

Abstract
Rationale
Cryptogenic strokes, those of unknown cause, have been estimated as high as 30–40% of strokes. Inflammation has been suggested as a critical etiological factor. However, there is lack of experimental evidence.
Objective
In this study, we investigated inflammation associated stroke etiology using a mouse model that developed spontaneous stroke due to myeloid deficiency of TGFβ signaling.
Methods and Results
We report that mice with deletion of Tgfbr2 in myeloid cells (Tgfbr2Myeko) developed cerebrovascular inflammation in the absence of significant pathology in other tissues, culminating in stroke and severe neurological deficits with 100% penetrance. The stroke phenotype can be transferred to syngeneic wild type mice via Tgfbr2Myeko bone marrow transplant, and can be rescued in Tgfbr2Myeko mice with wild-type bone marrow. The underlying mechanisms involved an increased type 1 inflammation, and cerebral endotheliopathy, characterized by elevated NFκB activation and TNF production by myeloid cells. A high fat diet accelerated stroke incidence. Anti-TNF treatment, as well as metformin and methotrexate, which are associated with decreased stroke risk in population studies, delayed stroke occurrence.
Conclusions
Our studies show that TGFβ signaling in myeloid cells is required for maintenance of vascular health, and provide insight into inflammation-mediated cerebrovascular disease and stroke.
Keywords: Inflammation, cerebrovascular disease/stroke, mouse, model, prevention
Subject Terms: Animal Models of Human Disease, Inflammation, Cerebrovascular Disease/Stroke
INTRODUCTION
Stroke is the second leading cause of death worldwide, behind heart disease, and is a leading cause of disability1. Estimates of cryptogenic stroke, of unknown cause, have been estimated as high as 30–40%2. Cerebrovascular or peripheral inflammation is a contributory factor when the cause of the stroke is known, as in atherosclerosis, but is often inferred even when no “smoking gun” can be found3. However, there is little experimental evidence. Current animal models utilize germline mutations or artery stenosis5, and do not provide etiological insight.
Chronic inflammation increases with aging and contributes to cerebrovascular disease, which is a risk factor for stroke3. Transforming growth factor beta (TGFβ) is a critical mediator in immune/inflammation homeostasis4, and alterations of this pathway are implicated in vascular diseases5,6. We previously reported that deletion of the gene encoding TGFβ receptor II (TβRII) in myeloid cells (Tgfbr2Myeko) decreased production of type 2 cytokines and cancer metastasis7,8. However, we show here that this type 1 skewed immune environment resulted in spontaneous stroke with 100% penetrance in C57BL/6 mice at 9–15 months of age, corresponding to middle age in humans. Characterization of these mice revealed defects in motor function, an increase in inflammatory cytokines, cerebral endotheliopathy, and ischemic brain lesions. Importantly, a high fat diet accelerated stroke incidence in mice analogous to poor diet as a risk factor for human stroke. Both metformin and methotrexate, which are associated with decreased stroke risk in population studies, as well as anti-TNF treatment, delayed stroke occurrence. Our data suggest Tgfbr2Myeko mice could be used to model risk factors and prevention of stroke.
METHODS
A number of experimental approaches were used including assessment of neurological impairment, MRI, bone marrow transplant, ex vivo and in vitro culture, molecular and cellular biology, immune/inflammation assays, statistical analysis etc. An expanded Materials and Methods section is available in the Online Data Supplement.
Animal experiments
All animal protocols were approved by the Animal Care and Use Committee at the NIH. Myeloid specific deletion of Tgfbr2 was accomplished through breeding of Tgfbr2 floxed mice with Lysozyme 2 promoter-driven Cre recombinase (LysM-Cre) mice as previously reported7. All experiments used homozygous floxed Tgfbr2 mice and control littermate in a C57BL/6 background. Euthanasia criteria were head tilt, circling behavior, trembling, paresis and/or paralysis, inability to obtain food or water, or greater than 20% body weight loss. For cytokine array, 6 month-old mice were used. For stroke risk assessment studies (Bio-plex), 8–12 month-old pre-stroke mice were used. For stroke prevention, treatments began at 6 months of age. High fat diet was initiated at 6 weeks of age. For bone marrow transplants, bone marrow was isolated from mice and injected into the tail vein of irradiated recipient mice.
In vitro experiments
Cytokine arrays and Bio-plex assays were done using plasma from mice at indicated ages. Myeloid cells were isolated from peripheral blood of 3–6 month-old mice using fluorescence activated cell sorting for endothelial co-culture experiments. RNA was purified using Qiagen columns. Mouse macrophage cell line RAW264.7, with shRNA-mediated knockdown of Tgfbr2, was used as a surrogate for Tgfbr2Myeko myeloid cells in western blot and chromatin immunoprecipitation experiments. Immunofluorescence was performed on frozen sections or cytospun cells from blood.
RESULTS
Neurological impairment and spontaneous stroke in Tgfbr2Myeko mice
In the course of breeding mice with myeloid deletion of Tgfbr2 (Tgfbr2Myeko) in a C57BL/6 background, it became apparent that Tgfbr2Myeko older than 6 months occasionally died of unknown causes. The median lifespan was 12 months, with 0% survival by 20 months of age for both males and females (Figure 1A). No mortality was observed in wt (either Tgfbr2fl/fl without LysM-cre, or LysM-cre alone) or LysM-cre, Tgfbr2fl/+ (Tgfbr2 myeloid heterozygous, Tgfbr2Myehet) over the course of the study. Tgfbr2Myeko mice showed signs of neurological impairment that presented as abnormal limb reflexes when lifted by the tail, head tilt, trembling, hiccups, abnormal gait, paresis and/or paralysis and weight loss (Online Figure IA and Movies 1–3). No neurological impairment or weight loss was observed in littermate wt or Tgfbr2Myehet mice. Most Tgfbr2Myeko mice in the lifespan cohort were euthanized for humane reasons due to above signs. All mice showed signs of neurological impairment at sacrifice. Tgfbr2Myeko mice showed motor function deficits on the ladder traversal test, where Tgfbr2Myeko mice had a high number of foot slips, and had a significantly higher ataxia coefficient with Digigait analysis (Figure 1B). Monthly rotarod testing revealed a precipitous decline in motor function over a short period, suggesting an event-related alteration in motor function rather than progressive neurodegeneration (Figure 1B).
Figure 1. Tgfbr2myeko mice develop severe neurodeficits and spontaneous stroke.
