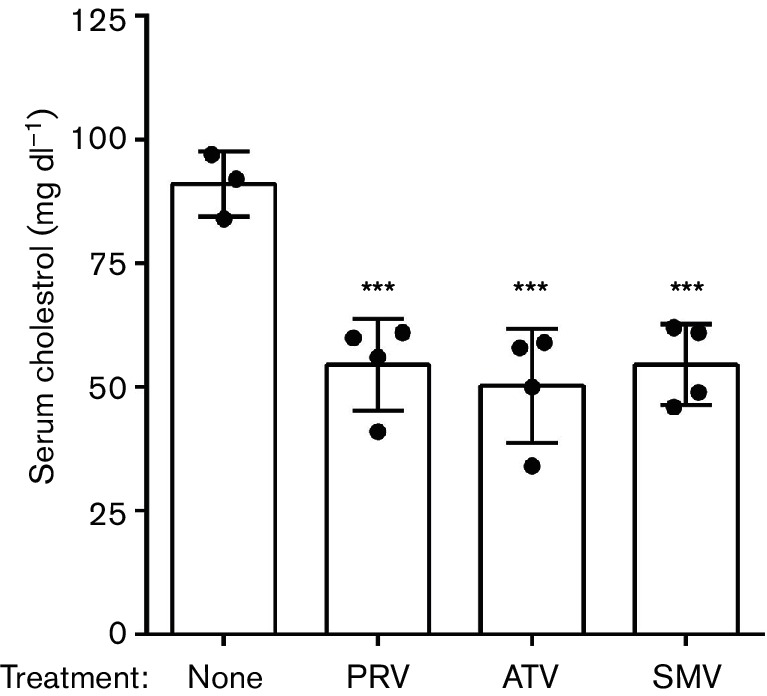
Mouse serum cholesterol levels with simvastatin (SMV), pravastatin (PRV), or atorvastatin (ATV) treatment. Total serum cholesterol was measured in statin-treated and untreated mice after 150 days of consuming statin-supplemented or control chow. There was a ≥40 % reduction in the average cholesterol in all three statins tested relative to control, indicating an effect on the mevalonate pathway. Each dot represents a single mouse. Error bars indicate one standard deviation. *** P value ≤0.001 by one-way ANOVA relative to untreated.
Scrapie disease in mice treated with statins
Mice inoculated with scrapie strain 22L and treated with statins were compared with untreated scrapie-inoculated control mice to assess whether treatment with pravastatin, atorvastatin, or simvastatin altered scrapie pathogenesis or disease tempo. None of the statins tested showed differences between drug-treated and untreated mice in the time for developing advanced clinical disease requiring euthanasia (Fig. 2a, b, c and Table 1). In addition, immunoblot analysis of representative brain homogenates of mice from this study demonstrated that the amounts of protease-resistant PrP (PrPres) in the drug-treated and untreated mice were similar (Fig. 2d). Therefore, statin treatment did not affect either the tempo of the prion disease or the brain accumulation of PrPres in infected mice.
Fig. 2.

Euthanasia plots and immunoblot detection of PrPres in 22L-infected mice treated with simvastatin, pravastatin, or atorvastatin. The time until euthanasia for untreated control scrapie-infected C57BL/10 mice was compared to that for mice treated with simvastatin (a), pravastatin (b), or atorvastatin (c). Mice were inoculated intracerebrally with scrapie strain 22L, and the x-axis shows the day post-inoculation (p.i.) when animals were euthanized due to advanced signs of clinical scrapie (see the Methods section for details). Curves were statistically analysed using the Mantel–Cox log rank. The number of mice per group (n) is indicated in each graph. (d) Brain protein samples from representative end-stage 22L-infected C57BL/10 mice treated with simvastatin, pravastatin, or atorvastatin, or untreated, were separated by SDS-PAGE. All samples were treated with proteinase K prior to separation as described in the Methods section. Each lane was loaded with 0.36 mg whole-brain equivalents and probed with anti-PrP antibody D13. The 21 and 31 kiloDalton protein standards are indicated to the left of the immunoblot.
Table 1. Survival times of statin-treated and untreated scrapie-infected C57BL/10 mice.
| Treatment | Survival times (day p.i.)* | Median day p.i.±sd |
|---|---|---|
| Simvastatin (n†=12) | 147, 152, 152, 152, 154, 158, 159, 160, 161, 164, 164, 166 | 158.5±6.0 |
| Atorvastatin (n=12) | 152, 152, 153, 153, 154, 154, 154, 158, 164, 164, 164, 164 | 154.0±5.3 |
| Pravastatin (n=10) | 140, 143, 147, 153, 153, 154, 154, 154, 161, 161 | 153.5±6.8 |
| Untreated (n=10) | 143, 152, 152, 152, 152, 153, 154, 154, 156, 161 | 152.5±4.5 |
*Number of days post-infection when individual mice were euthanized due to presence of advanced clinical signs.
†Number of mice in the study group.
Neuropathology and assessment of gliosis and PrPSc by immunohistochemistry
H and E staining of brain sections showed a similar spongiform change in the cortex of all the scrapie-infected mice, regardless of statin treatment (Fig. 3a, last column). To evaluate the amount of reactive gliosis and PrPSc accumulation in the brains of scrapie-infected mice, histological sections were probed with antibodies that specifically recognized GFAP, IBA-1, or PrP. The immunoreactive staining for GFAP, IBA-1, or PrP appeared similar in terminally infected mice, regardless of statin treatment (Fig. 3a). Furthermore, quantifiable pixel intensity analysis of individual sagittal sections of whole brain indicated that increases of GFAP, IBA-1 and PrP reactivity in all scrapie-infected cohorts were similar and significantly greater than for identically probed uninfected control brain sections (Fig. 3b). Thus, treatment with simvastatin, pravastatin, or atorvastatin did not substantially change the amount of gliosis in the brain. Furthermore, histology showed that PrPSc deposition was equivalent in drug-treated and untreated infected mice. This agreed with our earlier findings by immunoblot (Fig 2d).
Fig. 3.
