

Abstract
Transmission of Zika virus (ZIKV) in the Americas was first confirmed in May 2015 in northeast Brazil1. Brazil has had the highest number of reported ZIKV cases worldwide (more than 200,000 by 24 December 20162) and the most cases associated with microcephaly and other birth defects (2,366 confirmed by 31 December 20162). Since the initial detection of ZIKV in Brazil, more than 45 countries in the Americas have reported local ZIKV transmission, with 24 of these reporting severe ZIKV-associated disease3. However, the origin and epidemic history of ZIKV in Brazil and the Americas remain poorly understood, despite the value of this information for interpreting observed trends in reported microcephaly. Here we address this issue by generating 54 complete or partial ZIKV genomes, mostly from Brazil, and reporting data generated by a mobile genomics laboratory that travelled across northeast Brazil in 2016. One sequence represents the earliest confirmed ZIKV infection in Brazil. Analyses of viral genomes with ecological and epidemiological data yield an estimate that ZIKV was present in northeast Brazil by February 2014 and is likely to have disseminated from there, nationally and internationally, before the first detection of ZIKV in the Americas. Estimated dates for the international spread of ZIKV from Brazil indicate the duration of pre-detection cryptic transmission in recipient regions. The role of northeast Brazil in the establishment of ZIKV in the Americas is further supported by geographic analysis of ZIKV transmission potential and by estimates of the basic reproduction number of the virus.
Previous phylogenetic analyses have indicated that the ZIKV epidemic was caused by the introduction of an Asian genotype lineage into the Americas around late 2013, at least one year before its detection there4. An estimated 100 million people in the Americas are predicted to be at risk of acquiring ZIKV once the epidemic has reached its full extent5. However, little is known about the genetic diversity and transmission history of the virus in Brazil6. Reconstructing the spread of ZIKV from case reports alone is challenging because symptoms (typically fever, headache, joint pain, rashes, and conjunctivitis) overlap with those caused by co-circulating arthropod-borne viruses7 and owing to a lack of nationwide ZIKV-specific surveillance in Brazil before 2016.
We undertook a collaborative investigation of the molecular epidemiology of ZIKV in Brazil, including results from a mobile genomics laboratory that travelled through northeast Brazil during June 2016 (the ZiBRA project; http://www.zibraproject.org). Of five regions of Brazil (Fig. 1a), the northeast region has the most notified ZIKV cases (40% of Brazilian cases) and the most confirmed microcephaly cases (76% of Brazilian cases, as of 31 December 20162), raising questions about why the region has been so severely affected8. Furthermore, northeast Brazil is the most populous region of Brazil that also has potential for year-round ZIKV transmission9. With support from the Brazilian Ministry of Health and other institutions (see Acknowledgements), the ZiBRA laboratory screened 1,330 samples (almost exclusively serum or blood) from patients in 82 municipalities across 5 federal states (Fig. 1, Extended Data Table 1a). Samples provided by the public health laboratories of each state (LACEN) and the Fundação Oswaldo Cruz (FIOCRUZ) were screened for the presence of ZIKV by real-time quantitative PCR (RT-qPCR).
Fig. 1. Geographic and temporal distribution of ZIKV in Brazil.

a. Sampling location of genome sequences from Brazil and the Americas. Federal states in Brazil are coloured according to 5 geographic regions (lower inset). A red line surrounds the states surveyed by the ZiBRA mobile lab in 2016. State codes are PA=Pará, MA=Maranhão, CE=Ceará, TO=Tocantins, RN=Rio Grande do Norte, PB=Paraíba, PE=Pernambuco, AL=Alagoas, BA=Bahia, RJ=Rio de Janeiro, SP=São Paulo. Underlined states represent those from which sequences in this study were generated (upper inset). Publicly available sequences were also collated from non-underlined states. b. Confirmed and notified ZIKV cases in NE Brazil. Upper panel shows the temporal distribution of RT-qPCR+ cases detected during ZiBRA fieldwork. Only samples with known collection dates are included (n=138 out of 181 confirmed cases). Lower panel shows notified ZIKV cases in NE Brazil between 01 Jan 2015 and 19 Nov 2016 (n=122,779). The dashed line represents the average climatic vector suitability score for NE Brazil (Methods). The vertical arrow indicates date of ZIKV confirmation in NE Brazil/Americas1. c. Notified ZIKV cases in the Centre-West, Southeast, North, and South regions of Brazil (clockwise from top left). The dashed lines represent the average climatic vector suitability score for each region.
On average, ZIKV viraemia persists for 10 days after infection; symptoms develop after about 6 days and can last for 1–2 weeks10. In line with previous observations in Colombia11, we found that RT-qPCR-positive samples from northeast Brazil were, on average, collected only 2 days after the onset of symptoms. The median RT-qPCR cycle threshold (Ct) value of positive samples was correspondingly high, at 36 (Extended Data Fig. 1a, b). For northeast Brazil, the time series of RT-qPCR+ cases was positively correlated with the number of weekly notified cases (Pearson’s = 0.62; Fig. 1b).
The ability of the mosquito vector Aedes aegypti to transmit ZIKV is determined by ecological factors that affect adult survival, viral replication, and infective periods12. To investigate the receptivity of Brazilian regions to ZIKV transmission we used a measure of vector climatic suitability, derived from monthly temperature, relative humidity, and precipitation data13. Using linear regression we noted that, for each Brazilian region, there is a strong association between estimated climatic suitability and weekly notified cases (Fig. 1b, c; adjusted R2 > 0.84, P < 0.001; Extended Data Table 1b). Similar to previous findings from dengue virus outbreaks14,15, notified ZIKV cases lag climatic suitability by about 4–6 weeks in all regions, except northeast Brazil, where no time lag is evident. Despite these associations, numbers of notified cases should be interpreted cautiously because co-circulating dengue and chikungunya viruses exhibit symptoms similar to ZIKV, and the Brazilian case reporting system has evolved through time (see Methods). We estimated basic reproductive numbers (R0) for ZIKV in each Brazilian region from the weekly notified case data and found that R0 was high in northeast Brazil (R0 around 3 for both epidemic seasons; Extended Data Table 1c). Although our R0 values are approximate, in part owing to spatial variation in transmission across the large regions analysed here, they are consistent with estimates from other approaches16,17.
Encouraged by the utility of portable genomic technologies during the West African Ebola virus epidemic18 we used our open protocol19 to sequence ZIKV genomes directly from clinical material using MinION DNA sequencers. We were able to generate virus sequences within 48h of the mobile laboratory’s arrival at each LACEN. In pilot experiments using a cultured ZIKV reference strain20 we recovered 98% of the virus genome (Extended Data Fig. 1c). However, owing to low viral copy numbers in clinical samples (Extended Data Fig. 1a), many sequences exhibited incomplete genome coverage and required additional sequencing efforts in static labs once fieldwork had been completed. Whereas average genome coverage was typically high for samples with lower Ct values (85% for Ct<33; Fig. 2a, Extended Data Table 2), samples with higher Ct values had variable coverage (mean 72% for Ct>33; Fig. 2a). Unsequenced genome regions were non-randomly distributed (Fig. 2b), suggesting that the efficiency of PCR amplification varied among primer pair combinations. We generated 36 near-complete or partial genomes from the northeast, southeast and northern regions of Brazil, supplemented by nine sequences from samples from Rio de Janeiro municipality. To further reconstruct Zika virus transmission in the Americas, we include five new complete ZIKV genomes from Colombia and four from Mexico. In addition, we append to our dataset 115 publicly available sequences and 85 additional genomes from ref. 21. The final dataset comprised 254 ZIKV sequences, 241 of which were sampled in the Americas (see Methods).
Fig. 2. Zika virus genetic diversity and sequencing statistics.

a. The percentage of ZIKV genome sequenced plotted against RT-qPCR Ct-value, for each sample. Each circle represents a sequence recovered from an infected individual in Brazil and is coloured by sampling location. b. Illustration of sequencing coverage across the ZIKV genome for the ZiBRA sequences, including data generated by both mobile and static laboratories. c. Regression of sequence sampling dates against root-to-tip genetic distances in a maximum likelihood phylogeny of the Asian-ZIKV lineage. Extended Data Fig. 2b contains a comparable analysis that also includes P6-740 (the oldest Asian-ZIKV strain collected in 1966). d. Average pairwise genetic diversity of the PreAm-ZIKV strains (grey line) and of the Am-ZIKV lineage (black line), calculated using a sliding window of 300 nucleotides with a step size of 50 nucleotides.
