

Abstract
Hepatoblastoma (HB) is one of the most common hepatic malignancies in the pediatric population. HB are composed of a variety of tumors, which derived from different origins and had varying clinical outcomes. However, the unclear underlying mechanisms of HB limited exploring novel biomarkers and effective therapeutic targets. We searched microarray datasets on Gene Expression Omnibus (GEO) database and selected GSE75271 and GSE75283 datasets for comprehensive analysis. Weighted gene correlation network analysis (WGCNA) was employed to identify genes which were associated with tumor malignant phenotypes, including HB subtypes, Cairo classification and tumor stage. Coexpression analysis of identified genes was also performed and lncRNA-miRNA-mRNA network was finally conducted. Our results showed that a total of 22 lncRNAs, 13 miRNAs and 66 mRNAs were identified to be associated with tumor malignant phenotypes. Mechanistically, these molecules might promote the malignant phenotypes via regulating metabolic pathways. Among of them, 6 miRNAs (hsa-miR-106b, hsa-miR-130b, hsa-miR-19a, hsa-miR-19b, hsa-miR-20a and hsa-miR-301a), 8 lncRNAs (NR_102317, XR_245338, XR_428373, XR_924945, XR_929728, XR_931611, XR_935074 and XR_946696), and 6 mRNAs (EGFR, GAREM, INSIG1, KRT81, SAR1B and SDC1) were selected to conduct a lncRNA-miRNA-mRNA network. Taken together, our findings provide evidence for exploring molecular mechanisms of HB. Those identified malignant phenotype-associated molecules might be potential biomarkers and anti-cancer therapeutic targets in future.
Keywords: hepatoblastoma, lncRNA, miRNA, mRNA, malignant phenotype
INTRODUCTION
Hepatoblastoma (HB) is the most common malignant liver tumor in children, accounting for approximately 50% of pediatric hepatic-related cancers [1]. The incidence of HB in children with age <15 is about one per million (1/1,000,000), and nearly 20% of those patients already have a synchronous metastasis at the first diagnosis. Despite recent advances in treatment, such as surgical resection, adjuvant chemotherapy, and liver transplantation, the prognosis in advanced HB stages still remains poor [2, 3]. It has been accepted that HB are composed of a variety of tumors deriving from different immature liver precursors, including hepatocytes, biliary, and other epithelial or mesenchymal cells, which caused significant tumor heterogeneity [4, 5]. For decades, researchers have observed the varying clinical outcomes in patients with different histological subgroups [5–9]. In addition, other parameters, such as tumor stage, distant metastasis, multifocality, patient age and birth weight, have also been reported to be associated with prognosis [10, 11]. However, the underlying mechanisms remains unknown. A thorough understanding of molecular mechanisms regarding tumor progression is essential for exploring effective therapeutic targets against HB.
Studies have revealed that only a small proportion (1%–2%) of the genome encodes proteins, and the majority of the mammalian genome encodes plenty of non-coding RNAs [12–14]. As an important member of the non-coding RNAs, microRNAs (miRNAs), referred to 18-25 nucleotides, have been clearly demonstrated to regulate a variety of cellular processes via recognizing the 3’-untranslated regions of specific mRNAs and suppressing the expression of target genes [15]. Long non-coding RNAs (lncRNAs), another sort of non-coding RNAs with more than 200 nucleotides, are found to play pivotal roles in chromatin remodeling, transcription regulation and post-transcriptional mRNA processing [16–19]. Recently, increasing studies reported that lncRNAs could also function as competing endogenous RNAs (ceRNAs) by competitively binding to miRNAs through their miRNA response elements (MRE) [20]. These ceRNAs usually share MRE with other coding transcripts and therefore act as sponges for that cluster of miRNAs, protecting the targeted mRNA transcripts from degradation [21]. Although the functional mechanism that lncRNAs act as ceRNAs leading to miRNA deregulation has been observed in multiple malignancies, the lncRNA-miRNA-mRNA network in HB is far from being fully investigated.
In the present study, we performed a comprehensive analysis based on mRNA, lncRNA and miRNA expression profiling data derived from 50 HB patients and 5 controls, and identified differentially expressed mRNA, lncRNA and miRNA in HB. Next, we employed weighted gene correlation network analysis (WGCNA) and coexpression analysis to select the modules which are associated with tumor malignant phenotypes. Based on above results, we finally conducted several potential lncRNA-miRNA-mRNA networks in HB.