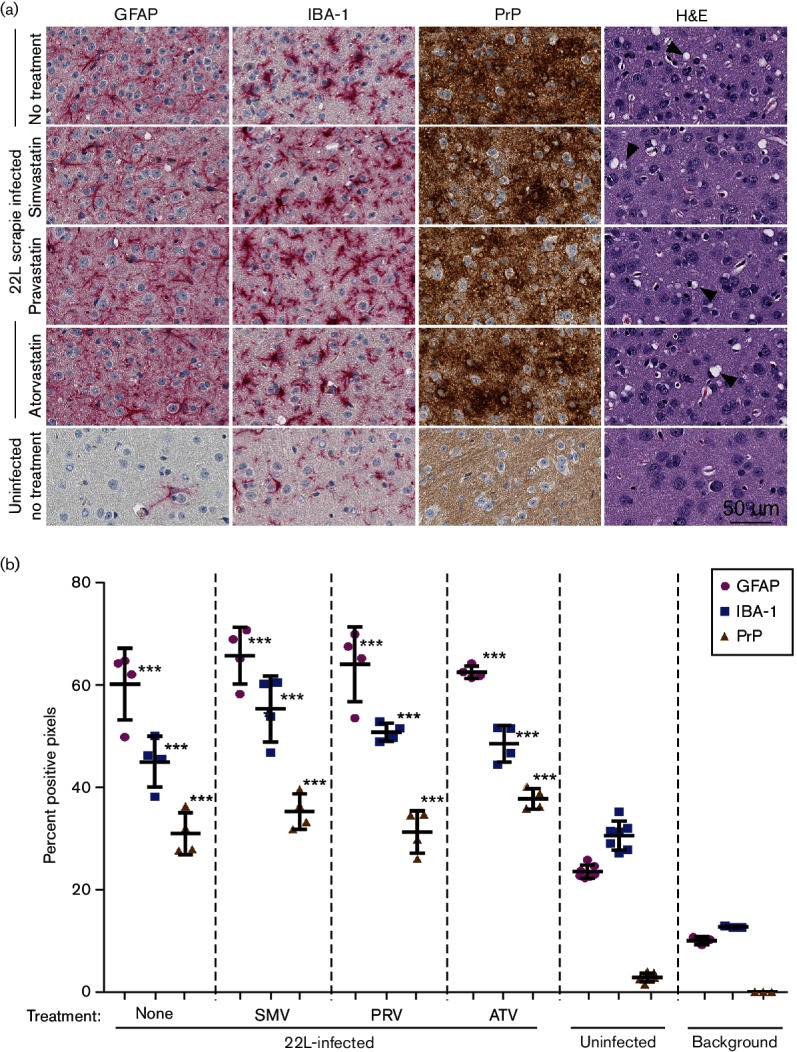
Neuropathology and immunohistochemical assessment of gliosis and PrP deposition in cortex brain sections from 22L-infected mice treated with simvastatin, pravastatin, or atorvastatin. (a) Brain sections from 22L-infected mice treated with simvastatin, pravastatin, atorvastatin, or untreated, were probed with antibodies specific for GFAP, IBA-1, or PrP. A representative H and E stained section shows that similar amounts of spongiform change (arrow heads) were observed in all scrapie-infected mice. Sections from the brain of untreated uninfected mice were similarly probed as a negative control. Representative images of the cerebral cortex are shown for all at the same scale as indicated. (b) Sagittal sections from brains of 22L-infected mice treated with statins or untreated as indicated were scanned to quantitatively assess the percentage of positive pixels after probing with antibodies specific for GFAP, IBA-1, or PrP. Uninfected mice were included as a negative control. The background represents several brain sections from 22L-infected mice that were analysed with the primary antibody omitted. Each character represents an independent sagittal section from a single mouse. Bars indicate the mean, and error bars indicate one standard deviation. GFAP, IBA-1 and PrP were not decreased in the statin-treated infected mice versus the untreated infected mice, indicating that statin treatment was not highly effective at reducing the presence of activated microglia or astroglia, or in reducing the amount of PrPSc observed. All 22L-infected mice, regardless of treatment, showed significantly higher staining for GFAP, IBA-1 and PrP relative to the uninfected controls. Data were analysed by one-way ANOVA. ***P≤0.0001.
Effect of statin treatment on neuroinflammation during scrapie infection
To evaluate whether statin treatment altered the expression of neuroinflammatory markers during scrapie infection, we assayed the transcription levels of 84 genes associated with inflammation by qRT-PCR array. Only 10 of the 84 genes assayed changed in expression with statin therapy (Table 2). Treatment with pravastatin reduced the expression of the greatest number of inflammatory genes relative to infected untreated mice (Table 3), but the expression of three of these reduced genes (Csf3, IL3 and Tnfsf4) is typically unchanged during scrapie infection [5]. Conversely, pravastatin therapy increased the expression of Cxcl13. In contrast, treatment with simvastatin did not reduce the expression of any of the 84 proinflammatory genes tested, but Ccl2 and Cxcl13 were slightly elevated during scrapie infection in simvastatin-treated mice (Table 3). Atorvastatin treatment decreased the expression of only one gene, Cxcr3, during scrapie infection (Table 3). Most of the inflammatory genes that we previously reported to be elevated in scrapie-infected brain (i.e. Cxcl10, IL1rn, Ccl4 and Ccl5) [5, 6, 40] were unchanged in statin-treated mice relative to the untreated infected controls (Tables 2, S1 and S2, available in the online Supplementary Material). Although the statin treatments varied in their ability to alter the expression of a small number of the 84 inflammatory genes tested, therapy did not affect time until euthanasia due to clinical disease, PrPres accumulation, or gliosis in the scrapie-infected brain.
Table 2. Comparison of the effects of scrapie infection and statin treatment on the expression of neuroinflammatory genes in the brain.
| Changed with statin therapy | Unchanged with statin therapy | ||
|---|---|---|---|
| Reduced expression | Increased expression | ||
| Changed with scrapie infection | 5* | 2 | 32† |
| Unchanged with scrapie infection | 3 | 0 | 42‡ |
*Number of neuroinflammatory genes is shown in each box. Data for change in expression with scrapie infection are from [5, 6]. Data for change in expression with statin treatment are from the present paper.
†Genes can be found in Table S1.
