

ABSTRACT
The inositol 1,4,5-trisphosphate receptor (IP3R) is one of two Ca2+ channels that gates Ca2+ release from ER-stores. The ligand IP3, generated upon specific G-protein coupled receptor activation, binds to IP3R to release Ca2+ into the cytosol. IP3R also mediates ER-store Ca2+ release into the mitochondria, under basal as well as stimulatory conditions; an activity that influences cellular bioenergetics and thus, cellular growth and proliferation. In Drosophila neuroendocrine cells expressing a hypomorphic mutant of IP3R, we observed reduced protein translation levels. Here, we discuss the possible molecular mechanism for this observation. We hypothesize that the cellular energy sensor, AMPK connects IP3R mediated Ca2+ release into the mitochondria, to protein translation, via the TOR pathway.
KEYWORDS: AMPK, larvae, starvation, TOR
Introduction
Ionic Ca2+ plays an important role in various cellular reactions and in order to utilize it for specific functions in a spatio-temporal manner, cells carefully manage Ca2+ levels in the cytosol as well as various organelles. Under resting conditions, the endoplasmic reticulum (ER) typically is the major store for Ca2+ with concentrations in the range 100–500 μM, while cytosolic Ca2+ is maintained at ∼100 nM.1 This large concentration gradient is maintained by the Sarco-Endoplasmic Reticulum Calcium ATP-ase (SERCA), which continuously pumps Ca2+ from the cytosol into the ER. Release of ER-store Ca2+ can be mediated by two known intracellular Ca2+ channels, the ryanodine receptor (RyR) and the inositol 1,4,5-trisphosphate receptor (IP3R). Although the two receptor share structural homology, they are activated by unique ligands and mediate distinct effects inside the cell. The physiological ligand for IP3R is IP3, which is generated by the action of phospholipase Cβ (PLCβ) on the lipid phosphatidylinositol 4,5-bisphosphate (PIP2). PLCβ in turn is activated by G-protein coupled receptors (GPCRs), typically by the Gαq subunit. Agonist mediated GPCR activation thus results in increased cytosolic Ca2+, via IP3R mediated Ca2+ release from the ER-store. This cytosolic Ca2+ potentiates a dazzling array of cellular processes, depending on the cell type and context.2 Release of ER-Store Ca2+ is also known to activate the Store-operated Ca2+ entry pathway that brings Ca2+ from the extracellular milieu into the cytosol.3 In certain cell types, IP3R can also be activated by receptor tyrosine kinases (RTKs) via PLC-γ. However in Drosophila, no genetic interaction has been observed between small wings (sl), the gene locus for the sole fly PLC-γ, and IP3R.4
While mammals have three IP3R isoforms coded by three distinct genes, in Drosophila melanogaster there is only one IP3R gene, which shares 57–60% sequence homology to mammalian IP3R1 and broad structural domain homology. Importantly, the rat IP3R1 can functionally complement Drosophila IP3R at the cellular as well as organismal level.5 There are three domains recognized on the IP3R: an N-terminal IP3/ligand binding domain, a central regulatory domain, followed by a C-terminal channel/pore forming domain. IP3R functions as a tetrametric channel, and a recently published cryo-EM structure predicts a major role for the N-terminal domain in gating channel properties despite a large number of residues between the two.6
To elucidate how IP3R function in distinct cellular contexts can influence systemic animal functioning, our lab generated Drosophila single point mutants for IP3R using an EMS mutagenesis based approach.7 Several point mutants were recovered, but none were homozygous viable. We have thus been utilizing IP3R hypomorphic heteroallelic mutant combinations to uncover systemic phenotypes and then tracking down the cellular basis for the manifestations of such phenotypes. One such study utilized a heteroallelic mutant with a combination that mutates S224F in the ligand binding domain, and G1891S in the regulatory domain. This mutant IP3R strain is referred to as itprku. In this strain, IP3R protein is expressed to the same levels as WT,8 whereas functionally, larval neurons exhibit attenuated Store-operated Ca2+ entry.9
