

ABSTRACT
As environments and pathogen landscapes shift, host defenses must evolve to remain effective. Due to this selection pressure, among-species comparisons of genetic sequence data often find immune genes to be among the fastest evolving genes across the genome. The full extent and nature of these immune adaptations, however, remain largely unexplored. In a recent study, we analyzed patterns of selection within distinct components of the Drosophila melanogaster immune pathway. While we found evidence of positive selection within some immune processes, immune genes were not universally characterized by signatures of strong selection. On the contrary, we even found that some immune functions show greater than expected constraint. Overall these results highlight 2 major factors that appear to play an outsize role in determining a gene's evolutionary rate: the type of pathogen the gene targets and the gene's position within the immune network. These results join a growing body of literature that highlight the complexity of immune adaptation. Rather than there being uniformly strong selection across all immune genes, a combination of pathogen-specificity and host genetic constraints appear to play key roles in determining each immune gene's individual evolutionary trajectory.
KEYWORDS: evolution, host-pathogen interactions, innate immunity, local adaptation, population genetics
Introduction
Drosophila has long been at the forefront of evolutionary genetic studies,1 and recent genome sequencing projects are pushing our knowledge even further. The past decade has seen the release of numerous Drosophila species genomes,2,3 and additional sequencing efforts are elucidating natural variation within single species, particularly D. melanogaster.4 These data sets provide valuable information that can be leveraged to understand the genetic basis of traits5 and the selection pressures acting upon them.6
Studies of local adaptation commonly rely on genome scans to identify genes that have experienced strong spatially variable selection. These tests look for genomic signatures that result from selection favoring a particular allele: changes in allele frequency,7 increases in linkage disequilibrium,8 and greater population differentiation.9 By comparing these values across the entire genome, selection scans identify outlier genes that show the greatest likelihood of having undergone adaptive evolution. Such approaches are powerful ways of locating rapidly evolving loci. For instance in Drosophila, selection scans have identified numerous genes that associate with the fly's new cosmopolitan habitat.10 In addition, genome scans can be combined with ecological data to investigate how organisms adapt to novel environments.11 Over the past several decades, multiple phenotypic characteristics have been described as varying clinally across multiple continents in D. melanogaster. The sequencing of genomes across these clines has additionally identified genetic variants that preferentially associate with temperate vs. tropical climates.12-14 Repeated temporal sampling within locations has further provided insight into how selection pressures vary seasonally as well as geographically.15
Due to the strength of selection they experience, genes identified by genome-wide scans are generally presumed to carry a large phenotypic effect. Mutations with large effect sizes, however, do not account for the full extent of adaptive evolution. Instead, many traits evolve through a process of polygenic adaptation whereby phenotypic change is the result of small alterations across numerous genes. This type of adaptation is largely invisible in an outlier approach.16 Gene-enrichment analysis partially addresses this knowledge gap by highlighting gene classes with a disproportionate number of rapidly adapting genes. Results from such analyses, however, must be interpreted with caution.17 Additionally, such enrichment studies generally rely on a binary categorization of genes and so fail to include information about genes that are not initially identified as evolutionary outliers. Therefore, while genome-wide scans robustly identify what genes are under selection, they are not ideal for identifying what traits are under selection.
Immune adaptation in D. melanogaster
The immune response is a prime example of how our knowledge of individual genes may diverge from our understanding of the complete trait. In Drosophila, both genome-wide scans and individual gene studies have found evidence of strong selection occurring in immune genes.12,13,18-21 Such results are echoed across other species, where there exist numerous canonical examples of host-pathogen interactions leading to strong selection. Although anecdotal, these results often lead us to assume that immune systems are universally rapidly evolving. Is this a valid assumption? Are immune processes generally characterized by rapid adaptation? Or are these textbook examples outliers, even among immune genes?
In a recent study, we investigated these questions using a set of lines sampled from 5 locations that span the ancestral and newly colonized range of D. melanogaster: Zimbabwe; the Netherlands; Beijing, China; Ithaca, NY, USA; and Tasmania, Australia.22 This set of 84 inbred lines, which we call the Global Diversity Lines, has been fully sequenced, allowing for concomitant investigation of phenotypic variation and population genetic patterns.23 Since our goal was to understand immune adaptation, we chose to use a targeted approach that examined all known immune genes in D. melanogaster. Instead of being studied individually, genes were grouped based on their function and analyzed en masse as a way of detecting smaller signatures of selection distributed across a larger number of genes. In other words, this approach down-weighted the contribution of outlier genes and allowed us to determine whether evolution across a full set of immune genes was elevated compared with the genomic average. One potential caveat to keep in mind with this approach, however, is the incompleteness of our understanding of immune gene function; any inaccuracies in these functional assignments will clearly weaken the ultimate conclusions.