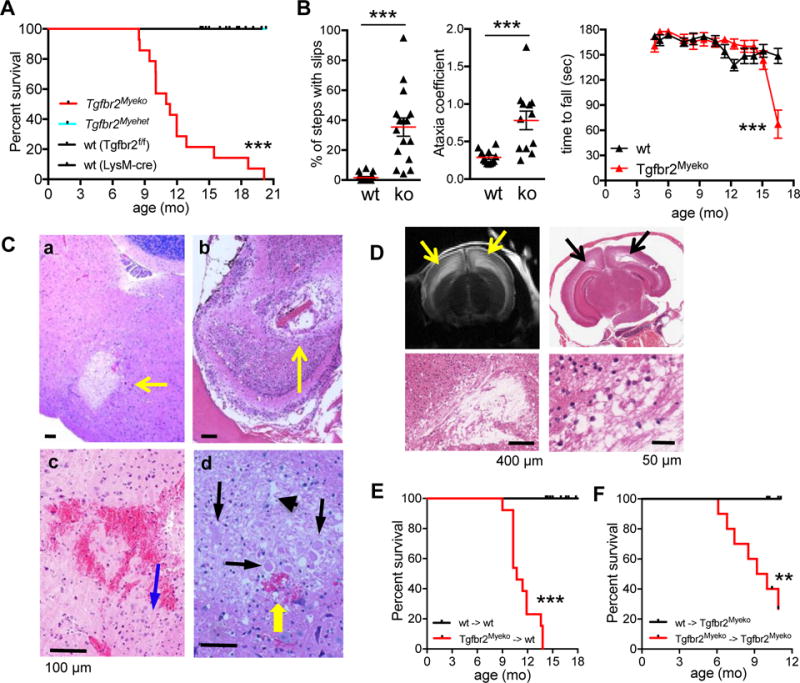
(A) Decreased overall survival of Tgfbr2Myeko (n=19) and compared with wt (Tgfbr2fl/fl, n=25 and LysM-cre, n=3) and heterozygous (Tgfbr2Myehet, n=9) littermates. Mice were sacrificed for humane endpoints, which included head tilt, abnormal gait, severe trembling, paresis or paralysis, or weight loss of >20% due to severe neurological impairments. p, Tgfbr2Myeko vs wt. (B) Decreased motor function and gait disturbance in Tgfbr2Myeko (ko) mice. Foot slips on ladder traversal test (n=4, left panel) and ataxia coefficient from Digigait analysis (n=3, center panel) in 9 mo-old pre-stroke Tgfbr2Myeko mice and wt littermates. Each data point is from one foot from one mouse, 4 points per mouse. Serial rotarod testing of young mice until decline in performance (n=5, right panel). (C) Post-ischemic brain lesions in Tgfbr2Myeko mice. H&E staining of brain sections from Tgfbr2Myeko stroke mice with neurological impairment: a, focal sub-acute infarction in brainstem; b, focal infarction in olfactory bulb; c, micro-hemorrhage in cortex; d, dead and degenerating neurons (black arrows); eosinophilic neurons with neuronal shrinkage (blue arrow); and neuropil vacuolation (arrow head); micro-hemorrhage (yellow arrow). Scale bars for all panels are 100 μm. (D) T2-weighted MRI of brain from Tgfbr2Myeko mouse with signs of stroke (upper left panel), with hyperintensities (yellow arrows); H&E from same mouse in corresponding regions on MRI, (black arrows) showing loss of brain tissue. Lower subpanels, zoom. (E) Tgfbr2Myeko bone marrow causes stroke in wt mice, (n=13 for Tgfbr2Myeko donors, n=4 for wt donors). wt->wt denotes donor->recipient mice. (F) wt bone marrow rescues stroke in Tgfbr2Myeko mice (n=10 per group). For E-F, tick marks are censored subjects (alive at termination of experiment). **p<0.01, ***p<0.001
Large vessel occlusion, or decreased blood flow, such as that typically seen in the vascular territory distal to a critical stenosis, was not detected on magnetic resonance angiography (Online Figure II). Atrial fibrillation, the most common cause of cardioembolic stroke, was not detected on electrocardiograms (data not shown). Rather histological analysis of brains from affected Tgfbr2Myeko mice revealed neuronal damage, small areas of focal ischemia, similar to human lacunar stroke9, and micro-hemorrhage (Figure 1C), as well as decreased cellularity in the olfactory lobes, brainstem and/or cortex in all affected animals (Online Figure IB). No lesions were observed in brains from 8 month-old wt and unaffected Tgfbr2Myeko mice (Online Figure IB). T2-weighted MRI showed larger brain lesions (Figure 1D upper left) that were consistent with neuronal tissue loss observed on histology (Figure 1D). Positive staining for glial fibrillary acidic protein (GFAP), indicated astrogliosis, which is typical in ischemia (Online Figure IC). Neither amyloid plaques nor demyelination outside of stroke lesions were observed, which excluded neurological diseases comparable with Alzheimer’s disease and multiple sclerosis (Online Figure ID). From behavioral and histological findings, the cause of neurological impairment and premature death in Tgfbr2Myeko mice was attributed to spontaneous stroke.
Bone marrow from Tgfbr2Myeko mice could recapitulate the phenotype in wt mice, while wt bone marrow could rescue the phenotype of Tgfbr2Myeko mice (Figure 1E,F). Wt mice that received Tgfbr2Myeko bone marrow exhibited mortality (Figure 1E) and brain microhemorrhages (Online Figure IF) similar to Tgfbr2Myeko mice. These data exclude the contribution of brain residing microglia10,11 and other cell types reported to be targets of LysM-cre12. Together, these data suggest that deficiency of TGFβ signaling in myeloid cells leads to decreased lifespan due to spontaneous stroke.
Inflammatory cerebrovascular lesions in brains of Tgfbr2Myeko mice
Risk factors for human stroke were not found in Tgfbr2Myeko mice, including high blood pressure, high blood glucose (Online Figure IE,F, Online Table I), high cholesterol or triglycerides (Online Table I), or elevated body weight (data not shown). Atherosclerosis in the heart and aorta was not observed by H&E or Oil Red O staining on a cereal diet, consistent with no elevation in plasma cholesterol. There also is no evidence for cerebral atherosclerosis (data not shown). Brains of affected mice showed infiltration of lymphocytes and macrophages around the cerebral arteries in areas of the brain otherwise devoid of immune cells (Figure 2A,B, Online Figure IIA,B). Tgfbr2Myeko mice, as well as wt mice that received Tgfbr2Myeko bone marrow, showed narrowed cerebral vessel lumens (Online Figure IIB,C). Some arterial lesions showed fibrotic scars that were consistent with thrombosis, fibrin deposition, vascular occlusion, and recanalization (Online Figure IIB). Inflamed vascular lesions were not observed in the kidneys, liver or spleen, consistent with blood chemistry that did not indicate damage to major organs (Online Table I). This might be analogous to the specific relationship between circulating immune cells and cerebral blood vessels in neurodegenerative diseases such Alzheimer’s disease13. There was no difference in brain immune cell profile by flow cytometry prior to stroke (Online Figure IID). No Evans blue leakage was found in the brain parenchyma of pre-stroke Tgfbr2Myeko mice, or in the majority of mice with signs of stroke (Online Figure IIE). These studies indicate an unlikely widespread breach of the blood brain barrier in Tgfbr2Myeko mice.
Figure 2. Vascular inflammation and endotheliopathy in Tgfbr2Myeko mice.