In our recent report, itprku larvae were observed to have lower rates of protein synthesis.10 Genetic studies revealed that loss of IP3R in dimm+ neuroendocrine (NE) cells partially phenocopied the mutant. Interestingly, systemic phenotypes were also recapitulated at the cellular level: dimm+ NE cells from itprku were observed to have lower levels of protein translation over a 30-minute time period, similar to levels seen in NE cells treated with the protein synthesis inhibitor cyclohexamide.10 dimm is a transcription factor expressed by many, but not all Drosophila neuroendocrine cells.11 Dimm regulates neuroendocrine cell fate as well as the scaling up of the protein machinery required for the regulated secretory pathway.12 dimm+ NE cells synthesize and secrete a large variety of neuropeptides (NPs), which directly or indirectly, influence development and physiology profoundly.13 Given the important role for NPs in metabolism, and that certain external stimuli can require immediate and robust over-expression as well as secretion of NPs,14 an effect of IP3R on protein translation in NE cells might hold regulatory significance. Indeed, loss of IP3R in dimm+ NE cells reduces pupariation of larvae subject to late–stage nutritional deprivation. As over-expression of WT IP3R in itprku NE cells was sufficient to restore basal cellular protein translation to levels as seen in the control, as well as improve systemic protein levels in itprku larvae,10 it appears that IP3R is a positive regulator of protein synthesis. This commentary speculates on the molecular mechanism that may link IP3R to protein translation.
In previous studies, depletion of ER-Ca2+ store in mammalian cell lines or non-neuronal primary cultures, either by chemicals that cause ER stress, or by activation of IP3R by hormones, has been reported to reduce protein synthesis, via eIF2α phosphorylation.15 This is opposite to what we observe, and may perhaps be explained on the basis of two major differences between these studies and the experiments we performed. First, we observe this effect specifically in NE cells, and not in neurons, a distinct population not previous explored in the context of IP3R functioning and protein translation. Perhaps this cell-specific effect of IP3R is restricted to cells with large protein synthetic requirements. Second, we observed protein translation defects in the absence of ER-Store Ca2+ depletion i.e., under basal non-stimulatory conditions, while previous studies have been undertaken in the presence of ER stress by addition of inhibitors such as thapsigargin or ionomycin, or stimulation of IP3R Ca2+ release by hormones. This may be significant in the context of a study by Foskett et al., that reported a role for low level constitutive Ca2+ leak by IP3R in cellular bioenergetics.16
IP3R and AMPK
IP3R mediated transfer of Ca2+ from the ER to the mitochondria is now a well established cellular process, studied mainly for its role in apoptosis and autophagy.17 However, it is also well known that there is a basal requirement for Ca2+ inside the mitochondria for the production of ATP. For example, Ca2+ increases the activity of dehydrogenases of the TCA cycle.18 Transport of Ca2+ relies on local areas of ER-mitochondria contact points called Mitochondrial Associated Membranes, which are enriched with several proteins including IP3R.19 In effect, IP3R mediated Ca2+ release affects mitochondrial functioning. Fitting this view is the observation that under normal, nutrient-rich conditions, genetic knockout of the three mammalian IP3R genes or inhibition of IP3Rs by XestosponginB (XeB) compromised ATP production in the mitochondria.16 Increased AMP/ADP:ATP ratio is sensed by AMP-activated protein kinase (AMPK). Consequently, AMPK is phosphorylated (pAMPK) and, in effect reduction of IP3R, via pAMPK results in higher levels of basal autophagy.16 Interestingly, loss of IP3R in many different cell lines results in increased basal autophagy and pAMPK.16,20
As a central regulator of cellular energy homeostasis pAMPK exerts a strong effect on protein and lipid biosynthesis. Because it is activated under low ATP conditions, pAMPK activates biosynthetic pathways that generate energy like fatty acid β oxidation, and inhibits high-energy consuming activities like protein synthesis and lipogenesis.21 Thus, AMPK may link IP3R Ca2+ release activity with protein translation.