RESULTS
Identification of the malignant phenotype-associated mRNAs and lncRNAs in HB
GSE75271 was selected for identify differentially expressed mRNA and lncRNAs in HB patients [22]. As shown in Figure 1A, Then, we performed a probe level analysis of GSE75271 by linear models for microarray data (LIMMA). Those probes with P-value less than 0.05 and foldchange more than 2.0 were identified as differentially expressed probes. One hundred ninety-four probes were primary selected, which were all down-regulated in HB patients (Figure 1B). Next, 194 probes were annotated by Affymetrix microarray annotation files according to previous reported method [23]. Finally, we identified a total of 61 lncRNAs and 133 mRNAs from 194 differentially expressed probes (Figure 1C).
Figure 1. Identification of differentially expressed mRNA and lncRNAs from the HB dataset.

(A) Normalization of dataset GSE75271. (B) A total of 194 downregulated probes were picked up from HB patients. (C) Within the 194 downregulated probes, 133 mRNAs and 61 lncRNAs were identified.
To further explore the malignant phenotype-associated mRNAs and lncRNAs, we performed a weighted gene correlation network analysis (WGCNA) (Figure 2A) and divided 194 differentially expressed probes into eight module eigengenes (ME), including MEgrey, MEred, MEblue, MEgreen, MEbrown, MEmagenta and MEblack (Figure 2B). Next, we employed module-trait relationship analysis to conduct the association between eight MEs and 13 clinical traits (including race, sex, age, HB subtypes, Cairo classification, tumor stage, CTNNB1, NFE2L2 and TERT mutation status, histological types, prognosis, tumor recurrent status and survival time). Interestingly, we found that one module (MEblue) was negatively associated with HB subtypes, Cairo classification and tumor stage, with P-values of 1x10-7, 2x10-8 and 1x10-4, respectively (Figure 2C). The blue module contained a total of 66 mRNAs and 22 lncRNAs, which were selected as malignant phenotype-associated mRNAs and lncRNAs for the further analysis (Table 1).
Figure 2. The WGCNA analysis of the malignant phenotype-associated mRNAs and lncRNAs.

(A) The cluster dendrogram of differentially expressed mRNAs and lncRNAs derived from the dataset GSE75271. (B) The cluster dendrogram of module eigengenes. (C) The module-trait relationship analysis between the 8 odules and clinical characteristics. ME: module eigengenes.
Table 1. Identification of HB-associated mRNAs, miRNAs and lncRNAs.
| HB-associated mRNAs | HB-associated miRNAs |
HB-associated lncRNAs |
||
|---|---|---|---|---|
| AC005523.3 | GPRC5C | REPS2 | NR_045387 | hsa-miR-106b |
| ADHFE1 | GSTA1 | RET | NR_024548 | hsa-miR-130a |
| AKR7A3 | GSTZ1 | RIPK4 | NR_027005 | hsa-miR-130b |
| ALDH6A1 | HAO2 | RNF144B | NR_040515 | hsa-miR-17 |
| AMBP | INSIG1 | RPS6KA4 | NR_102317 | hsa-miR-18a |
| APBB1IP | KMO | SAR1B | NR_102357 | hsa-miR-19a |
| APOA1 | KNG1 | SDC1 | XR_171896 | hsa-miR-19b |
| CBS | KRT81 | SEBOX | XR_245338 | hsa-miR-20a |
| CD53 | LCAT | SLC22A7 | XR_427361 | hsa-miR-25 |
| CDC37L1 | LGR4 | SLC7A2 | XR_428373 | hsa-miR-301a |
| CRAT | LPA | SMARCA4 | XR_922928 | hsa-miR-301b |
| CTAG2 | MGAT5 | STS | XR_923052 | hsa-miR-449a |
| CTAGE15 | MIR6778 | SYDE1 | XR_924107 | hsa-miR-451 |
| CYP1A2 | MUT | TTC39C | XR_924945 | |
| CYP2C18 | NEUROG3 | UGT1A1 | XR_924990 | |
| CYP4F2 | NFKBIZ | VNN3 | XR_929728 | |
| DHTKD1 | OAF | WFDC3 | XR_931611 | |
| EGFR | PCCB | ZFYVE28 | XR_931899 | |
| ETS2 | PIGC | ZG16 | XR_933428 | |
| GAREM | PTCRA | ZNF584 | XR_935074 | |
| GLYAT | RARG | ZSCAN5A | XR_946696 | |
| GPLD1 | RDH16 | ZYG11A | XR_949848 | |
Identification of the malignant phenotype-associated miRNAs in HB
We also identified tumor-associated miRNAs using WGCNA method based on GSE75283, which was miRNA profiling dataset derived from the same HB patients as GSE75271 (Figure 3A). Similarly, we divided 887 miRNAs into ten MEs, including MEgreen, MEpink, MEblack, MEtan, MEgreenyellow, MEmidnoghtblue, MEpurple, MEsalmon, MEblue and MEmagenta (Figure 3B). Interestingly, the module-trait relationship analysis revealed that MEmidnoghtblue was positively associated with HB subtypes, Cairo classification and tumor stage, with P-values of 2x10-4, 3x10-4 and 2x10-4, respectively (Figure 3C). The midnoghtblue module contained 13 miRNAs, which were identified as malignant phenotype-associated miRNAs (Table 1).