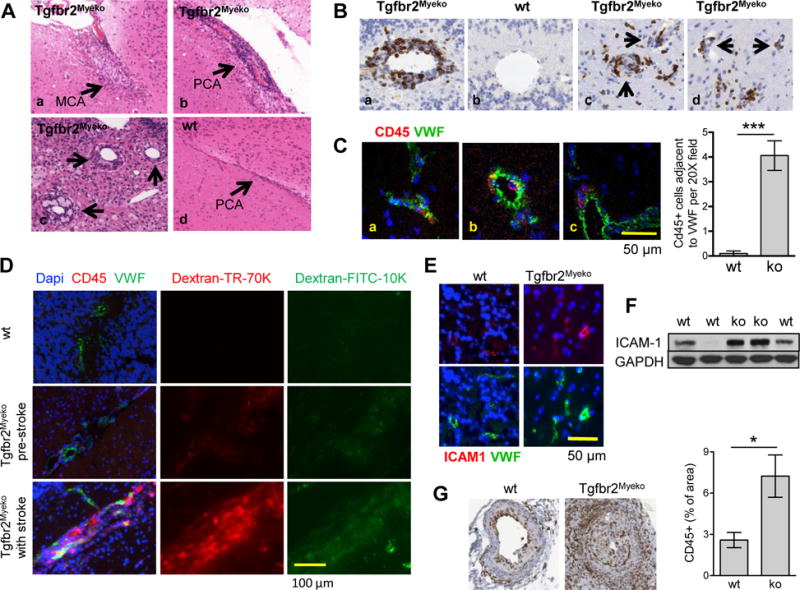
(A) H&E showing vascular inflammation in brain sections from Tgfbr2Myeko mice with signs of stroke. (a) middle cerebral artery (MCA); (b,d) posterior cerebral artery (PCA) (c) inflammation surrounding small vessels in the brainstem of Tgfbr2Myeko and wt mice. Arrows indicate inflammatory vascular lesions, or normal PCA for panel 4. (B) Inflammatory cerebral vascular lesions (CD45 immune cells, brown) in brain from Tgfbr2Myeko mice with stroke. Medium sized vessel of the cerebellum (a) compared with wt (b); Immune infiltrate surrounding small vessels in Tgfbr2Myeko brains (c,d). (C) Immune infiltration surrounding small and medium vessels in Tgfbr2Myeko mice pre-stroke. Von Willebrand factor (VWF, green) and immune cells (CD45, red) in brain of Tgfbr2Myeko mice. Nuclei in blue (DAPI). CD45+ cells within a small blood vessel, notice elongated shape (a). Localization of CD45 outside of blood vessels and adjacent to VWF (all Tgfbr2Myeko shown). Quantitative data on the right. (D) Breach of the blood brain barrier in areas of cerebrovascular inflammation in pre-stroke and post-stroke Tgfbr2Myeko mice. Mice were sacrificed 1h after IV injection of fluorescent dextrans. (E) Increased endothelial activation in brains of Tgfbr2Myeko mice pre-stroke. Inflammatory endothelium (ICAM1, red) and blood vessels (VWF, green). (F) ICAM1 Western blot of brain protein extracts from Tgfbr2Myeko mice and wt controls. (G) Increased inflammatory infiltrate surrounding femoral arteries of Tgfbr2Myeko mice following experimental wire injury. Immune cell IHC staining (CD45, brown) of tissue sections from femoral arteries 13 days after wire injury of Tgfbr2Myeko mice compared with wt littermates (n=3). Shown are representative images. Quantitative data at right, ***p<0.001, *p<0.05.
Recruitment of inflammatory cells to blood vessels in normal brain adjacent to stroke lesions (Figure 2B) indicated a possible causal role of vascular inflammation in stroke occurrence. In brains from 6 month-old pre-stroke Tgfbr2Myeko mice, there was significant co-localization of immune cells with the vasculature (Figure 2C). Leakage of fluorescent Dextrans into the brain parenchyma revealed focal breakdown of the blood brain barrier in regions of the brain co-staining for immune cells, but not in areas of the brain without immune infiltrate (Figure 2D). Leakage of fluorescent dextrans was more severe and widespread in mice with signs of stroke. Elevated brain expression of endothelial activation markers, intercellular adhesion molecule 1 (ICAM1) and Von Willebrand factor (VWF) was observed in pre-stroke Tgfbr2Myeko mice (Figure 2E–F). To further examine whether Tgfbr2Myeko mice might have enhanced susceptibility for vascular inflammation, femoral arteries of Tgfbr2Myeko and wt mice were subjected to wire injury in vivo. This model is widely used in cardiovascular studies in mice, and produces a significant inflammatory response that is not observed with carotid wire injury14. At 13 days post injury, there was an increase in infiltrating CD45+ inflammatory cells surrounding the arteries in Tgfbr2Myeko mice compared with wt littermates (Figure 2G). These data suggest the involvement of inflammatory cells in the cerebral vasculature in the etiology of cerebrovascular disease in Tgfbr2Myeko mice.
Mechanisms of vascular inflammation
Proinflammatory cytokines cause vascular damage under pathological conditions15. To investigate whether an inflammatory cytokine environment plays a role in stroke etiology, cytokines were investigated in plasma from 6 month-old pre-stroke Tgfbr2Myeko and wt mice. There was a significant elevation of pro-inflammatory cytokines such as TNF, CXCL10 and CCL2 (Figure 3A, Online Figure IIIA). Representative type 2 cytokines such as IL4 and IL10 were not detected in plasma from either wt or Tgfbr2Myeko mice (Figure 3A). This enhanced type 1, and deficient type 2 polarization is also observed in vascular inflammation and atherosclerotic plaque formation20,21.
Figure 3. Increased inflammatory cytokines and mediators resulting from deletion of myeloid specific TGFβ signaling.