AMPK is a highly conserved Ser/Thr kinase heterotrimer consisting of a catalytic α subunit and, regulatory β and γ subunits. While there are multiple genes for each subunit in mammals, Drosophila contains a single gene for α, β and γ respectively. There is ∼60% homology between mammalian and Drosophila AMPK, but importantly there is functional conservation.22 Firstly, at the structural level, AMPK functions as a heterotrimer in both systems; Second, the key site for AMPK phosphorylation on the so called “activation loop” of the α subunit (T172 in humans, T184 in flies) is conserved and necessary for AMPK activity; Third, both are activated by AMP, although the degree of stimulation of the Drosophila kinase is to a lower level (4.5-fold compared with 22-fold)23; Fourth, downstream targets of AMPK activity such as acetyl-CoA carboxylase (ACC) are conserved. One major difference between the fly and mammalian AMPK is lipidation: Gly2 of the β subunit in mammalian systems is myristoylated and this plays a key role in tethering AMPK to the plasma membrane, which further allows it to be phosphorylated by LKB1. However, lack of the myristoylation does not prevent the activation of AMPK by LKB1 as in an LKB Drosophila mutant, low but detectable levels of pAMPK was observed.24 Further, even in mammalian systems, basal pAMPK was detectable in the absence of LKB1.25 Another well known activator of AMPK is Ca2+ calmodulin-dependent protein kinase kinase 2 (CaMKK2), a cytosolic kinase that is activated upon increased Ca2+ concentrations. A Drosophila homologue (CG17698) for CaMKK2 has been annotated, but as yet there is no biochemical evidence regarding its role and therefore, it is not discussed further.
In a preliminary experiment (Fig. 1), whole larval lysates were probed for pAMPK levels, as well two markers to indicate activity of the insulin/TOR signaling pathway, pAkt and pS6K. While levels of pAKT were unchanged between control and itprku, pS6K was reduced. Interestingly, pAMPK was increased in itprku. Thus, at the systemic level AMPK appears overactive in the IP3R mutant, and in agreement with reports that pAMPK inhibits the TOR signaling pathway (discussed in more detail below), pS6K levels were lowered. Another link that suggests that AMPK function is affected in itprku stems from the observation that itprku has higher levels of TAGs (Triacylglycerides) both in larval as well as adult stages.10,26 While at a cellular level activated AMPK promotes lipolysis, at a systemic level it can have an opposite effect. For example, activation of AMPK in mice POMC (Pro-opiomelanocortin) neurons causes an increase in NPs, Neuropeptide Y and Agouti Related Peptide, which leads to more feeding, and a concomitant increase in body weight.27 This is similar to itprku which as adults display hyperphagia and increased body weight, both of which are rescued by over-expression of IP3R in NE cells,28 some of which are functionally analogous to human POMC neurons. Also, in a Drosophila AMPK hypomorphic mutant, decreased levels of TAGs were observed24 which suggests that up-regulation of AMPK may promote a systemic increase in TAGs.
Figure 1.
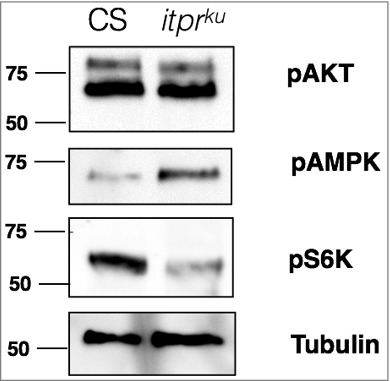
Western blot of whole larval lysates. 30 μg of lysates prepared from 3rd instar larvae were probed with 1:1000 pAKT (Cell Signalling), 1:1000 pAMPK (Cell Signalling), 1:1000 pS6K (Cell Signalling) and 1:5000 tubulin (DHSB).