Figure 3. The WGCNA analysis of the malignant phenotype-associated miRNAs.

(A) The cluster dendrogram of differentially expressed miRNAs derived from the dataset GSE75283. (B) The cluster dendrogram of module eigengenes. (C) The module-trait relationship analysis between the 10 modules and clinical characteristics. ME: module eigengenes.
Coexpression analysis of malignant phenotype-associated molecules
To explore the possible relationship between malignant phenotype-associated molecules, including mRNAs, lncRNAs and miRNAs, we performed coexpression analysis. As shown in Figure 4, we found that the lncRNAs were positively associated with nearly almost mRNAs. Interestingly, we also found that identified miRNAs were negatively associated with both mRNAs and lncRNAs. These results suggested that these mRNAs, lncRNAs and miRNAs might form lncRNA-miRNA-mRNA network, which affected the progression of HB patients. In order to predict biological function of above identified lncRNA-miRNA-mRNA network, we performed Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. Biological process analysis suggested these molecules were mainly involved in metabolic biological processes, such as lipoprotein metabolic process and oxidation-reduction process (Figure 5A). Cellular components analysis showed that they were located at endoplasmic reticulum (ER) membrane, the latter of which played a critical role in liver metabolism (Figure 5B). Besides, molecular function analysis also showed these molecules mostly belonged to heme-binding and oxygen-binding protein families (Figure 5C). KEGG pathway analysis further revealed that these malignant phenotype-associated molecules might be involved in regulating the metabolic related pathways to influence carcinogenesis and tumor progression (Figure 5D).
Figure 4. Coexpression analysis of malignant phenotype-associated molecules.

The heatmap was presented by correlation coefficient between each pair of malignant phenotype-associated molecules. Range of colors (red to blue) shows the correlation coefficient (high to low).
Figure 5. Function annotation of malignant phenotype-associated molecules.

Gene ontology analysis of biological processes (A), cellular components (B) and molecular function (C) of those molecules. (D) The KEGG pathway enrichment analysis.
Construction of lncRNA-miRNA-mRNA network in HB
We further conducted lncRNA-miRNA-mRNA network in HB based on the coexpression analysis of malignant phenotype-associated molecules. The construction of lncRNA-miRNA-mRNA network included three steps: (a) candidate malignant phenotype-associated lncRNAs, miRNAs and mRNAs were selected according to WGCNA results; (b) the correlation coefficients between lncRNAs, miRNAs and mRNAs were calculated based on their corresponding profiling data; (c) targets of miRNAs were predicted from Targetscan (http://www.targetscan.org/). Finally, a total of 6 miRNAs (hsa-miR-106b, hsa-miR-130b, hsa-miR-19a, hsa-miR-19b, hsa-miR-20a and hsa-miR-301a), 8 lncRNAs (NR_102317, XR_245338, XR_428373, XR_924945, XR_929728, XR_931611, XR_935074 and XR_946696), and 6 mRNAs (EGFR, GAREM, INSIG1, KRT81, SAR1B and SDC1) were selected to conduct lncRNA-miRNA-mRNA network. Heatmap was conducted to present the correlation coeffecient between molecules (Figure 6A), and Cytoscape were used for visualization of lncRNA-miRNA-mRNA network (Figure 6B).
Figure 6. Construction of lncRNA-miRNA-mRNA network in HB.