‡Genes can be found in Table S2.
Table 3. Inflammatory genes significantly altered with statin treatment of 22L-infected mice compared to untreated 22L-infected control mice.
| Simvastatin | Pravastatin | Atorvastatin | |||
|---|---|---|---|---|---|
| Gene | Increased in scrapie [5] | FC† | FC | FC | Description |
| Ccl2 | Yes | 2.5* | 1.5 | 1.7 | Chemokine (C-C motif) ligand 2 |
| Cxcl11 | Yes | 1.6 | −2.1* | −1.1 | Chemokine (C-X-C motif) ligand 11 |
| Cxcl13 | Yes | 2.3* | 2.0* | 1.6 | Chemokine (C-X-C motif) ligand 13 |
| Cxcr3 | Yes | −1.6 | −1.9 | −4.0* | Chemokine (C-X-C motif) receptor 3 |
| Cxcr5 | Yes | 1.1 | −2.7* | −1.8 | Chemokine (C-X-C motif) receptor 5 |
| Osm | Yes | −1.1 | −4.3* | −1.5 | Oncostatin M |
| Tnf | Yes | 1.2 | −2.5* | −1.5 | Tumour necrosis factor |
| Csf3 | No | 1.2 | −2.7*** | −1.2 | Colony stimulating factor 3 |
| Il3 | No | −1.1 | −2.8* | −1.8 | Interleukin 3 |
| Tnfsf4 | No | 1.1 | −2.3* | −1.5 | Tumour necrosis factor superfamily, member 4 |
Bold numerals represent significant changes that met our criteria of a minimum twofold change.
Underlined bold numerals are changes where treatment increased chemokine expression.
†Fold change in gene expression in infected statin-treated versus infected untreated mice.
* P value ≤ 0.05, ***P value ≤0.001.
Discussion
In the present blinded study, we tested the ability of simvastatin, pravastatin and atorvastatin to influence scrapie pathogenesis in a mouse-adapted scrapie model. All three of these statins have been shown to penetrate the blood–brain barrier in mice [41, 42]. However, in our experiments there was no increase in the time of survival after long-term oral treatment with atorvastatin, simvastatin, or pravastatin. This was surprising because both simvastatin and pravastatin treatment were previously shown to extend survival time after scrapie infection by 10 to 20 days (Table 4) in a disease model that has a typical duration ranging from 140 to 180 days. However, in these earlier studies, the investigators gave no indication that they were blinded to the experimental cohorts of mice [36–39]. This may have influenced the results because of the difficulty in objectively determining the time until euthanasia in clinical prion disease models. Our use of a blinded protocol to reduce experimental bias may be a factor in explaining why we did not see the same beneficial effect of statin therapy reported by others.
Table 4. Comparison of published studies performed with statins on mice inoculated intracerebrally with scrapie.
| Mok et al. 2006* | Kempster et al. 2007 | Haviv et al. 2008 | Vetrugno et al. 2009 | This study | |
|---|---|---|---|---|---|
| Statin† (dose‡) | SMV (100) |
SMV (1.0) |
SMV (20, 10, 2) |
PRV (200) |
SMV (100) PRV (200) ATV (50) |
| Route of statin delivery | Chow | Drinking water | Drinking water | Drinking water | Chow |
| Start of treatment | At 100 days p.i. | At inoculation | At 41 or 72 days p.i. | At inoculation | At inoculation |
| Mouse strain | C57BL/6 | C57BL/6 j | FVB/N | C57BL/6 | C57BL/10 |
| Scrapie strain | 139A | ME7 | RML | 139A | 22L |
| Inoculum | 20 ul of 0.1 % or 0.01 % BH | 20 ul of 10 % BH | 30 ul of 1.0 % BH | ? ul of 1.0 % BH | 30 ul of 1.0 % BH |
| Cytokines | nd | nd | Assessed 94§ | nd | Assessed 84 (see Tables 2 and S1) |
| Average survival time improvement | 0.1 %=16 day 0.01 %=20 day |
10 days | 14 days (Cumulative) | 17 days | None |
| PrP-res deposition | Unaffected | Unaffected | Increased in treatment group | Unaffected | Unaffected |
BH, brain homogenate; nd, not determined.
*Author and year of publication.
†SMV, simvastatin; PRV, pravastatin; ATV, atorvastatin.
‡mg kg−1 day−1
§With treatment: increase in Ccl2, Ccl5, IL1b and Cxcr3, and decrease in Cxcl11 and Cox2.
Differences between the scrapie strains used in our study and those used in the previous studies may also have been a confounding factor when comparing these experiments. We infected our mice with scrapie strain 22L, while previous studies used strains ME7 (38), RML [37], or 139A [36, 39] (Table 4). However, in our earlier studies of the neuroinflammatory response in scrapie-infected brain, mice infected with strains ME7, 22L and RML all showed upregulation of similar neuroinflammatory genes [6]. Thus, the neuroinflammatory response to scrapie infection in mice does not appear to be unique for these scrapie strains. Therefore, the use of different scrapie strains in these statin treatment experiments is unlikely to be responsible for the lack of effectiveness of the treatment in our experiments.
In the previous studies using simvastatin to treat scrapie-infected mice, various drug doses were used, ranging from 1.0 to 100 mg kg−1 day−1 [38, 39]. Furthermore, Haviv et al. treated mice with three different doses (2, 10 and 20 mg kg−1 day−1) [37]. Oddly, similar changes in scrapie incubation period were seen among the studies (Table 4), regardless of the dose. In our studies, we used the highest of these doses and still saw no effect on incubation period and no reduction in pro-inflammatory gene expression with treatment.
There were notable differences between our gene expression results and those of Haviv et al. [37] when comparing simvastatin-treated mice. Although we saw a similar increase in Ccl2 expression with simvastatin treatment of scrapie-infected mice to Haviv et al., we did not see increases in the expression of Ccl5, Cxcr3, or IL1b, even though we treated with up to 5 times more drug. These discrepancies could be due to differences in the mice used, since the FVB/N mice used by Haviv et al. [37] have a mutation in the C5 complement allele that has been associated with immunological hyper-responsiveness in asthma disease models [43, 44]. This might account for the increased expression of several inflammatory genes in their study [37]. Furthermore, the 2 week difference in time until euthanasia in the statin-treated and untreated mice of Haviv et al. could also account for the slight increase in a few genes, since neuroinflammatory gene expression is known to increase with time during scrapie infection [5, 6]. Nonetheless, we both assessed over 80 genes associated with inflammation and found that most these genes were unchanged with simvastatin treatment of scrapie-infected mice.