The American ZIKV epidemic comprises a single founder lineage4,22,23 (hereafter termed Am-ZIKV) derived from Asian genotype viruses (hereafter termed PreAm-ZIKV) from southeast Asia and the Pacific4. A sliding window analysis of pairwise genetic diversity along the ZIKV genome shows that the diversity of PreAm-ZIKV strains is on average about two-fold greater than that of Am-ZIKV viruses (Fig. 2d), reflecting a longer period of ZIKV circulation in Asia and the Pacific than in the Americas. The genetic diversity of Am-ZIKV strains will increase in the future and updated diagnostic assays are recommended to guarantee RT-qPCR sensitivity24.
It has been suggested that recent ZIKV epidemics may be linked causally to a higher apparent evolutionary rate for the Asian genotype than the African genotype25,26. However, such comparisons are confounded by an inverse relationship between the timescale of observation and estimated evolutionary rates27. Regression of sequence sampling dates against root-to-tip genetic distances indicates that molecular clock models can be applied reliably to the Asian ZIKV lineage (Fig. 2c, Extended Data Figs 2, 3). We estimate the whole-genome evolutionary rate of Asian ZIKV to be 1.12 × 10−3 substitutions per site per year (95% Bayesian credible interval (BCI) 0.97–1.27 × 10−3), consistent with other estimates for this lineage4,26. We found no significant differences in evolutionary rates among ZIKV genome regions (Extended Data Table 3a). The estimated ratio of divergence at nonsynonymous and synonymous sites (dN/dS) of the Am-ZIKV lineage is low (0.11, 95% confidence interval 0.10–0.13), as observed for other vector-borne flaviviruses28, but is higher than that of PreAm-ZIKV viruses (0.061, 0.047–0.077), probably owing to the raised probability of observing slightly deleterious changes in short-term datasets, as observed during previous epidemics29.
We used two phylogeographic approaches with different assumptions30,31 to reconstruct the origins and spread of ZIKV in Brazil and the Americas. We dated the common ancestor of ZIKV in the Americas (node B, Fig. 3) to Jan 2014 (95% BCI October 2013–April 2014; Extended Data Tables 3b, c), in line with previous estimates4,26. We find evidence that northeast Brazil played a central role in the establishment and dissemination of Am-ZIKV. Although northeast Brazil is the most probable location of node B (location posterior support 0.83, Fig. 3), the current data do not allow us to exclude the hypothesis that node B was in the Caribbean (Fig. 3 dashed branches) owing to the presence of two sequences from Haiti in one of its descendant lineages. More importantly, most Am-ZIKV sequences descend from a radiation of lineages (node C and its immediate descendants; Fig. 3) dated to late February 2014 (95% BCIs of node C, November 2013–May 2014). Node C is more strongly inferred to have existed in northeast Brazil (location posterior support 0.99, Fig. 3). All 20 replicate analyses performed on subsampled datasets place node C in Brazil, and 14 of them place node C in northeast Brazil (Extended Data Fig. 4). Consequently, we conclude that node C reflects the crucial turning point in the emergence of ZIKV in the Americas. If further data show that node B did exist in Haiti, then it is likely that Haiti acted as an intermediate ‘stepping stone’ for the arrival and establishment of Am-ZIKV in Brazil, from where the virus subsequently spread to other regions. This perspective is consistent with the lower population size of Haiti compared to Brazil. We infer that node C was present in northeast Brazil several months before three notable events, each of which also occurred in northeast Brazil: (i) the retrospective identification of a cluster of suspected but unconfirmed ZIKV cases in December 20141; (ii) the collection of the oldest ZIKV genome sequence from Brazil, reported here, sampled in February 2015; and (iii) confirmation of cases of ZIKV transmission in northeast Brazil in March 201532,33.
Fig. 3. Phylogeography of ZIKV in the Americas.
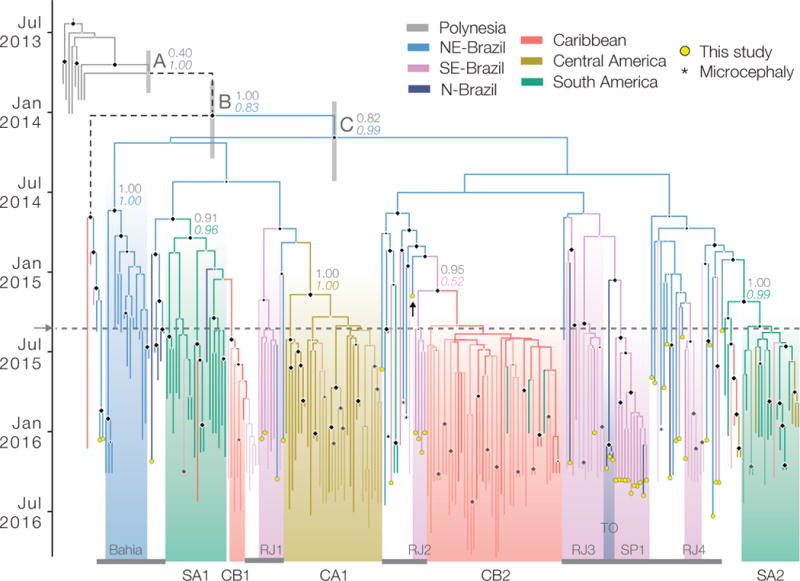
Maximum clade credibility phylogeny, estimated from complete and partial Am-ZIKV genomes using a molecular clock phylogeographic approach (Methods). Terminal branches with yellow circles indicate sequences reported in this study. Terminal branches with no circles and reduced opacity are those reported in a companion paper20. Thin vertical grey boxes indicate statistical uncertainty of estimated dates of nodes A, B and C (Extended Data Table 3c). Branch colours indicate the most probable ancestral lineage locations. Diamonds at internal nodes are sized in proportion to clade posterior probabilities. For selected nodes, coloured numbers show the posterior probabilities of ancestral locations and numbers in grey are clade posterior probabilities. Asterisks indicate the three available genomes from microcephaly cases. A black arrow indicates the oldest Brazilian ZIKV sequence. The grey arrow and dotted line denotes when ZIKV was first confirmed in the Americas1. Nodes A and B are equivalent to the nodes named identically in4. Text labels along the bottom of the figure denote clades of sequences from regions outside of NE Brazil. RJ1 to RJ4 are clades from Rio de Janeiro state, TO from Tocantins, and SP1 from São Paulo state. Clades from outside Brazil are denoted CB1 and CB2 (Caribbean), SA1 and SA2 (South America excluding Brazil), and CA1 (Central America). Thin grey horizontal lines along the bottom of the figure denote sequences from Brazil.
Our results further indicate that viruses from northeast Brazil were important for the continental spread of ZIKV. Within Brazil, we find instances of virus lineage movement from northeast to southeast Brazil; most of these events are dated to the second half of 2014 and led to onwards transmission in Rio de Janeiro (RJ1–RJ4; Fig. 3) and São Paulo states (SP1; Fig. 3). We infer that ZIKV lineages disseminated from northeast Brazil to elsewhere in Central America, the Caribbean, and South America. Most Am-ZIKV strains sampled outside Brazil fall into four well-supported phylogenetic groups (Fig. 3); three (SA1/CB1, CA1 and SA2) are inferred to have been exported from northeast Brazil between July 2014 and April 2015, whereas the Caribbean clade CB2 appears to have originated from southeast Brazil around March 2015 (Figs 3, 4). Each viral lineage export occurred during a period of climatic suitability for vector transmission in the recipient location (Fig. 4). For the earliest exports to Central America (CA1) and South America (SA1), there is an estimated 11–12-month gap between the date of export and the date of ZIKV detection in the recipient location, suggesting a complete season of undetected transmission. These periods of cryptic transmission are relevant to studies of spatiotemporal trends in reported microcephaly, because they help to define the appropriate timeframe for baseline (pre-ZIKV) microcephaly in each region.
Fig. 4. Establishment of Am-ZIKV in the Americas.

The earliest inferred dates of lineage export to non-Brazilian regions, represented by box-and-whisker plots. Each plot corresponds to the earliest movement between a pair of locations with well-supported virus lineage migration. The first exports to South America outside Brazil (SA1 in Fig. 3), to Central America (CA1) and to the Caribbean (CB1) are shown in panels a–c, respectively. Box and whisker plots were generated in ggplot2, with boxes representing the median and interquartile ranges of the estimated date of earliest movement. In each of a–c, dashed lines show the estimated climatic vector suitability score for each recipient region, averaged across the countries for which sequence data is available (see Methods). In each of a-c, the bar plots show available notified ZIKV case data (plots adapted from PAHO) for the countries with the earliest confirmed cases (Colombia61 in panel a, Mexico62 in b, and Puerto Rico63 in c). Coloured arrows indicate the earliest confirmation of ZIKV autochthonous cases in each non-Brazilian region. The vertical dashed line represents the date of ZIKV confirmation in the Americas.