Drosophila possesses an innate immune system, but while it lacks the type of specificity conferred through vertebrate adaptive immunity, it contains several distinct pathways that preferentially respond to specific pathogen classes. Certain pathogen types might show greater spatial variability than others, leading to variability in the selection pressures imposed on these individual pathways. By annotating each immune gene based on the type of pathogen it targets, we could therefore investigate whether specific pathogen classes exert stronger selection pressures than others. Similarly, we considered another source of variation in adaptation: pathway position. Proteins at the network periphery (e.g., recognition receptors and effector molecules) are more likely to directly interface with pathogens. This direct contact might drive rapid adaptation. Alternatively, external environmental forces might impose selection pressures on general pathway functioning, which would preferentially target signaling genes. To study this source of variability, we additionally classified each gene based on its pathway role: recognition receptor, signaling molecule, or effector molecule. Overall, this categorization approach allowed us to test hypotheses about evolution in (1) the immune system as a whole, (2) immune responses to particular classes of pathogens, and (3) genes at different positions within the immune network.
For each gene, we acquired polymorphism data from the Global Diversity Lines as well as divergence data between D. melanogaster and D. simulans. With these data, we were able to compare patterns of long-term, species-level evolution to patterns of more recent, within-species adaptation. In all these cases, we did not try to quantify an absolute adaptive rate but rather asked about the rate of adaptation in immune genes relative to non-immune genes. We therefore compared the evolutionary patterns of the immune genes with those of a set of control genes with no immune function. These control genes were chosen to resemble the immune genes in several other evolutionarily important characteristics including length and genomic position.
Can species-level divergence predict local adaptation?
Our comparison of D. melanogaster and D. simulans genes found that immune gene categories showed extensive heterogeneity in their rates of adaptation, an observation that is consistent with past studies.18,19 Several categories differed from the background rate of adaptive evolution that we calculated with our control gene set (Fig. 1). Certain gene categories—including encapsulation genes, Toll pathway genes, and viral defense genes—showed evidence of heightened levels of adaptive evolution compared with the control category. Conversely, we also found gene categories—such as phagocytosis and epithelial defense genes—that had fewer adaptive changes than expected.
Figure 1.
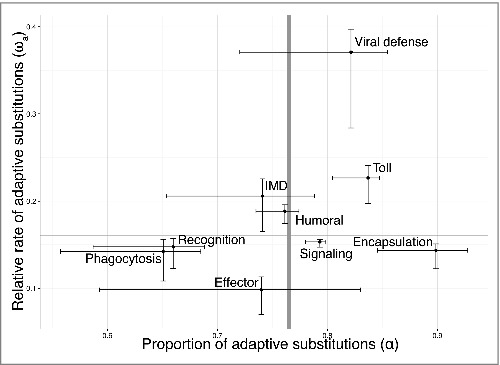
Proportion of adaptive substitutions (α) and the rate of adaptive substitutions (ωa) in a subset of key immune gene classes. The statistic α compares the neutral and adaptive divergence between 2 species to the neutral and adaptive polymorphism currently observed within one of those species. The statistic ωa similarly considers both adaptive divergence and adaptive polymorphism, and gives the adaptive substitution rate relative to the neutral one. Both metrics were calculated with the program DFE-alpha40 using D. melanogaster-D. simulans nucleotide divergence and polymorphism data from the Zimbabwe D. melanogaster population. The 95% confidence intervals were calculated with jackknife resampling. The gray bars mark the α and ωa values calculated with a set of non-immune control genes.
A priori, we expected that the variation in species-level divergence rates would be reflected on the population level. This, however, was not the case. Instead, at the population level we found that, by and large, immune categories were statistically indistinguishable from each other and from their set of matched control genes. Of our 20 immune gene categories, only 3 showed strong signs of an unusual rate of local adaptation at the protein-coding level: viral defense genes, phagocytosis genes, and recognition receptors (Fig. 2). Interestingly, only viral defense genes showed evidence of increased adaptation at both the species and population level.
Figure 2.