pAMPK and protein translation via Insulin/TOR signaling
itprku is deficient in its ability to pupariate under nutritional stress.10,29 Over-expression of wild type IP3R or genes of the Insulin/ TOR signaling pathways, in NE cells, was sufficient to rescue the pupariation deficiency of itprku.10 Further, in NE cells, loss of InR (Insulin Receptor), a key regulator of the insulin pathway, resulted in larvae which were deficient in pupariation under nutritional stress; a deficiency that could be over-come when wild type IP3R was over-expressed in NE cells. Together, these genetic experiments suggested that IP3R and the Insulin/TOR pathway can functionally compensate for each other, and are sufficient to ultimately affect a systemic, organism-level adaptation to nutrient stress. The Insulin and TOR pathways are well known growth signaling pathways that link nutritional status to protein synthesis either through growth factors such as insulin, or nutrients themselves, such as amino acids. Notably, many components of the two pathways are conserved between Drosophila and mammals.30 There is also considerable cross-talk between the two pathways – for example, pAKT can inhibit TSC2, alleviating the repression of Rheb and thus promoting TOR activation.30
In mammalian systems, pAMPK is known to suppress protein synthesis by its action at two points in the TOR signaling pathway: 1) It can phosphorylate TSC2, which inactivates TOR, leading to a reduction in pS6K and an increase in p4eBP31, and 2) it can phosphorylate Raptor, a key scaffold of the mTORC1 complex, thereby preventing the complex from recruiting S6K and 4eBP.32 Information regarding regulation of TOR signaling by AMPK in Drosophila is scarce. In the adult fly gut, loss of AMPK did not change pS6K levels and over-expression of TSC1/2, or TORDN did not rescue muscle phenotypes of an AMPK hypomorphic mutant.24 However, specific over-expression of AMPK in adult fly neurons led to increased pAMPK and decreased pS6K.33 This is similar to mouse cortical neurons, where over-activation of AMPK reduces axonal growth and specification, via its inhibitory effect on the mTOR pathway.34 Thus AMPK functionality in the context of TOR signaling appears to be cell-specific.
Future directions
At present, the link between IP3R and AMPK has been established only in mammalian cells, where the physiological significance remains to be understood. However, tools are available to investigate their connection in Drosophila. In mammals, the Hsp70 family chaperone GRP75 (Glucose-regulated protein 75) binds IP3R and places it in close juxtaposition to VDAC1 (Voltage Dependent Anion Channel), located on the mitochondrial outer membrane. Ca2+ uptake into the inner mitochondrial membrane is then facilitated by the Mitochondrial Calcium Uniporter, MCU.35 There are Drosophila homologues for Hsp70 and VDAC family of proteins, but no conserved genetic homologue of GRP75 or VDAC1. MCU is significantly conserved; a recent paper describes Drosophila MCU (CG18769) having evolutionarily conserved Ca2+ transporter function as well as genetic interaction with IP3R.36 Notably, oxidative stress by application of tert-butyl hydroperoxide on larval muscles resulted in increased [Ca2+]mito, that was abrogated upon IP3R knockdown.36 Moreover, in Drosophila larval neuroblasts, ER-mitochondria contact sites were proposed to be stabilized by the rhoGTPase Miro, which promotes mitochondrial Ca2+ uptake, and positively regulates mitochondrial metabolism.37 Loss of IP3R or porin (a Drosophila homologue of the VDAC family) decreased basal mitochondrial Ca2+ levels in Miro over-expressing NBs, suggesting a functional connection between IP3R, Porin and Miro.37
Taken together, a molecular mechanism linking IP3R to protein translation via AMPK is proposed (Fig. 2). This mechanism is likely to be relevant in cells/ tissues where the IP3R affects protein translation. Increased levels of pAMPK in whole larval lysates suggest that the IP3R may affect protein translation in other cell types, in addition to NE cells. This aspect needs to be addressed in future studies. Validation of the proposed mechanism will require loss-of-function and gain-of-function studies with Porin, Miro and AMPK in Drosophila NE cells, in the context of protein translation, at the cellular as well as systemic level. The proposed hypothesis predicts that loss of IP3R Ca2+ channel activity would activate AMPK, which in turn would inhibit TOR signaling, thereby reducing protein translation. Under nutrient-rich conditions, nutrients and insulin signaling may over-ride this inhibitory effect, by providing positive regulation of the insulin/TOR signaling pathway, to produce a small net decrease in protein translation, resulting in a small systemic effect. Hence, protein synthesis in itprku is lower, but not low enough to be detrimental to organismal survival in nutrient-rich conditions. Under conditions of nutrient deprivation, insulin/TOR signaling is turned off, and therefore, these pathways are unable to compensate for reduced protein translation in cells where IP3R mediated Ca2+ release is also compromised. Particularly in cells like NE cells, that need to scale their secretory process and NP synthesis in response to starvation, i.e., increase their protein synthesis under starvation, the loss of IP3R mediated Ca2+ release may have stronger systemic consequences, such as the inability of larvae to pupariate under nutrient stress. We are currently in the process of identifying NPs that operate in the context of IP3R signaling and are required for adaptation to nutrient stress.