Large-scale surveillance of ZIKV is challenging because many cases may be asymptomatic, and ZIKV co-circulates in some regions with other arthropod-borne viruses that have overlapping symptoms (for example, dengue, chikungunya, Mayaro, and Oropouche viruses). However combining virus genomic and epidemiological data can generate insights into vector-borne virus transmission. A system of continuous and structured virus sequencing in Brazil, integrated with surveillance data, could provide timely information to inform effective responses against Zika and other viruses, including the recently re-emerged yellow fever virus34.
Methods
Sample collection
Between the 1st and 18th June 2016, 1330 samples from cases notified as ZIKV infected were tested for ZIKV infection in the Northeast region of Brazil (NE Brazil). During this period, 4 of the 5 laboratories in the region visited by the ZiBRA project were in the process of implementing molecular diagnostics for ZIKV. The ZiBRA team spent 2–3 days in each state central public health laboratory (LACEN). The samples analysed had been previously collected from patients who had attended a municipal or state public health facility, presenting maculopapular rash and at least two of the following symptoms: fever, conjunctivitis, polyarthralgia, or periarticular edema. The majority of samples were linked to a digital record that collated epidemiological and clinical data: date of sample collection, location of residence, demographic characteristics, and date of onset of clinical symptoms (when available).
The ZiBRA project was supported by the Brazilian Ministry of Health (MoH) as part of the emergency public health response to Zika. Samples had been previously obtained for routine diagnostic purposes from persons visiting local clinics by the Brazilian National Health Surveillance network as part of Zika virus surveillance activities. In these cases, we used samples without informed consent with the approval of the Brazilian Ministry of Health. Specifically, residual anonymized clinical diagnostic samples, with no or minimal risk to patients, were provided for research and surveillance purposes within the terms of Resolution 510/2016 of CONEP (Comissão Nacional de Ética em Pesquisa, Ministério da Saúde; National Ethical Committee for Research, Ministry of Health). For samples obtained from patients engaged in longitudinal studies of Zika virus in São Paulo and Tocantins states, informed consent was obtained (IRB CAAE 53153916.7.0000.0065). Samples from patients followed in Salvador and Feira de Santana were analysed under institutional approval from CPqGM/FioCruz/BA (1.184.454). Urine and plasma samples from Rio de Janeiro were obtained from patients at the Fiocruz Viral Hepatitis Ambulatory (Oswaldo Cruz Institute, Rio de Janeiro, Brazil) with Institutional Review Board approval (IRB142/01) from the Oswaldo Cruz Institute. RNA was extracted at the Paul-Ehrlich-Institut and sequenced at the University of Birmingham, UK.
Nucleic acid isolation and RT-qPCR
Serum, blood and urine samples were obtained from patients 0 to 228 days after first symptoms (Extended Data Table 1a). Viral RNA was isolated from 200 µl Zika-suspected samples using either the NucliSENS easyMag system (BioMerieux, Basingstoke, UK) (Ribeirão Preto samples), the ExiPrep Dx Viral RNA Kit (BIONEER, Republic of Korea) (Rio de Janeiro samples) or the QIAamp Viral RNA Mini kit (QIAGEN, Hilden, Germany) (all other samples) according to the manufacturer’s instructions. Ct values were determined for all samples by probe-based RT-qPCR against the prM target (using 5′ FAM as the probe reporter dye) as previously described34. RT-qPCR assays were performed using the QuantiNova Probe RT-qPCR Kit (20 ul reaction volume; QIAGEN) with amplification in the Rotor-Gene Q (QIAGEN) following the manufacturer’s protocol. Primers/probe were synthesised by Integrated DNA Technologies (Leuven, Belgium). The following reaction conditions were used: reverse transcription (50°C, 10 min), reverse transcriptase inactivation and DNA polymerase activation (95°C, 20 sec), followed by 40 cycles of DNA denaturation (95°C, 10 secs) and annealing-extension (60°C, 40 sec). Positive and negative controls were included in each batch; however, due to the large number of samples tested in a short time it was possible only to run each sample without replication.
Whole genome sequencing
Sequencing was attempted on all positive samples obtained from NE Brazil regardless of Ct value. All samples collected in Brazil that are reported in this study were sequenced with the Oxford Nanopore MinION. Sequencing statistics can be found in Extended Data Table 2. The protocol employed cDNA synthesis with random primers followed by gene specific multiplex PCR and is presented in detail in Quick et al. 18. In brief, extracted RNA was converted to cDNA using the Protoscript II First Strand cDNA synthesis Kit (New England Biolabs, Hitchin, UK) and random hexamer priming. ZIKV genome amplification by multiplex PCR was attempted using the ZikaAsianV1 primer scheme and 40 cycles of PCR using Q5 High-Fidelity DNA polymerase (NEB) as described in Quick et al.18. PCR products were cleaned-up using AmpureXP purification beads (Beckman Coulter, High Wycombe, UK) and quantified using fluorimetry with the Qubit dsDNA High Sensitivity assay on the Qubit 3.0 instrument (Life Technologies). PCR products for samples yielding sufficient material were barcoded and pooled in an equimolar fashion using the Native Barcoding Kit (Oxford Nanopore Technologies, Oxford, UK). Sequencing libraries were generated from the barcoded products using the Genomic DNA Sequencing Kit SQK-MAP007/SQK-LSK208 (Oxford Nanopore Technologies). Sequencing libraries were loaded onto a R9/R9.4 flowcell and data was collected for up to 48 hours but generally less. As described18, consensus genome sequences were produced by alignment of two-direction reads to a Zika virus reference genome (strain H/PF/2013, GenBank Accession number: KJ776791) followed by nanopore signal-level detection of single nucleotide variants. Only positions with ≥20x genome coverage were used to produce consensus alleles. Regions with lower coverage, and those in primer-binding regions were masked with N characters. Validation of our sequencing approach on the MinION platform was undertaken by using the MinION platform to sequence a WHO reference strain of Zika virus that was also sequenced using the Illumina Miseq platform19; identical consensus sequences were recovered regardless of the MinION chemistry version employed (R7.3, R9 and R9.4) (Extended Data Fig. 1c).
Collation of genome-wide data sets
Our complete and partial genome sequences were appended to a global data set of all available published ZIKV genome sequences (up until January 2017) using an in-house script that retrieves updated GenBank sequences on a daily basis. In addition to the genomes generated from samples collected in NE Brazil during ZiBRA fieldwork, samples were sent directly to University of São Paulo and elsewhere for sequencing. Thirteen genomes from Ribeirão Preto, São Paulo state (SP; SE-Brazil region) and seven genomes from Tocantins (TO; N-Brazil region) were sequenced at University of São Paulo. Nine genomes from Rio de Janeiro (RJ; SE-Brazil region) were sequenced in Birmingham, UK, and added to our dataset. All these genomes were generated using the same primer scheme as the ZiBRA samples collected in NE Brazil18. In addition to these 45 sequences from Brazil, we further included in analysis 9 genomes from ZIKV strains sampled outside of Brazil in order to contextualise the genetic diversity of Brazilian ZIKV, giving rise to a final data set of 54 sequences. Specifically, we included 5 genomes from samples collected in Colombia and 4 new genomes from Mexico, which were generated using the protocols described in refs. 35 and 22, respectively.
GenBank sequences belonging to the African genotype of ZIKV were identified using the Arboviral genotyping tool (http://bioafrica2.mrc.ac.za/rega-genotype/typingtool/aedesviruses) and excluded from subsequent analyses, as our focus of study was the Asian genotype of ZIKV, and the Am-ZIKV lineage in particular. To assess the robustness of molecular clock dating estimates to the inclusion of older sequences, analyses were performed both with and without the P6-740 strain, the oldest known strain of the ZIKV-Asian genotype (sampled in 1966 in Malaysia). Our final alignment comprised the sequences reported in this study (n=54) plus publicly available ZIKV-Asian genotype sequences, as of 1st March 2017 (n=115). We also included in our analysis 85 additional genomes from a companion paper20. The dataset used for analysis therefore included sequences from 254 Zika virus isolates, 241 of which were from the Americas. Unpublished but publicly available genomes were included in our analysis only if we had written permission from those who generated the data (see Acknowledgments).
Maximum likelihood analysis and recombination screening
Preliminary maximum likelihood (ML) trees were estimated with ExaMLv336 using a per-site rate category model and a gamma distribution of among site rate variation. For the final analyses, ML trees were estimated using PhyML37 under a GTR nucleotide substitution model38, with a gamma distribution of among site rate variation, as selected by jModeltest.v.239. Branch support was inferred using 100 bootstrap replicates37. Final ML trees were estimated with NNI and SPR heuristic tree search algorithms; equilibrium nucleotide frequencies and substitution model parameters were estimated using ML37 (see Extended Data Fig. 3).