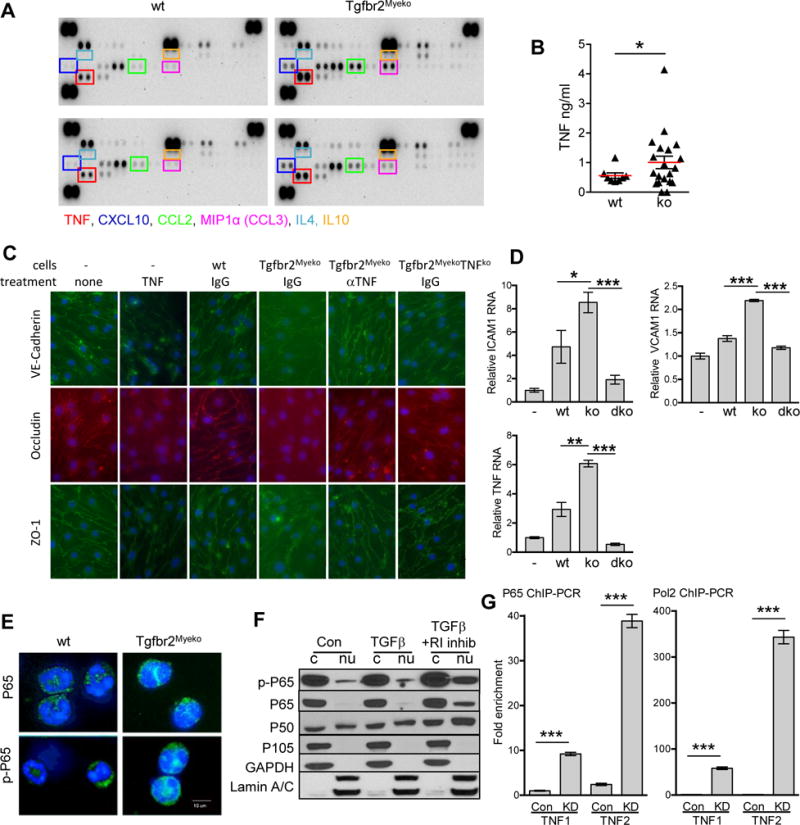
(A) Cytokine antibody array of plasma from one male and one females 6 month-old pre-stroke Tgfbr2Myeko mice, compared one male and one female wt (n=2 for each group). Boxed cytokines are indicated with same color text below. (B) TNF ELISA of plasma from 6–8 month wt and Tgfbr2Myeko mixed male and female mice pre-stroke (n=10 wt and n=20 ko). (C) Tgfbr2Myeko myeloid cells cause interruption in adherens junctions and tight junctions between co-cultured C57BL/6 mouse brain microvascular endothelial cells. Co-culture with peripheral blood myeloid cells (cells) from wt, Tgfbr2Myeko or Tgfbr2MyekoTNF−/− (ko) mice. Some samples were treated with TNF, aTNF neutralizing antibody or IgG isotype antibody. Notice disruption in the endothelial junctions with TNF treatment and by Tgfbr2Myeko myeloid cells. (D) C57BL/6 brain microvascular endothelial cells have elevated RNA for markers of endothelial damage (top panels) and TNF (lower panel) following co-culture with Tgfbr2Myeko (ko) myeloid cells that can be reduced when myeloid cells lack TNF also (dko, Tgfbr2MyekoTNF−/−). (E) Increased NFkB transcription factor subunit P65 (encoded by Rela) and active, phosphorylated P65 in myeloid cells from Tgfbr2Myeko mice. P65 and p-P65 (green), nuclei (blue, Dapi). Shown are representative pictures. (F) Increased nuclear NFkB subunits P65 and P50 (encoded by Nfkb1) and active p-P65 upon blockade of TGFb signaling using a TbR1 (R1) inhibitor. Western blots of nuclear (nu) and cytoplasmic (c) fractions from RAW264.7 cells. (G) Increased binding of NFkB to the TNF promoter after knock-down (KD) of Tgfbr2. Fold enrichment of TNF from P65 Chromatin immunoprecipitation (ChIP) of RAW264.7 cells. TNF1 or TNF2 indicates two different regions in the TNF promoter (see Online Figure 3C).
Increased blood TNF is particularly interesting as it occurs with aging, vascular inflammation, and cardiovascular disease16. Wt mice had TNF levels that fell into a narrow range, while many Tgfbr2Myeko mice had elevated levels (Figure 3B). Two mouse macrophage cell lines and one human myeloid cell line expressed lower levels of TNF RNA after treatment with TGFβ (Online Figure IIIB), suggesting that TGFβ signaling in myeloid cells is required for repression of TNF.
TNF is known to cause endothelial cell damage in vitro and in vivo16. Unlike wt myeloid cells, Tgfbr2Myeko myeloid cells caused disruption of adherens and tight junctions of C57BL/6 mouse brain microvascular endothelial cells (MBMEC) in co-culture, as visualized by fragmented border staining of VE-Cadherin, ZO-1 and Occludin (Figure 3C). A TNF neutralizing antibody or deletion of TNF also in myeloid cells prevented endothelial damage mediated by Tgfbr2Myeko myeloid cells (Figure 3C). This result was further validated using human dermal microvascular endothelial cells (Online Figure IIIC). Increased TNF, and increased markers for endothelial damage, ICAM-1 and VCAM-1 RNA, were observed in MBMEC co-cultured with Tgfbr2Myeko myeloid cells, which was prevented by deletion of TNF in Tgfbr2Myeko myeloid cells (Figure 3D). These results suggest a cross talk of myeloid cells with endothelial cells through TNF. Taken together, these data suggest that myeloid-produced TNF due to deficiency of TGFβ signaling is critical for endothelial damage, consistent with roles of TNF in stroke initiation and progression17.
Transcription factor NFκB is a master regulator of inflammatory programs, and is activated in aged hematopoietic stem cells18. Active, phosphorylated-p65 (encoded by Rela) nuclear translocation was observed in myeloid cells sorted from Tgfbr2Myeko mice (Figure 3E). Blockade of TGFβ signaling in RAW264.7 cells using a TβRI inhibitor increased phosphorylation of p65, and nuclear translocation of p65 and p50 (Figure 3F). Of note, the TNF promoter region contains 5 putative NFκB binding sites (Online Figure IIID). Chromatin Immunoprecipitation (ChIP) for p65 or RNA polymerase 2 (POLR2a) showed enrichment of TNF promoter binding by TβRII knockdown in RAW264.7 cells (Figure 3G, Online Figure IIIE). Together, our data suggest that TβRII knockdown activated a NFκB-TNF inflammatory program that contributes to the endotheliopathy in Tgfbr2Myeko mice.
Stroke risk assessment and prevention
The events prior to stroke are particularly interesting and important in understanding stroke etiology and stroke risk assessment, which remain one of the toughest challenges19. To compile a stroke risk profile, 32 plasma cytokines were measured from a young and old wt, young and old (prestroke), and old post-stroke Tgfbr2Myeko mice. Tgfbr2Myeko post- stroke mice showed a unique cytokine profile that overlapped with that from pre-stroke mice (Figure 4A). A human RNA dataset of peripheral blood mononuclear cells (PBMC) from patients 6 months post ischemic stroke20, who are at high risk for subsequent stroke21, revealed increased expression of inflammatory cytokines, including TNF (Figure 4B). Ingenuity pathway analysis (IPA) of significantly altered pro-inflammatory genes from the human dataset, revealed striking similarity to our mouse pre-stroke cytokine signature (Online Figure IV). This suggests that Tgfbr2Myeko mice model inflammatory processes important for stroke in human patients.
Figure 4. Increased inflammatory plasma cytokines, human correlation, and modeling risk/prevention.