Figure 2.
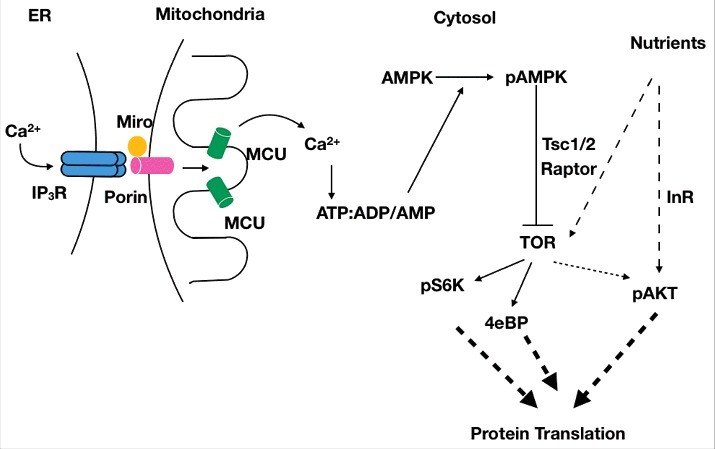
Hypothetical schematic connecting basal IP3R mediated Ca2+ release to protein translation, via AMPK and the TOR signalling pathway. Basal transfer of Ca2+ from the ER to the inner mitochondria via Porin and Mitochondrial Calcium Uniporter (MCU), promotes ATP synthesis and inhibits phosphorylation of AMP-activated kinase (AMPK). Reduced IP3R mediated Ca2+ release decreases ATP production, leading to increased levels of AMP and ADP, that activates AMPK, which inhibits protein synthesis via its negative regulation of the TOR pathway. Thus, even under nutrient-rich conditions, overall protein synthesis levels would be lower in cells with reduced IP3R function. However, cell survival is not affected as nutrients can keep the insulin/TOR signalling pathway active, thereby over-riding the inhibitory effects of pAMPK on protein synthesis. In conditions where both nutrients are withdrawn as well as IP3R function is compromised, the net reduction in protein synthesis assumes physiological significance.
The regulation of protein translation by IP3R has not been previously reported in NE cells. It would be interesting to understand if similar mechanisms are at play in mammalian NE cells. Given the far-reaching consequences that products of some neuroendocrine cells, such as POMC neurons, have on integrating nutritional status to feeding and metabolism, our studies in Drosophila provide the basis to investigate regulation of protein synthesis by IP3R mediated Ca2+ release in a novel context.
Disclosure of potential conflicts of interest
The authors report no conflict of interest.
Funding
This work was supported by The Wellcome Trust/DBT India Alliance under Grant IA/E/12/1/500742 to M and NCBS-TIFR core funds to GH.