Recombination may impact evolutionary estimates40 and has been shown to be present in the ZIKV-African genotype41. In addition to restricting our analysis to the Asian genotype of ZIKV, we employed the 12 recombination detection methods available in RDPv442 and the Phi-test approach43 available in SplitsTree44 to further search for evidence of recombination in the ZIKV-Asian lineage. No evidence of recombination was found.
Analysis of the temporal molecular evolutionary signal in our ZIKV alignments was conducted using TempEst45. In brief, collection dates in the format yyyy-mm-dd (ISO 8601 standard) were regressed against root-to-tip genetic distances obtained from the ML phylogeny. When precise sampling dates were not available, a precision of 1 month or 1 year in the collection dates was taken into account.
To compare the pairwise genetic diversity of PreAm-ZIKV strains from Asia and the Pacific with Am-ZIKV viruses from the Americas, we used a sliding window approach with 300 nt wide windows and a step size of 50 nt. Sequence gaps were ignored; hence the average pairwise difference per window was obtained by dividing the total pairwise nucleotide differences by the total number of pairwise comparisons.
Molecular clock phylogenetics and gene-specific dN/dS estimation
To estimate Bayesian molecular clock phylogenies, analyses were run in duplicate using BEASTv.1.8.446 for 30 million MCMC steps, sampling parameters and trees every 3000 steps. We employed a model selection procedure using both path-sampling and stepping stone models47 to estimate the most appropriate combination of molecular clock and coalescent models for Bayesian phylogenetic analysis. The best fitting combination was a Bayesian skyline tree prior and a relaxed molecular clock model, with log-normally distributed variation in rates among branches (Extended Data Table 3b). A non-informative continuous time Markov chain reference prior49 on the molecular clock rate was used. Convergence of MCMC chains was checked with Tracer v.1.6. After removal of burn-in, posterior tree distributions were combined and subsampled to generate an empirical distribution of 1,500 molecular clock trees.
To estimate rates of evolution per gene we partitioned the alignment into 10 genes (3 structural genes C, prM, E, and 7 non-structural genes NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) and employed a SDR06 substitution model48 and a strict molecular clock model, using an empirical distribution of molecular clock phylogenies. To estimate the ratio of nonsynonymous to synonymous substitutions per site (dN/dS) for the PreAm-ZIKV and the Am-ZIKV lineages, we used the single likelihood ancestor counting (SLAC) method50 implemented in HyPhy51. This method was applied to two distinct codon-based alignments and their corresponding ML trees which comprised the PreAm-ZIKV and Am-ZIKV sequences, respectively.
Phylogeographic analysis
We investigated virus lineage movements using our empirical distribution of phylogenetic trees and the sampling location of each ZIKV sequence. The sampling location of sequences collected from returning travellers was set to the travel destination in the Americas where infection likely occurred. We discretised sequence sampling locations in Brazil into the geographic regions defined in the main text. The number of sequences per region available for analysis was 10 for N Brazil, 41 for NE Brazil and 54 for SE Brazil. No viral genetic data was available for the Centre-West (CW) and the South (S) Brazilian regions. We similarly discretised the locations of ZIKV sequences sampled outside of Brazil. These were grouped according to the United Nations M49 coding classification of macro-geographical regions. Our analysis included 53 sequences from the Caribbean, 38 from Central America, 17 from Polynesia, 37 from South America (excluding Brazil), 3 from Southeast Asia and 1 from Micronesia. To account for the possibility of sampling bias arising from a larger number of sequences from particular locations, we repeated all phylogeographic analyses using (i) the full dataset (n=254) and (ii) ten jackknife resampled datasets (n=74) in which taxa from each location (except for Southeast Asia and Micronesia) were randomly sub-sampled to 10 sequences (the number of sequences available for N-Brazil).
Phylogeographic reconstructions were conducted using two approaches; (i) using the asymmetric52 discrete trait evolution models implemented in BEASTv1.8.446 and (ii) using the Bayesian structured coalescent approximation (BASTA)29 implemented in BEAST2v.2. The latter has been suggested to be less sensitive to sampling biases53. For both approaches, maximum clade credibility trees were summarized from the MCMC samples using TreeAnnotator after discarding 10% as burn-in. The posterior estimates of the location of nodes A, B and C (depicted in Fig. 3) from these two analytical approaches (applied to both the complete and jackknifed data sets) can be found in Extended Data Fig. 4.
For the discrete trait evolution approach, we counted the expected number of transitions among each pair of locations (net migration) using the robust counting approach54,55 available in BEASTv1.8.446. We then used those inferred transitions to identify the earliest estimated ZIKV introductions into new regions. These viral lineage movement events were statistically supported (with Bayes factors > 3) using the BSSVS (Bayesian stochastic search variable selection) approach implemented in BEASTv.1.8.430. Box plots for node ages were generated using the ggplot256 package in R software57.
Epidemiological analysis
Weekly suspected ZIKV data per Brazilian region were obtained from the Brazilian Ministry of Health (MoH). Cases were defined as suspected ZIKV infection when patients presented maculopapular rash and at least two of the following symptoms: fever, conjunctivitis, polyarthralgia or periarticular edema. Because notified suspected ZIKV cases are based on symptoms and not molecular diagnosis, it is possible that some notified cases represent other co-circulating viruses with related symptoms, such as dengue and Chikungunya viruses. Further, case reporting may have varied among regions and through time. Data from 2015 came from the pre-existing MoH sentinel surveillance system that comprised 150 reporting units throughout Brazil, which was eventually standardised in Feb 2016 in response to the ZIKV epidemic. We suggest that these limitations should be borne in mind when interpreting the ZIKV notified case data and we consider the R0 values estimated here to be approximate. That said, our time series of RT-qPCR+ ZIKV diagnoses from NE Brazil qualitatively match the time series of notified ZIKV cases from the same region (Fig. 1b). To estimate the exponential growth rate of the ZIKV outbreak in Brazil, we fit a simple exponential growth rate model to each stage of the weekly number of suspected ZIKV cases from each region separately:
| (1) |
where Iw is the number of cases in week w. As described in main text, the Brazilian regions considered here were NE Brazil, N-Brazil, S-Brazil, SE-Brazil, and CW-Brazil. The time period over which exponential growth occurs was determined by plotting the log of Iw and selecting the period of linearity (Extended Data Fig. 5). A linear model was then fitted to this period to estimate the weekly exponential growth rate rW:
| (2) |
Let g(.) be the probability density distribution of the epidemic generation time (i.e. the duration between the time of infection of a case and the mean time of infection of its secondary infections). The following formula can be used to derive the reproduction number R from the exponential growth rate r and density g(.)58.
| (3) |
In our baseline analysis, following Ferguson et al.59 we assume that the ZIKV generation time is Gamma-distributed with a mean of 20.0 days and a standard deviation (SD) of 7.4 days. In a sensitivity analysis, we also explored scenarios with shorter mean generation times (10.0 and 15.0 days) but unchanged coefficient of variation SD/mean=7.4/20=0.37 (Extended Data Table 1c).
Association between Aedes aegypti climatic suitability and ZIKV notified cases
To account for seasonal variation in the geographical distribution of the ZIKV vector Aedes aegypti in Brazil we fitted high-resolution maps60 to monthly covariate data. Covariate data included time-varying variables, such as temperature-persistence suitability, relative humidity, and precipitation, as well as static covariates such as urban versus rural land use. Maps were produced at a 5km × 5km resolution for each calendar month and then aggregated to the level of the five Brazilian regions used in this study (Extended Data Fig. 6). For consistency, we rescaled monthly suitability values so that the sum of all monthly maps equalled the annual mean map9.
We then assessed the correlation between monthly Aedes aegypti climatic suitability and the number of weekly ZIKV notified cases in each Brazilian region, to test how well vector suitability explains the variation in the number of ZIKV notified cases. To account for the correlation in each Brazilian region we fit a linear regression model with a lag and two breakpoints. As there may be a lag between trends in suitability and trends in notified cases, we include a temporal term in the model to allow for a shift in the respective curves. Thus for each region, different sets of the constant and linear terms are fitted to different time periods. More formally,
| (4) |
where yi represents notified cases in a particular region in month i, xi is the climatic suitability in that region in month i, l is the time lag that yields the highest correlation between yi and xi and T is the set of time indexes in the correlated region.
We then find the values of T and l that provide the highest adjusted-R2 by stepwise iterative optimisation. For each value of T evaluated, the optimal value of l (i.e. that which gives the highest adjusted-R2 for the model above) is found by the optim function in R57. Climatic suitability values were only calculated for each month, so to calculate suitability values for any given point in time we interpolated between the monthly values using a linear function. We found no significant effect of residual autocorrelation in our data (Extended Data Fig. 7).