Most of the genes that we previously described as being highly increased in the brains of scrapie-infected mice (i.e. Ccl4, Ccl5, Ccl8, Ccl12, Cxcl9, Cxcl10, IL1 and IL1rn) [5, 6] were unaffected by simvastatin, pravastatin, or atorvastatin therapy (Table S1). This was contrary to our original hypothesis that the expression of genes involved in neuroinflammation in scrapie-infected mice might be reduced with long-term statin therapy. We were especially surprised at the lack of immunomodulation with atorvastatin treatment in our scrapie-infected mice, since atorvastatin treatment significantly decreased expression of Cxcl10 in cerebral malaria [45], IL1 and Tnf in AD [30], and Tnf in EAE [31] rodent models. Possibly the inflammatory response in prion disease differs from the neuroinflammation in cerebral malaria, AD and EAE, as these diseases demonstrate substantial leukocyte transmigration into the brain [46–49]. In contrast, infiltration of leukocytes is rare or low-level in prion diseases [50–52]. The neuroinflammation in the CNS in response to scrapie is likely the direct response of glia to neuronal damage, with limited involvement of the peripheral immune response. This might explain why the anti-neuroinflammatory effects of statins, which reduce the penetration of peripheral immune cells into the CNS [32, 33, 45, 53], were not effective in our scrapie experiments.
Although statin treatment in many animal models of neuroinflammation has shown promise, its efficacy in human clinical trials remains controversial. Some clinical investigations reported that statin therapy reduced the incidence of Parkinson’s disease [54–56], but others concluded that statins are ineffective in halting progression, risk, or associated dementia in Parkinson’s disease [57, 58]. Clinical trials to assess the effectiveness of statins on AD progression have also produced mixed findings, with some groups reporting that statin therapy improved cognition and enhanced memory in AD patients [22, 24, 59, 60], but others reporting no benefit from statin treatment [22, 61–63]. Likewise, the findings from clinical trials with MS patients [64–68] led investigators to conclude that statin treatment may offer little benefit in MS.
Based on our results, simvastatin, pravastatin, or atorvastatin therapy alone were not effective in reducing the neuroinflammation or increasing the survival time associated with scrapie infection with strain 22L. In animal models the treatment of prion disorders has been difficult, and only treatments near the time of infection or prior to clinical signs have slowed disease progression [69–71]. Perhaps future experiments should investigate the combination of drugs with different mechanisms, such as statins or other anti-inflammatory drugs to target neuroinflammation, given together with drugs capable of delaying prion replication or blocking neurotoxicity. Such combinations have been useful in the treatment of cancer and various infectious diseases, and might also be useful in treating prion diseases.
Methods
Ethics statement
All mice were housed at the Rocky Mountain Laboratory (RML) in an AAALAC-accredited facility in compliance with guidelines provided by the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research Council). Experimentation followed RML Animal Care and Use Committee approved protocol 2015-034.
Mice and scrapie inoculations
The C57BL/10 (C57) mice were originally obtained from Jackson Laboratories and have been inbred at RML for several years. They were gender and age matched. All experimental mice were inoculated i.c. in the left hemisphere with 30 µl of a 1.0 % (w/v) strain 22L scrapie brain homogenate stock (final of 6.0×105 LD50) in phosphate-buffered balanced saline with 2 % foetal bovine serum on the same day. Mice were scored weekly for clinical signs beginning at 116 days p.i. and biweekly once clinical signs became more prominent. Observers were blinded to the drug treatment groups. A single score was assigned to each individual mouse based on the extent and level of clinical signs observed. Scorers paid particular attention to the severity of somnolence, kyphosis, gait abnormalities and decrease in body condition [72]. Scores were as follows: 0 = normal; 1 = subtle clinical signs; 2 = moderate clinical signs; and 3 = advanced clinical signs. Mice were euthanized when they reached a score of 3. Brain tissue was dissected for future use in histology, Western blot, or RNA gene expression assays. Survival curves were created using GraphPad Prism 7, and statistical analysis was performed on the curves by Mantel–Cox log rank.
Oral statin administration
The pharmaceutical-grade statins used in this study were manufactured by Accord BioPharma, Inc. (simvastatin, National Drug Code (NDC) # 16729-006-17); Glenmark Pharmaceuticals, Inc. (pravastatin, NDC # 68462-197-90); and Mylan Institutional, Inc. (atorvastatin, NDC # 0378-3952-09). The statins were formulated into rodent chow (Teklad Diets, Envigo) to deliver approximately 100, 200, or 50 mg kg−1 day−1 respectively for simvastatin, pravastatin and atorvastatin based on an average consumption of 5 g day−1 and an average weight of 25 g/mouse. The drug doses for simvastatin and pravastatin were chosen to mimic the levels achieved in mice by Mok et al. and Ventrugna et al. [36, 39]. Atorvastatin has a half-life of ~14 h, which is longer than that of simvastatin (~3 h) or pravastatin (~2 h) and allows drug accumulation to occur. To achieve equal effectiveness based on pharmacokinetics, the dose relative to simvastatin was halved [35]. Treated mice were fed drug-supplemented chow from the time of scrapie inoculation until the experimental endpoint. Control mice were fed identical chow without drug. Each treatment group consisted of 10 to 12 mice. For the duration of the study, investigators were blinded as to the treatment of each group of mice to avoid potential experimental bias.
Serum cholesterol analysis
Whole blood (approximately 0.5 ml) was collected from age-matched, uninfected control mice that were treated (or mock-treated) for 150 days with the same statin-medicated chows that were fed to the scrapie-infected mice. Blood samples were allowed to clot and then centrifuged for 7 min at 650 g. Serum was removed for cholesterol analysis. Cholesterol was measured using an IDEXX VetTest 8008 chemistry analyser and cholesterol slides (IDEXX product code 98-20365), while the investigator was blinded to the treatment of the mouse cohorts. Data were evaluated using GraphPad Prism 7 and statistical significance was assessed by one-way analysis of variance (ANOVA).