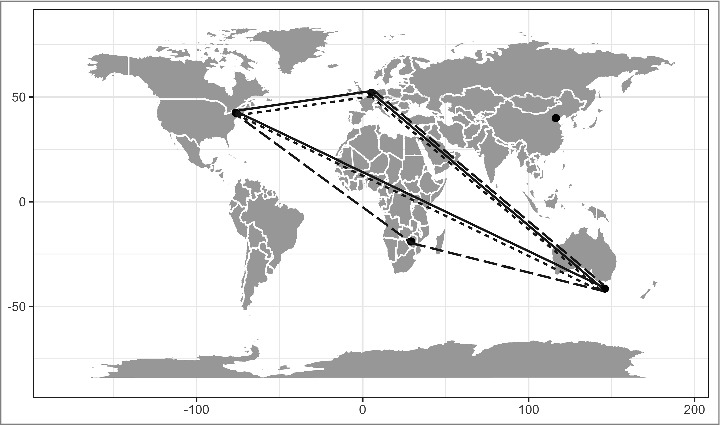
Pairwise population comparisons showing elevated population differentiation (FST) within immune classes. Three immune gene classes showed evidence of increased pairwise population differentiation between populations: phagocytosis genes (solid lines); viral defense or RNAi genes (dashed lines); and recognition receptors (dotted lines). This suggests that these 3 gene groups are experiencing divergent selection pressures in different populations, leading to changes in their respective allele frequencies. The presence of elevated FST was determined by comparing values in the immune gene set to those in the matched control.
Pathogen classes exert different selective pressures
The results from this study suggest that pathogen classes differ in the degree to which they have shaped the D. melanogaster genome. Prior to our study, the impact of viral pathogens had been recognized, and ample evidence had pointed to the existence of an arm's race—or at least a one-sided arm's race24—between Drosophila and its viral pathogens.19,25,26 By additionally considering the immune processes that target other pathogens, our study suggests that the fly's evolutionary relationship with viruses may be unique. The pattern we observed in Toll pathway genes is one interesting example. At the population level, we found that Toll pathway genes trended toward displaying lower—not higher—rates of adaptation. Other immune pathways, like the encapsulation process that targets the eggs of parasitoid wasps, were statistically indistinguishable from their matched control genes.
One explanation for this pattern might be the extent of host-specificity found among viruses. While not all known Drosophila viral pathogens are species-specific,27 viral Drosophila pathogens do display a certain degree of host-specificity. There is also evidence that viruses can rapidly invade populations,28 which might drive the complete or partial selective sweeps observed in viral defense genes. Conversely, the majority of sampled bacterial and fungal pathogens appear to be largely generalist or opportunistic. Still, viruses are not the only specialist pathogens that flies confront. Certain parasitoid wasps are also known to specialize on Drosophila species. Why then do we not detect strong signatures of local adaptation in parasitoid-defense genes? One answer might relate to the tempo of adaptation. On a species-divergence level, the proportion of adaptive substitutions (α) was relatively high for genes involved in encapsulation, one of Drosophila's key defenses against parasitoids. The overall rate of adaptation (ωa), however, was below the genome average. Parasitoid wasp defense may therefore evolve slowly but surely, leading to only small differences between populations that are not manifested as widespread changes throughout the set of encapsulation genes that we analyzed. In this regard, it is worth noting that 3 parasitoid response genes were in the top 5% of genes based on global population differentiation (FST) at nonsynonymous sites. The gene with the second highest global FST was lectin-24A, a gene that is upregulated in flies following wasp attack and that is hypothesized to be involved in recognition of wasp eggs.29 Our analytic approach was designed to detect numerous small adaptive changes throughout a pathway. It is possible that the encapsulation response has instead locally adapted through a few genetic changes of large effect.
A second explanation for these patterns rests not with the pathogens but with the pathways themselves. Does the varied genetic architecture of these diverse pathways influence their relative rates of adaptation? Notably, certain pathways—for example, Toll and JNK—contain numerous highly pleiotropic genes that perform pivotal functions in both immunity and basic development. Conversely, the majority of anti-viral RNAi genes evolved from gene duplications that allowed for rapid, unconstrained neo-functionalization.30 Further, while our analysis necessarily treated each pathway as unique, there are numerous examples of cross talk between them.31,32 This inter-dependence could create further constraints on the evolution of certain pathogen-specific defenses. Our definition of a pathway's role therefore simplifies important aspects of fly biology that no doubt impact evolutionary rate. Exploring these intricacies will be key for understanding the mechanisms behind immune adaptation. Regarding the source of evolutionary pressure, however, such discoveries will only open further questions: is the greater independence and higher evolvability of certain immune pathways a byproduct of genetic architecture or is it itself an adaptation to the selective pressure imposed by specific pathogens?