Data availability
Sequences of the primers and probes used here have been available at http://www.zibraproject.org since the beginning of the project. XML files and datasets analysed in this study are available from the same website. New Brazilian sequences are available in GenBank under accession numbers KY558989 to KY559032 and KY817930. New Colombian and Mexican sequences are available under accession numbers KY317936-40 and KY606271-4, respectively. See Extended Data Table 2 for further details.
Extended Data
Extended Data Figure 1.

a. The distribution of CT-values for the RT-qPCR+ samples tested during the ZiBRA journey in Brazil (n=181 samples; median CT = 35.96). b. shows the distribution of the temporal lag between the date of onset of clinical symptoms and the date of sample collection of RT-qPCR+ samples (median lag = 2 days). Red dashed lines represent the median of the distributions. (c) Validation of sequencing approaches. A phylogeny of the ZIKV Asian genotype estimated using PhyML37 is shown. The expanded clade highlighted in blue contains the WHO reference ZIKV sequence19 (accession number KX369547), which was generated using Illumina MiSeq. Sequences generated using MinION chemistries R9.4 2D, R9.4 1D, R9 1D, R9 2D and R7.3 2D contain no nucleotide differences and hence were also placed in this clade. Scale bars represent expected nucleotide substitutions per site (s/s). Am-ZIKV=American Zika virus lineage.
Extended Data Figure 2.

Temporal signal of the ZIKV Asian genotype. The correlation between sampling dates and genetic distances from the tips to the root of a maximum likelihood (ML) tree, estimated using PhyML37, was explored using TempEst45. a. Estimates for the dataset used in the phylogenetic analysis presented in Fig. 3c, and b. estimates for the same dataset with the addition of the P6-740 strain sampled in 1966 (accession number HQ234499).
Extended Data Figure 3.

A non-clock maximum likelihood phylogeny of our ZIKV data set. Bootstrap branch support values are shown at each node. The phylogeny was estimated using PhyML37. Sequences generated in this study are highlighted in red. Scale bar represents expected nucleotide substitutions per site.
Extended Data Figure 4.

Ancestral node location posterior probabilities (ANLPP), for nodes A, B and C, estimated using the complete dataset (top row) and ten replicate subsampled data sets (other rows). See Methods for details. ANLPPs were calculated using two approaches: DTA=discrete trait analysis method30 (left side columns) and BASTA=Bayesian structured coalescent approximation method29 (right side columns). For each method, we employed an asymmetric model of location exchange to estimate ancestral node locations and to infer patterns of virus spread among regions.
Extended Data Figure 5.
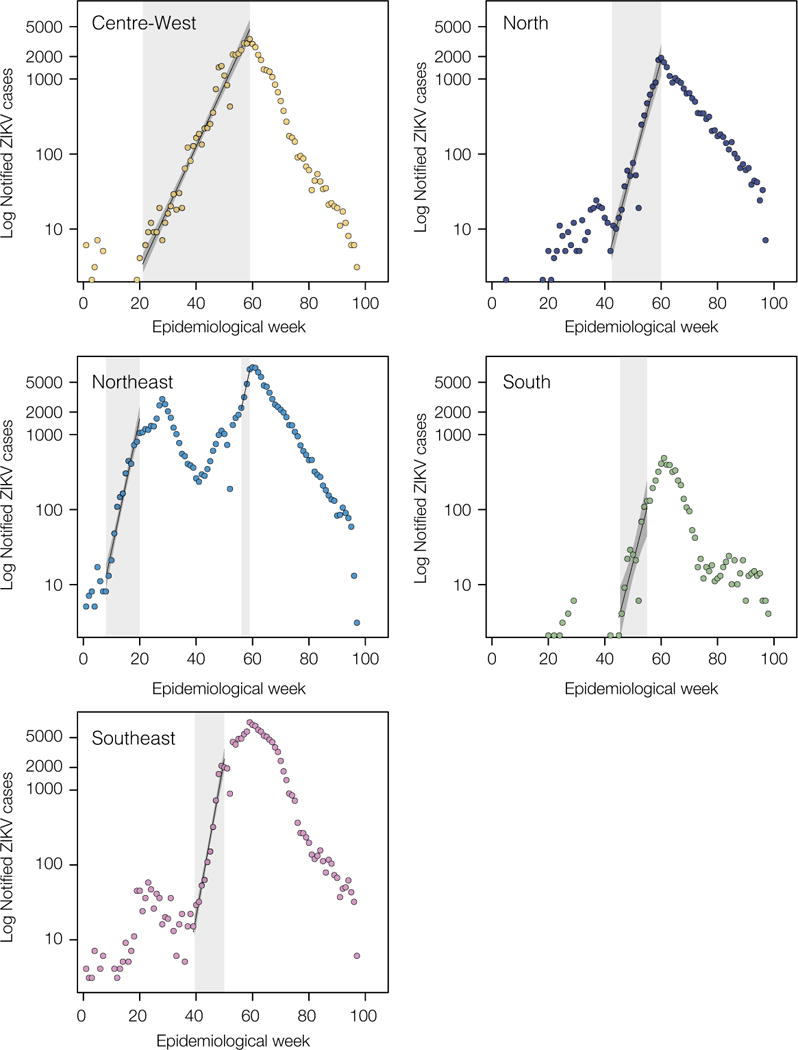
Epidemic growth rates estimated from weekly ZIKV notified cases in Brazil. Time series show the number of ZIKV notified cases in each region of Brazil. Periods from which exponential growth were estimated are highlighted in grey.
Extended Data Figure 6.

Seasonal suitability for ZIKV transmission in the Americas. These maps were estimated by collating data on Aedes mosquitoes, temperature, relative humidity and precipitation, and are the basis of the trends in suitability for different regions shown in main text Figs. 1 and 4. For method details, see 9,60.
Extended Data Figure 7.

Partial autocorrelation functions for the linear model associating climatic suitability and ZIKV notified cases in each geographic region in Brazil. The residuals for the North, Northeast, Centre-West and Southeast regions show no autocorrelation, while a small amount of autocorrelation cannot be excluded for the South region.
Extended Data Table 1.
a. Summary of the clinical samples tested (n=1330, of which 181 were RT-qPCR+) by the ZiBRA mobile lab in June 2016, NE Brazil. 84% of samples with known collection dates (n=698 of 826) were from 2016. ZIKV notified cases were confirmed using RT-qPCR (see Methods). Collection lag represents the median time interval (in days) between the date of onset of clinical symptoms and date of sample collection (both dates available for n=219) for all samples (including those that subsequently tested RT-qPCR negative). Federal states are RN: Rio Grande do Norte, PB: Paraíba, PE: Pernambuco, AL: Alagoas, BA: Bahia. Sample numbers in the FioCruz, PE row include RT-PCR+ cases from Pernambuco generated at Fiocruz Pernambuco. b. Parameters of the model measuring the link between climatic vector suitability and notified ZIKV cases in different Brazilian regions (CW: Centre-West, N: North, NE: Northeast, SE: Southeast, S: South). For each region, the table provides the estimated correlated time period (T), P-value of the linear term of suitability in T, adjusted-R2 of the model, and time lag (l). c. For each region, estimates of the basic reproductive number (R) of ZIKV are shown for several values of generation time (g) parameter, together with the corresponding estimates of exponential growth rate (r) (per day) obtained from notified ZIKV case counts (see Extended Data Fig. 7). 1st: epidemic wave in 2015; 2nd: epidemic wave in 2016.
| (a) | |||
|---|---|---|---|
| Laboratory, Federal state | No. Positives/Tested (%) | Ct value (mean, min-max) | Collection lag (median, min-max) |
| LACEN, RN | 27/335 (8.1%) | 35.9 (18.6–39.1) | 5 (4–16) |
| LACEN, PB | 26/276 (9.4%) | 35.7 (30.7–37.0) | 6 (0–88) |
| FioCruz, PE | 95/315 (30%) | 34.6 (24.1–38.3) | 2.5 (0–33) |
| LACEN, AL | 16/140 (11%) | 34.1 (27.1–40.2) | 2 (0–3) |
| FioCruz, BA | 17/264 (6.4%) | 35.8 (24.7–39.2) | 4 (0–228) |
| (b) | |||||
|---|---|---|---|---|---|
| N | NE | CW | S | SE | |
| Correlated time period | 12/2015 to 10/2016 | 7/2015 to 10/2016 | 9/2015 to 8/2016 | 6/2015 to 5/2016 | 11/2015 to 9/2016 |
| P-value | <0.0001 | 0.00013 | <0.0001 | <0.0001 | <0.0001 |
| Adjusted-R2 | 0.929 | 0.8448 | 0.987 | 0.9543 | 0.953 |
| Time lag (months) | 1.27 | 0 | 1.12 | 1.19 | 1.33 |
| (c) | ||||
|---|---|---|---|---|
| Region | R (mean, CI), g =20 days | R (mean, CI), g =15 days | R (mean, CI), g=10 days | Growth rate (r, CI) |
| CW | 1.71 (1.65–1.78) | 1.46 (1.20–1.77) | 1.29 (1.13–1.46) | 0.027 (0.02–0.03) |
| N | 2.48 (2.19–2.81) | 1.98 (1.80–2.18) | 1.58 (1.48–1.69) | 0.046 (0.04–0.05) |
| NE, 1st | 3.12 (2.69–3.60) | 2.36 (2.11–2.63) | 1.78 (1.65–1.91) | 0.06 (0.05–0.07) |
| NE, 2nd | 3.03 (2.74–3.36) | 2.31 (2.14–2.49) | 1.75 (1.66–1.84) | 0.06 (0.05–0.06) |
| SE | 3.85 (3.35–4.42) | 2.77 (2.49–3.07) | 1.98 (1.84–2.12) | 0.07 (0.06–0.076) |
| S | 2.57 (1.72–3.82) | 2.04 (1.50–2.75) | 1.61 (1.31–1.97) | 0.05 (0.04–0.07) |
Extended Data Table 2.