RNA isolation and qRT-PCR analysis
Mouse brain halves were homogenized in 3.0 ml ZR RNA buffer (Zymo Research) and stored for up to 5 days at −80 °C before processing. Total RNA was isolated using the Quick-RNA MidiPrep (Zymo Research) and treated with 4 units of DNase I (Ambion) for 1 h at 37 °C. RNA was stored with 1 x RNase inhibitor (SUPERase-In; Ambion) at −80 °C until use. For quantitative analysis of changes in transcription using qRT-PCR arrays, 400 ng of high-quality RNA from each sample was reverse-transcribed to synthesize cDNA using the RT2 First Stand kit per the manufacturer’s instructions (Qiagen). Each cDNA reaction was mixed with 2× RT2 SYBR Green Mastermix purchased from Qiagen with RNase-free water to a final volume of 1.3 ml. Ten microlitres of the mixture was then added to each well of a 384-well format plate of the mouse inflammatory cytokine and receptors super array PAMM-011ZE (Qiagen). The analysis was carried out on an Applied Biosystems ViiA 7 real-time PCR system with a 384-well block using the following conditions: 1 cycle at 10 min, 95 °C; 40 cycles at 15 s, 95 °C; and then 1 min, 60 °C with fluorescence data collection. Melting curves were generated at the end of the completed run to determine the quality of the reaction products. Raw threshold cycle (Ct) data were collected with a Ct of 35 as the cutoff. Ct data were analysed using the web-based RT² Profiler PCR array data analysis (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php). All Ct values were normalized to the average of the Ct values for the housekeeping genes Actb, Gapdh and Hsp90ab1. Changes in transcription were calculated by the software using the ΔΔCt-based method. Statistical analysis was performed using the unpaired Student’s t-test to compare the replicate 2−ΔCt values for each gene in the infected group versus the infected statin-treated groups. A mean of ≥2.0-fold change and a P-value of ≤0.05 was considered significant. Each treatment and control group consisted of a minimum of three independent RNA samples.
Immunohistochemistry and quantitation
After the mice were euthanized, the brains were removed and half of each brain placed in 3.7 % phosphate-buffered formalin for 3 to 5 days before dehydration and embedding in paraffin. Serial 5 µm sections were cut using a standard Leica microtome, placed on positively charged glass slides, and dried overnight at 56 °C. Slides were stained with a standard protocol of hematoxylin and eosin (H and E) for observation of the overall pathology. For the detection of microglia, sections were probed with a 1 : 2000 dilution of polyclonal rabbit antibodies against ionized calcium-binding adapter-1 (IBA-1) provided by Dr John Portis. For detection of astrocytes, sections were probed with a 1 : 3500 dilution of polyclonal rabbit antibodies to glial fibrillary acidic protein (GFAP) from Dako (Z0334). The secondary antibody, biotinylated goat anti-rabbit (Biogenex Ready-to-use Super Sensitive Rabbit Link) was applied undiluted to the slides. Detection was performed using the REDMap detection kit (Ventana #760–123). Slide processing was completed in a Discovery XT slide stainer (Ventana,) and read, with the treatments blinded to the investigator, by microscopy as previously described [40].
Scrapie-associated PrP (PrPSc) antigens were exposed by incubation in CC1 buffer (Ventana) containing Tris-borate-EDTA, pH 8.0, for 100 min at 95 °C as previously described [6]. PrP was stained with the human anti-PrP monoclonal antibody D13, where D13 culture supernatant was used at a dilution of 1 : 100 for 2 h at 37 °C. The secondary antibody was biotinylated goat anti-human IgG at a 1 : 250 dilution (Jackson ImmunoResearch), and streptavidin–biotin peroxidase was used with 3, 3′-diaminobenzidine (DAB) as the chromogen (DAB map kit; Ventana). Hematoxylin was used as a counterstain for all slides.
Sections stained for GFAP, IBA-1, and PrP were scanned with an Aperio ScanScope XT (Aperio Technologies, Inc.) and quantified using the ImageScope positive pixel count algorithm (version 9.1). This algorithm interprets the darkness (brown or red hue) of the pixel and parcels the positive pixel intensities into three categories based on designated intensity ranges: weak–positive (176–220), positive (101–175), and strong–positive (0–100). For each stained brain section, a 5-micron thick median sagittal section representing approximately 55 mm2 was evaluated at 1× magnification with the treatments blinded to the investigator. For the GFAP and IBA-1 stained sections all three pixel intensity categories were used to calculate the percentage of positive pixels (positive pixels/total pixels × 100). For the PrP-stained sections, only strong positive pixel intensities were counted to calculate the percentage of positive pixels. This adjustment was made to minimize the contribution of positive pixels detected due to normal PrP-sen staining. Control slides of brain sections from scrapie-infected mice were taken through the antibody staining procedure with the primary antibody omitted to assess background due to the secondary antibody and chromogenic substrate deposition. Data were analysed using GraphPad Prism 7 and statistical analysis was performed by one-way ANOVA.
SDS-PAGE and immunoblotting
Brains were homogenized (20 % w/v) using a Mini Bead Beater (BioSpec Products) as previously described [7] in ice-cold cell lysis buffer (Bio-Rad) supplemented with 2× Complete EDTA-free protease inhibitor cocktail (Roche) and 1× cell lysis factors 1 and 2 (Bio-Rad) consisting of sodium orthovanadate and sodium fluoride to prevent the dephosphorylation of proteins. For the detection of PrPres by immunoblot, tissue samples were analysed as described previously [40, 73]. Briefly, 0.36 mg of whole-brain equivalents were treated with proteinase K, separated by SDS-PAGE, transferred to polyvinylidene difluoride and probed with a 1 : 100 dilution of monoclonal human anti-PrP antibody D13. The secondary antibody was peroxidase-conjugated anti-human IgG used at 1 : 5000 (Sigma), and immunoreactive bands were visualized using an enhanced chemiluminescence detection system (Thermo Scientific).