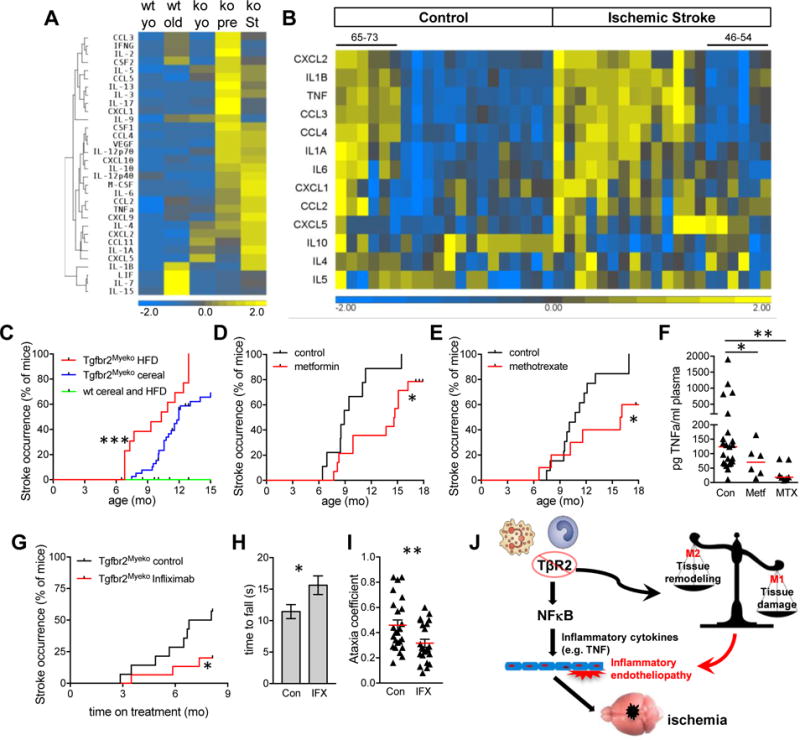
(A) Increased inflammatory cytokines in plasma from Tgfbr2Myeko mice pre-stroke, and following signs of stroke. Heatmap of plasma inflammatory cytokines from Tgfbr2Myeko (ko) young (yo) and older mice pre-stroke (pre) and after signs of stroke (St), compared with young and older wt mice. Each lane is the average of all mice within each group: Young wt and ko (n=4 for each, 6 weeks old); Old wt (10 months old, n=4); Old ko pre-stroke (7–11 months old, n=17); Old ko with stroke (7–12 months old, n=10). (B) Increased inflammatory cytokine RNA in PBMC from patients 6 months after acute stroke, who are at high risk for subsequent stroke. Heatmap of human dataset (GSE22255) from stroke patients, compared with non-stroke controls. Each lane is one patient. 65–73 in control group are older males in this group that were more similar to the ischemic stroke group. 46–54 in the ischemic stroke group are younger females that were more similar to the control group. (C) High fat diet (HFD) accelerates stroke occurrence in Tgfbr2Myeko mice (n=13–17). (D) Metformin delays stroke occurrence in Tgfbr2Myeko mice. Female Tgfbr2Myeko mice were given Metformin in the chow starting at 6 months of age (n=14), compared with female Tgfbr2Myeko given chow without metformin (n=14). (E) Methotrexate delays stroke occurrence in Tgfbr2Myeko mice. Male and female Tgfbr2Myeko mice that received weekly Methotrexate injections starting at 6 months of age (n=10), compared to Tgfbr2Myeko mice injected with vehicle (n=13). Tick marks on survival curves are censored subjects (alive at termination of experiment). Stoke criteria are in the online methods. (F) Plasma TNF levels in mice treated with metformin (Metf) or methotrexate (MTX), control, n=23 males and females; metf, n=6 females; MTX, n=8 males and females. (G) Delayed stroke occurrence with anti-TNF therapy. Tgfbr2Myeko mice were injected with Infliximab or saline (control) weekly beginning at 6–10 months of age (n=4–5 females plus 9–10 males/group). Mice in each group are age matched. (H–I) Infliximab improves neuromuscular function in Tgfbr2Myeko mice. (H) Time to fall on inverted screen test for mice treated with Infliximab (IFX) or saline (Con) for 6 mo. n=4–5 females and 9–10 males per group. (I) Decreased Ataxia coefficient in mice treated with IFX for 8 mo (Digigait analyis, n=6 males/group). Stroke occurrence was assessed by head tilt, abnormal gait, severe trembling, paresis or paralysis, or weight loss of >20% due to severe neurological impairment. (J) Schematic hypothesis for inflammatory endotheliopathy and stroke in Tgfbr2Myeko mice. Tgfbr2 deletion in myeloid cells led to increased production of type 1 inflammatory cytokines including TNF, which is mediated through increased NFkB transcriptional activity. Increased circulating TNF leads to damage of cerebral blood vessels, and subsequent recruitment of inflammatory cells to the cerebral endothelium. Increased type 1 and decreased type 2 inflammatory responses lead to endothelial damage, resulting in vascular stenosis/occlusion, and ultimately ischemic stroke. ***p<0.001, **p<0.01, *p<0.05.
Spontaneous stroke has been achieved in several mouse models through germline mutation22–24, but is not representative of the clinical conditions when specific genetic alterations are not a factor. Tgfbr2Myeko mice were used to model human stroke risk factors and prevention. High fat diet (HFD), a major stroke risk factor25, accelerated stroke occurrence in Tgfbr2Myeko mice (Figure 4C). High fat diet increased blood cholesterol in both wt and Tgfbr2Myeko mice to a similar extent, but did not significantly increase blood glucose or blood pressure in either group (Online Figure VA). Prevention with anti-inflammatory drugs was started at 6 months of age, just prior to earliest stroke occurrence, but when Tgfbr2Myeko mice show increased inflammatory cytokines (Figure 3A). Metformin, a drug widely used for diabetes, and which decreased stroke risk in diabetics in epidemiological studies26, delayed stroke occurrence (Figure 4D). Treatment with low dose Methotrexate, a drug used to treat auto-inflammatory diseases such as rheumatoid arthritis and severe psoriasis27, also delayed stroke occurrence (Figure 4E). Both methotrexate and metformin treatments led to a significant decrease in plasma TNF (Figure 4F). Importantly, anti-TNF therapy with Infliximab delayed stroke occurrence in Tgfbr2Myeko mice, and led to improved neuromuscular function in pre-stroke Tgfbr2Myeko mice (Figure 4G–I). Tgfbr2Myeko mice with whole body deletion of TNF also showed a trend toward delayed stroke occurrence (Online Figure VB). Of interest, PTGS2 (the gene encoding cyclooxygenase 2, COX2) was elevated and suggested to be a signaling node in the human stroke risk dataset (Online Figure IV). However, Tgfbr2Myeko mice given the COX2 inhibitor, Celecoxib, in the diet showed significantly accelerated stroke (Online Figure VC), consistent with the increased risk for stroke with Celecoxib use in humans28. These data provide evidence that Tgfbr2Myeko mice can be used to model stroke risk factors and prevention.
DISCUSSION
We provide evidence using a novel mouse model, of a cause and effect relationship between inflammation, and cerebrovascular disease and stroke. In this model, myeloid cells that lack Tgfbr2, create a type 1 polarized inflammatory immune environment characterized by elevated inflammatory cytokines. In Tgfbr2Myeko mice, stroke leads to decreased motor control, and, in particular, we show gait disturbances, which are a common cause of disability in stroke patients.