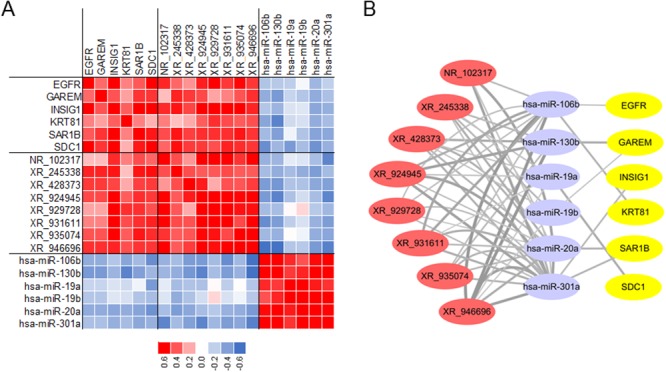
(A) Selected 6 mRNAs, 8 lncRNAs and 6 miRNAs for network. (B) Predicted lncRNA-miRNA-mRNA network in HB.
DISCUSSION
HB is a common malignant hepatic tumor in children and the prognosis varies among different categories. To date, there are limited effective methods to treat HB. Hence, it’s essential to explore underlying molecular mechanism of HB. In the present study, we for the first time identified the malignant phenotype-associated lncRNAs, mRNAs and miRNAs, which were associated with HB subtypes, Cairo Classification and tumor stage. Next, we conducted the lncRNA-miRNA-mRNA network based on their expression patterns to provide a molecular explanation for HB patients.
With the advancement of whole-genomic sequencing technologies, lncRNAs have attracted increasing attention [24]. Amount studies have reported that aberrant lncRNA expressions could serve as novel molecular biomarkers in the cancer diagnosis and prognosis prediction [25, 26]. LncRNAs could function as oncogenes, regulating alternation of key signaling pathways, and promoting tumor growth and metastasis [27, 28]. Recently, the miRNA sponge role of lncRNAs and lncRNA-miRNA-mRNA network have been widely accepted [20]. For instance, lncRNAs SPRY4-IT1 could sponge miR-101-3p and consequently increase EZH2 expression to promote cellular proliferation and metastasis of bladder cancer cells [29]. LncRNA-UCC was found to promote colorectal cancer progression by sponging miR-143 [30], and lncRNA-CCAT1 promotes hepatocellular carcinoma progression by sponging let-7 [31]. However, whether the lncRNA-miRNA-mRNA network plays an important role in HB patients have not been fully explored. A recent study reported that TUG1/miR-34a-5p/VEGFA network was involved in regulating hypervascularity and hepatoblastoma progression [32]. In the present study, we, for the first time, explored malignant phenotype-associated lncRNAs, miRNAs and mRNAs in HB by using WGCNA methods, respectively. A total of 22 lncRNAs, 13 miRNAs and 66 mRNAs were identified, which were strongly associated with HB subtypes, Cairo classification and tumor stage. These findings may provide us potential biomarkers and/or anti-cancer targets in future.
One hallmark of cancer is the metabolic reprogramming in cancer cells, including glycometabolism, lipid metabolism and amino acid metabolism [33–35]. In HB cell lines HepG2, Hep3B and HuH-6, the metabolic reprogramming has been defined as one important hallmark and contributes to tumor progression [36]. Several driver genes of HB, such as β-catenin, YAP and c-Myc [37, 38], could also affect intracellular energy metabolism. Moreover, it was also be found that that molecular targeting of mitochondrial metabolism holds promise as a novel and effective therapeutic approach for HB [39]. In the present study, we also performed pathway enrichment analysis of above identified molecules. Our results revealed that most molecules associated with HB classification and tumor stage belonged to metabolic pathway, indicating a close interaction between metabolic pathways and HB malignant phenotypes.
In this study, we also conducted the lncRNA-miRNA-mRNA network consisting of 6 miRNAs, 8 lncRNAs and 6 mRNAs, which was associated with HB classification and tumor stage. In this conducted network, we identified two genes in epidermal growth factor (EGF) signaling, EGFR and SDC1 [40], acting as effector genes. Recently, a whole transcriptome analysis based on HB patients suggested that aberrant EGF signaling was associated with HB classification [41]. The loss of EGFR signaling members were shown to be more present in less differentiated embryonal and undifferentiated small cells subtypes of HB. Therefore, our findings might also provide evidence for explaining the underlying molecular variation of different HB subtypes.