Funding information
This work was supported by the National Institute of Allergy and Infectious Diseases, Division of Intramural Research.
Acknowledgements
We thank Jeffrey Severson for assistance with animal husbandry; Nancy Kurtz and Lori Lubke for assistance with immunohistochemical procedures; and Drs Clayton Winkler, Sue Priola and Byron Caughey for critical reading of the manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
References
- 1.Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases) Brain Pathol. 1995;5:459–466. doi: 10.1111/j.1750-3639.1995.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 2.Jeffrey M, González L. Classical sheep transmissible spongiform encephalopathies: pathogenesis, pathological phenotypes and clinical disease. Neuropathol Appl Neurobiol. 2007;33:373–394. doi: 10.1111/j.1365-2990.2007.00868.x. [DOI] [PubMed] [Google Scholar]
- 3.Scallet AC, Ye X. Excitotoxic mechanisms of neurodegeneration in transmissible spongiform encephalopathies. Ann N Y Acad Sci. 1997;825:194–205. doi: 10.1111/j.1749-6632.1997.tb48429.x. [DOI] [PubMed] [Google Scholar]
- 4.Black SA, Stys PK, Zamponi GW, Tsutsui S. Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity. Front Cell Dev Biol. 2014;2:45. doi: 10.3389/fcell.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll JA, Striebel JF, Race B, Phillips K, Chesebro B. Prion infection of mouse brain reveals multiple new upregulated genes involved in neuroinflammation or signal transduction. J Virol. 2015;89:2388–2404. doi: 10.1128/JVI.02952-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll JA, Striebel JF, Rangel A, Woods T, Phillips K, et al. Prion strain differences in accumulation of PrPSc on neurons and glia are associated with similar expression profiles of neuroinflammatory genes: comparison of three prion strains. PLoS Pathog. 2016;12:e1005551. doi: 10.1371/journal.ppat.1005551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tribouillard-Tanvier D, Striebel JF, Peterson KE, Chesebro B. Analysis of protein levels of 24 cytokines in scrapie agent-infected brain and glial cell cultures from mice differing in prion protein expression levels. J Virol. 2009;83:11244–11253. doi: 10.1128/JVI.01413-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aiken JM, Williamson JL, Marsh RF. Evidence of mitochondrial involvement in scrapie infection. J Virol. 1989;63:1686–1694. doi: 10.1128/jvi.63.4.1686-1694.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi SI, Ju WK, Choi EK, Kim J, Lea HZ, et al. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol. 1998;96:279–286. doi: 10.1007/s004010050895. [DOI] [PubMed] [Google Scholar]
- 10.Park JH, Kim BH, Park SJ, Jin JK, Jeon YC, et al. Association of endothelial nitric oxide synthase and mitochondrial dysfunction in the hippocampus of scrapie-infected mice. Hippocampus. 2011;21:319–333. doi: 10.1002/hipo.20753. [DOI] [PubMed] [Google Scholar]
- 11.Sisková Z, Mahad DJ, Pudney C, Campbell G, Cadogan M, et al. Morphological and functional abnormalities in mitochondria associated with synaptic degeneration in prion disease. Am J Pathol. 2010;177:1411–1421. doi: 10.2353/ajpath.2010.091037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi HS, Choi YG, Shin HY, Oh JM, Park JH, et al. Dysfunction of mitochondrial dynamics in the brains of scrapie-infected mice. Biochem Biophys Res Commun. 2014;448:157–162. doi: 10.1016/j.bbrc.2014.04.069. [DOI] [PubMed] [Google Scholar]
- 13.Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, et al. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- 14.Liberski PP, Sikorska B, Bratosiewicz-Wasik J, Gajdusek DC, Brown P. Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: from apoptosis to autophagy. Int J Biochem Cell Biol. 2004;36:2473–2490. doi: 10.1016/j.biocel.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Hope J, Shearman MS, Baxter HC, Chong A, Kelly SM, et al. Cytotoxicity of prion protein peptide (PrP106-126) differs in mechanism from the cytotoxic activity of the Alzheimer's disease amyloid peptide, Aβ25-35. Neurodegeneration. 1996;5:1–11. doi: 10.1006/neur.1996.0001. [DOI] [PubMed] [Google Scholar]
- 16.Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, et al. Inflammation in neurodegenerative diseases– an update. Immunology. 2014;142:151–166. doi: 10.1111/imm.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 18.Valera E, Spencer B, Masliah E. Immunotherapeutic approaches targeting Amyloid-β, α-Synuclein, and tau for the treatment of neurodegenerative disorders. Neurotherapeutics. 2016;13:179–189. doi: 10.1007/s13311-015-0397-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zilka N, Korenova M, Novak M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol. 2009;118:71–86. doi: 10.1007/s00401-009-0499-y. [DOI] [PubMed] [Google Scholar]
- 20.Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, et al. Who fans the flames of Alzheimer's disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012;9:47. doi: 10.1186/1742-2094-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiss AB, Wirkowski E. Statins in neurological disorders: mechanisms and therapeutic value. Scientific World J. 2009;9:1242–1259. doi: 10.1100/tsw.2009.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q, Yan J, Chen X, Li J, Yang Y, et al. Statins: multiple neuroprotective mechanisms in neurodegenerative diseases. Exp Neurol. 2011;230:27–34. doi: 10.1016/j.expneurol.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Bedi O, Dhawan V, Sharma PL, Kumar P. Pleiotropic effects of statins: new therapeutic targets in drug design. Naunyn Schmiedebergs Arch Pharmacol. 2016;389:695–712. doi: 10.1007/s00210-016-1252-4. [DOI] [PubMed] [Google Scholar]