Our data suggest that inflammation is required for myeloid-mediated cerebrovascular disease that leads to stroke, and as such may be an early event in stroke etiology. Our findings are consistent with the recent identification of DADA2 (deficiency of ADA2) an autoinflammatory disease caused by loss of function mutations in ADA2, an adenosine deaminase almost exclusively produced and secreted by myeloid cells. Affected children suffer from recurrent fever early onset stroke and small vessel vasculitis29–31. Human monocytes and macrophages with mutant ADA2 are skewed towards a proinflammatory state, which causes endothelial cell damage in vitro and in vivo29. Anti-TNF treatment of these patients leads to dramatic clinical improvement of the inflammatory features, and reduction in recurrent strokes32. Although there is no mouse homolog of CECR1 (encoding ADA2), DADA2 patients provide valuable insight into myeloid cell function, inflammation and stroke risk. In fact there is increasing evidence for roles of inflammation in stroke etiology33,34. In particular, a recent clinical trial using anti-inflammatory therapy targeting interleukin-1β led to a significant decrease in strokes in cardiovascular patients with high C-reactive protein35. Our studies, together with others offer insight for cryptogenic strokes, approximately 30% of all strokes. Identification of high stroke risk patients without currently known risk factors, such as with a plasma cytokine profile, would allow intervention.
Decreased expression of Tgfbr2 in PBMC with age has been reported recently36, and reduced TGFß signaling was observed in aged hematopoietic stem cells37. Mutation or polymorphism in TGFß pathway genes, TGFB1, TGFBR1 and TGFBR2 are associated with vascular disorders that have high stroke risk and inflammation such as Loeys Dietz syndrome6,38, Kawasaki disease39,40, and Moyamoya disease41,42. Tgfbr2 polymorphism is also associated with intracerebral hemorrhage43. Genetic attenuation of TGFβ signaling in myeloid cells thus constitutes human relevance, and is an experimental approach to investigate inflammation associated stroke etiology. We propose the consequential upregulation of NFκB and enhanced production of TNF is one significant mechanism, consistent with the association of TNF with vascular diseases, atherosclerosis and stroke15–17. Anti-TNF therapy appears to be efficacious in prevention of cardiovascular events in rheumatoid arthritis44,45. Anti-TNF therapy has also been shown to decrease motor impairment when given to patients greater than one year after stroke, presumably by decreasing persistent neuroinflammation46. Comparably, we found that anti-TNF therapy of Tgfbr2Myeko mice with Infliximab delayed stroke occurrence, and decreased neuromuscular deficits in Tgfbr2Myeko pre-stroke mice. Deletion of TNF in Tgfbr2Myeko mice also trended toward delayed stroke occurrence, supporting our assertion that TNF is an important factor leading to stroke in Tgfbr2Myeko mice. However, the immune system is a very complex web of cell types and mechanisms that are intricately entwined. Given the scope of inflammatory cytokine elevation, there may be more than one mechanism or cytokine critical for the stroke phenotype in Tgfbr2Myeko mice.
We have demonstrated the utility of Tgfbr2Myeko mice in modeling inflammation-mediated vascular risk factors for the prevention of stroke. Tgfbr2Myeko mice showed delayed stroke occurrence with anti-inflammation treatments, and accelerated stroke occurrence when adding a risk factor, HFD. Although there is limited data regarding methotrexate as a prevention for stroke, the cardiovascular inflammation reduction trial (CIRT) is addressing this question in hopes of shedding light on the “inflammatory hypothesis of atherothrombosis”47. If high stroke risk patients could be identified, such as with a plasma cytokine profile, treatment with drugs identified using Tgfbr2Myeko mice might prevent or delay strokes, prolong life and decrease disability.
In summary, we propose that deficiency in myeloid TGFβ signaling activates an NFκB-TNF inflammatory program leading to cerebral inflammatory endotheliopathy and cerebrovascular atherosclerosis, culminating in spontaneous stroke (Figure 4J). Tgfbr2Myeko mice developed smaller lesions more closely resembling lacunar strokes, which are thought to result from cerebral vascular lesions, as in Tgfbr2Myeko mice9. We anticipate that our studies provide insight into inflammation and vascular disease-associated stroke etiology. Myeloid TGFβ signaling could be exploited for intervention of these diseases.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Stroke is a leading cause of disability and death worldwide.
Approximately 30% of ischemic strokes are cryptogenic (of unknown cause).
Common stroke risk factors such as hypertension, poor diet, smoking, physical inactivity, and aging are associated with systemic inflammation, although no direct cause and effect relationship has been demonstrated.
What New Information Does This Article Contribute?
Inflammation, mediated by myeloid cells deficient in TGFß receptor 2 (Tgfbr2), leads to cerebrovascular disease, neurological deficits, and stroke in 100% of mice, at approximately middle age, in the absence of any other stroke risk factors.
Inflammatory plasma cytokines, most notably Tumor Necrosis Factor (TNF), are elevated prior to stroke in these mice, and are largely conserved after stroke, similar to what has been observed in post-stroke patients.
Metformin and low dose methotrexate lower plasma TNF levels, and similar to anti-TNF therapy, can delay stroke occurrence in these mice.
In this study, we report that spontaneous stroke in mice leads to disability and death, in the absence of exogenous treatment. This is the only animal model of stroke caused by inflammation that provides evidence for a cause and effect relationship between inflammation and stroke. Mice lacking Tgfbr2 in myeloid cells (Tgfbr2Myeko) developed stroke in approximately middle age, in the absence of any known stroke risk factors. Tgfbr2Myeko mice had high plasma levels of inflammatory cytokines prior to stroke, and after stroke, similar to what has been observed in post-stroke patients. Cerebrovascular inflammation, and associated focal breach of the blood brain barrier was observed prior to stroke in these mice. Modeling stroke prevention, using metformin (anti-diabetic), low dose methotrexate (anti-inflammatory) and Infliximab (anti-TNF antibody), reduced inflammation and delayed stroke occurrence in mice. These data suggest that patients with specific cytokine profiles might benefit from anti-inflammatory treatment for stroke prevention. Tgfbr2Myeko mice thus provide a valuable tool to investigate the etiology and progression of vascular inflammation to cerebral vascular disease to stroke.
Acknowledgments
We thank Drs. Giorgio Trinchieri and David Wink, NCI for critical reading of the manuscript. We thank Karen Wolcott, Subhadra Banerjee for technical assistance on FACS. We appreciate technical assistance of Neuroscience core staff for Ladder traversal and Gait analysis. We thank Dr. Hong San for the wire injury procedure and Dr. Suman Vodnala for technical assistance on bone marrow transplantation.
SOURCES OF FUNDING
This work is supported by NCI intramural funding to Dr. Li Yang.