In conclusion, we identified malignant phenotype-associated lncRNAs, miRNAs and mRNAs by employing WGCNA method. Mechanistically, these molecules might promote the malignant phenotypes of HB via regulating metabolic pathways. Moreover, we also conducted a lncRNAs-miRNAs-mRNAs network based on above findings. To our best knowledge, this is the first comprehensive study investigating lncRNA-miRNA-mRNA network in HB. If validated, our findings might provide evidence for exploring anti-cancer target in HB patients.
MATERIALS AND METHODS
Microarray data
Gene and miRNA profiling data of HB patients were searched from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo), which is a public database containing freely available profiling datasets. We finally selected two datasets for bioinformatic analysis, including GSE75271 (mRNA profiling data based on Affymetrix Human Genome U133 Plus 2.0 Array platform) and GSE75283 (miRNA profiling data based on Agilent-029297 Human miRNA Microarray v15 platform), which were derived from the same 50 HB patients and 5 controls [22]. All raw data were downloaded from GEO database.
Data normalization and probe annotation
The raw data of GSE75271 was stored as probe-level CEL files, and was quantile normalized using Robust Multi-array Average (RMA) method. After data normalization, expression level of each probe in GSE75271 was obtained. The probe sequences with corresponding Affymetrix probe IDs were downloaded from the Affymetrix website (http://www.affymetrix.com). The annotation of lncRNA transcripts were then performed according to previous reported method [23]. A total of 8240 lncRNA transcripts were generated with RefSeq transcript IDs. The probe ID-centric gene expression profile was also generated according to Affymetrix annotation files.
Weighted gene correlation network analysis
WGCNA is an algorithm for constructing a co-expression network, defined by the similarity of gene co-expression [42]. In data processing, the genome-wide gene expression data was initially filtrated with measuring the consistency of gene expression profiles by Pearson correlation, then we utilized the power adjacent function to Pearson correlation matrix to transform data into weighted gene co-expression networks. Network module represents a cluster of closely interconnected genes. Finally, the adjacency matrix, a measurement of topology similarity, is converted into the topological overlap matrix (TOM), and modules are detected by cluster analysis [43].
Pathway enrichment analysis
GO analysis is freely available for users in the annotation and biological properties of genes, gene products and sequences [44]. KEGG is a knowledge base for systematic analysis of gene functions in terms of the networks of genes and molecules [45]. We used GO analysis and KEGG analysis to identify the function of the aberrantly expressed candidate lncRNAs-associated genes. The P-value of each enriched pathway was assigned with -log10 transformation. P<0.05 (-log10(P-value)=1.30) was considered as statistically significant.
Statistical analysis
All data were analyzed by R software 3.4.1 (https://www.r-project.org/). For the pair of molecule i and j, Pearson correlation coefficient was computed. P < 0.05 was considered statistically significant.
Abbreviations
- ceRNAs
competing endogenous RNAs
- ER
endoplasmic reticulum
- EGF
epidermal growth factor
- GEO
Gene Expression Omnibus
- GO
Gene ontology
- HB
Hepatoblastoma
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LIMMA
linear models for microarray data
- lncRNA
long non-coding RNAs
- miRNA
microRNAs
- MRE
miRNA response elements
- ME
module eigengenes
- RMA
Robust Multi-array Average
- TOM
topological overlap matrix
- WGCNA
weighted gene correlation network analysis
Footnotes
Author contributions
Kai Qu and Shunbin Dong: Designed the research and analyzed the data; Sida Liu and Fujing Xie: Collected and analyzed data; Sinan Liu: Constructed figures; Xiaohong Xiang and Kai Qu: Drafted and revised the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by National Science Foundation of China (No. 81201549); Natural Science Basic Research Plan in Shaanxi Province of China (No. 2017JM8039); Fundamental Research Fund for the Central Universities (No. 2016qngz05); the Clinical Research Award of the First Affiliated Hospital of Xi'an Jiaotong University, China (No. XJTU1AF-CRF-2015-011).
REFERENCES
- 1.Bosman F, Carneiro F, Hruban R, Theise N. World Health Organization Classification of Tumours of the Digestive System. 2012.