Sequencing statistics. Accession numbers, sample IDs, sequencing coverage, RT-qPCR values and epidemiological information for the samples from Brazil generated in this study. For the sequences from RJ state, alignments were performed against version 2 (KJ776791.2) of the genome reference; all other sequences used version 1 (KJ776791.1).
| Accession Number | Sample ID | Aligned Reads | Consensus nucleotide bases (% of reference) | RT-qPCR Ct | Collection Date | Municipality | State |
|---|---|---|---|---|---|---|---|
| KY558989 | ZBRA105 | 58128 | 9846 (92) | 29.5 | 2015-02-23 | João Câmara | RN |
| KY558990 | ZBRC14 | 19111 | 8612 (81) | 32.81 | 2016-01-15 | Recife | PE |
| KY558991 | ZBRC16 | 9161 | 7178 (67) | 34.94 | 2016-01-19 | Garanhuns | PE |
| KY558992 | ZBRC18 | 7183 | 7459 (70) | 35.14 | 2016-01-06 | Caetes | PE |
| KY558993 | ZBRC25 | 20533 | 5688 (53) | 35.89 | 2016-01-18 | Sanharo | PE |
| KY558994 | ZBRC28 | 7905 | 8987 (84) | 36.02 | 2016-01-18 | Limoeiro | PE |
| KY558995 | ZBRC301 | 20826 | 9843 (92) | 31.99 | 2015-05-13 | Paulista | PE |
| KY558996 | ZBRC302 | 26331 | 10007 (94) | 30.78 | 2015-05-13 | Paulista | PE |
| KY558997 | ZBRC303 | 12575 | 5873 (55) | 32.81 | 2015-05-14 | Olinda | PE |
| KY558998 | ZBRC313 | 16530 | 9478 (89) | 30.77 | 2015-06-15 | Paulista | PE |
| KY558999 | ZBRC319 | 17316 | 10565 (99) | 24.07 | 2016-07-10 | Olinda | PE |
| KY559000 | ZBRC321 | 11434 | 8647 (81) | 30.62 | 2015-08-09 | Paulista | PE |
| KY559001 | ZBRD103 | 13192 | 8380 (78) | 29.09 | 2015-08-20 | Murici | AL |
| KY559002 | ZBRD107 | 77118 | 7415 (69) | 30.31 | 2015-09-09 | Maceió | AL |
| KY559003 | ZBRD116 | 21211 | 9785 (92) | 27.13 | 2015-08-28 | Arapiraca | AL |
| KY559004 | ZBRE69 | 2313 | 6866 (64) | 24.72 | 2016-04-16 | Feira de Santana | BA |
| KY559005 | ZBRX1 | 21267 | 10559 (99) | 25 | 2016-04-18 | Ribeirão Preto | SP |
| KY559006 | ZBRX2 | 24105 | 9961 (93) | 32 | 2016-04-18 | Ribeirão Preto | SP |
| KY559007 | ZBRX4 | 14722 | 10563 (99) | 26 | 2016-04-18 | Ribeirão Preto | SP |
| KY559008 | ZBRX6 | 12516 | 6893 (64) | 33 | 2016-04-19 | Ribeirão Preto | SP |
| KY559009 | ZBRX7 | 10981 | 8563 (80) | 33 | 2016-04-19 | Ribeirão Preto | SP |
| KY559010 | ZBRX8 | 7445 | 8702 (81) | 33 | 2016-04-19 | Ribeirão Preto | SP |
| KY559011 | ZBRX11 | 21214 | 9379 (88) | 31 | 2016-04-19 | Ribeirão Preto | SP |
| KY559012 | ZBRX12 | 19838 | 10305 (97) | 31 | 2016-04-19 | Ribeirão Preto | SP |
| KY559013 | ZBRX13 | 11809 | 10564 (99) | 21 | 2016-04-24 | Ribeirão Preto | SP |
| KY559014 | ZBRX14 | 5873 | 7469 (70) | 33 | 2016-04-24 | Ribeirão Preto | SP |
| KY559015 | ZBRX15 | 20190 | 10563 (99) | 27 | 2016-04-24 | Ribeirão Preto | SP |
| KY559016 | ZBRX16 | 9698 | 9027 (85) | 32 | 2016-04-25 | Ribeirão Preto | SP |
| KY559017 | ZBRX100 | 5976 | 9609 (90) | 28.5 | 2016-05-19 | Ribeirão Preto | SP |
| KY559018 | ZBRX102 | 13990 | 9508 (89) | 33.91 | 2016-02-25 | Porto Nacional | TO |
| KY559019 | ZBRX103 | 17635 | 9514 (89) | 36.76 | 2016-05-24 | Araguaina | TO |
| KY559020 | ZBRX106 | 29877 | 8458 (79) | 32.36 | 2016-03-07 | Palmas | TO |
| KY559021 | ZRBX127 | 18914 | 10066 (94) | 29.6 | 2016-03-10 | Palmas | TO |
| KY559022 | ZRBX128 | 18480 | 8650 (81) | 28.79 | 2016-03-13 | Palmas | TO |
| KY559023 | ZBRX130 | 16667 | 9914 (93) | 29.06 | 2016-03-22 | Palmas | TO |
| KY559024 | ZBRX137 | 15895 | 9767 (91) | 34.83 | 2016-03-03 | Palmas | TO |
| KY559025 | ZBRY1 | 41036 | 8941 (84) † | 33.53 | 2016-01 | Rio de Janeiro | RJ |
| KY559026 | ZBRY4 | 27865 | 8433 (79) † | 34.21 | 2016-01 | Rio de Janeiro | RJ |
| KY559027 | ZBRY6 | 11779 | 10300 (97) † | 22.66 | 2016-01 | Rio de Janeiro | RJ |
| KY559028 | ZBRY12 | 4980 | 3061 (28) † | 33.66 | 2016-01 | Rio de Janeiro | RJ |
| KY559029 | ZBRY11 | 18530 | 5873 (55) † | 31.11 | 2016-01 | Rio de Janeiro | RJ |
| KY559030 | ZBRY10 | 14067 | 5712 (53) † | 30.84 | 2016-01 | Rio de Janeiro | RJ |
| KY559031 | ZBRY8 | 5708 | 9184 (86) † | 30.96 | 2016-01 | Rio de Janeiro | RJ |
| KY559032 | ZBRY7 | 7749 | 9018 (84) † | 28.07 | 2016-01 | Rio de Janeiro | RJ |
| KY817930 | ZBRY14 | 8040 | 5389 (50) † | 34.2 | 2016-02-15 | Rio de Janeiro | RJ |
Extended Data Table 3.
a. Estimated per-gene rates of evolution (mean and 95% Bayesian credible intervals=BCIs) are shown in units of 10−3 substitutions per site per year. b. Log-marginal likelihood estimates using the path-sampling (PS) and Stepping-Stone (SS) model selection approaches47. The overall ranking of the models is shown in parentheses for each estimator and the best-fitting combination is underscored. Two molecular clock models were tested here. SC: Strict clock model, UCLN: uncorrelated relaxed clock with lognormal distribution46. c. Estimated dates of nodes A, B and C (Fig. 3) under various different molecular clock and coalescent model combinations. TMRCA: time of the most recent common ancestor, BCI: Bayesian credible interval, SC: strict molecular clock model, UCLN: uncorrelated clock with lognormal distribution.