- 25.Kumar A, Sharma N, Gupta A, Kalonia H, Mishra J. Neuroprotective potential of atorvastatin and simvastatin (HMG-CoA reductase inhibitors) against 6-hydroxydopamine (6-OHDA) induced Parkinson-like symptoms. Brain Res. 2012;1471:13–22. doi: 10.1016/j.brainres.2012.06.050. [DOI] [PubMed] [Google Scholar]
- 26.Xu YQ, Long L, Yan JQ, Wei L, Pan MQ, et al. Simvastatin induces neuroprotection in 6-OHDA-lesioned PC12 via the PI3K/AKT/caspase 3 pathway and anti-inflammatory responses. CNS Neurosci Ther. 2013;19:170–177. doi: 10.1111/cns.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE, et al. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci. 2009;29:13543–13556. doi: 10.1523/JNEUROSCI.4144-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selley ML. Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res. 2005;1037:1–6. doi: 10.1016/j.brainres.2004.02.083. [DOI] [PubMed] [Google Scholar]
- 29.Zhao L, Chen T, Wang C, Li G, Zhi W, et al. Atorvastatin in improvement of cognitive impairments caused by amyloid β in mice: involvement of inflammatory reaction. BMC Neurol. 2016;16:18. doi: 10.1186/s12883-016-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YY, Fan YC, Wang M, Wang D, Li XH. Atorvastatin attenuates the production of IL-1β, IL-6, and TNF-α in the hippocampus of an amyloid β1-42-induced rat model of Alzheimer's disease. Clin Interv Aging. 2013;8:103–110. doi: 10.2147/CIA.S40405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Youssef S, Stüve O, Patarroyo JC, Ruiz PJ, Radosevich JL, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 32.Stanislaus R, Singh AK, Singh I. Lovastatin treatment decreases mononuclear cell infiltration into the CNS of Lewis rats with experimental allergic encephalomyelitis. J Neurosci Res. 2001;66:155–162. doi: 10.1002/jnr.1207. [DOI] [PubMed] [Google Scholar]
- 33.Greenwood J, Walters CE, Pryce G, Kanuga N, Beraud E, et al. Lovastatin inhibits brain endothelial cell Rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. Faseb J. 2003;17:905–907. doi: 10.1096/fj.02-1014fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanislaus R, Pahan K, Singh AK, Singh I. Amelioration of experimental allergic encephalomyelitis in Lewis rats by lovastatin. Neurosci Lett. 1999;269:71–74. doi: 10.1016/S0304-3940(99)00414-0. [DOI] [PubMed] [Google Scholar]
- 35.Klotz U. Pharmacological comparison of the statins. Arzneimittelforschung. 2003;53:605–611. doi: 10.1055/s-0031-1297156. [DOI] [PubMed] [Google Scholar]
- 36.Vetrugno V, di Bari MA, Nonno R, Puopolo M, D'Agostino C, et al. Oral pravastatin prolongs survival time of scrapie-infected mice. J Gen Virol. 2009;90:1775–1780. doi: 10.1099/vir.0.009936-0. [DOI] [PubMed] [Google Scholar]
- 37.Haviv Y, Avrahami D, Ovadia H, Ben-Hur T, Gabizon R, et al. Induced neuroprotection independently from PrPSc accumulation in a mouse model for prion disease treated with simvastatin. Arch Neurol. 2008;65:762–775. doi: 10.1001/archneur.65.6.762. [DOI] [PubMed] [Google Scholar]
- 38.Kempster S, Bate C, Williams A. Simvastatin treatment prolongs the survival of scrapie-infected mice. Neuroreport. 2007;18:479–482. doi: 10.1097/WNR.0b013e328058678d. [DOI] [PubMed] [Google Scholar]
- 39.Mok SW, Thelen KM, Riemer C, Bamme T, Gültner S, et al. Simvastatin prolongs survival times in prion infections of the central nervous system. Biochem Biophys Res Commun. 2006;348:697–702. doi: 10.1016/j.bbrc.2006.07.123. [DOI] [PubMed] [Google Scholar]
- 40.Tribouillard-Tanvier D, Race B, Striebel JF, Carroll JA, Phillips K, et al. Early cytokine elevation, PrPres deposition, and gliosis in mouse scrapie: no effect on disease by deletion of cytokine genes IL-12p40 and IL-12p35. J Virol. 2012;86:10377–10383. doi: 10.1128/JVI.01340-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen C, Lin J, Smolarek T, Tremaine L. P-glycoprotein has differential effects on the disposition of statin acid and lactone forms in mdr1a/b knockout and wild-type mice. Drug Metab Dispos. 2007;35:1725–1729. doi: 10.1124/dmd.107.015677. [DOI] [PubMed] [Google Scholar]
- 42.Johnson-Anuna LN, Eckert GP, Keller JH, Igbavboa U, Franke C, et al. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J Pharmacol Exp Ther. 2005;312:786–793. doi: 10.1124/jpet.104.075028. [DOI] [PubMed] [Google Scholar]
- 43.Wong K, Bumpstead S, van der Weyden L, Reinholdt LG, Wilming LG, et al. Sequencing and characterization of the FVB/NJ mouse genome. Genome Biol. 2012;13:R72. doi: 10.1186/gb-2012-13-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sellers RS. Translating mouse models: immune variation and efficacy testing. Toxicol Pathol. 2017;45:134–145. doi: 10.1177/0192623316675767. [DOI] [PubMed] [Google Scholar]
- 45.Wilson NO, Solomon W, Anderson L, Patrickson J, Pitts S, et al. Pharmacologic inhibition of CXCL10 in combination with anti-malarial therapy eliminates mortality associated with murine model of cerebral malaria. PLoS One. 2013;8:e60898. doi: 10.1371/journal.pone.0060898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mcmanus RM, Mills KH, Lynch MA. T cells-protective or pathogenic in Alzheimer's disease? J Neuroimmune Pharmacol. 2015;10:547–560. doi: 10.1007/s11481-015-9612-2. [DOI] [PubMed] [Google Scholar]
- 47.Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, et al. On the pathogenic role of brain-sequestered αβ CD8+ T cells in experimental cerebral malaria. J Immunol. 2002;169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- 48.Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183:7169–7177. doi: 10.4049/jimmunol.0901906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol. 1981;11:195–199. doi: 10.1002/eji.1830110307. [DOI] [PubMed] [Google Scholar]