- 2.Spector LG, Birch J. The epidemiology of hepatoblastoma. Pediatr Blood Cancer. 2012;59:776–779. doi: 10.1002/pbc.24215. [DOI] [PubMed] [Google Scholar]
- 3.Dong R, Jia D, Xue P, Cui X, Li K, Zheng S, He X, Dong K. Genome-wide analysis of long noncoding RNA (lncRNA) expression in hepatoblastoma tissues. PLoS One. 2014;9:e85599. doi: 10.1371/journal.pone.0085599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowland JM. Hepatoblastoma: assessment of criteria for histologic classification. Med Pediatr Oncol. 2002;39:478–483. doi: 10.1002/mpo.10171. [DOI] [PubMed] [Google Scholar]
- 5.Stocker JT. Hepatoblastoma. Semin Diagn Pathol. 1994;11:136–143. [PubMed] [Google Scholar]
- 6.Kremer N, Walther AE, Tiao GM. Management of hepatoblastoma: an update. Curr Opin Pediatr. 2014;26:362–369. doi: 10.1097/MOP.0000000000000081. [DOI] [PubMed] [Google Scholar]
- 7.Czauderna P, Lopez-Terrada D, Hiyama E, Haberle B, Malogolowkin MH, Meyers RL. Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr. 2014;26:19–28. doi: 10.1097/MOP.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 8.McCarville MB, Roebuck DJ. Diagnosis and staging of hepatoblastoma: imaging aspects. Pediatr Blood Cancer. 2012;59:793–799. doi: 10.1002/pbc.24221. [DOI] [PubMed] [Google Scholar]
- 9.Malogolowkin MH, Katzenstein HM, Meyers RL, Krailo MD, Rowland JM, Haas J, Finegold MJ. Complete surgical resection is curative for children with hepatoblastoma with pure fetal histology: a report from the Children's Oncology Group. J Clin Oncol. 2011;29:3301–3306. doi: 10.1200/JCO.2010.29.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maibach R, Roebuck D, Brugieres L, Capra M, Brock P, Dall'Igna P, Otte JB, De Camargo B, Zsiros J, Zimmermann A, Aronson D, Childs M, Scopinaro M, et al. Prognostic stratification for children with hepatoblastoma: the SIOPEL experience. Eur J Cancer. 2012;48:1543–1549. doi: 10.1016/j.ejca.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 11.Aronson DC, Schnater JM, Staalman CR, Weverling GJ, Plaschkes J, Perilongo G, Brown J, Phillips A, Otte JB, Czauderna P, MacKinlay G, Vos A. Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol. 2005;23:1245–1252. doi: 10.1200/JCO.2005.07.145. [DOI] [PubMed] [Google Scholar]
- 12.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 14.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 15.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 16.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spitale RC, Tsai MC, Chang HY. RNA templating the epigenome: long noncoding RNAs as molecular scaffolds. Epigenetics. 2011;6:539–543. doi: 10.4161/epi.6.5.15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arunkumar G, Murugan AK, Prasanna Srinivasa Rao H, Subbiah S, Rajaraman R, Munirajan AK. Long non-coding RNA CCAT1 is overexpressed in oral squamous cell carcinomas and predicts poor prognosis. Biomed Rep. 2017;6:455–462. doi: 10.3892/br.2017.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumazin P, Chen Y, Trevino LR, Sarabia SF, Hampton OA, Patel K, Mistretta TA, Zorman B, Thompson P, Heczey A, Comerford S, Wheeler DA, Chintagumpala M, et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology. 2017;65:104–121. doi: 10.1002/hep.28888. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Sun S, Pu JK, Tsang AC, Lee D, Man VO, Lui WM, Wong ST, Leung GK. Long non-coding RNA expression profiles predict clinical phenotypes in glioma. Neurobiol Dis. 2012;48:1–8. doi: 10.1016/j.nbd.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng J, Li P, Zhang Q, Yang Z, Fu S. A four-long non-coding RNA signature in predicting breast cancer survival. J Exp Clin Cancer Res. 2014;33:84. doi: 10.1186/s13046-014-0084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Y, Chen HY, Yu CY, Xu J, Wang JL, Qian J, Zhang X, Fang JY. A long non-coding RNA signature to improve prognosis prediction of colorectal cancer. Oncotarget. 2014;5:2230–2242. doi: 10.18632/oncotarget.1895. https://doi.org/10.18632/oncotarget.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu MT, Hu JW, Yin R, Xu L. Long noncoding RNA: an emerging paradigm of cancer research. Tumour Biol. 2013;34:613–620. doi: 10.1007/s13277-013-0658-6. [DOI] [PubMed] [Google Scholar]