Conclusion
Currently, we lack ideal measures of local adaptation or recent evolution. In the coming years, the enhanced sensitivity of increased genome sampling4 and novel analytical methods33 will no doubt allow the detection of more subtle differences among the various immune processes explored in our study. Additionally, as we expand our understanding of Drosophila immunology—particularly in under-studied areas like parasitoid wasp defense—we may find novel examples of adaptation not included here. Still, these results demonstrate that the majority of immune genes are not extreme evolutionary outliers at the amino acid level. In fact, a key subset of immune genes shows a high level of conservation among species, among populations, or both. This result may appear surprising, but it joins a growing number of studies that have reached similar conclusions about the evolution of immunity in general and innate immunity in particular. While the most studied branch of human immunity—the adaptive immune system—contains numerous examples of strong directional selection, the innate immune system is characterized by purifying selection.34-36 Non-model organisms like newts,37 sticklebacks,38 and mallards39 have also shown signals of purifying selection or a lack of local adaptation. These results suggest that Drosophila is not an anomaly, but differences between these organisms exist, showing that even our more nuanced conclusions are not universal. Whereas these studies agree that evolution is not uniform across all immune genes, the categories experiencing higher conservation or more rapid adaption differ among species. Does this variation reflect true biologic differences in how species combat pathogens? Or is it an artifact created by disparities in analytical approaches and data availability? Further studies across a diverse array of organisms are needed as we refine the framework with which we construct our expectations regarding immune system evolution. As we move forward, however, it seems likely that both adaptation and conservation of genes will warrant attention as we seek to understand the evolution of complex host-pathogen interactions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the members of the Clark laboratory and Aquadro laboratory for helpful comments and discussions on this project. The comments of two anonymous reviewers improved this manuscript.
Funding
This work was supported by the National Institutes of Health, grant number R01 AI-064950 to A.G.C. and Brian Lazzaro.
References
- [1].Morgan TH. A Critique of the Theory of Evolution. Princeton: Princeton University Press; 1916. 197 p [Google Scholar]
- [2].St. Pierre SE, Ponting L, Stefancsik R, McQuilton P. FlyBase 102—advanced approaches to interrogating FlyBase. Nucleic Acids Res 2014; 42(D1):D780-8; PMID:24234449; https://doi.org/ 10.1093/nar/gkt1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Celniker SE, Dillon LAL, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al.. Unlocking the secrets of the genome. Nature 2009; 459(7249):927-30; PMID:19536255; https://doi.org/ 10.1038/459927a [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lack JB, Lange JD, Tang AD, Corbett-Detig RB, Pool JE. A Thousand Fly Genomes: An Expanded Drosophila Genome Nexus. Mol Biol Evolution 2016; 33(12):3308-13; PMID:27687565; https://doi.org/ 10.1093/molbev/msw195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al.. The Drosophila melanogaster genetic reference panel. Nature 2012; 482(7384):173-8; PMID:22318601; https://doi.org/ 10.1038/nature10811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lawrie DS, Petrov DA. Comparative population genomics: power and principles for the inference of functionality. Trends Genetics 2014; 30(4):133-9; PMID:24656563; https://doi.org/ 10.1016/j.tig.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res 2005; 15(11):1566-75; PMID:16251466; https://doi.org/ 10.1101/gr.4252305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, Gabriel SB, Platko J V, Patterson NJ, McDonald GJ, et al.. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002; 419(6909):832-7; PMID:12397357; https://doi.org/ 10.1038/nature01140 [DOI] [PubMed] [Google Scholar]