Nonstandard Abbreviations and Acronyms
- ChIP
chromatin immunoprecipitation
- Cox2
Cyclooxygenase (protein encoded by PTGS2)
- GFAP
glial fibrillary acidic protein
- ICAM1
Intercellular Adhesion Molecule 1
- LysM-cre
Lysozyme 2 promoter driven Cre recombinase
- MRI
magnetic resonance imaging
- NFκB
nuclear factor-κB
- PBMC
peripheral blood mononuclear cells
- Polr2a
polymerase (RNA) II (DNA directed) polypeptide A
- Pre-stroke
mice prior to discernable signs of stroke
- Post-stroke
mice that showed obvious signs of stroke as detailed in Methods
- PTGS2
prostaglandin-endoperoxide synthase 2
- TβRI
transforming growth factor beta receptor 1 (protein)
- TβRII
Transforming growth factor beta receptor 2 (protein)
- Tgfbr2Myeko
Transforming growth factor receptor 2, myeloid knockout (Tgfbr2−/−)
- TGFβ
Transforming growth factor beta
- TNF
tumor necrosis factor
- VWF
Von Willebrand factor
- wt
wild type
Footnotes
DISCLOSURES
None
References
- 1.Writing Group Members. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després J-P, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics Committee, Stroke Statistics Subcommittee Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Saver JL. CLINICAL PRACTICE. cryptogenic stroke. N Engl J Med. 2016;374:2065–2074. doi: 10.1056/NEJMcp1503946. [DOI] [PubMed] [Google Scholar]
- 3.Fu Y, Liu Q, Anrather J, Shi F-D. Immune interventions in stroke. Nat Rev Neurol. 2015;11:524–535. doi: 10.1038/nrneurol.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol. 2014;32:51–82. doi: 10.1146/annurev-immunol-032713-120257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beaufort N, Scharrer E, Kremmer E, Lux V, Ehrmann M, Huber R, Houlden H, Werring D, Haffner C, Dichgans M. Cerebral small vessel disease-related protease HtrA1 processes latent TGF-β binding protein 1 and facilitates TGF-β signaling. Proc Natl Acad Sci U S A. 2014;111:16496–16501. doi: 10.1073/pnas.1418087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 7.Pang Y, Gara SK, Achyut BR, Li Z, Yan HH, Day C-P, Weiss JM, Trinchieri G, Morris JC, Yang L. TGF-β signaling in myeloid cells is required for tumor metastasis. Cancer Discov. 2013;3:936–951. doi: 10.1158/2159-8290.CD-12-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, Geng Y, Gray JW, Moses HL, Yang L. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70:6139–6149. doi: 10.1158/0008-5472.CAN-10-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caplan LR. Lacunar infarction and small vessel disease: pathology and pathophysiology. J Stroke. 2015;17:2–6. doi: 10.5853/jos.2015.17.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 11.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orthgiess J, Gericke M, Immig K, Schulz A, Hirrlinger J, Bechmann I, Eilers J. Neurons exhibit Lyz2 promoter activity in vivo: Implications for using LysM-Cre mice in myeloid cell research. Eur J Immunol. 2016;46:1529–1532. doi: 10.1002/eji.201546108. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz M, Kipnis J, Rivest S, Prat A. How do immune cells support and shape the brain in health, disease, and aging? J Neurosci. 2013;33:17587–17596. doi: 10.1523/JNEUROSCI.3241-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moser J, van Ark J, van Dijk MC, Greiner DL, Shultz LD, van Goor H, Hillebrands J-L. Distinct Differences on Neointima Formation in Immunodeficient and Humanized Mice after Carotid or Femoral Arterial Injury. Sci Rep. 2016;6:35387. doi: 10.1038/srep35387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9:731–740. doi: 10.1038/nrrheum.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Park Y, Wu J, Chen X ping, Lee S, Yang J, Dellsperger KC, Zhang C. Role of TNF-alpha in vascular dysfunction. Clin Sci. 2009;116:219–230. doi: 10.1042/CS20080196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nat Med. 2002;8:1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 18.Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13:376–389. doi: 10.1038/nri3433. [DOI] [PubMed] [Google Scholar]
- 19.Sabayan B, Gussekloo J, de Ruijter W, Westendorp RGJ, de Craen AJM. Framingham stroke risk score and cognitive impairment for predicting first-time stroke in the oldest old. Stroke. 2013;44:1866–1871. doi: 10.1161/STROKEAHA.113.001460. [DOI] [PubMed] [Google Scholar]
- 20.Krug T, Gabriel JP, Taipa R, Fonseca BV, Domingues-Montanari S, Fernandez-Cadenas I, Manso H, Gouveia LO, Sobral J, Albergaria I, Gaspar G, Jiménez-Conde J, Rabionet R, Ferro JM, Montaner J, Vicente AM, Silva MR, Matos I, Lopes G, Oliveira SA. TTC7B emerges as a novel risk factor for ischemic stroke through the convergence of several genome-wide approaches. J Cereb Blood Flow Metab. 2012;32:1061–1072. doi: 10.1038/jcbfm.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohan KM, Wolfe CDA, Rudd AG, Heuschmann PU, Kolominsky-Rabas PL, Grieve AP. Risk and cumulative risk of stroke recurrence: a systematic review and meta-analysis. Stroke. 2011;42:1489–1494. doi: 10.1161/STROKEAHA.110.602615. [DOI] [PubMed] [Google Scholar]
- 22.Wallays G, Nuyens D, Silasi-Mansat R, Souffreau J, Callaerts-Vegh Z, Van Nuffelen A, Moons L, D’Hooge R, Lupu F, Carmeliet P, Collen D, Dewerchin M. Notch3 Arg170Cys knock-in mice display pathologic and clinical features of the neurovascular disorder cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Arterioscler Thromb Vasc Biol. 2011;31:2881–2888. doi: 10.1161/ATVBAHA.111.237859. [DOI] [PubMed] [Google Scholar]
- 23.Iida S, Baumbach GL, Lavoie JL, Faraci FM, Sigmund CD, Heistad DD. Spontaneous stroke in a genetic model of hypertension in mice. Stroke. 2005;36:1253–1258. doi: 10.1161/01.str.0000167694.58419.a2. [DOI] [PubMed] [Google Scholar]