- 28.Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339:159–166. doi: 10.1016/j.canlet.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 29.Liu D, Li Y, Luo G, Xiao X, Tao D, Wu X, Wang M, Huang C, Wang L, Zeng F, Jiang G. LncRNA SPRY4-IT1 sponges miR-101-3p to promote proliferation and metastasis of bladder cancer cells through up-regulating EZH2. Cancer Lett. 2017;388:281–291. doi: 10.1016/j.canlet.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Huang FT, Chen WY, Gu ZQ, Zhuang YY, Li CQ, Wang LY, Peng JF, Zhu Z, Luo X, Li YH, Yao HR, Zhang SN. The novel long intergenic noncoding RNA UCC promotes colorectal cancer progression by sponging miR-143. Cell Death Dis. 2017;8:e2778. doi: 10.1038/cddis.2017.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng L, Yang SB, Xu FF, Zhang JH. Long noncoding RNA CCAT1 promotes hepatocellular carcinoma progression by functioning as let-7 sponge. J Exp Clin Cancer Res. 2015;34:18. doi: 10.1186/s13046-015-0136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong R, Liu GB, Liu BH, Chen G, Li K, Zheng S, Dong KR. Targeting long non-coding RNA-TUG1 inhibits tumor growth and angiogenesis in hepatoblastoma. Cell Death Dis. 2016;7:e2278. doi: 10.1038/cddis.2016.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Currie E, Schulze A, Zechner R, Walther TC, Farese RV., Jr Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang F, Du G. Dysregulated lipid metabolism in cancer. World J Biol Chem. 2012;3:167–174. doi: 10.4331/wjbc.v3.i8.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byun JK, Choi YK, Kang YN, Jang BK, Kang KJ, Jeon YH, Lee HW, Jeon JH, Koo SH, Jeong WI, Harris RA, Lee IK, Park KG. Retinoic acid-related orphan receptor alpha reprograms glucose metabolism in glutamine-deficient hepatoma cells. Hepatology. 2015;61:953–964. doi: 10.1002/hep.27577. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Lu J, Edmunds LR, Kulkarni S, Dolezal J, Tao J, Ranganathan S, Jackson L, Fromherz M, Beer-Stolz D, Uppala R, Bharathi S, Monga SP, et al. Coordinated activities of multiple Myc-dependent and Myc-independent biosynthetic pathways in hepatoblastoma. J Biol Chem. 2016;291:26241–26251. doi: 10.1074/jbc.M116.754218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng K, Cubero FJ, Nevzorova YA. c-MYC-making liver sick: role of c-MYC in hepatic cell function, homeostasis and disease. Genes (Basel) 2017;8:123. doi: 10.3390/genes8040123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glushakova LG, Lisankie MJ, Eruslanov EB, Ojano-Dirain C, Zolotukhin I, Liu C, Srivastava A, Stacpoole PW. AAV3-mediated transfer and expression of the pyruvate dehydrogenase E1 alpha subunit gene causes metabolic remodeling and apoptosis of human liver cancer cells. Mol Genet Metab. 2009;98:289–299. doi: 10.1016/j.ymgme.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, Jin H, Rapraeger AC. Syndecan-1 and Syndecan-4 capture epidermal growth factor receptor family members and the alpha3beta1 integrin via binding sites in their ectodomains: novel synstatins prevent kinase capture and inhibit alpha6beta4-integrin-dependent epithelial cell motility. J Biol Chem. 2015;290:26103–26113. doi: 10.1074/jbc.M115.679084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ranganathan S, Ningappa M, Ashokkumar C, Higgs BW, Min J, Sun Q, Schmitt L, Subramaniam S, Hakonarson H, Sindhi R. Loss of EGFR-ASAP1 signaling in metastatic and unresectable hepatoblastoma. Sci Rep. 2016;6:38347. doi: 10.1038/srep38347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J, Yu L, Zhang S, Chen X. Network analysis-based approach for exploring the potential diagnostic biomarkers of acute myocardial infarction. Front Physiol. 2016;7:615. doi: 10.3389/fphys.2016.00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao H, Cai W, Su S, Zhi D, Lu J, Liu S. Screening genes crucial for pediatric pilocytic astrocytoma using weighted gene coexpression network analysis combined with methylation data analysis. Cancer Gene Ther. 2014;21:448–455. doi: 10.1038/cgt.2014.49. [DOI] [PubMed] [Google Scholar]
- 44.Gene Ontology Consortium The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006;34:D322–326. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]