- [9].Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nature Rev Genetics 2009; 10(9):639-50; PMID:19687804; https://doi.org/ 10.1038/nrg2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pool JE, Corbett-Detig RB, Sugino RP, Stevens KA, Cardeno CM, Crepeau MW, Duchen P, Emerson JJ, Saelao P, Begun DJ, et al.. Population Genomics of Sub-Saharan Drosophila melanogaster: African Diversity and Non-African Admixture. PLoS Genetics 2012; 8(12):e1003080; PMID:23284287; https://doi.org/ 10.1371/journal.pgen.1003080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Günther T, Coop G. Robust identification of local adaptation from allele frequencies. Genetics 2013; 195(1):205-20; https://doi.org/ 10.1534/genetics.113.152462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kolaczkowski B, Kern AD, Holloway AK, Begun DJ. Genomic differentiation between temperate and tropical australian populations of Drosophila melanogaster. Genetics 2011; 187(1):245-60; PMID:21059887; https://doi.org/ 10.1534/genetics.110.123059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fabian DK, Kapun M, Nolte V, Kofler R, Schmidt PS, Schlötterer C, Flatt T. Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Mol Ecol 2012; 21(19):4748-69; PMID:22913798; https://doi.org/ 10.1111/j.1365-294X.2012.05731.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Reinhardt JA, Kolaczkowski B, Jones CD, Begun DJ, Kern AD. Parallel Geographic Variation in Drosophila melanogaster. Genetics 2014; 197(1):361-73; PMID:24610860; https://doi.org/ 10.1534/genetics.114.161463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bergland AO, Behrman EL, O'Brien KR, Schmidt PS, Petrov DA. Genomic evidence of rapid and stable adaptive oscillations over seasonal time scales in Drosophila Bolnick D, editor. PLoS Genetics 2014; 10(11):e1004775; PMID:25375361; https://doi.org/ 10.1371/journal.pgen.1004775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stephan W. Signatures of positive selection: from selective sweeps at individual loci to subtle allele frequency changes in polygenic adaptation. Mol Ecol 2016; 25(1):79-88; PMID:26108992; https://doi.org/ 10.1111/mec.13288 [DOI] [PubMed] [Google Scholar]
- [17].Pavlidis P, Jensen JD, Stephan W, Stamatakis A. A critical assessment of storytelling: Gene ontology categories and the importance of validating genomic scans. Mol Biol Evolution 2012; 29(10):3237-48; PMID:22617950; https://doi.org/ 10.1093/molbev/mss136 [DOI] [PubMed] [Google Scholar]
- [18].Sackton TB, Lazzaro BP, Schlenke TA, Evans JD, Hultmark D, Clark AG. Dynamic evolution of the innate immune system in Drosophila. Nat Genetics 2007; 39(12):1461-8; PMID:17987029; https://doi.org/ 10.1038/ng.2007.60 [DOI] [PubMed] [Google Scholar]
- [19].Obbard DJ, Welch JJ, Kim K-W, Jiggins FM, Sackton T, Lazzaro B, Schlenke T, Evans J, Hultmark D, Schlenke T, et al.. Quantifying adaptive evolution in the Drosophila immune system Begun DJ, editor. PLoS Genetics 2009; 5(10):e1000698; PMID:19851448; https://doi.org/ 10.1371/journal.pgen.1000698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Juneja P, Lazzaro BP. Haplotype structure and expression divergence at the Drosophila cellular immune Gene eater. Mol Biol Evolution 2010; 27(10):2284-99; PMID:20444883; https://doi.org/ 10.1093/molbev/msq114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hübner S, Rashkovetsky E, Kim YB, Oh JH, Michalak K, Weiner D, Korol AB, Nevo E, Michalak P. Genome differentiation of Drosophila melanogaster from a microclimate contrast in Evolution Canyon, Israel. Proc Natl Acad Sci U S A 2013; 110(52):21059-64; PMID:24324170; https://doi.org/ 10.1073/pnas.1321533111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Early AM, Arguello JR, Cardoso-Moreira M, Gottipati S, Grenier JK, Clark AG. Survey of global genetic diversity within the drosophila immune system. Genetics 2017; 205(1):353-366; PMID:27815361; https://doi.org/ 10.1534/genetics.116.195016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Grenier JK, Arguello JR, Moreira MC, Gottipati S, Mohammed J, Hackett SR, Boughton R, Greenberg AJ, Clark AG. Global diversity lines - a five-continent reference panel of sequenced Drosophila melanogaster strains. G3 (Bethesda, Md.) 2015; 5(4):593-603; PMID:25673134; https://doi.org/ 10.1534/g3.114.015883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Obbard DJ, Dudas G. The genetics of host–virus coevolution in invertebrates. Curr Opin Virol 2014; 8:73-8; PMID:25063907; https://doi.org/ 10.1016/j.coviro.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kolaczkowski B, Hupalo DN, Kern AD. Recurrent adaptation in RNA interference genes across the drosophila phylogeny. Mol Biol Evolution 2011; 28(2):1033-42; PMID:20971974; https://doi.org/ 10.1093/molbev/msq284 [DOI] [PubMed] [Google Scholar]