- 50.Williams AE, Lawson LJ, Perry VH, Fraser H. Characterization of the microglial response in murine scrapie. Neuropathol Appl Neurobiol. 1994;20:47–55. doi: 10.1111/j.1365-2990.1994.tb00956.x. [DOI] [PubMed] [Google Scholar]
- 51.Betmouni S, Perry VH, Gordon JL. Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience. 1996;74:1–5. doi: 10.1016/0306-4522(96)00212-6. [DOI] [PubMed] [Google Scholar]
- 52.Lewicki H, Tishon A, Homann D, Mazarguil H, Laval F, et al. T cells infiltrate the brain in murine and human transmissible spongiform encephalopathies. J Virol. 2003;77:3799–3808. doi: 10.1128/JVI.77.6.3799-3808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neuhaus O, Strasser-Fuchs S, Fazekas F, Kieseier BC, Niederwieser G, et al. Statins as immunomodulators: comparison with interferon-β1b in MS. Neurology. 2002;59:990–997. doi: 10.1212/WNL.59.7.990. [DOI] [PubMed] [Google Scholar]
- 54.Undela K, Gudala K, Malla S, Bansal D. Statin use and risk of Parkinson's disease: a meta-analysis of observational studies. J Neurol. 2013;260:158–165. doi: 10.1007/s00415-012-6606-3. [DOI] [PubMed] [Google Scholar]
- 55.Friedman B, Lahad A, Dresner Y, Vinker S. Long-term statin use and the risk of Parkinson's disease. Am J Manag Care. 2013;19:626–632. [PubMed] [Google Scholar]
- 56.Gao X, Simon KC, Schwarzschild MA, Ascherio A. Prospective study of statin use and risk of Parkinson disease. Arch Neurol. 2012;69:380–384. doi: 10.1001/archneurol.2011.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang X, Alonso A, Guo X, Umbach DM, Lichtenstein ML, et al. Statins, plasma cholesterol, and risk of Parkinson's disease: a prospective study. Mov Disord. 2015;30:552–559. doi: 10.1002/mds.26152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tison F, Nègre-Pagès L, Meissner WG, Dupouy S, Li Q, et al. Simvastatin decreases levodopa-induced dyskinesia in monkeys, but not in a randomized, placebo-controlled, multiple cross-over ("n-of-1") exploratory trial of simvastatin against levodopa-induced dyskinesia in Parkinson's disease patients. Parkinsonism Relat Disord. 2013;19:416–421. doi: 10.1016/j.parkreldis.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Sparks DL, Sabbagh M, Connor D, Soares H, Lopez J, et al. Statin therapy in Alzheimer's disease. Acta Neurol Scand Suppl. 2006;185:78–86. doi: 10.1111/j.1600-0404.2006.00689.x. [DOI] [PubMed] [Google Scholar]
- 60.Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer's disease. Ther Adv Neurol Disord. 2013;6:19–33. doi: 10.1177/1756285612461679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74:956–964. doi: 10.1212/WNL.0b013e3181d6476a. [DOI] [PubMed] [Google Scholar]
- 62.Simons M, Schwärzler F, Lütjohann D, von Bergmann K, Beyreuther K, et al. Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol. 2002;52:346–350. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- 63.Trompet S, van Vliet P, de Craen AJ, Jolles J, Buckley BM, et al. Pravastatin and cognitive function in the elderly. Results of the PROSPER study. J Neurol. 2010;257:85–90. doi: 10.1007/s00415-009-5271-7. [DOI] [PubMed] [Google Scholar]
- 64.Pihl-Jensen G, Tsakiri A, Frederiksen JL. Statin treatment in multiple sclerosis: a systematic review and meta-analysis. CNS Drugs. 2015;29:277–291. doi: 10.1007/s40263-015-0239-x. [DOI] [PubMed] [Google Scholar]
- 65.Birnbaum G, Cree B, Altafullah I, Zinser M, Reder AT. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology. 2008;71:1390–1395. doi: 10.1212/01.wnl.0000319698.40024.1c. [DOI] [PubMed] [Google Scholar]
- 66.Lanzillo R, Orefice G, Quarantelli M, Rinaldi C, Prinster A, et al. Atorvastatin combined to interferon to verify the efficacy (ACTIVE) in relapsing-remitting active multiple sclerosis patients: a longitudinal controlled trial of combination therapy. Mult Scler. 2010;16:450–454. doi: 10.1177/1352458509358909. [DOI] [PubMed] [Google Scholar]
- 67.Togha M, Karvigh SA, Nabavi M, Moghadam NB, Harirchian MH, et al. Simvastatin treatment in patients with relapsing-remitting multiple sclerosis receiving interferon beta 1a: a double-blind randomized controlled trial. Mult Scler. 2010;16:848–854. doi: 10.1177/1352458510369147. [DOI] [PubMed] [Google Scholar]
- 68.Sorensen PS, Lycke J, Erälinna JP, Edland A, Wu X, et al. Simvastatin as add-on therapy to interferon β-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): a placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011;10:691–701. doi: 10.1016/S1474-4422(11)70144-2. [DOI] [PubMed] [Google Scholar]
- 69.Demaimay R, Adjou KT, Beringue V, Demart S, Lasmézas CI, et al. Late treatment with polyene antibiotics can prolong the survival time of scrapie-infected animals. J Virol. 1997;71:9685–9689. doi: 10.1128/jvi.71.12.9685-9689.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pocchiari M, Schmittinger S, Masullo C. Amphotericin B delays the incubation period of scrapie in intracerebrally inoculated hamsters. J Gen Virol. 1987;68:219–223. doi: 10.1099/0022-1317-68-1-219. [DOI] [PubMed] [Google Scholar]
- 71.Priola SA, Raines A, Caughey WS. Porphyrin and phthalocyanine antiscrapie compounds. Science. 2000;287:1503–1506. doi: 10.1126/science.287.5457.1503. [DOI] [PubMed] [Google Scholar]
- 72.Striebel JF, Race B, Carroll JA, Phillips K, Chesebro B. Knockout of fractalkine receptor Cx3cr1 does not alter disease or microglial activation in prion-infected mice. J Gen Virol. 2016;97:1481–1487. doi: 10.1099/jgv.0.000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meade-White K, Race B, Trifilo M, Bossers A, Favara C, et al. Resistance to chronic wasting disease in transgenic mice expressing a naturally occurring allelic variant of deer prion protein. J Virol. 2007;81:4533–4539. doi: 10.1128/JVI.02762-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.