References
- [1].Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235-49. doi: 10.1016/S0143416002001823. PMID:12543086. [DOI] [PubMed] [Google Scholar]
- [2].Clapham DE, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD, Takemori H, et al.. Calcium signaling. Cell [Internet]. 2007;131:1047-58. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18083096. doi: 10.1016/j.cell.2007.11.028. [DOI] [Google Scholar]
- [3].Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev. 2015;95:1383-436. doi: 10.1152/physrev.00020.2014. PMID:26400989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Banerjee S, Joshi R, Venkiteswaran G, Agrawal N, Srikanth S, Alam F, Hasan G. Compensation of inositol 1,4,5-trisphosphate receptor function by altering sarco-endoplasmic reticulum calcium ATPase activity in the Drosophila flight circuit. J Neurosci [Internet]. 2006;26:8278-88. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16899722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chakraborty S, Hasan G. Functional Complementation of Drosophila itpr Mutants by Rat Itpr1. J Neurogenet [Internet]. 2012;26:328-37. Available from: http://www.tandfonline.com/doi/full/10.3109/01677063.2012.697501. doi: 10.3109/01677063.2012.697501. [DOI] [PubMed] [Google Scholar]
- [6].Fan G, Baker ML, Wang Z, Baker MR, Sinyagovskiy PA, Chiu W, Ludtke SJ, Serysheva II. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature [Internet]. 2015;527:336-41. Available from: doi: 10.1038/nature15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Joshi R, Venkatesh K, Srinivas R, Nair S, Hasan G. Genetic dissection of itpr gene function reveals a vital requirement in aminergic cells of Drosophila larvae. Genetics [Internet]. 2004;166:225-36. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15020420. doi: 10.1534/genetics.166.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chakraborty S, Deb BK, Chorna T, Konieczny V, Taylor CW, Hasan G. Mutant IP3 receptors attenuate store-operated Ca2+ entry by destabilizing STIM-Orai interactions in Drosophila neurons. J Cell Sci. 2016;129(20):3903-3910. doi: 10.1242/jcs.191585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Venkiteswaran G, Hasan G. Intracellular Ca2+ signaling and store-operated Ca2+ entry are required in Drosophila neurons for flight. Proc Natl Acad Sci U S A [Internet]. 2009;106:10326-31. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19515818. doi: 10.1073/pnas.0902982106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Megha Hasan G. IP3R-mediated Ca2+ release regulates protein metabolism in Drosophila neuroendocrine cells: implications for development under nutrient stress. Development. 2017;144:1484-9. doi: 10.1242/dev.145235. PMID:28289132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Park D, Veenstra JA, Park JH, Taghert PH. Mapping peptidergic cells in Drosophila: where DIMM fits in. PLoS One [Internet]. 2008;3:e1896 Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18365028. doi: 10.1371/journal.pone.0001896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hamanaka Y, Park D, Yin P, Annangudi SP, Edwards TN, Sweedler J, Meinertzhagen IA, Taghert PH. Transcriptional orchestration of the regulated secretory pathway in neurons by the bHLH protein DIMM. Curr Biol [Internet]. 2008;20:9-18. Available from:\ http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20045330. doi: 10.1016/j.cub.2009.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nassel DR, Winther AM. Drosophila neuropeptides in regulation of physiology and behavior. Prog Neurobiol [Internet]. 2010;92:42-104. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20447440. doi: 10.1016/j.pneurobio.2010.04.010. [DOI] [PubMed] [Google Scholar]
- [14].Taghert PH, Nitabach MN. Peptide neuromodulation in invertebrate model systems. Neuron. 2012;76:82-97. doi: 10.1016/j.neuron.2012.08.035. PMID:23040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brostrom MA, Brostrom CO. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium. 2003;34:345-63. doi: 10.1016/S0143-4160(03)00127-1. PMID:12909081. [DOI] [PubMed] [Google Scholar]
- [16].Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung K-H, Yang J, Parker I, et al.. Essential Regulation of Cell Bioenergetics By Constitutive InsP(3) Receptor Ca(2+) Transfer to Mitochondria. Cell [Internet]. 2010;142:270-83. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2911450/. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Decuypere J-P, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB. The IP3 receptor–mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta – Mol Cell Res [Internet]. 2011;1813:1003-13. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0167488910003095. doi: 10.1016/j.bbamcr.2010.11.023. [DOI] [PubMed] [Google Scholar]
- [18].Lee D, Michalak M. Calcium and bioenergetics: from endoplasmic reticulum to mitochondria. Animal Cells Syst (Seoul) [Internet]. 2012;16:269-73. Available from: http://www.tandfonline.com/doi/abs/10.1080/19768354.2012.685181. doi: 10.1080/19768354.2012.685181. [DOI] [Google Scholar]