- 24.Jeanne M, Jorgensen J, Gould DB. Molecular and Genetic Analyses of Collagen Type IV Mutant Mouse Models of Spontaneous Intracerebral Hemorrhage Identify Mechanisms for Stroke Prevention. Circulation. 2015;131:1555–1565. doi: 10.1161/CIRCULATIONAHA.114.013395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, Rangarajan S, Islam S, Pais P, McQueen MJ, Mondo C, Damasceno A, Lopez-Jaramillo P, Hankey GJ, Dans AL, Yusoff K, Truelsen T, Diener H-C, Sacco RL, Ryglewicz D, Czlonkowska A, Weimar C, Wang X, Yusuf S, INTERSTROKE investigators Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. The Lancet. 2010;376:112–123. doi: 10.1016/S0140-6736(10)60834-3. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y-Y, Leu H-B, Chen T-J, Chen C-L, Kuo C-H, Lee S-D, Kao C-L. Metformin-inclusive therapy reduces the risk of stroke in patients with diabetes: a 4-year follow-up study. J Stroke Cerebrovasc Dis. 2014;23:e99–105. doi: 10.1016/j.jstrokecerebrovasdis.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P, Siu S, Kraft J, Lynde C, Pope J, Gulliver W, Keeling S, Dutz J, Bessette L, Bissonnette R, Haraoui B. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74:480–489. doi: 10.1136/annrheumdis-2014-206624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, Stone DL, Chae JJ, Rosenzweig SD, Bishop K, Barron KS, Kuehn HS, Hoffmann P, Negro A, Tsai WL, Cowen EW, Pei W, Milner JD, Silvin C, Heller T, Chin DT, Patronas NJ, Barber JS, Lee C-CR, Wood GM, Ling A, Kelly SJ, Kleiner DE, Mullikin JC, Ganson NJ, Kong HH, Hambleton S, Candotti F, Quezado MM, Calvo KR, Alao H, Barham BK, Jones A, Meschia JF, Worrall BB, Kasner SE, Rich SS, Goldbach-Mansky R, Abinun M, Chalom E, Gotte AC, Punaro M, Pascual V, Verbsky JW, Torgerson TR, Singer NG, Gershon TR, Ozen S, Karadag O, Fleisher TA, Remmers EF, Burgess SM, Moir SL, Gadina M, Sood R, Hershfield MS, Boehm M, Kastner DL, Aksentijevich I. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370:911–920. doi: 10.1056/NEJMoa1307361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, Zlotogorski A, Berkun Y, Press JJ, Mukamel M, Voth I, Hashkes PJ, Harel L, Hoffer V, Ling E, Yalcinkaya F, Kasapcopur O, Lee MK, Klevit RE, Renbaum P, Weinberg-Shukron A, Sener EF, Schormair B, Zeligson S, Marek-Yagel D, Strom TM, Shohat M, Singer A, Rubinow A, Pras E, Winkelmann J, Tekin M, Anikster Y, King M-C, Levy-Lahad E. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370:921–931. doi: 10.1056/NEJMoa1307362. [DOI] [PubMed] [Google Scholar]
- 31.Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, Conti G, Marchetti F, Picco P, Tommasini A, Martino S, Malattia C, Gallizi R, Podda RA, Salis A, Falcini F, Schena F, Garbarino F, Morreale A, Pardeo M, Ventrici C, Passarelli C, Zhou Q, Severino M, Gandolfo C, Damonte G, Martini A, Ravelli A, Aksentijevich I, Ceccherini I, Gattorno M. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. 2017;76:1648–1656. doi: 10.1136/annrheumdis-2016-210802. [DOI] [PubMed] [Google Scholar]
- 32.The Deficiency of Adenosine Deaminase Type 2 (DADA2)—Results of Anti-TNF Treatment in a Cohort of Patients with a History of Stroke - ACR Meeting Abstracts [Internet] [cited 2017 Sep 25]; Available from: http://acrabstracts.org/abstract/the-deficiency-of-adenosine-deaminase-type-2-dada2-results-of-anti-tnf-treatment-in-a-cohort-of-patients-with-a-history-of-stroke/
- 33.Boehme AK, Esenwa C, Elkind MSV. Stroke risk factors, genetics, and prevention. Circ Res. 2017;120:472–495. doi: 10.1161/CIRCRESAHA.116.308398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esenwa CC, Elkind MS. Inflammatory risk factors, biomarkers and associated therapy in ischaemic stroke. Nat Rev Neurol. 2016;12:594–604. doi: 10.1038/nrneurol.2016.125. [DOI] [PubMed] [Google Scholar]
- 35.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, CANTOS Trial Group Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017 doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 36.Halper B, Hofmann M, Oesen S, Franzke B, Stuparits P, Vidotto C, Tschan H, Bachl N, Strasser E-M, Quittan M, Wagner K-H, Wessner B. Influence of age and physical fitness on miRNA-21, TGF-β and its receptors in leukocytes of healthy women. Exerc Immunol Rev. 2015;21:154–163. [PubMed] [Google Scholar]
- 37.Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, Gu H, Bock C, Meissner A, Göttgens B, Darlington GJ, Li W, Goodell MA. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guerrerio AL, Frischmeyer-Guerrerio PA, Huang C, Wu Y, Haritunians T, McGovern DPB, MacCarrick GL, Brant SR, Dietz HC. Increased Prevalence of Inflammatory Bowel Disease in Patients with Mutations in Genes Encoding the Receptor Subunits for TGFβ. Inflamm Bowel Dis. 2016;22:2058–2062. doi: 10.1097/MIB.0000000000000872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu C, Jain S, Davila S, Hibberd ML, Lin KO, Molkara D, Frazer JR, Sun S, Baker AL, Newburger JW, Rowley AH, Shulman ST, Burgner D, Breunis WB, Kuijpers TW, Wright VJ, Levin M, Eleftherohorinou H, Coin L, Popper SJ, Relman DA, Fury W, Lin C, Mellis S, Tremoulet AH, Burns JC. Transforming growth factor-beta signaling pathway in patients with Kawasaki disease. Circ Cardiovasc Genet. 2011;4:16–25. doi: 10.1161/CIRCGENETICS.110.940858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi K, Oharaseki T, Yokouchi Y. Update on etio and immunopathogenesis of Kawasaki disease. Curr Opin Rheumatol. 2014;26:31–36. doi: 10.1097/BOR.0000000000000010. [DOI] [PubMed] [Google Scholar]
- 41.Mejia-Munne JC, Ellis JA, Feldstein NA, Meyers PM, Connolly ES. Moyamoya and Inflammation. World Neurosurg. 2017;100:575–578. doi: 10.1016/j.wneu.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 42.Roder C, Peters V, Kasuya H, Nishizawa T, Takehara Y, Berg D, Schulte C, Khan N, Tatagiba M, Krischek B. Polymorphisms in TGFB1 and PDGFRB are associated with Moyamoya disease in European patients. Acta Neurochir (Wien) 2010;152:2153–2160. doi: 10.1007/s00701-010-0711-9. [DOI] [PubMed] [Google Scholar]
- 43.Lim YH, Jeong YS, Kim SK, Kim DH, Yun DH, Yoo SD, Kim H-S, Baik HH. Association between TGFBR2 gene polymorphism (rs2228048, Asn389Asn) and intracerebral hemorrhage in Korean population. Immunol Invest. 2011;40:569–580. doi: 10.3109/08820139.2011.559498. [DOI] [PubMed] [Google Scholar]
- 44.Tocci G, Goletti D, Marino V, Matucci A, Milano GM, Cantini F, Scarpa R. Cardiovascular outcomes and tumour necrosis factor antagonists in chronic inflammatory rheumatic disease: a focus on rheumatoid arthritis. Expert Opin Drug Saf. 2016;15:55–61. doi: 10.1080/14740338.2016.1218469. [DOI] [PubMed] [Google Scholar]
- 45.McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol. 2009;6:410–417. doi: 10.1038/nrcardio.2009.57. [DOI] [PubMed] [Google Scholar]
- 46.Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs. 2012;26:1051–1070. doi: 10.1007/s40263-012-0013-2. [DOI] [PubMed] [Google Scholar]
- 47.Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J, Zaharris E, Gupta M, Clearfield M, Libby P, Hasan AAK, Glynn RJ, Ridker PM. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199–207.e15. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.