- [26].Obbard DJ, Jiggins FM, Bradshaw NJ, Little TJ. Recent and recurrent selective sweeps of the Antiviral RNAi Gene Argonaute-2 in three species of Drosophila. Mol Biol Evolution 2011; 28(2):1043-56; PMID:20978039; https://doi.org/ 10.1093/molbev/msq280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Webster CL, Waldron FM, Robertson S, Crowson D, Ferrari G, Quintana JF, Brouqui J-M, Bayne EH, Longdon B, Buck AH, et al.. The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. Plos Biol 2015; 13(7):e1002210; PMID:26172158; https://doi.org/ 10.1371/journal.pbio.1002210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Carpenter JA, Obbard DJ, Maside X, Jiggins FM. The recent spread of a vertically transmitted virus through populations of Drosophila melanogaster. Mol Ecol 2007; 16(18):3947-54; PMID:17725574; https://doi.org/ 10.1111/j.1365-294X.2007.03460.x [DOI] [PubMed] [Google Scholar]
- [29].Keebaugh ES, Schlenke TA. Adaptive evolution of a novel Drosophila Lectin induced by Parasitic Wasp attack. Mol Biol Evolution 2012; 29(2):565-77; PMID:21873297; https://doi.org/ 10.1093/molbev/msr191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Obbard DJ, Gordon KH, Buck AH, Jiggins FM. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc Lond B Biol Sci 2009; 364(1513):99-115; https://doi.org/ 10.1098/rstb.2008.0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Park JM, Brady H, Ruocco MG, Sun H, Williams D, Lee SJ, Kato T, Richards N, Chan K, Mercurio F, et al.. Targeting of TAK1 by the NF-kappa B protein Relish regulates the JNK-mediated immune response in Drosophila. Genes Dev 2004; 18(5):584-94; https://doi.org/ 10.1101/gad.1168104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tanji T, Hu X, Weber ANR, Ip YT. Toll and IMD pathways synergistically activate an innate immune response in Drosophila melanogaster. Mol Cell Biol 2007; 27(12):4578-88; PMID:17438142; https://doi.org/ 10.1128/MCB.01814-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Field Y, Boyle EA, Telis N, Gao Z, Gaulton KJ, Golan D, Yengo L, Rocheleau G, Froguel P, McCarthy MI, et al.. Detection of human adaptation during the past 2000 years. Science 2016; 354(6313):760-764; PMID:27738015; https://doi.org/ 10.1126/science.aag0776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mukherjee S, Sarkar-Roy N, Wagener DK, Majumder PP. Signatures of natural selection are not uniform across genes of innate immune system, but purifying selection is the dominant signature. Proc Natl Acad Sci U S A 2009; 106(17):7073-8; PMID:19359493; https://doi.org/ 10.1073/pnas.0811357106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Deschamps M, Laval G, Fagny M, Itan Y, Abel L, Casanova J-L, Patin E, Quintana-Murci L. Genomic signatures of selective pressures and introgression from Archaic Hominins at human innate immunity genes. A J Hum Genetics 2016; 98(1):5-21; PMID:26748513; https://doi.org/ 10.1016/j.ajhg.2015.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mukherjee S, Ganguli D, Majumder PP. Global footprints of purifying selection on Toll-Like Receptor genes primarily associated with response to bacterial infections in humans. Genome Biol Evolution 2014; 6(3):551-8; PMID:24554585; https://doi.org/ 10.1093/gbe/evu032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fijarczyk A, Dudek K, Babik W. Selective landscapes in newt immune genes Inferred from patterns of nucleotide variation. Genome Biol Evolution 2016; 8(11):3417-32; PMID:27702815; https://doi.org/ 10.1093/gbe/evw236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Robertson S, Bradley JE, MacColl ADC. No evidence of local adaptation of immune responses to Gyrodactylus in three-spined stickleback (Gasterosteus aculeatus). Fish Shellfish Immunol 2017; 60:275-81; https://doi.org/ 10.1016/j.fsi.2016.11.058 [DOI] [PubMed] [Google Scholar]
- [39].Chapman JR, Hellgren O, Helin AS, Kraus RHS, Cromie RL, Waldenström J. The evolution of innate immune genes: Purifying and balancing selection on β-Defensins in Waterfowl. Mol Biol Evolution 2016; 33(12):3075-87; PMID:27524825; https://doi.org/ 10.1093/molbev/msw167 [DOI] [PubMed] [Google Scholar]
- [40].Eyre-Walker A, Keightley PD. Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evolution 2009; 26(9):2097-108; PMID:19535738; https://doi.org/ 10.1093/molbev/msp119 [DOI] [PubMed] [Google Scholar]