- [19].Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F, et al.. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun Signal [Internet]. 2011;9:19 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3198985/. doi: 10.1186/1478-811X-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, Smith N, Molgo J, Diehl JA, et al.. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep.2016;15:219-20. doi: 10.1016/j.celrep.2016.03.045. PMID:27050774. [DOI] [PubMed] [Google Scholar]
- [21].Mihaylova MM, Shaw RJ. The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy, & metabolism. Nat Cell Biol [Internet]. 2011;13:1016-23. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3249400/. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sinnett SE, Brenman JE. The Role of AMPK in Drosophila melanogaster. EXS. 2016;107:389-401. PMID:27812989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].PAN DA, HARDIE DG. A homologue of AMP-activated protein kinase in Drosophila melanogaster is sensitive to AMP and is activated by ATP depletion. Biochem J [Internet]. 2002;367:179 LP-186 Available from: http://www.biochemj.org/content/367/1/179.abstract. doi: 10.1042/bj20020703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bland ML, Lee RJ, Magallanes JM, Foskett JK, Birnbaum MJ. AMPK supports growth in Drosophila by regulating muscle activity and nutrient uptake in the gut. Dev Biol [Internet]. 2010;344:293-303. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20478298. doi: 10.1016/j.ydbio.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, Hardie DG. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem. 1995;270:27186-91. doi: 10.1074/jbc.270.45.27186. PMID:7592975. [DOI] [PubMed] [Google Scholar]
- [26].Subramanian M, Metya SK, Sadaf S, Kumar S, Schwudke D, Hasan G. Altered lipid homeostasis in Drosophila InsP3 receptor mutants leads to obesity and hyperphagia. Dis Model Mech [Internet]. 2013;6:734-44. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3634656&tool=pmcentrez&rendertype=abstract. doi: 10.1242/dmm.010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Minokoshi Y, Alquier T, Furukawa N, Kim Y-B, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, et al.. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature [Internet]. 2004;428:569-74. Available from: https://doi.org/ 10.1038/nature02440. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- [28].Subramanian M, Jayakumar S, Richhariya S, Hasan G. Loss of IP3 receptor function in neuropeptide secreting neurons leads to obesity in adult Drosophila. BMC Neurosci [Internet]. 2013;14:157 Available from: http://www.ncbi.nlm.nih.gov/pubmed/24350669. doi: 10.1186/1471-2202-14-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jayakumar S, Richhariya S, Reddy OV, Texada MJM, Hasan G, Richariya S, Reddy VO, Texada MJM, Hasan G. Drosophila larval to pupal switch under nutrient stress requires IP3R/Ca2+ signalling in glutamatergic interneurons. Elife [Internet]. 2016;2016;5:e17 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27494275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Das R, Dobens LL. Conservation of gene and tissue networks regulating insulin signalling in flies and vertebrates. Biochem Soc Trans [Internet]. 2015;43(5):1057-1062. Available from: http://www.biochemsoctrans.org/content/43/5/1057. [DOI] [PubMed] [Google Scholar]
- [31].Inoki K, Zhu T, Guan K-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell [Internet]. 2003;115:577-90. Available from: http://www.sciencedirect.com/science/article/pii/S0092867403009292. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- [32].Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214-26. doi: 10.1016/j.molcel.2008.03.003. PMID:18439900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ulgherait M, Rana A, Rera M, Graniel J, Walker DW. AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep [Internet]. 2014;8:1767-80. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25199830. doi: 10.1016/j.celrep.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Williams T, Courchet J, Viollet B, Brenman JE, Polleux F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc Natl Acad Sci [Internet]. 2011;108:5849-54. Available from: http://www.pnas.org/content/108/14/5849.abstract. doi: 10.1073/pnas.1013660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Krols M, Bultynck G, Janssens S. ER–Mitochondria contact sites: A new regulator of cellular calcium flux comes into play. J Cell Biol [Internet]. 2016;214(4):367-370. Available from: http://jcb.rupress.org/content/214/4/367/tab-figures-data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Choi S, Quan X, Bang S, Yoo H, Kim J, Park J, Park K-S, Chung J. Mitochondrial calcium uniporter in Drosophila transfers calcium between the endoplasmic reticulum and mitochondria in oxidative stress-induced cell death. J Biol Chem [Internet]. 2017;292:14472-14485. Available from: http://www.jbc.org/lookup/doi/10.1074/jbc.M116.765578. doi: 10.1074/jbc.M116.765578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lee S, Lee K-S, Huh S, Liu S, Lee D-Y, Hong SH, Yu K, Lu B. Polo Kinase Phosphorylates Miro to Control ER-Mitochondria Contact Sites and Mitochondrial Ca(2+) Homeostasis in Neural Stem Cell Development. Dev Cell [Internet]. 2016;37:174-89. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27093086. doi: 10.1016/j.devcel.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]