| (a) | |||
|---|---|---|---|
| Gene | Mean | Lower BCI | Upper BCI |
| C | 0.86 | 0.65 | 1.06 |
| prM | 0.98 | 0.85 | 1.12 |
| E | 1.04 | 0.87 | 1.24 |
| NS1 | 0.97 | 0.83 | 1.12 |
| NS2A | 0.98 | 0.83 | 1.13 |
| NS2B | 1.12 | 0.93 | 1.34 |
| NS3 | 0.93 | 0.75 | 1.11 |
| NS4A | 0.87 | 0.74 | 1.01 |
| NS4B | 1.11 | 0.9 | 1.35 |
| NS5 | 1.35 | 0.87 | 1.12 |
| (b) | |||
|---|---|---|---|
| Clock | Coalescent | PS | SS |
| UCLN | Skyline | −32090.664 | −32116.195 |
| SC | Skyline | −32117.581 | −32148.760 |
| UCLN | Exponential | −32193.426 | −32218.348 |
| UCLN | Constant | −32206.219 | −32234.196 |
| SC | Constant | −32229.262 | −32257.900 |
| SC | Exponential | −32244.500 | −32270.815 |
| (c) | ||||
|---|---|---|---|---|
| Clock model | Coalescent prior |
Node A TMRCA (95% BCIs) |
Node B TMRCA (95% BCIs) |
Node C TMRCA (95% BCIs) |
| SC | Constant | 2013.59 (2013.4,2013.77) |
2013.83 (2013.6,2014.05) |
2013.90 (2013.65,2014.12) |
| SC | Exponential | 2013.59 (2013.38,2013.77) |
2013.82 (2013.58,2014.04) |
2013.89 (2013.65,2014.11) |
| SC | Skyline | 2013.66 (2013.48,2013.81) |
2013.93 (2013.74,2014.14) |
2013.99 (2013.75,2014.18) |
| UCLN | Constant | 2013.65 (2013.42,2013.84) |
2013.91 (2013.63,2014.2) |
2014.04 (2013.73,2014.32) |
| UCLN | Exponential | 2013.66 (2013.45,2013.84) |
2013.88 (2013.64,2014.13) |
2014 (2013.73,2014.25) |
| UCLN | Skyline | 2013.71 (2013.54,2013.85) |
2014.03 (2013.76,2014.26) |
2014.16 (2013.89,2014.41) |
Acknowledgments
We are deeply grateful to Fundação Oswaldo Cruz in Bahia and Pernambuco states, University of São Paulo, Instituto Evandro Chagas, and the Brazilian Zika virus surveillance network for their essential contributions. We thank the following for giving us permission to use their unpublished genomes available on GenBank: Robert Lanciotti (CDC, USA), John Lednicky (University of Florida, USA), Antoine Enfissi (Institut Pasteur de la Guyane), F. Baldanti (Pavia University, Italy), Reed Shabman (ATCC, USA), Brett Picket (JCVI, USA), Raymond Schinazi (Emory University, USA), Myrna Bonaldo (Instituto Oswaldo Cruz, Rio de Janeiro, Brazil), Michael Gale (University of Washington, USA), Maria Capobianchi and Catilletti Concetta (INMI “L Spallanzani”, Italy), Mariana Leguia (NAMRU6, Peru), José Alberto Diaz (InDRE, Mexico), Edgar Sevilla-Reyes (INER, Mexico), Alexander Franz (University of Missouri, USA), Mariano Garcia-Blanco (Duke University, USA), MJ van Hemert (LUMC, Netherlands). We thank Pedro Fernando da Costa Vasconcelos, Sueli Guerreiro Rodrigues, Jedson Cardoso, Janaina Vasconcelos, João Vianez Junior (Instituto Evandro Chagas, Brazil), Juliana Gil Melgaço (FIOCRUZ, Rio de Janeiro, Brazil), Johannes Blumel (Paul-Ehrlich-Institut, Langen, Germany), Marcia Cristina Brito Lobato, Liliana Nunes Fava (Tocantins State Department of Health, Brazil), Constância Ayres (Instituto Aggeu Magalhães, Brazil) and Filipa Campos. LCJA thanks QIAGEN for reagents and equipment, MRTN thanks FERPEL for consumables. We thank Oxford Nanopore for technical support, particularly Rosemary Dokos, Zoe McDougall, Simon Cowan, Gordon Sanghera, and Oliver Hartwell. This work was supported by a MRC/Wellcome Trust/Newton Fund Zika Rapid Response grant (MC_PC_15100/ZK/16-078) and by the USAID Emerging Pandemic Threats Program-2 PREDICT-2 (Cooperative Agreement AID-OAA-A-14-00102). NJL is supported by a MRC Bioinformatics Fellowship. NRF is funded by a Sir Henry Dale Fellowship (grant 204311/Z/16/Z). CNPq contributed to trip expenses (grant 457480/2014-9). ACC was supported by FAPESP #2012/03417-7 and MRTN by CNPq grant no. 302584/2015-3. AB and TB were supported by NIH award R35 GM119774. AB is supported by NSF Graduate Research Fellowship Program (grant DGE-1256082). TB is a Pew Biomedical Scholar. CYC is partially supported by NIH grant R01 HL105704 and an award from Abbott Laboratories, Inc. EH is supported by a National Health and Medical Research Council Australia Fellowship (GNT1037231). C.-H.W. is supported by MRC and CRUK (ANR00310) and by Wellcome Trust and Royal Society (grant 101237/Z/13/Z). SCH is supported by the Wellcome Trust. This research received funding from the ERC under grant agreements 614725-PATHPHYLODYN and 278433-PREDEMICS, and from EU Horizon 2020 under agreements 643476-COMPARE and 734548-ZIKAlliance. TJ and ETJM acknowledge funding from IDAMS, DENFREE, DengueTools, and PPSUS-FACEPE (project APQ-0302-4.01/13). RFF received funding from FACEPE (APQ-0044.2.11/16 and APQ-0055.2.11/16) and from CNPq (439975/2016-6). SAB was supported by the Sicherheit von Blut und Geweben hinsichtlich der Abwesenheit von Zikaviren from the German Ministry of Health.
Competing Financial Interests: NJL received speaking fees from Oxford Nanopore Technologies (ONT) and has received free-of-charge reagents in support of the ZiBRA project from ONT. OGP receives consultancy income from Metabiota Inc, CA, USA. CYC is the director of the UCSF-Abbott Viral Diagnostics and Discovery Center and receives research support from Abbott Laboratories, Inc.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions: NRF, LCJA, MRTN, ECS, NL and OGP designed the study. NRF, JQ, NL, IM, JGJ, MG, SCH, AB, ACdC, LCF, SPS, TB, PSL, BLN, HAOM, MRTN, and LCJA undertook fieldwork and experiments. NRF, JT, C-HW, OGP, JR and LdP performed genetic analyses. NRF, MUG, OGP and SC performed epidemiological analyses. NRF, JQ, MUGK, NL and OGP wrote the manuscript. ECH, AR, TB, MRTN, ECS and LCJA edited the manuscript. Other authors were critical for coordination, collection, processing, sequencing and bioinformatics of samples. All authors read and approved the contents of the manuscript.
Author Information: Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Kindhauser MK, Allen T, Frank V, Santhana RS, Dye C. Zika: the origin and spread of a mosquito-borne virus. Bulletin of the World Health Organization. 2016;94:675–686C. doi: 10.2471/BLT.16.171082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ministério da Saúde. Boletins Epidemiológicos—Secretaria de Vigilância em Saúde. 2017 http://portalsaude.saude.gov.br/index.php/o-ministerio/principal/secretarias/svs/boletim-epidemiologico.
- 3.WHO. Situation Report - Zika virus, microcephaly, Guillain-Brarré syndrome. 2017 Jan 18; ( http://apps.who.int/iris/bitstream/10665/253604/1/zikasitrep20Jan17-eng.pdf?ua=1, 2017)
- 4.Faria NR, et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science. 2016;352:345–349. doi: 10.1126/science.aaf5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alex Perkins T, Siraj AS, Ruktanonchai CW, Kraemer MU, Tatem AJ. Model-based projections of Zika virus infections in childbearing women in the Americas. Nat Microbiol. 2016;1:16126. doi: 10.1038/nmicrobiol.2016.126. [DOI] [PubMed] [Google Scholar]
- 6.Lessler J, et al. Assessing the global threat from Zika virus. Science. 2016;353:aaf8160. doi: 10.1126/science.aaf8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vasconcelos PF, Calisher CH. Emergence of Human Arboviral Diseases in the Americas, 2000–2016. Vector Borne and Zoonotic Diseases. 2016;16:295–301. doi: 10.1089/vbz.2016.1952. [DOI] [PubMed] [Google Scholar]
- 8.Vogel G. One year later, Zika scientists prepare for a long war. Science. 2016;354:1088–1089. doi: 10.1126/science.354.6316.1088. [DOI] [PubMed] [Google Scholar]
- 9.Bogoch II, et al. Potential for Zika virus introduction and transmission in resource-limited countries in Africa and the Asia-Pacific region: a modelling study. The Lancet Infectious Diseases. 2016;16:1237–1245. doi: 10.1016/S1473-3099(16)30270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lessler JT, Ott CT, Carcelen AC, Konikoff JM, Williamson J, Bi Q, et al. Times to key events in the course of Zika infection and their implications: a systematic review and pooled analysis [Submitted] Bull World Health Organ. 2016 doi: 10.2471/BLT.16.174540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pacheco O, et al. Zika Virus Disease in Colombia - Preliminary Report. The New England JKournal of Medicine. 2016 doi: 10.1056/NEJMoa1604037. [DOI] [PubMed] [Google Scholar]
- 12.Liu-Helmersson J, Stenlund H, Wilder-Smith A, Rocklov J. Vectorial capacity of Aedes aegypti: effects of temperature and implications for global dengue epidemic potential. PloS One. 2014;9:e89783. doi: 10.1371/journal.pone.0089783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuong HQ, et al. Quantifying the emergence of dengue in Hanoi, Vietnam: 1998–2009. PLoS Negl Trop Dis. 2011;5:e1322. doi: 10.1371/journal.pntd.0001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gharbi M, et al. Time series analysis of dengue incidence in Guadeloupe, French West Indies: forecasting models using climate variables as predictors. BMC Infectious Diseases. 2011;11:166. doi: 10.1186/1471-2334-11-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caminade C, et al. Global risk model for vector-borne transmission of Zika virus reveals the role of El Nino 2015. PNAS. 2017;114:119–124. doi: 10.1073/pnas.1614303114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rocklov J, et al. Assessing Seasonal Risks for the Introduction and Mosquito-borne Spread of Zika Virus in Europe. EBioMedicine. 2016;9:250–256. doi: 10.1016/j.ebiom.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quick J, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530:228–232. doi: 10.1038/nature16996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quick J, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature Protocols. 2017 doi: 10.1038/nprot.2017.066. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trosemeier JH, et al. Genome Sequence of a Candidate World Health Organization Reference Strain of Zika Virus for Nucleic Acid Testing. Genome Announcements. 2016;4 doi: 10.1128/genomeA.00917-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metsky HC, et al. Genome sequencing reveals Zika virus diversity and spread in the Americas. bioRxiv. 2017 https://doi.org/10.1101/109348.
- 21.Giovanetti M, et al. Zika virus complete genome from Salvador, Bahia, Brazil. Infection, Genetics and Evolution. 2016;41:142–145. doi: 10.1016/j.meegid.2016.03.030. [DOI] [PubMed] [Google Scholar]
- 22.Naccache SN, et al. Distinct Zika Virus Lineage in Salvador, Bahia, Brazil. Emerging Infectious Diseases. 2016;22 doi: 10.3201/eid2210.160663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corman VM, et al. Assay optimization for molecular detection of Zika virus. Bulletin of the World Health Organization. 2016;94:880–892. doi: 10.2471/BLT.16.175950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, et al. From discovery to outbreak: the genetic evolution of the emerging Zika virus. Emerg Microbes Infect. 2016;5:e111. doi: 10.1038/emi.2016.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pettersson JHO, Eldholm V, Seligmna SJ, Lundkvist A, Falconar AK, Gaunt MW, Musso D, Nougairede A, Charrel R, Gould EA, Lamballerie X. How Did Zika Virus Emerge in the Pacific Islands and Latin America? mBio. 2016;7:201239–201216. doi: 10.1128/mBio.01239-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holmes EC, Dudas G, Rambaut A, Andersen KG. The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature. 2016;538:193–200. doi: 10.1038/nature19790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holmes EC. Patterns of intra- and interhost nonsynonymous variation reveal strong purifying selection in dengue virus. Journal of Virology. 2003;77:11296–11298. doi: 10.1128/JVI.77.20.11296-11298.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park DJ, et al. Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone. Cell. 2015;161:1516–1526. doi: 10.1016/j.cell.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Maio N, Wu CH, O’Reilly KM, Wilson D. New Routes to Phylogeography: A Bayesian Structured Coalescent Approximation. PLoS Genetics. 2015;11:e1005421. doi: 10.1371/journal.pgen.1005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Computational Biology. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campos GS, Bandeira AC, Sardi SI. Zika Virus Outbreak, Bahia, Brazil. Emerging Infectious Diseases. 2015;21:1885–1886. doi: 10.3201/eid2110.150847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zanluca C, et al. First report of autochthonous transmission of Zika virus in Brazil. Memorias do Instituto Oswaldo Cruz. 2015;110:569–572. doi: 10.1590/0074-02760150192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paules CI, Fauci AS. Yellow Fever — Once Again on the Radar Screen in the Americas. The New England Journal of Medicine. 2017 doi: 10.1056/NEJMp1702172. [DOI] [PubMed] [Google Scholar]
- 34.Lanciotti RS, et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerging Infectious Diseases. 2008;14:1232–1239. doi: 10.3201/eid1408.080287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grubaugh ND, et al. multiple introductions of Zika virus into the United States revealed through genomic epidemiology. bioRxiv. 2017 doi: 10.1038/nature22400. https://doi.org/10.1101/104794. [DOI] [PMC free article] [PubMed]
- 36.Kozlov AM, Aberer AJ, Stamatakis A. ExaML version 3: a tool for phylogenomic analyses on supercomputers. Bioinformatics. 2015;31:2577–2579. doi: 10.1093/bioinformatics/btv184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 38.Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 39.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schierup MH, Hein J. Consequences of recombination on traditional phylogenetic analysis. Genetics. 2000;156:879–891. doi: 10.1093/genetics/156.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faye O, et al. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl Trop Dis. 2014;8:e2636. doi: 10.1371/journal.pntd.0002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1:vev003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172:2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 45.Rambaut A, Lam TT, Fagundes de Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen) Virus Evolution. 2016;2 doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baele G, Li WL, Drummond AJ, Suchard MA, Lemey P. Accurate model selection of relaxed molecular clocks in bayesian phylogenetics. Molecular Biology and Evolution. 2013;30:239–243. doi: 10.1093/molbev/mss243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shapiro B, Rambaut A, Drummond AJ. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Molecular Biology and Evolution. 2006;23:7–9. doi: 10.1093/molbev/msj021. [DOI] [PubMed] [Google Scholar]
- 49.Ferreira MAR, Suchard MA. Bayesian analysis of elapsed times in continuous-time Markov chains. Can J Stat. 2008;36:355–368. [Google Scholar]
- 50.Kosakovsky Pond SL, Frost SD. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Molecular Biology and Evolution. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 51.Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 52.Edwards CJ, et al. Ancient hybridization and an Irish origin for the modern polar bear matriline. Current Biology : CB. 2011;21:1251–1258. doi: 10.1016/j.cub.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouckaert R, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Computational Biology. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minin VN, Suchard MA. Fast, accurate and simulation-free stochastic mapping. Philos Trans R Soc Lond B Biol Sci. 2008;363:3985–3995. doi: 10.1098/rstb.2008.0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Brien JD, Minin VN, Suchard MA. Learning to count: robust estimates for labeled distances between molecular sequences. Molecular Biology and Evolution. 2009;26:801–814. doi: 10.1093/molbev/msp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wickham H. ggplot2: elegant graphics for data analysis. Springer; New York: p. 2009. [Google Scholar]
- 57.R: A Language and Environment for Computing. R Foundation for Statistical Computing; Vienna, Austria: p. 2014. [Google Scholar]
- 58.Cori A, Ferguson NM, Fraser C, Cauchemez S. A new framework and software to estimate time-varying reproduction numbers during epidemics. American Journal of Epidemiology. 2013;178:1505–1512. doi: 10.1093/aje/kwt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferguson NM, et al. EPIDEMIOLOGY. Countering the Zika epidemic in Latin America. Science. 2016;353:353–354. doi: 10.1126/science.aag0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kraemer MU, et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife. 2015;4:e08347. doi: 10.7554/eLife.08347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.PAHO/WHO. Zika Epidemiological Update - Colombia (21 Dec 2016) Washington, D. C.: 2016. [Google Scholar]
- 62.PAHO/WHO. Zika Epidemiological Update - Mexico (20 Dec 2016) Washington, D. C.: 2016. [Google Scholar]
- 63.PAHO/WHO. Zika Epidemiological Update - Puerto Rico (20 Dec 2016) Washington, D. C.: 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequences of the primers and probes used here have been available at http://www.zibraproject.org since the beginning of the project. XML files and datasets analysed in this study are available from the same website. New Brazilian sequences are available in GenBank under accession numbers KY558989 to KY559032 and KY817930. New Colombian and Mexican sequences are available under accession numbers KY317936-40 and KY606271-4, respectively. See Extended Data Table 2 for further details.